

Abstract
The purpose of this study was to determine whether metrics of brain tissue integrity derived from postmortem MRI are associated with late life cognitive decline, independent of cerebral vessel disease. Using data from 554 older adults, we employed voxelwise regression to identify regions where the postmortem MRI transverse relaxation rate constant R2 was associated with the rate of decline in global cognition. We then used linear mixed models to investigate the association between a composite R2 measure and cognitive decline, controlling for neuropathology including three indices of vessel disease: atherosclerosis, arteriolosclerosis, and cerebral amyloid angiopathy. This composite R2 measure was associated with the rate of decline (0.049 unit annually per R2 unit, p<0.0001) and accounted for 6.1% of its variance, beyond contributions from vessel disease indices and other prominent age-related neuropathologies. Thus, postmortem brain R2 reflects disease processes underlying cognitive decline that are not captured by vessel disease indices or other standard neuropathologic indices and may provide a measure of brain tissue integrity that is complementary to histopathologic evaluation.
Keywords: Transverse relaxation, R2, voxelwise, atherosclerosis, arteriolosclerosis, cerebral amyloid angiopathy
1. INTRODUCTION
Cognitive decline, one of the most common and feared experiences of late life (Cutler, 2002), is a costly phenomenon that will increasingly strain healthcare resources as the population ages (Hayden et al., 2011, Zhu et al., 2013). Despite being the focus of intense research efforts, gaps in our understanding of the underlying neurobiology of late life cognitive decline impede the development of effective interventions to prevent or delay its onset. In particular, although common age-related neuropathologies are important determinants of cognitive impairment and decline, they account for less than half of the heterogeneity observed in rates of decline (Boyle et al., 2013), suggesting that other factors are involved. This highlights a pressing need to identify novel factors that influence late life cognitive decline.
MRI is a powerful tool that may detect important brain changes related to cognitive decline. Recently, we reported that a metric of tissue integrity derived from postmortem MRI, the transverse relaxation rate constant R2 (inverse of T2), accounted for approximately 10% of the variation in the rate of late life cognitive decline, beyond the contributions of neuropathologic indices of the three most common causes of dementia – Alzheimer’s disease (AD), cerebrovascular disease (CVD), and Lewy body disease (LBD) (Dawe et al., 2016). While the specific mechanisms linking lower R2 values to faster cognitive decline are unknown, one plausible hypothesis is that hypoxic-ischemic events are particularly injurious to myelinating oligodendrocytes and their progenitors (Back et al., 2002, Pantoni et al., 1996), and that demyelination stemming from loss of these cells impairs cognition by disrupting communication within various brain networks (Felts et al., 1997, Pievani et al., 2011). This type of damage is likely accessible to postmortem R2 imaging because the tissue’s R2 value depends largely on the ratio of free water molecules, which exhibit a slower transverse relaxation (smaller R2 value), to those that are trapped between myelin layers and therefore exhibit a faster transverse relaxation (larger R2 value) (Laule et al., 2006). Thus, brain tissue whose free water content increases due to some form of degeneration is expected to have a smaller R2 imaging metric. Although previous analyses examining the relation of R2 with cognitive decline controlled for micro- and gross infarcts, other vascular pathologies may also result in ischemic conditions leading to demyelination or axonal loss. In particular, pathologies underlying cerebral vessel disease, namely atherosclerosis, arteriolosclerosis, and cerebral amyloid angiopathy (CAA), are recognized as prominent contributors to hypoxic-ischemia (Bijanki et al., 2013, Chung et al., 2009, Gurol et al., 2013, Kalback et al., 2004, Román et al., 2002).
In the current study, we extended our previous work on the association between postmortem R2 and late life cognitive decline by examining whether this association is independent of cerebral vessel disease. Further, because cognitive decline tends to accelerate in the years just prior to death (i.e., terminal decline), we also investigated whether R2 is primarily associated with the relatively early, gradual decline in cognition or the more rapid terminal decline phase (Wilson et al., 2010). Participants were 554 individuals from the Rush Memory and Aging Project (MAP) and the Religious Orders Study (ROS) who underwent at least two annual cognitive evaluations. Upon death, one cerebral hemisphere from each individual underwent MRI and neuropathologic evaluation. Using voxelwise regression analyses, we first delineated the brain regions where R2 exhibited significant association with the rate of cognitive decline. Using linear mixed models, we quantified the degree to which R2 in these regions accounted for the variance in cognitive decline after adjustment for three hallmark cerebral vessel diseases: atherosclerosis, arteriolosclerosis, and CAA. Finally, in a subset of participants who had at least five cognitive evaluations, we employed random changepoint models to examine specific components of the cognitive trajectory (i.e., preterminal and terminal slopes), to elucidate the temporal associations of cognitive decline with R2.
2. METHODS
2.1. Participants and Specimens
Participants were individuals enrolled in two longitudinal clinical pathologic studies of aging and dementia, MAP and ROS (Bennett et al., 2012a, Bennett et al., 2012b), with harmonized cognitive, neuropathologic, and postmortem MRI data collection procedures. The Institutional Review Board of Rush University Medical Center approved these studies, which are carried out in accordance with the Declaration of Helsinki. At enrollment, participants are free of known dementia, sign an anatomical gift act, and agree to brain donation after death. At the time of analyses, a total of 3202 individuals had been enrolled (1858 MAP, 1344 ROS), 1535 were deceased, and 1327 had undergone autopsy. Since 2006 when postmortem MRI began, 610 autopsied brain specimens had undergone postmortem R2 imaging and had passed quality control and post-processing. Of these decedents, 554 had undergone at least two cognitive evaluations during life, forming the sample of the current work.
2.2. Cognitive Evaluation and Clinical Diagnosis
Participants underwent an annual battery of tests to assess cognitive abilities, including the MMSE (used for descriptive purposes only) and 17 other tests that are common between the two studies: Logical Memory I for immediate recall, Logical Memory II for delayed recall, East Boston immediate recall, East Boston delayed recall, word list memory, word list recall, word list recognition, Boston naming, category fluency, National Adult Reading, digit span forward, digit span backward, digit ordering, Symbol Digits Modalities, number comparison, judgment of line orientation, and progressive matrices tests. The individual scores for the 17 tests were converted to z-scores based on the mean and standard deviation of the combined MAP-ROS cohort at baseline, which were then averaged to form a composite score for global cognition. A clinical diagnosis of AD or MCI was rendered by a clinician at every assessment based on these cognitive test scores and clinical judgment by a neuropsychologist. At death, a neurologist with expertise in dementia reviewed all available clinical data (but remained blinded to all postmortem data) and rendered a summary diagnostic opinion of the most likely clinical diagnosis at the time of death.
2.3. Postmortem MRI and R2 Image Processing
Postmortem MRI procedures have been described previously (Dawe et al., 2014). Upon death of a participant, the brain was removed and hemisected during rapid autopsy. The cerebral hemisphere with more visible pathology was immersed in 4% paraformaldehyde solution and refrigerated at 4 degrees Celsius. At approximately one month postmortem, this hemisphere was imaged using a 3-Tesla MRI scanner. R2 maps were generated from the fast spin echo images using in-house software to fit an exponential decay of the form S=S0·exp(−R2·TE) to the data in each voxel, where S is the magnitude of the MRI signal measured at known echo times of TE. R2 maps were spatially registered to a cerebral hemisphere template, first using linear and then nonlinear transformations as previously described (Dawe et al., 2016). To minimize differences in R2 stemming from the use of four different MRI scanners throughout the postmortem MRI data collection, we carried out a voxel-by-voxel normalization of R2 values within each group of hemispheres imaged on a given scanner by subtracting the median and dividing by the interquartile range of R2 for that scanner, to guard against undue influence from outliers.
2.4. Neuropathologic Indices
The imaged hemisphere was then sliced into 1-cm thick coronal slabs, and neuropathologic indices were derived for AD, CVD, LBD, hippocampal sclerosis (HS), and TDP-43 as previously described (Boyle et al., 2013, Nag et al., 2015, Wilson et al., 2015) and summarized here. AD: Amyloid load and the density of paired helical filament tau tangles were quantified using antibody-specific immunohistochemistry. We computed summary measures for each by averaging the observed values across seven cortical regions and the hippocampus and taking the square root to account for skewness in the distributions. CVD: Chronic gross infarcts were coded as present or absent. We did not include microinfarcts because previous work demonstrated no association of microinfarcts with cognitive decline. LBD: Lewy bodies were coded as present if they were identified in any of seven brain regions using a monoclonal antibody to alpha-synuclein. HS: Typical hippocampal sclerosis was coded as present or absent based on the observation of severe neuronal loss and gliosis in CA1 or the subiculum. TDP-43: A rat phosphorylated monoclonal TAR5P-1D3 TDP-43 antibody (pS409/410, 1:100, Ascenion, Munich, Germany) applied to six brain regions provided a four-level staging of the extent of TDP-43 pathology: a value of 0 for no TDP-43 or stages of 1 to 3 corresponding to TDP-43 present only in the amygdala, also extending to other limbic structures, and in neocortical regions as well.
Indices of cerebral large and small vessel disease were derived as previously described. Atherosclerosis was rated on a semiquantitative scale based on the degree of occlusion of arteries in the Circle of Willis, with values 0-3 corresponding to none, mild, moderate, or severe (Arvanitakis et al., 2016). Similarly, arteriolosclerosis was graded on a semiquantitative scale with values 0-3 corresponding to no, mild, moderate, or severe occlusion arterioles of the anterior basal ganglia (Arvanitakis et al., 2016). For CAA, meningeal and parenchymal vessels in four neocortical regions were graded for amyloid deposition on a semiquantitative scale, which were then averaged and converted to a summary measure with values 0-3 corresponding to no, mild, moderate, and severe CAA (Boyle et al., 2015).
2.5. Statistical Analyses
We first conducted a voxelwise regression analysis in which the R2 value for each voxel served as the explanatory term of interest and the person-specific slope of global cognitive decline estimated from linear mixed models was the outcome. This voxelwise analysis controlled for demographics and all available neuropathologic indices, namely AD pathology, CVD, LBD, HS, TDP-43, atherosclerosis, arteriolosclerosis, and CAA. Correction for multiple testing (approximately 400,000 tissue-containing voxels) was accomplished by using a false discovery rate (FDR) of 0.05 and also performing spatial clustering (minimum cluster size of 100 voxels), as previously described (Dawe et al., 2016). The mean R2 value for each of the resultant clusters was computed for all brain specimens, and their correlations were examined using Pearson correlation and principal component analysis before combining them into a composite R2 measure for use in subsequent analyses.
We then carried out a series of linear mixed models implemented in SAS 9.4 (SAS Institute, Inc.) to assess the contribution of the composite R2 measure to the variance in the rate of global cognitive decline when controlling for different types of neuropathology. Scores of global cognition from all available assessments served as the longitudinal outcome in these models. The reference model included only an intercept and a term for time in years prior to death, which estimated the mean rate of decline (i.e., slope). We sequentially added explanatory terms for demographics, the three common causes of dementia (AD, CVD, and LBD) and two other neuropathologies (HS and TDP-43), and finally the three indices of cerebral vessel disease (atherosclerosis, arteriolosclerosis, and CAA). We conducted these models first without and then with the composite R2 measure. For each model, we computed the reduction in variance relative to a previous model, thereby quantifying the contribution of the most recently entered explanatory terms to the variation in rate of cognitive decline.
Random changepoint models capture the nonlinear trajectory of cognitive decline and facilitate testing for differential associations of R2 with decline during preterminal and terminal phases (Wilson et al., 2010). In these models, the cognitive trajectory is modeled using four parameters: a first slope, which represents the rate of cognitive decline during the preterminal phase; the changepoint, which represents the time when the rate of cognitive decline accelerates; the second slope, which represents the rate of decline during the terminal phase; and the intercept, which represents the level of cognitive function proximate to death. To fit changepoint models, we required that participants have at least five cognitive evaluations with the last evaluation occurring within two years of death, narrowing the sample to 419 individuals. We included demographic terms, the nine available neuropathologic indices (amyloid, tangles, CVD, LBD, HS, TDP-43, atherosclerosis, arteriolosclerosis, and CAA), and the composite R2 measure as predictors, with global cognition as the outcome. The changepoint models were implemented in OpenBugs software using a Bayesian Monte Carlo Markov Chain approach.
3. RESULTS
3.1. Descriptive Data
Demographic and clinical characteristics of the sample (n=554) are shown in Table 1. Participants were on average 90.4 years of age (SD=6.0, range=65-108) at the time of death and had completed 9.3 annual cognitive exams (SD=4.6, range=2-22). Based on neuropathologic evaluation, 379 individuals (68.4%) met the NIA-Reagan intermediate or high likelihood criteria for a pathologic diagnosis of AD, 189 (34.1%) had one or more gross infarcts (the index of CVD), Lewy bodies were present in 127 (22.9%), HS in 67 (12.1%), and some degree of TDP-43 in 314 (56.7%). Concerning the indices of cerebral vessel disease, moderate to severe atherosclerosis was noted in 152 individuals (27.4%), arteriolosclerosis in 146 (26.4%), and CAA in 195 (35.2%). Correlations among demographics and pathologies are shown in Table 2.
Table 1.
Demographic, Cognitive, and Pathologic Characteristics of Participants
| Variable | Mean (SD) or N (%) |
|---|---|
| Total Participants | 554 (100%) |
| MAP | 286 (51.6%) |
| ROS | 268 (48.4%) |
| Demographic | |
| Age at Death (years) | 90.4 (6.0) |
| Female | 395 (71.3%) |
| Education (years) | 15.8 (3.6) |
| White, Non-Hispanic | 536 (96.9%) |
| Clinical Diagnosis | |
| Cognitive Impairment at Baseline | |
| Mild Cognitive Impairment (MCI) | 151 (27.3%) |
| Alzheimer’s Disease (AD) | 33 (6.0%) |
| Cognitive Impairment at Death | |
| Mild Cognitive Impairment (MCI) | 143 (26.3%) |
| Alzheimer’s Disease (AD) | 230 (42.4%) |
| Cognitive | |
| Number of Annual Cognitive Evaluations | 9.3 (4.6) |
| Baseline MMSE (score out of 30) | 27.7 (2.8) |
| Proximate to Death MMSE | 20.5 (9.2) |
| Global Cognition, Baseline (composite of 17 z-scores) | −0.07 (0.59) |
| Global Cognition, Proximate to Death | −0.85 (1.11) |
| Global Cognition, Estimated Linear Rate of Change (per year) | −0.10 (0.10) |
| Pathologic | |
| Postmortem Interval (hours) | 8.7 (5.8) |
| Amyloid Load (square root transform) | 2.04 (1.19) |
| Tangles (square root transform) | 2.28 (2.12) |
| Gross Infarcts (present) | 189 (34.1%) |
| Lewy Bodies (present) | 127 (22.9%) |
| Hippocampal sclerosis (present) | 67 (12.1%) |
| TDP-43 (stage 0-3) | 1.11 (1.13) |
| Cerebral amyloid angiopathy (semiquantitative 0-3) | 1.13 (1.05) |
| Atherosclerosis (semiquantitative 0-3) | 1.11 (0.84) |
| Arteriolosclerosis (semiquantitative 0-3) | 1.03 (0.86) |
Table 2.
Correlations among Demographics and Pathologic Indices
| Amyloid | Tangles | CVD | LBD | HS | TDP-43 | Athero | Arteriolo | CAA | |
|---|---|---|---|---|---|---|---|---|---|
| Age | 0.12c | 0.21e | 0.12c | −0.03 | 0.07 | 0.20e | 0.16d | 0.06 | 0.15d |
| Sex | −0.08a | −0.16e | 0.01 | 0.04 | −0.02 | −0.10b | −0.02 | −0.09b | −0.06 |
| Education | −0.10b | −0.07a | −0.12c | −0.02 | 0.04 | −0.01 | −0.06 | −0.08a | 0.04 |
| Amyloid | 0.50e | 0.05 | 0.09b | 0.05 | 0.09b | 0.01 | 0.02 | 0.33e | |
| Tangles | 0.04 | 0.10b | −0.01 | 0.24e | 0.04 | 0.07a | 0.34e | ||
| CVD | 0.01 | 0.05 | 0.03 | 0.20e | 0.13c | 0.02 | |||
| LBD | 0.05 | 0.04 | −0.02 | 0.00 | 0.03 | ||||
| HS | 0.38e | 0.02 | 0.03 | 0.03 | |||||
| TDP-43 | 0.05 | 0.05 | 0.10b | ||||||
| Athero | 0.31e | 0.02 | |||||||
| Arteriolo | 0.01 |
p<0.10,
p<0.05,
p<0.01,
p<0.001,
p<0.0001.
3.2. Heterogeneity in Rates of Cognitive Decline
We first employed linear mixed models to estimate the rate of decline in global cognition, which was 0.10 unit per year (SE = 0.004, p<0.0001), with substantial heterogeneity observed across individuals. The variance of person specific rates of decline was 0.0083 (p<0.0001).
3.3. Brain Regions in which R2 is Associated with Rate of Cognitive Decline
Next we sought to identify specific brain regions in which R2 was most strongly associated with cognitive decline, independent of known pathology. In voxelwise models controlling for pathologic indices, R2 was associated with the rate of global cognitive decline in six regions located primarily within white matter, as illustrated in Figure 1 and summarized in Table 3. The regression coefficients of R2 in these regions were all positive, meaning that persons exhibiting slower (smaller) R2 values tended to decline more rapidly (a more negative slope of decline). The mean R2 values of the regions exhibited moderate to strong correlation with each other (Table 3) and principal component analysis revealed a single dominant eigenvalue accounting for 59% of their total variance. Therefore, we averaged the R2 values from all six regions into a single composite R2 measure, in order to maximize parsimony of subsequent analyses.
Figure 1.
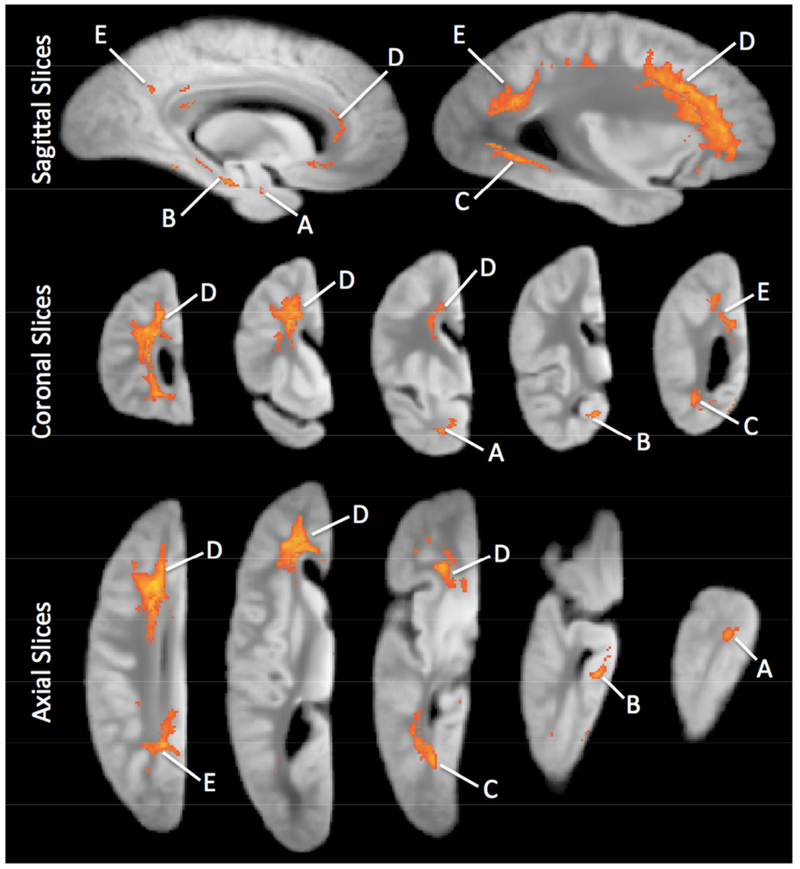
Sagittal, coronal, and axial views of clusters of voxels for which R2 was associated with the estimated slope of decline in global cognitive ability. These images were generated by a voxelwise analysis controlling for demographics, AD pathology , CVD, LBD, HS, TDP-43, and the indices of atherosclerosis, arteriosclerosis, and CAA. The region identifiers A-F correspond to those in Table 3.
Table 3.
Summary of Brain Regions in which R2 is Associated with Rate of Cognitive Decline
| Region Identifier | Location | Volume (mm3) | Correlations with R2 of other Regions (Pearson’s r) | ||||
|---|---|---|---|---|---|---|---|
| B | C | D | E | F | |||
| Region A | Anterior Temporal Lobe WM | 134 | 0.62 | 0.58 | 0.46 | 0.43 | 0.36 |
| Region B | Hippocampus/Parahippocampal WM | 191 | - | 0.52 | 0.44 | 0.38 | 0.32 |
| Region C | Posterior Temporal Lobe WM | 1,094 | - | 0.62 | 0.74 | 0.48 | |
| Region D | Majority of Frontal Lobe WM | 13,243 | - | 0.73 | 0.46 | ||
| Region E | Parietal/Occipital Lobe WM | 3,613 | - | 0.50 | |||
| Region F | Parietal Lobe GM | 140 | - | ||||
All correlations significant at p<0.0001.
3.4. Variance in Rate of Cognitive Decline Accounted for by Neuropathology and R2
We conducted a series of linear mixed models to assess the variance in rate of cognitive decline accounted for by the composite R2 measure (Table 4). In models that controlled for demographics, amyloid, tangles, CVD, LBD, HS, and TDP-43, the addition of terms for the composite R2 measure led to an increase in the portion of the random slope variance accounted for by the model from 41.3% to 48.1% (Models 1a and 1b). Thus, R2 explained 6.7% of the total variance in rate of cognitive decline, beyond the contributions from demographics and neuropathologic indices other than cerebral vessel diseases. After further adjustment for indices of atherosclerosis, arteriolosclerosis, and CAA, R2 explained 6.1% of the total variance (Models 2a and 2b).
Table 4.
Contributions to Variance in Cognitive Decline from Demographics, Neuropathologic Indices, and R2
| Variables | Model Identifier | |||
|---|---|---|---|---|
| 1a | 1b | 2a | 2b | |
| Time | −0.015a (0.010) | −0.0041 (0.0099) | 0.037d (0.011) | 0.015 (0.011) |
| Age | 0.0026e (0.0006) | 0.0029e (0.0006) | 0.0029e (0.0006) | 0.0032e (0.0006) |
| Sex | 0.011 (0.008) | 0.0087 (0.0077) | 0.010 (0.0080) | 0.0082 (0.0077) |
| Education | 0.0019a (0.0010) | 0.0019b (0.0009) | 0.0016 (0.0010) | 0.0017a (0.0009) |
| Amyloid | −0.0042 (0.0035) | −0.0043 (0.0033) | −0.0047 (0.0035) | −0.0045 (0.0034) |
| Tangles | −0.036e (0.004) | −0.030e (0.003) | −0.035e (0.0035) | −0.030e (0.0035) |
| Gross Infarcts | −0.034e (0.008) | −0.025d (0.007) | −0.027d (0.008) | −0.019b (0.008) |
| Lewy Bodies | −0.020b (0.009) | −0.020b (0.008) | −0.021b (0.009) | −0.021b (0.008) |
| Hippocampal Sclerosis | −0.066e 0.012 | −0.055e (0.012) | −0.067e (0.012) | −0.056e (0.012) |
| TDP-43 | −0.0062a (0.0036) | −0.0040 (0.0035) | −0.0059 (0.0036) | −0.0039 (0.0035) |
| Atherosclerosis | −0.018e (0.005) | −0.017d (0.005) | ||
| Arteriolosclerosis | −0.0057 (0.0044) | −0.0030 (0.0043) | ||
| Cerebral Amyloid Angiopathy | −0.0004 (0.0036) | −0.0018 (0.0035) | ||
| Composite R2 | 0.051e (0.007) | 0.049e (0.007) | ||
| % of Variance Accounted for by all Effects | 41.3% | 48.1% | 43.7% | 49.8% |
| % of Variance Accounted for only by Effects in Boldface | - | 6.7% | 2.4% | 6.1% |
| Model Identifier for Comparison | - | 1a | 1a | 2a |
p<0.10,
p<0.05,
p<0.01,
p<0.001,
p<0.0001.
The variables listed in the first column represent model terms consisting of the interaction of that variable with time. Effect sizes for those terms are shown, along with their standard errors in parentheses.
3.5. Association of R2 with preterminal and terminal phases of decline
Changepoint models provide an indication of the time-dependent relations of R2 with decline in global cognition. In this analysis, which included data from 419 individuals who had at least five cognitive exams and adjusted for all available neuropathologies including indices of cerebral vessel diseases, the composite R2 measure was associated with the slope of the preterminal phase of decline (0.012 cognitive unit per year per standard deviation of R2, 95% credible interval = 0.005 to 0.020) and also with the location of the changepoint (0.74 year per standard deviation of R2, 95% credible interval = 0.51 to 0.97), but we did not observe R2 association with the terminal slope. Thus, an individual with a composite R2 value at the 10th percentile (i.e., a relatively low, or slow, R2) would typically exhibit a 0.016-unit per year faster rate of preterminal decline and a transition to the terminal phase (i.e., changepoint) 0.98 year earlier than an individual with R2 equal to the sample median (Fig. 2).
Figure 2.
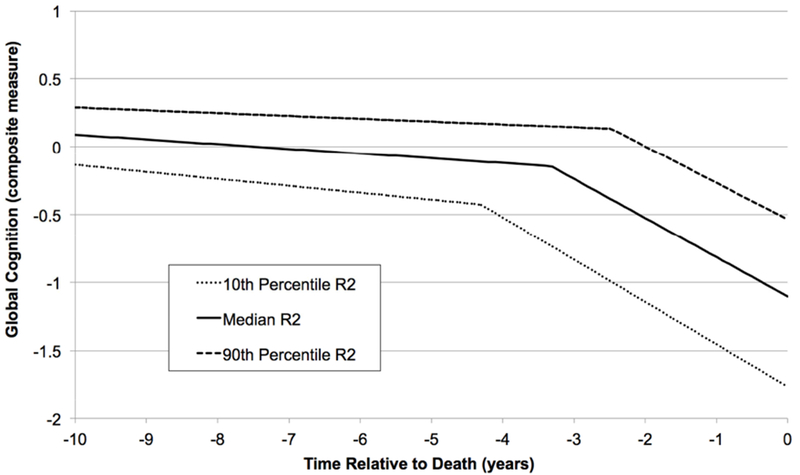
Changepoint trajectories illustrating the association of the composite postmortem R2 measure with a nonlinear model of cognitive decline. The solid line corresponds to the typical decline trajectory for an individual with R2 and AD pathology equal to the sample medians. The dotted and dashed lines represent decline trajectories for individuals with R2 at the 10th and 90th percentiles, respectively. The displayed trajectories also control for age, sex, education, amyloid, tangles, CVD, LBD, HS, TDP-43, atherosclerosis, arteriolosclerosis, and CAA.
4. DISCUSSION
We investigated the association of postmortem brain R2 with late life cognitive decline after controlling for several common neuropathologic indices including three cerebral vessel diseases, namely atherosclerosis, arteriolosclerosis, and CAA. In voxelwise analyses of MR images from over 500 participants, quantitative R2 was associated with the linear rate of cognitive decline in several white matter brain regions, the most extensive of which encompassed much of the frontal lobe white matter. A composite measure of R2 formed from these regions accounted for approximately 6% of the variance in decline beyond that attributable to neuropathologic indices. The variance accounted for by R2 remained similar when cerebral vessel disease indices were included in the analysis. Further, changepoint models showed that R2 was related to preterminal changes in global cognitive function and the timing of the onset of terminal decline. These findings highlight the independent association of postmortem R2 with late life cognitive decline, particularly in the preterminal phase of decline, and have implications for the clinical translation of results generated from postmortem R2 studies.
Considerable evidence links different forms of vessel disease to multiple neuroimaging measures, such as white matter hyperintensities on T2-weighted or fluid-attenuated inversion recovery (FLAIR) sequences (Erten-Lyons et al., 2013, Gurol et al., 2013, Holland et al., 2008, Moroni et al., 2016, Pico et al., 2002) and lower fractional anisotropy of white matter on diffusion tensor imaging (DTI) (Bijanki et al., 2013, Bos et al., 2012, Jolly et al., 2013, Salat et al., 2006). A related line of research links cerebral vessel disease indices to dementia and lower performance in certain cognitive domains in late life (Arvanitakis et al., 2011, Arvanitakis et al., 2016, Boyle et al., 2015, Greenberg et al., 2004, Reijmer et al., 2016, Smith, 2017, Thal et al., 2003, van Oijen et al., 2007). In addition, our own prior work showed a strong relationship between postmortem R2 (the inverse of T2) in white matter and cognitive decline (Dawe et al., 2016). A logical hypothesis unifying these bodies of research is that cerebral vessel diseases contribute to late life cognitive decline via tissue damage that manifests on MRI as alterations to quantitative R2 (Abraham et al., 2016, Banerjee et al., 2016, Prins and Scheltens, 2015). According to this hypothesis, associations of R2 with late life cognitive decline would be expected to weaken substantially after controlling for indices of vessel disease, which plausibly contribute to myelin degradation or axonal loss thought to be responsible for both a decrease in the tissue’s R2 and cognitive decline. Instead, the portion of the variance in the rate of cognitive decline accounted for by R2 was essentially unchanged (6.1% vs. 6.7%) when indices of atherosclerosis, arteriolosclerosis, and CAA were included in the analysis. The effect size of the R2 composite also remained nearly constant (0.049 vs. 0.051 cognitive unit per year per standard deviation of R2). Thus, the relation of R2 to cognitive decline was largely independent of the three vessel disease indices considered in this work.
One explanation for these findings is that the R2 alterations associated with cognitive decline reflect tissue damage stemming from conditions separate from those pathologies that were controlled for in the current work, including cerebral vessel diseases. This does not necessarily rule out a variety of other vascular etiologies, such as brain hypoperfusion (Brickman et al., 2009) stemming from, for example, decreased cardiac output (Alosco et al., 2013, van der Velpen, Isabelle F et al., 2017) or obstructive sleep apnea (Avci et al., 2017, Macey et al., 2008). Quantitative R2 imaging may therefore be a valuable tool for assessment of damage to white matter inflicted by a variety of pathologies or conditions that are not captured by standard neuropathologic evaluation, but future work is needed to examine this issue.
Another possibility is that R2 alterations indeed stem from cerebral vessel disease or other pathologies already considered in our analyses, but that R2 probes an additional facet of the disease process that accounts for variation in the rate of decline beyond the pathologic indices themselves. For example, although atherosclerosis, arteriolosclerosis, and CAA all disrupt cerebral hemodynamics, the severity and chronicity of the resulting hypoperfusion may also depend on factors like blood pressure and autoregulation (van der Velpen, Isabelle F et al., 2017) and the extent of collateral blood supply to the tissue (Campbell et al., 2013). Neuroimaging markers such as R2, it seems, may provide a summary measure of the tissue damage resulting from the altered hemodynamics brought about by cerebral vessel diseases, such as myelin degradation, axonal loss, or edema (Gouw et al., 2010). Because of their more direct link to the disruption of action potentials, these types of tissue damage may have a stronger influence on cognition than do the vessel disease indices themselves, which are envisioned to reside upstream in the causal chain (Banerjee et al., 2016, Weller et al., 2009). In this regard, postmortem R2 imaging can provide powerful measures of tissue integrity that are complementary to traditional histopathologic evaluation of cerebral vessel disease and other neuropathologies.
This study also extends prior work by examining for the first time the temporal association of R2 with cognitive decline. This was elucidated in the current work using changepoint models, which account for the nonlinearity of late life cognitive decline by fitting a cognitive trajectory that has distinct preterminal and terminal phases with differing slopes. We observed an association of the composite R2 measure with the rate of preterminal decline and also with the temporal location of the changepoint, which marks the onset of the more rapid terminal decline phase. Therefore, just as hallmark AD neuropathology may begin accumulating in the brain as early as the fifth decade of life (Braak et al., 2011), the underlying contributors to alterations in postmortem R2 appear to be influencing the cognitive trajectory well before death, not just in the final few years. Future interventions targeting those contributors are therefore promising in their ability to decrease the rate of cognitive decline during the preterminal period and also to delay the onset of the more severe terminal decline that immediately precedes death, resulting in compression of morbidity.
This study has strengths as well as limitations. One strength is the combination of autopsy data, brain MRI, and up to 22 annual cognitive evaluations in older adults, which is rare and allows for investigation of the associations between neuroimaging and cognition independent of known neuropathology. This is most feasibly achieved in large, community-based studies, but such studies have some limitations, including that the cohort is composed of willing participants who may not be representative of the general population. In addition, while the postmortem approach to MRI employed in this study was the most viable means of collecting the several hundred imaging datasets with matching autopsy data, this technique may be regarded as a limitation because of the uncertainty of translating results derived from postmortem imaging to the antemortem case. However, recent work demonstrating a strong correlation between antemortem and postmortem R2 values suggests that such a translation is promising, if not yet assured. Another limitation of the current work is related to the voxelwise analysis technique, which provides excellent spatial specificity, but at the expense of power. Work is underway to advance adaptive region-based approaches that improve statistical power without meaningful sacrifices to spatial resolution. This postmortem study is also limited in that we did not account for participants’ cause of death in analyses. Finally, we did not account for white matter hyperintensities (WMH) that are commonly visible in the brains of older adults on fluid-attenuated inversion recovery (FLAIR) MRI pulse sequences. FLAIR data collection is ongoing, and refined WMH segmentation methods are being developed toward the goal of elucidating the associations among R2, WMH, and cognitive decline.
HIGHLIGHTS.
Postmortem MRI transverse relaxation (R2) is associated with the linear rate of global cognitive decline in late life and accounts for more than 5% of its variance.
The association of R2 with cognitive decline persists after accounting for indices of cerebral vessel disease, namely atherosclerosis, arteriolosclerosis, and cerebral amyloid angiopathy.
R2 is also associated with specific components of a nonlinear model of cognitive decline.
R2 reflects a dimension of brain tissue integrity not captured by current histopathologic indices and therefore may highlight important targets for future interventions against late life cognitive decline.
ACKNOWLEDGEMENTS
We are grateful to the participants of the Rush Memory and Aging Project and the Religious Orders study, as well as the faculty and staff of the Rush Alzheimer’s Disease Center. This work was supported by the National Institute on Aging (P30AG10171, R01AG15819, R01AG33678, R01AG34374, R01AG17917), the National Institute of Neurological Disorders and Stroke (UH2NS100599), and the Illinois Department of Public Health. More information regarding obtaining MAP data for research use can be found at the RADC Research Resource Sharing Hub (www.radc.rush.edu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abraham HMA, Wolfson L, Moscufo N, Guttmann CR, Kaplan RF, White WB, 2016. Cardiovascular risk factors and small vessel disease of the brain: blood pressure, white matter lesions, and functional decline in older persons. 36, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alosco ML, Brickman AM, Spitznagel MB, Garcia SL, Narkhede A, Griffith EY, Raz N, Cohen R, Sweet LH, Colbert LH, Josephson R, Hughes J, Rosneck J, Gunstad J, 2013. Cerebral Perfusion is Associated With White Matter Hyperintensities in Older Adults With Heart Failure. 19, E34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA, 2016. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. 15, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA, 2011. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann.Neurol. 69, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci AY, Avci S, Lakadamyali H, Can U, 2017. Hypoxia and inflammation indicate significant differences in the severity of obstructive sleep apnea within similar apnea-hypopnea index groups, 1–9. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM, 2002. Selective vulnerability of late oligodendrocyte progenitors to hypoxia–ischemia. 22, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee G, Wilson D, Jger HR, Werring DJ, 2016. Novel imaging techniques in cerebral small vessel diseases and vascular cognitive impairment. 1862, 926–938. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS, 2012a. Overview and findings from the religious orders study. 9, 628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS, 2012b. Overview and findings from the rush Memory and Aging Project. 9, 646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijanki KR, Arndt S, Magnotta VA, Nopoulos P, Paradiso S, Matsui JT, Johnson HJ, Moser DJ, 2013. Characterizing white matter health and organization in atherosclerotic vascular disease: a diffusion tensor imaging study. 214, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos D, Vernooij MW, Elias-Smale SE, Verhaaren BF, Vrooman HA, Hofman A, Niessen WJ, Witteman JC, van der Lugt A, Ikram MA, 2012. Atherosclerotic calcification relates to cognitive function and to brain changes on magnetic resonance imaging. 8, S111. [DOI] [PubMed] [Google Scholar]
- Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, Bennett DA, 2013. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann.Neurol. 74, 478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, Yu L, Nag S, Leurgans S, Wilson RS, Bennett DA, Schneider JA, 2015. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. 85, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K, 2011. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. 70, 960–969. [DOI] [PubMed] [Google Scholar]
- Brickman AM, Zahra A, Muraskin J, Steffener J, Holland CM, Habeck C, Borogovac A, Ramos MA, Brown TR, Asllani I, Stern Y, 2009. Reduction in cerebral blood flow in areas appearing as white matter hyperintensities on magnetic resonance imaging. 172, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BC, Christensen S, Tress BM, Churilov L, Desmond PM, Parsons MW, Barber PA, Levi CR, Bladin C, Donnan GA, 2013. Failure of collateral blood flow is associated with infarct growth in ischemic stroke. 33, 1168–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Kim J, Kim K, Ahn K, 2009. Hypoperfusion and ischemia in cerebral amyloid angiopathy documented by 99mTc-ECD brain perfusion SPECT. 50, 1969–1974. [DOI] [PubMed] [Google Scholar]
- Cutler NE, 2002. American perceptions of aging in the 21st Century: A myths and realities of aging chartbook. National Council on the Aging. [Google Scholar]
- Dawe RJ, Bennett DA, Schneider JA, Leurgans SE, Kotrotsou A, Boyle PA, Arfanakis K, 2014. Ex vivo T 2 relaxation: associations with age-related neuropathology and cognition. Neurobiol.Aging. 35, 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe RJ, Yu L, Leurgans SE, Schneider JA, Buchman AS, Arfanakis K, Bennett DA, Boyle PA, 2016. Postmortem MRI: a novel window into the neurobiology of late life cognitive decline. Neurobiol.Aging. 45, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erten-Lyons D, Silbert L, Mattek N, Dodge H, Green S, Tran H, Quinn J, Wild K, Oken B, Woltjer R, 2013. Neuropathologic Basis of White Matter Hyperintensity Accumulation (S44. 001). 80, S44. 001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts PA, Baker TA, Smith KJ, 1997. Conduction in segmentally demyelinated mammalian central axons. 17, 7267–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw AA, Seewann A, Van Der Flier, Wiesje M, Barkhof F, Rozemuller AM, Scheltens P, Geurts JJ, 2010. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations, jnnp. 2009.204685. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE, 2004. Amyloid angiopathy-related vascular cognitive impairment. 35, 2616–2619. [DOI] [PubMed] [Google Scholar]
- Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, Vashkevich A, Ayres AM, Auriel E, Etten E, 2013. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann.Neurol. 73, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden KM, Reed BR, Manly JJ, Tommet D, Pietrzak RH, Chelune GJ, Yang FM, Revell AJ, Bennett DA, Jones RN, 2011. Cognitive decline in the elderly: an analysis of population heterogeneity. Age Ageing. 40, 684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland CM, Smith EE, Csapo I, Gurol ME, Brylka DA, Killiany RJ, Blacker D, Albert MS, Guttmann CR, Greenberg SM, 2008. Spatial distribution of white-matter hyperintensities in Alzheimer disease, cerebral amyloid angiopathy, and healthy aging. 39, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly TA, Bateman GA, Levi CR, Parsons MW, Michie PT, Karayanidis F, 2013. Early detection of microstructural white matter changes associated with arterial pulsatility. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalback W, Esh C, Castao EM, Rahman A, Kokjohn T, Luehrs DC, Sue L, Cisneros R, Gerber F, Richardson C, 2004. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease. Neurol.Res. 26, 525–539. [DOI] [PubMed] [Google Scholar]
- Laule C, Leung E, Li DK, Traboulsee AL, Paty DW, MacKay AL, Moore GR, 2006. Myelin water imaging in multiple sclerosis: quantitative correlations with histopathology. 12, 747–753. [DOI] [PubMed] [Google Scholar]
- Macey PM, Kumar R, Woo MA, Valladares EM, Yan-Go FL, Harper RM, 2008. Brain structural changes in obstructive sleep apnea. 31, 967–977. [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Ammirati E, Magnoni M, D’Ascenzo F, Anselmino M, Anzalone N, Rocca MA, Falini A, Filippi M, Camici PG, 2016. Carotid atherosclerosis, silent ischemic brain damage and brain atrophy: a systematic review and meta-analysis. Int.J.Cardiol. 223, 681–687. [DOI] [PubMed] [Google Scholar]
- Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA, Schneider JA, 2015. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann.Neurol. 77, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH, Gutierrez JA, 1996. Cerebral white matter is highly vulnerable to ischemia. 27, 1641–1647. [DOI] [PubMed] [Google Scholar]
- Pico F, Dufouil C, Lvy C, Besanon V, de Kersaint-Gilly A, Bonithon-Kopp C, Ducimetire P, Tzourio C, Alprovitch A, 2002. Longitudinal study of carotid atherosclerosis and white matter hyperintensities: the EVA-MRI cohort. 14, 109–115. [DOI] [PubMed] [Google Scholar]
- Pievani M, de Haan W, Wu T, Seeley WW, Frisoni GB, 2011. Functional network disruption in the degenerative dementias. 10, 829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins ND, Scheltens P, 2015. White matter hyperintensities, cognitive impairment and dementia: an update. 11, 157–165. [DOI] [PubMed] [Google Scholar]
- Reijmer YD, van Veluw SJ, Greenberg SM, 2016. Ischemic brain injury in cerebral amyloid angiopathy. 36, 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Román GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC, 2002. Subcortical ischaemic vascular dementia. 1, 426–436. [DOI] [PubMed] [Google Scholar]
- Salat DH, Smith EE, Tuch DS, Benner T, Pappu V, Schwab KM, Gurol ME, Rosas HD, Rosand J, Greenberg SM, 2006. White matter alterations in cerebral amyloid angiopathy measured by diffusion tensor imaging. 37, 1759–1764. [DOI] [PubMed] [Google Scholar]
- Smith EE, 2017. Cerebral Amyloid Angiopathy as a Cause of Neurodegeneration. J.Neurochem. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD, 2003. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. 62, 1287–1301. [DOI] [PubMed] [Google Scholar]
- van der Velpen Isabelle F, Yancy CW, Sorond FA, Sabayan B, 2017. Impaired Cardiac Function and Cognitive Brain Aging. CanJ.Cardiol. [DOI] [PubMed] [Google Scholar]
- van Oijen M, Jan de Jong F, Witteman J, Hofman A, Koudstaal PJ, Breteler M, 2007. Atherosclerosis and risk for dementia. Ann.Neurol. 61, 403–410. [DOI] [PubMed] [Google Scholar]
- Weller RO, Boche D, Nicoll JA, 2009. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 118, 87. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA, 2010. Neurodegenerative basis of age-related cognitive decline. 75, 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Boyle PA, Yu L, Segawa E, Sytsma J, Bennett DA, 2015. Conscientiousness, dementia related pathology, and trajectories of cognitive aging. Psychol.Aging. 30, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CW, Sano M, Ferris SH, Whitehouse PJ, Patterson MB, Aisen PS, 2013. Health-Related Resource Use and Costs in Elderly Adults with and without Mild Cognitive Impairment. J.Am.Geriatr.Soc 61, 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]