

Abstract
Substituted 1,3-dienes are valuable synthetic intermediates used in myriad catalytic transformations, yet modern catalytic methods for their preparation in a highly modular fashion using simple precursors are relatively few. We report here an aerobic boron Heck reaction with cyclobutene that forms exclusively linear 1-aryl-1,3-dienes using (hetero)arylboronic acids, or 1,3,5-trienes using alkenylboronic acids, rather than typical Heck products (i.e., substituted cyclobutenes). Experimental and computational mechanistic data support a pericyclic mechanism for C−C bond cleavage that enables the cycloalkene to circumvent established limitations associated with diene reagents in Heck-type reactions.
Graphical Abstract
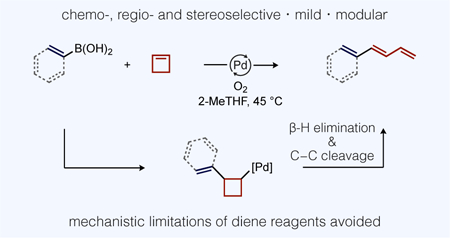
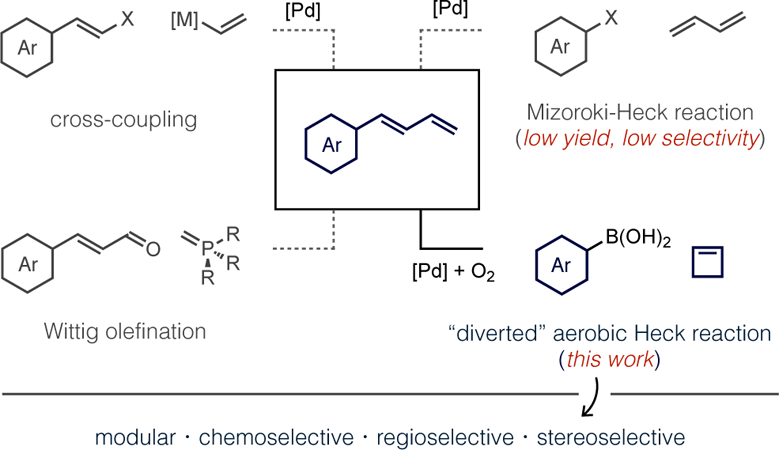
Substituted 1,3-dienes are common synthetic building blocks featured in a wide array of complexity-building catalytic transformations, including recently developed asymmetric hydrofunctionalizations,1 difunctionalizations,2 C−H functionalizations,3 cycloadditions,4 and cross-coupling.5 Preparations of 1,3-dienes, 1-aryl-1,3-dienes being a particularly prevalent subset in modern catalytic methods, classically involve disconnections at the central sigma bond of the diene,6 such as through Mizoroki-Heck reactions, cross-coupling,7,8 and ene-yne metathesis,9 or disconnection at the double bond in the case of Wittig-type olefinations (Scheme 1, left).10 Drawbacks of these approaches include functional group compatibility with strongly basic organometallic reagents or, more importantly, limited structural diversity in commercial starting materials (i.e., styrenyl halides or cinnamaldehydes). The development of single-step catalytic routes to substituted 1,3-dienes thus remains highly desirable, particularly if diverse and widely-available building blocks, such as boronic acids, could be used as substrates.11 We report here a mild Pd-catalyzed aerobic coupling of (hetero)arylboronic acids or alkenylboronic acids with cyclobutene to generate substituted 1,3-dienes or 1,3,5-trienes, respectively, in a regio- and stereoselective fashion.
Scheme 1.
Representative Routes to 1-Aryl-1,3-Dienes.
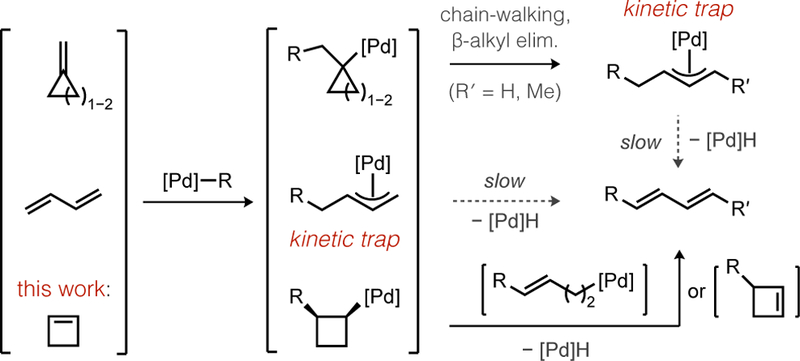
The focus on cyclobutene in this work was deliberate because direct synthesis of 1-aryl-1,3-dienes by arylation of butadiene suffers several established mechanistic limitations. The Pd-catalyzed reaction of aryl halides with butadiene (Scheme 1, upper right) was reported to occur in poor yields with competing formation of 1,4-diaryl-1,3-diene side products. Heck suggested this occurs because the immediate product (1-aryl-1,3-diene) is more reactive than butadiene in subsequent catalytic turnovers.12 Another problem occurs immediately following migratory insertion of butadiene, which forms a stabilized (π-allyl)Pd intermediate that is reluctant to release diene by β-H elimination (Scheme 2).13
Scheme 2.
Evolution of Intermediates Following Insertion of Butadiene or Related Alkenes.
The kinetic problems noted above could potentially be avoided by the use of a butadiene surrogate. Rupture of strained rings by β-alkyl elimination14 has been observed following migratory insertion of methylenecycloalkanes, which could provide an alternative path to diene formation (Scheme 2, top). While Larock did observe such β-alkyl elimination during reactions of anionic palladate complexes, facile “chain-walking”15 also occurred that shuttled Pd back to the thermodynamically most stable intermediate – a π-allyl complex.16 A ring opening reaction of cycloalkenes might nevertheless be a viable pathway to substituted dienes if chain-walking could be suppressed.
We hypothesized that electrophilic, rather than the electron-rich Pd complexes previously studied, could offer a potential solution because the former has been shown to form kinetic product distributions (i.e., no chain-walking) during oxidative Heck reactions.17,18 An electrophilic organo-Pd intermediate might then react with cyclobutene by either of two conceivable pathways for C−C cleavage (Scheme 2, bottom) to form 1-substituted 1,3-dienes without leading to (π-allyl)Pd intermediates. We thus studied the boron Heck reaction to test this idea.
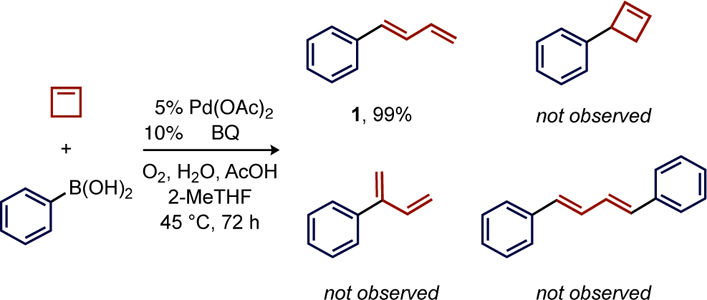
An optimization campaign identified suitable conditions for the aerobic reaction of phenylboronic acid with cyclobutene in the presence of Pd(OAc)2. In the best case using 5 mol% Pd with added acetic acid and water, a near quantitative yield (99%) of trans-1-phenyl-1,3-butadiene (1) was generated after 72 h at 45 °C (Table 1). The absence of detectable quantities of the typical Heck product (3-phenylcyclobutene) or 1,4-diphenyl-1,3-butadiene indicates surprisingly high selectivity in this catalytic process. Additionally, absence of 2-phenyl-1,3-butadiene highlights the complementary regioselectivity compared to (neocuproine)Pd-catalyzed aerobic Heck reactions developed by Stahl that favor branched products.19 Substitution of butadiene for cyclobutene led to complete suppression of reactivity (entry 1), which is consistent with the hypothesis that reaction pathways leading to (π-allyl)Pd intermediates are detrimental to catalysis. The use of lower O2 pressure in a balloon (14 psig) also generated 69% of 1, which should be attractive for applications without pressure equipment (entry 2). The use of 10% O2 in N2 mixture, close to the limiting oxygen concentration of 2-methyltetrahydrofuran (2-MeTHF),20 also produced 1 in 80% yield at the same oxygen partial pressure as the standard conditions (entry 3).
Table 1.
Aerobic Boron Heck Reaction with Cyclobutene.a
 | ||
|---|---|---|
| entry | deviation from the standard conditions | yield 1 (%)b |
| 1 | butadiene instead of cyclobutene | 0 |
| 2 | O2 balloon (14 psig) | 69 |
| 3c | 10% O2, balance N2 (500 psig) | 80 |
| 4 | PhBPin instead of PhB(OH)2 | 83 |
| 5 | PhBF3K instead of PhB(OH)2 | 79 |
| 6 | 1 mol% Pd | 43 |
| 7 | 1 mol% Pd, 96 h | 90 |
| 8 | omit BQ | 84 |
| 9 | omit AcOH | 72 |
| 10 | omit H2O | 43 |
| 11 | omit AcOH and H2O | 31 |
| 12 | omit Pd | 0 |
Conditions: boronic acid (0.25 mmol, 0.2 M), cyclobutene (2.7 equiv), AcOH (4 equiv), H2O (10 equiv), Pd(OAc)2, BQ, and BHT inhibitor (1000 ppm) in 2-MeTHF at 45 °C under O2 (50 psig).
Yield determined by NMR versus Bn2O as standard. Pin = pinacolato.
After additional heating at 75 °C for 6 h.
Several alternative boron reagents were also effective nucleophiles in the model reaction, such as the pinacol ester or trifluoroborate analogues of phenylboronic acid (entries 4 and 5), producing 1 in 83% and 79% yield, respectively. The inclusion of a radical inhibitor, butylated hydroxytoluene (BHT), was important in all cases for stabilizing the 1,3-diene products under the aerobic conditions. High yield of 1 (90%) is still possible with a five-fold reduction in catalyst loading with increased time (entries 6 and 7). Catalytic p-benzoquinone (BQ) enhances the yield of 1 but is not required for aerobic turnover (entry 8). The importance of added water and acetic acid on product yield are more pronounced (entries 9−11), which we speculate could promote transmetalation21 and/or catalyst turnover from a [Pd]−H or Pd0 species.22 The use of an industrially preferred solvent (2-MeTHF),23 molecular oxygen as terminal oxidant, and the generation of benign byproducts (e.g., boric acid and water) are several attractive features of this method. Organoboron reagents also complement the substrates used in existing methods to prepare dienes and trienes, such as aldehydes10–11 or haloarenes,11b while also avoiding the need for harsh oxidants or bases.
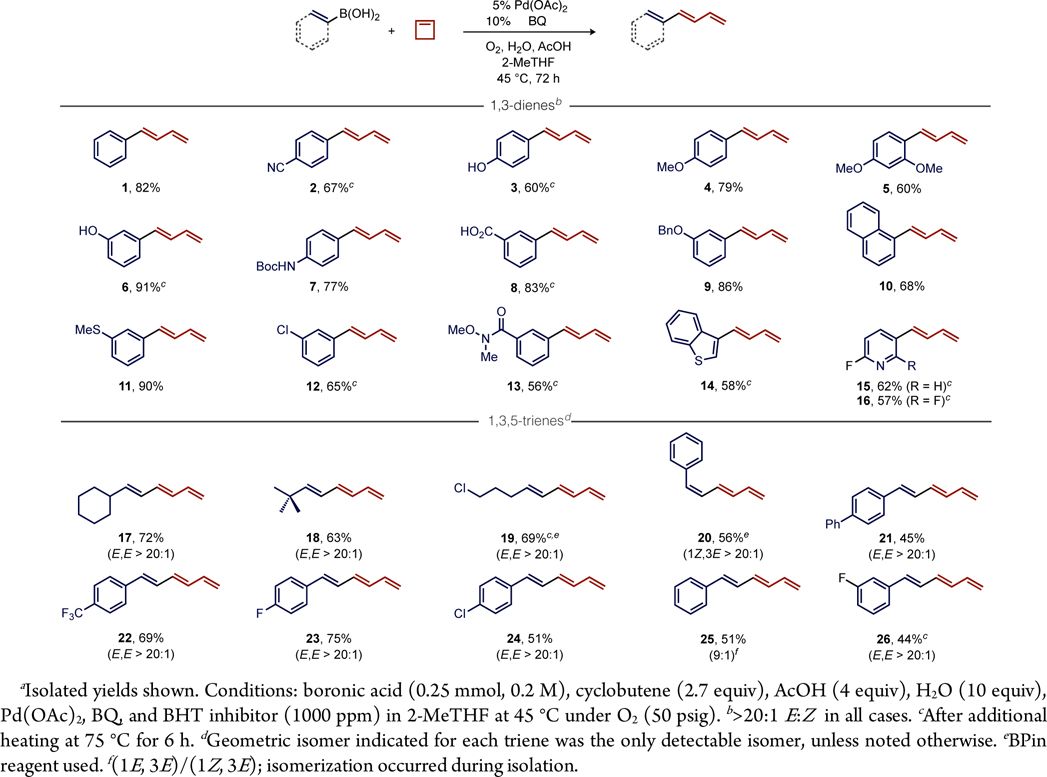
We next examined a series of other (hetero)arylboronic acids to establish the generality of this transformation. 1-Aryl-1,3-dienes derived from arylboronic acids with para- or meta-withdrawing substituents (2, 6, 8, 9, and 11−13) formed in good isolated yield (56%−91%). Arylboronic acids with electron-releasing substituents (3−5) were obtained in slightly reduced yet reasonable yields (60%−79%). The compatibility of the catalyst with free phenol, carboxylic acid, Weinreb’s amide, and coordinating thioether functional groups is very good. Reactions with 3-benzothienyl, 3-(2-fluoro)pyridyl, and 3-(2,6-difluoro)pyridyl boronic acids also generated 1-heteroaryl-1,3-dienes 14−16 in reasonable isolated yields (57%−62%). The fluoropyridine units in 15 and 16 are notable for their utility in medicinal chemistry for further elaboration by SNAr reactions.
We found the standard conditions used for arylboronic acid coupling with cyclobutene are also directly applicable to alkenylboronic acids (Table 2), which extend the π-conjugation of the products. Competing 6π-electrocyclization was not observed, which allowed the formation of a range of substituted 1,3,5-trienes by this diverted aerobic Heck reaction. Formation of cyclohexyl- (17), tert-butyl- (18), and chloropropyl- (19) substituted (1E,3E)-1,3,5-trienes occurred in good isolated yields (63%−72%) and as the only detectable stereoisomer. Alternatively, the use of a Z-alkenylboronic acid generated the 1Z-configured triene product 20 stereospecifically. Finally, a range of trans-styrenylboronic acids generated (1E,3E)-1-aryl-1,3,5-trienes 21−26 in 44%−75% isolated yields. Preliminary attempts using alkylboronic acids (e.g., Me, Bu, i-Pr, c-Pr) were not successful under the standard conditions.
Table 2.
Applicable (Hetero)Aryl and Alkenyl Boronic Acids to Aerobic Heck Reaction with Cyclobutene.a
 |
We conducted DFT calculations to establish a mechanistic rationalization for the formation of linear 1,3-dienes rather than branched isomers (e.g., 2-aryl-1,3-dienes) or normal Heck products (e.g., 3-arylcyclbutenes), the results of which are summarized in Figure 1. The phenyl-Pd species formed by transmetalation of PhB(OH)2 to Pd(II) initially forms 27 upon coordination of cyclobutene. Migratory insertion through a Cossee-Arlman mechanism (TS28) generates a cyclobutyl-Pd intermediate 29. This insertion reaction is more exoergic (−11 kcal/mol) than typical insertions of acyclic alkenes,24 which reflects a conformationally-enforced, stabilizing η2-arene interaction. Other plausible cyclobutyl-Pd species with alternative coordination modes of the acetate or 2-phenylcyclobutyl ligands were also evaluated (Figure S3), but these were less stable than 29 by 5.4−15.0 kcal/mol because the planar κ2-OAc in 29 minimizes steric interactions with the Ph group. Two postulated reactions pathways bifurcate from this point.
Figure 1.
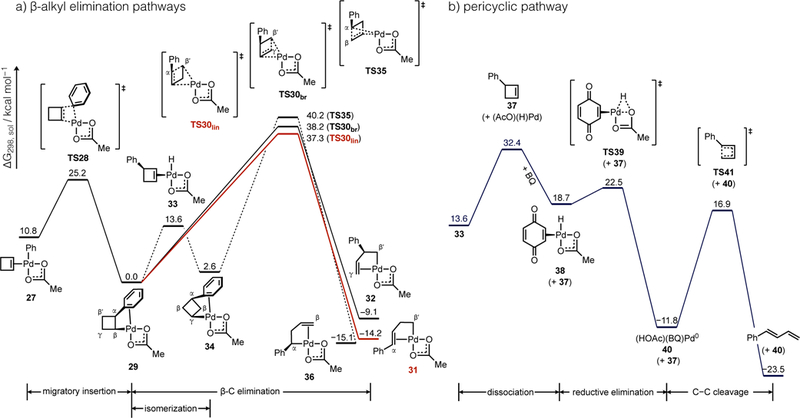
Potential energies of key steps in putative reaction pathways to 1-aryl-1,3-diene products involving C−C cleavage by either a (a) β-alkyl elimination or (b) pericyclic mechanism. Geometry optimizations were carried out at the B3LYP/LANL2DZ-6–31-G(d) and solvation corrections at the M06/SDD-6–311+G(d,p)/SMD(THF) level of theory.
One pathway to diene 1 from cyclobutyl-Pd complex 29 would proceed by C−C bond scission through β-C elimination, an elementary reaction that has ample precedent among the group 10 metals.16,25 Cleavage of the Cα−Cβ′ or Cβ′−Cγ cyclobutyl bond by this mechanism would generate new alkyl-Pd intermediates 31 or 32, respectively (Figure 1a). Formation of complex 31 is calculated to be exoergic by 14 kcal/mol and occurs through a lower energy transition state (TS30lin) than the competing pathway toward the branched diene (TS30br), possibly due to more favorable benzylic stabilization.26 Product 1 is then formed by β-H elimination from 31 (not shown).
An alternative pathway that could advance the common intermediate 29 to diene 1 could occur initially by formation of a [Pd]−H species (33) through β-H elimination followed by reinsertion with the opposite regioselectivity to generate a new symmetric cyclobutyl-Pd intermediate 34 (Figure 1a). While β-C elimination from 34 could only lead to the linear diene product, which could rationalize the experimentally observed selectivity, TS35 involving Cα−Cβ scission is calculated to proceed with a higher barrier than the alternative β-C elimination pathways. These mechanisms thus do not adequately account for the exclusive linear selectivity for formation of 1 over 2-phenyl-1,3-butadiene given the calculated ΔΔG‡ of ca. 1 kcal/mol between linear and branched product formation.
Another pathway to diene formation could begin from the intermediate 33 formed after β-H elimination (Figure 1b). Exchange of coordinated 3-phenylcyclobutene (37) for BQ occurs with a barrier of 32 kcal/mol by a dissociative mechanism. While associative mechanisms for release of product 37 might occur with lower barriers, the fact that the dissociative mechanism is lower in energy than TS30lin in the β-C elimination pathway is nonetheless informative. Subsequent BQ-promoted H−OAc reductive elimination to form Pd(0) is strongly exoergic and renders the process irreversible. Other pathways for oxidative turnover of Pd by O2 or BQ are possible but were not considered here.22,27 Linear product 1 can then be formed from free 37 by 4π-electrocyclic ring opening, which is calculated to occur with a considerable energy barrier (ΔG‡ = 29 kcal/mol).28 This significant barrier to product formation suggests 37 could accumulate during the course of the catalytic reaction. To test this, we conducted a reaction with phenylboronic acid using low pressure of O2 (14 psig) that facilitated periodic sampling for 1H NMR analysis. A kinetic profile generated from these data (Figure S2) indeed revealed early accumulation of intermediate 37, which peaks after ca. 12 h (92%). Product 1 grows in more slowly over 72 h to a final yield of 69%. With consideration of these computational and experimental mechanistic data, we conclude that the most likely reaction pathway involves an initial Heck process to generate a 3-substituted cyclobutene followed by pericyclic ring opening to reveal the final diene or triene product. Control of alkene geometry would be expected by this mechanism because C−C cleavage would be stereospecific through a pericyclic process. The high regioselectivity can also be rationalized because the Pd-catalyzed reaction can only form 3-substituted cyclobutenes by stereospecific syn-migratory insertion and syn-β-H elimination in the absence of chain walking.
In summary, a mild and modular route to synthetically versatile 1-aryl-1,3-dienes and substituted 1,3,5-trienes has been developed. The normal, aerobic Heck reaction in these cases diverts through a pathway involving C−C bond scission by pericyclic ring opening. This mechanism allows cyclobutene to function as a masked form of butadiene thereby circumventing mechanistic liabilities associated with the latter in Heck-type reactions. The reported method complements disconnections in classic synthetic routes to 1,3-dienes, such as by Wittig olefination or Pd-catalyzed cross-coupling, and also benefits from the wide availability of commercial organoboron reagents. The applicability of other nucleophiles and cycloalkenes to this reaction manifold will be the foci of future efforts.
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by Princeton University.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.0000000. Experimental procedures, computational data and characterization and spectral data for new compounds.
Notes
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Goldfogel MJ; Roberts CC; Meek SJ; Intermolecular Hydroamination of 1,3-Dienes Catalyzed by Bis(phosphine)carbodicarbene–Rhodium Complexes. J. Am. Chem. Soc 2014, 136, 6227–6230 [DOI] [PubMed] [Google Scholar]; (b) Saini V; O’Dair M; Sigman MS; Synthesis of Highly Functionalized Tri- and Tetrasubstituted Alkenes via Pd-Catalyzed 1,2-Hydrovinylation of Terminal 1,3-Dienes. J. Am. Chem. Soc 2015, 137, 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Roberts CC; Matías DM; Goldfogel MJ; Meek SJ; Lewis Acid Activation of Carbodicarbene Catalysts for Rh-Catalyzed Hydroarylation of Dienes. J. Am. Chem. Soc 2015, 137, 6488–6491 [DOI] [PubMed] [Google Scholar]; (d) Thullen SM; Rovis T; A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System. J. Am. Chem. Soc 2017, 139, 15504–15508 [DOI] [PubMed] [Google Scholar]; (e) Yang XH; Dong VM; Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 1774–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Adamson NJ; Hull E; Malcolmson SJ; Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd-PHOX Catalyst. J. Am. Chem. Soc 2017, 139, 7180–7183 [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gu L; Wolf LM; Zielinski A; Thiel W; Alcarazo M; alpha-Dicationic Chelating Phosphines: Synthesis and Application to the Hydroarylation of Dienes. J. Am. Chem. Soc 2017, 139, 4948–4953 [DOI] [PubMed] [Google Scholar]; (h) Gui Y-Y; Hu N; Chen X-W; Liao LL; Ju T; Ye J-H; Zhang Z; Li J; Yu D-G; Highly Regio- and Enantioselective Copper-Catalyzed Reductive Hydroxymethylation of Styrenes and 1,3-Dienes with CO2. J. Am. Chem. Soc 2017, 139, 17011–17014 [DOI] [PubMed] [Google Scholar]; (i) Marcum JS; Roberts CC; Manan RS; Cervarich TN; Meek SJ; Chiral Pincer Carbodicarbene Ligands for Enantioselective Rhodium-Catalyzed Hydroarylation of Terminal and Internal 1,3-Dienes with Indoles. J. Am. Chem. Soc 2017, 139, 15580–15583 [DOI] [PubMed] [Google Scholar]; (j) Adamson NJ; Wilbur KCE; Malcolmson SJ; Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc 2018, 140, 2761–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Bar GLJ; Lloyd-Jones GC; Booker-Milburn KI; Pd(II)-Catalyzed Intermolecular 1,2-Diamination of Conjugated Dienes. J. Am. Chem. Soc 2005, 127, 7308–7309 [DOI] [PubMed] [Google Scholar]; (b) Du H; Yuan W; Zhao B; Shi Y; Catalytic Asymmetric Diamination of Conjugated Dienes and Triene. J. Am. Chem. Soc 2007, 129, 11688–11689 [DOI] [PubMed] [Google Scholar]; (c) Liao L; Jana R; Urkalan KB; Sigman MS; A Palladium-Catalyzed Three-Component Cross-Coupling of Conjugated Dienes or Terminal Alkenes with Vinyl Triflates and Boronic Acids. J. Am. Chem. Soc 2011, 133, 5784–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu X; Lin H-C; Li M-L; Li L-L; Han Z-Y; Gong L-Z; Enantioselective 1,2-Difunctionalization of Dienes Enabled by Chiral Palladium Complex-Catalyzed Cascade Arylation/Allylic Alkylation Reaction. J. Am. Chem. Soc 2015, 137, 13476– 13479 [DOI] [PubMed] [Google Scholar]; (e) Liu Y; Xie Y; Wang H; Huang H; Enantioselective Aminomethylamination of Conjugated Dienes with Aminals Enabled by Chiral Palladium Complex-Catalyzed C–N Bond Activation. J. Am. Chem. Soc 2016, 138, 4314–4317 [DOI] [PubMed] [Google Scholar]; (f) Sardini SR; Brown MK; Catalyst Controlled Regiodivergent Arylboration of Dienes. J. Am. Chem. Soc 2017, 139, 9823– 9826 [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Huang Y; Smith KB; Brown MK; Copper-Catalyzed Borylacylation of Activated Alkenes with Acid Chlorides. Angew. Chem. Int. Ed. Engl 2017, 56, 13314–13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Liao L; Guo R; Zhao X; Organoselenium-Catalyzed Regioselective C-H Pyridination of 1,3-Dienes and Alkenes. Angew. Chem. Int. Ed. Engl 2017, 56, 3201–3205 [DOI] [PubMed] [Google Scholar]; (b) Bai L; Wang Y; Ge Y; Liu J; Luan X; Diastereoselective Synthesis of Dibenzo[b,d]azepines by Pd(II)-Catalyzed [5 + 2] Annulation of o-Arylanilines with Dienes. Org. Lett 2017, 19, 1734–1737 [DOI] [PubMed] [Google Scholar]; (c) Chen SS; Wu MS; Han ZY; Palladium-Catalyzed Cascade sp(2) C-H Functionalization/Intramolecular Asymmetric Allylation: From Aryl Ureas and 1,3-Dienes to Chiral Indolines. Angew. Chem. Int. Ed. Engl 2017, 56, 6641–6645. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hoyt JM; Schmidt VA; Tondreau AM; Chirik PJ; Iron-catalyzed intermolecular [2+2] cycloadditions of unactivated alkenes. Science 2015, 349, 960–963 [DOI] [PubMed] [Google Scholar]; (b) Kim H; Kim S; Kim J; Son JY; Baek Y; Um K; Lee PH; One-Pot Synthesis of Indolizines via Sequential Rhodium-Catalyzed [2 + 1]-Cyclopropanation, Palladium-Catalyzed Ring Expansion, and Oxidation Reactions from Pyridotriazoles and 1,3-Dienes. Org. Lett 2017, 19, 5677–5680 [DOI] [PubMed] [Google Scholar]; (c) Lang B; Zhu H; Wang C; Lu P; Wang Y; Rhodium-Catalyzed Cycloadditions between 3-Diazoindolin-2-imines and 1,3-Dienes. Org. Lett 2017, 19, 1630–1633 [DOI] [PubMed] [Google Scholar]; (d) Kim S; Kim H; Um K; Lee PH; Synthesis of Azepinoindoles via Rhodium-Catalyzed Formal Aza-[4 + 3] Cycloaddition Reaction of 3-Diazoindolin-2-imines with 1,3-Dienes in One-Pot. J. Org. Chem 2017, 82, 9808–9815. [DOI] [PubMed] [Google Scholar]
- (5).(a) Sargent BT; Alexanian EJ; Cobalt-Catalyzed Carbonylative Cross-Coupling of Alkyl Tosylates and Dienes: Stereospecific Synthesis of Dienones at Low Pressure. J. Am. Chem. Soc 2017, 139, 12438–12440 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen H-C; Wang P-S; Tao Z-L; Han Z-Y; Gong L-Z; An Enantioselective Multicomponent Carbonyl Allylation of Aldehydes with Dienes and Alkynyl Bromides Enabled by Chiral Palladium Phosphate. Adv. Synth. Catal 2017, 359, 2383–2389. [Google Scholar]
- (6).(a) De Paolis M; Chataigner I; Maddaluno J Recent Advances in Stereoselective Synthesis of 1,3-Dienes. In Stereoselective Alkene Synthesis; Wang J, Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012, p 87–146 [DOI] [PubMed] [Google Scholar]; (b) Mehta G; Rao HS Synthesis of Conjugated Dienes and Polyenes. In The Chemistry of Dienes and Polyenes; Rappoport Z, Ed.; Wiley: New York, 1997; Vol. 1, p 359–480 [Google Scholar]; (c) Vasil’ev A; P. Serebryakov E; Synthetic methodologies for carbo-substituted conjugated dienes. Russ. Chem. Rev 2001, 70, 735–776. [Google Scholar]
- (7).(a) Yamashita M; Hirano K; Satoh T; Miura M; Synthesis of α,ω-Diarylbutadienes and -Hexatrienes via Decarboxylative Coupling of Cinnamic Acids with Vinyl Bromides under Palladium Catalysis. Org. Lett 2010, 12, 592–595 [DOI] [PubMed] [Google Scholar]; (b) Wang G; Mohan S; Negishi E.-i.; Highly selective synthesis of conjugated dienoic and trienoic esters via alkyne elementometalation–Pd-catalyzed cross-coupling. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 11344–11349 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Molloy JJ; Seath CP; West MJ; McLaughlin C; Fazakerley NJ; Kennedy AR; Nelson DJ; Watson AJB; Interrogating Pd(II) Anion Metathesis Using a Bifunctional Chemical Probe: A Transmetalation Switch. J. Am. Chem. Soc 2018, 140, 126–130. [DOI] [PubMed] [Google Scholar]
- (8).Olivares AM; Weix DJ; Multimetallic Ni- and Pd-Catalyzed Cross-Electrophile Coupling To Form Highly Substituted 1,3-Dienes. J. Am. Chem. Soc 2018, 140, 2446–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Diver ST; Giessert AJ; Enyne Metathesis (Enyne Bond Reorganization). Chem. Rev 2004, 104, 1317–1382. [DOI] [PubMed] [Google Scholar]
- (10).(a) Barluenga J; Rodríguez F; Álvarez‐Rodrigo L; Fañanás FJ; Zirconium ‐ Mediated Cross ‐ Coupling of Terminal Alkynes and Vinyl Bromides: Selective Synthesis of Cyclobutene and 1,3 ‐Diene Derivatives. Chem. Eur. J 2004, 10, 101–108 [DOI] [PubMed] [Google Scholar]; (b) Zhu L; Wehmeyer RM; Rieke RD; The direct formation of functionalized alkyl(aryl)zinc halides by oxidative addition of highly reactive zinc with organic halides and their reactions with acid chlorides, .alpha.,.beta.-unsaturated ketones, and allylic, aryl, and vinyl halides. J. Org. Chem 1991, 56, 1445–1453 [Google Scholar]; (c) Chatterjee T; Dey R; Ranu BC; An easy access to styrenes: trans aryl 1,3-, 1,4- and 1,5-dienes, and 1,3,5- trienes by Hiyama cross-coupling catalyzed by palladium nanoparticles. New J. Chem 2011, 35, 1103–1110 [Google Scholar]; (d) Hatanaka Y; Hiyama T; Cross-coupling of organosilanes with organic halides mediated by a palladium catalyst and tris(diethylamino)sulfonium difluorotrimethylsilicate. J. Org. Chem 1988, 53, 918–920 [Google Scholar]; (e) Alacid E; Nájera C; Aqueous Sodium Hydroxide Promoted Cross-Coupling Reactions of Alkenyltrialkoxysilanes under Ligand-Free Conditions. J. Org. Chem 2008, 73, 2315–2322 [DOI] [PubMed] [Google Scholar]; (f) Lu G.-p.; Voigtritter KR; Cai C; Lipshutz BH; Ligand effects on the stereochemistry of Stille couplings, as manifested in reactions of Z-alkenyl halides. Chem. Commun 2012, 48, 8661–8663 [DOI] [PubMed] [Google Scholar]; (g) Stille JK; Groh BL; Stereospecific cross-coupling of vinyl halides with vinyl tin reagents catalyzed by palladium. J. Am. Chem. Soc 1987, 109, 813–817 [Google Scholar]; (h) Krasovskiy AL; Haley S; Voigtritter K; Lipshutz BH; Stereoretentive Pd-Catalyzed Kumada–Corriu Couplings of Alkenyl Halides at Room Temperature. Org. Lett 2014, 16, 4066–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Molander GA; Rivero MR; Suzuki Cross-Coupling Reactions of Potassium Alkenyltrifluoroborates. Org. Lett 2002, 4, 107–109 [DOI] [PubMed] [Google Scholar]; (j) Justyna SF; Aline R; Adrian F; Jędrzej W; Maciej K; Piotr P; A highly selective synthesis of 1‐substituted (E)‐buta‐1,3‐dienes with 4,4,5,5‐tetramethyl‐2‐vinyl ‐ 1,3,2 ‐ ioxaborolane as building block. Appl. Organomet. Chem 2014, 28, 137–139. [Google Scholar]
- (11).(a) Mundal DA; Lutz KE; Thomson RJ; Stereoselective Synthesis of Dienes from N-Allylhydrazones. Org. Lett 2009, 11, 465–468 [DOI] [PubMed] [Google Scholar]; (b) Nguyen VT; Dang HT; Pham HH; Nguyen VD; Flores-Hansen C; Arman HD; Larionov OV; Highly Regio- and Stereoselective Catalytic Synthesis of Conjugated Dienes and Polyenes. J. Am. Chem. Soc 2018, 140, 8434–8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mitsudo T; Fischetti W; Heck RF; Palladium-catalyzed syntheses of aryl polyenes. J. Org. Chem 1984, 49, 1640–1646. [Google Scholar]
- (13).Jeffery T; Palladium-catalysed Arylation of 1,3-Dienes : A Highly Chemo, Regio and Stereoselective Synthesis of (E,E) Conjugated Dienic Aromatics. Tetrahedron Lett 1992, 33, 1989–1992. [Google Scholar]
- (14).O’Reilly ME; Dutta S; Veige AS; β-Alkyl Elimination: Fundamental Principles and Some Applications. Chem. Rev 2016, 116, 8105–8145. [DOI] [PubMed] [Google Scholar]
- (15).Shultz LH; Brookhart M; Measurement of the Barrier to Beta-Hydride Elimination in Beta-Agostic Palladium-Ethyl Complex: A Model for the Energetics of Chain-Walking in (Alpha-Diimine)PdR+Olefin Polymerization Catalysts. Organometallics 2001, 20, 3975. [Google Scholar]
- (16).Larock RC; Varaprath S; Mercury in Organic Chemistry. 30. Synthesis of (π-Allyl)Palladium Compounds Via Organopalladium Additions to Alkenyl- and Methylenecyclopropanes and Alkenyl- and Methylenecyclobutanes. J. Org. Chem 1984, 49, 3432. [Google Scholar]
- (17).(a) Werner EW; Sigman MS; A Highly Selective and General Palladium Catalyst for the Oxidative Heck Reaction of Electronically Nonbiased Olefins. J. Am. Chem. Soc 2010, 132, 13981–13983 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Delcamp JH; Brucks AP; White MC; A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc 2008, 130, 11270–11271 [DOI] [PubMed] [Google Scholar]; (c) Su Y; Jiao N; Control of Chemo-, Regio-, and Stereoselectivities in Ligand-Free Pd-Catalyzed Oxidative Heck Reactions of Arylboronic Acids or Alkenylboronate with Allyl Esters. Org. Lett 2009, 11, 2980–2983. [DOI] [PubMed] [Google Scholar]
- (18).(a) Karimi B; Behzadnia H; Elhamifar D; Akhavan PF; Esfahani FK; Zamani A; Transition-Metal-Catalyzed Oxidative Heck Reactions. Synthesis 2010, 2010, 1399–1427 [Google Scholar]; (b) Lee AL; Enantioselective oxidative boron Heck reactions. Org. Biomol. Chem 2016, 14, 5357–5366. [DOI] [PubMed] [Google Scholar]
- (19).Zheng C; Wang D; Stahl SS; Catalyst-Controlled Regioselectivity in the Synthesis of Branched Conjugated Dienes via Aerobic Oxidative Heck Reactions. J. Am. Chem. Soc 2012, 134, 16496–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Osterberg PM; Niemeier JK; Welch CJ; Hawkins JM; Martinelli JR; Johnson TE; Root TW; Stahl SS; Experimental Limiting Oxygen Concentrations for Nine Organic Solvents at Temperatures and Pressures Relevant to Aerobic Oxidations in the Pharmaceutical Industry. Org. Process Res. Dev 2015, 19, 1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Siegmann K; Pregosin PS; Venanzi LM; Reaction of organoboron compounds with platinum(II) disolvento complexes. Organometallics 1989, 8, 2659–2664. [Google Scholar]
- (22).(a) Konnick MM; Gandhi BA; Guzei IA; Stahl SS; Reaction of Molecular Oxygen with a PdII – Hydride To Produce a PdII – Hydroperoxide: Acid Catalysis and Implications for Pd‐Catalyzed Aerobic Oxidation Reactions. Angew. Chem. Int. Ed 2006, 45, 2904–2907 [DOI] [PubMed] [Google Scholar]; (b) Konnick MM; Stahl SS; Reaction of Molecular Oxygen with a PdII- Hydride To Produce a PdII-Hydroperoxide: Experimental Evidence for an HX-Reductive-Elimination Pathway. J. Am. Chem. Soc 2008, 130, 5753–5762. [DOI] [PubMed] [Google Scholar]
- (23).Prat D; Pardigon O; Flemming H-W; Letestu S; Ducandas V; Isnard P; Guntrum E; Senac T; Ruisseau S; Cruciani P; Hosek P; Sanofi’s Solvent Selection Guide: A Step Toward More Sustainable Processes. Org. Process Res. Dev 2013, 17, 1517–1525. [Google Scholar]
- (24).(a) Xu L; Hilton MJ; Zhang X; Norrby P-O; Wu Y-D; Sigman MS; Wiest O; Mechanism, Reactivity, and Selectivity in Palladium-Catalyzed Redox-Relay Heck Arylations of Alkenyl Alcohols. J. Am. Chem. Soc 2014, 136, 1960–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dang Y; Qu S; Wang Z-X; Wang X; A Computational Mechanistic Study of an Unprecedented Heck-Type Relay Reaction: Insight into the Origins of Regio- and Enantioselectivities. J. Am. Chem. Soc 2014, 136, 986–998. [DOI] [PubMed] [Google Scholar]
- (25).(a) Noyori R; Takaya H; Reaction of Methylenecyclopropanes with Palladium Chloride. J. Chem. Soc. D 1969, 525; Green M; Hughes RP; Transition-Metal Complexes of Methylenecyclopropanes: Ring-Opening Reactions Promoted by Palladium(II). J. Chem. Soc., Chem. Commun 1974, 686; (b) Green M; Hughes RP; Reactions of Coordinated Ligands Part IX. Ring-Opening of Methylenecyclopropanes by Palladium(II)-Nucleofile Systems - Formation of Substituted η3-but-3-Enyl Complexes of Palladium(II). J. Chem. Soc., Dalton Trans 1976, 1880; (c) Hosokawa T; Maitlis PM; Model System for Acid and Base Reactions, Carbonylation, And β-Hydride Elimination in Organopalladium Chemistry. J. Am. Chem. Soc 1972, 94, 3238 [Google Scholar]; (d) Lehmkuhl H; Naydowski C; Benn R; Rufinska A; Schroth G; η5-Cyclopentadienyl-η2-Olefin-Alkylnickel. J. Organomet. Chem 1982, 228, C1 [Google Scholar]; (e) Thomson SK; Young GB; Thermolytic Rearrangement of Cis-Bis-(Phosphine)Bis(Trimethylsilyl)Methyl Platinum(II) Complexes Via Beta-Alkyl Transfer. Organometallics 1989, 8, 2068 [Google Scholar]; (f) Ankianiec BC; Christou V; Hardy DT; Thomson SK; Young GB; Mechanism of Thermolytic Rearrangment of Cis-Bis(Silylmethyl)Platinum(II) Complexes: Beta-Carbon Transfer Predominates over Hydrogen-Transfer. J. Am. Chem. Soc 1994, 116, 9963 [Google Scholar]; (g) Attig TG; Metal Hydride Induced Ring-Opening Reactions of Methylenecyclopropane Derivatives - Formation of Butenylplatinum(II) Complexes. Inorg. Chem 1978, 17, 3097 [Google Scholar]; (h) Phillips RL; Puddephatt RJ; Reactions of Methylenecyclopropane with Some Hydridoplatinum(II) Complexes. J. Chem. Soc., Dalton Trans 1978, 1736; (i) Phillips RL; Puddephatt RJ; A Cyclopropylplatinum to Π-Allylplatinum Rearrangement. J. Organomet. Chem 1977, 136, C52 [Google Scholar]; (j) Abo-Amer A; Puddephatt RJ; Reactivity and Mechanism in the Ring-Opening of Cyclopropylmethylplatinum(IV) Complexes. Inorg. Chem. Commun 2011, 14, 111 [Google Scholar]; (k) Flood TC; Statler JA; Synthesis, Characterization, and Rearragements of (1-Methylcyclbutyl)Methyl Platinum(II) Complexes: Very Mild Ring-Strain-Induced Carbon Carbon Activation. Organometallics 1984, 3, 1795 [Google Scholar]; (l) Flood TC; Bitler SP; Reversible Formal Alkene Insertion into a Chelated Platinum Alkyl Bond. J. Am. Chem. Soc 1984, 106, 6076 [Google Scholar]; (m) Ermer SP; Struck GE; Bitler SP; Richards R; Bau R; Flood TC; Kinetics and Conformation in the Reversible Insertion of an Alkene into a Platinum Carbon Bond in a Chelated (Pentenyl)Platinum Complex. Organometallics 1993, 12, 2634 [Google Scholar]; (n) Zhugralin AR; Kobylianskii IJ; Chen P; Experimental Gas-Phase and in Silico Investigation of β-Methyl Elimination from Cationic Palladium Alkyl Species. Organometallics 2015, 34, 1301. [Google Scholar]
- (26).(a) Doherty NM; Bercaw JE; Kinetics and mechanism of the insertion of olefins into transition metal-hydride bonds. J. Am. Chem. Soc 1985, 107, 2670–2682 [Google Scholar]; (b) Fristrup P; Le Quement S; Tanner D; Norrby P-O; Reactivity and Regioselectivity in the Heck Reaction: Hammett Study of 4-Substituted Styrenes. Organometallics 2004, 23, 6160–6165. [Google Scholar]
- (27).(a) Stahl SS; Thorman JL; Nelson RC; Kozee MA; Oxygenation of Nitrogen-Coordinated Palladium(0): Synthetic, Structural, and Mechanistic Studies and Implications for Aerobic Oxidation Catalysis. J. Am. Chem. Soc 2001, 123, 7188–7189 [DOI] [PubMed] [Google Scholar]; (b) Decharin N; Stahl SS; Benzoquinone-Promoted Reaction of O2 with a PdII−Hydride. J. Am. Chem. Soc 2011, 133, 5732–5735. [DOI] [PubMed] [Google Scholar]
- (28).Pomerantz M; Hartman PH; Thermal rearrangement of 3-phenylcyclobutene. Tetrahedron Lett 1968, 9, 991–993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.