

SUMMARY
Single-cell RNA sequencing is a powerful tool by which to characterize the transcriptional profile of low-abundance cell types, but its application to the inner ear has been hampered by the bony labyrinth, tissue sparsity, and difficulty dissociating the ultra-rare cells of the membranous cochlea. Herein, we present a method to isolate individual inner hair cells (IHCs), outer hair cells (OHCs), and Deiters’ cells (DCs) from the murine cochlea at any post-natal time point. We harvested more than 200 murine IHCs, OHCs, and DCs from post-natal days 15 (p15) to 228 (p228) and leveraged both short- and long-read single-cell RNA sequencing to profile transcript abundance and structure. Our results provide insights into the expression profiles of these cells and document an unappreciated complexity in isoform variety in deafness-associated genes. This refined view of transcription in the organ of Corti improves our understanding of the biology of hearing and deafness.
Graphical Abstract
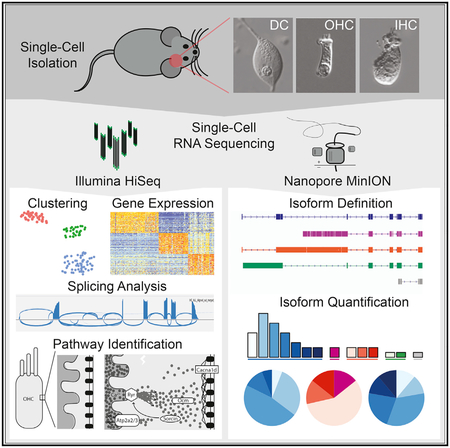
In Brief
Single-cell RNA-seq of inner and outer auditory hair cells facilitates the identification of cell type-defining genes across a range of expression levels. Full-length reverse transcription with long-read sequencing identifies novel exons and unappreciated splicing diversity among deafness-associated genes.
INTRODUCTION
The sensory cells of the mammalian auditory system are among the most highly specialized cell types in the human body. Subgrouped into inner hair cells (IHCs) and outer hair cells (OHCs), these cells are unique in their morphology and function, using common cellular components such as actin filaments, myosin motors, and ion channels in unique configurations to perform the delicate work of mechanosensation. In the human ear there are approximately 3,500 IHCs, which as the true sensory receptors in audition are responsible for 95% of afferent transduction, and 12,000 OHCs that act as motor units to amplify the acoustic signal (Pujol et al., 2016). The aggregate number, about 15,500 HCs per cochlea, is astonishingly small compared with the 4.6 million cones and 92 million rods in each human retina or the 50 million olfactory cells in the human nasal cavity (Curcio et al., 1990; Sarafoleanu et al., 2009).
Transcriptome profiling of HCs from both human and animal model tissue has proved difficult for several reasons beyond their scarcity. As part of the membranous labyrinth, these cells lie within the petrous portion of the temporal bone and are surrounded by a bony labyrinth that ranks as the hardest bone in the body (Figure 1A). The encased cochlear membranous labyrinth is gossamer thin and is suspended in perilymphatic fluid, making it challenging to dissect (Figure 1B). The estimated 415,000 cells in the murine membranous labyrinth fall into at least 16, and potentially more than 40, cell types (M.N. Nguyen et al., 2018, Assoc. Res. Otolaryngol., conference). Sensory HCs account for ~4,000 of these cells, thus representing <1% of the aggregate total (Figure S1) (Ehret and Frankenreiter, 1977).
Figure 1. Single-Cell Isolation from the Mature Murine Cochlea.

(A) Temporal bones were removed from p15–p228 C3Heb/FeJ WT mice and opened to remove the membranous labyrinth of the cochlea, which was divided into apical and basal halves.
(B) Cochlear tissues in (A) contain the organ of Corti and its characteristic three rows of OHCs and one row of IHCs, as seen in the scanning electron microscopy image. To facilitate isolation of individual cells, the membranous labyrinth is digested in collagenase and gently triturated using a p1000 pipette tip.
(C) Cartoon depiction of micropipette isolation, wash, and re-isolation with deposition of the isolated cell into 4 μL of RNase inhibitor containing lysis buffer.
(D) Representative isolated single cells. IHCs and OHCs exhibit distinct morphological features, including a cuticular plate and stereocilia that are clearly visible on the apical surface under a 20× differential interference contrast (DIC) objective. IHCs have a rounded, flask-like shape, while OHCs are more oblong and cylindrical (Liu et al., 2014). Scale bars indicate 5 μm.
(E) To begin unbiased clustering, principal-component analysis (PCA) was performed using Seurat to identify top differentially expressed genes among all p15 cells in the dataset (n = 132). PC 1 and PC 2 showed clearly defined blocks of differential expression and were carried forward into tSNE clustering.
(F) tSNE clustering shows three distinct clusters. To test concordance of these clusters with morphological cell type assignments, we have overlaid colors to represent morphological cell types (DCs, red; IHCs, green; OHCs, blue). We found 100% concordance between morphological cell type assignment and unbiased cluster assignment.
(G) Heatmap of the top 100 cluster-defining genes in each cluster. Although the blocks defined in this plot are distinct, the DC and OHC groups appear to be better defined than the IHC group, as some cells in the IHC group express DC- and OHC-defining genes.
The first transcriptome profiling experiment on cochlear tissue was performed Cho et al. (2002). Since then, a large number of studies have leveraged microarray, PCR, and RNA sequencing (RNA-seq) technologies to explore the auditory transcriptome (Schimmang and Maconochie, 2016). Low-input reverse transcription and RNA isolation technologies have revolutionized these studies, facilitating cell type-specific transcriptomics. To date, seven cell type-specific gene expression profiling experiments have been made publicly available through the University of Maryland’s gEAR and Harvard University’s SHIELD inner ear gene expression databases (Table S1) (Cai et al., 2015; Elkon et al., 2015). Most commonly, microfluidics chip- and droplet-based techniques have been coupled with fluorescence-activated cell sorting (FACS) to generate single-cell datasets from pools of IHCs and OHCs harvested at time points up to post-natal day 7 (p7), which is before the onset of hearing in mice (Cai et al., 2015; Elkon et al., 2015; Scheffer et al., 2015). To our knowledge, the only published report of RNA-seq data on individual, unpooled cochlear sensory cells sequenced 10 HCs at p1 (Table S1) (Burns et al., 2015).
These reports attest to the difficulty of applying single-cell isolation and sequencing to the organ of Corti. As an alternative to chip- and droplet-based techniques, we have modified the simple isolation technique described by Liu et al. (2014) and Li et al. (2018). Using pulled glass micropipettes, we isolated individual cells from the membranous labyrinth and defined cell type identity through unbiased cluster analysis. Differential expression analysis on more than 200 individual IHCs, OHCs, and Deiters’ cell (DCs) at mature time points ranging from p15 to p228 identified cell type-defining genes and pathways. Splicing analysis and long-read sequencing of these cells uncovered a staggering degree of splicing heterogeneity and revealed widespread expression of unannotated exons and isoforms. These data challenge many assumptions about gene structure derived from current annotations and suggest that cell-specific annotations will be required to accurately capture the expression profiles unique to the wide variety of auditory cell types.
RESULTS
Estimation of Cochlear Non-sensory Cell Abundance
Auditory HCs are low in abundance compared with the wide variety of supporting cell types in the mammalian cochlea. To estimate the number of HCs available for single-cell isolation in the murine cochlear membranous labyrinth, we assumed that there are ~765 IHCs in the mature murine cochlea (Ehret and Franken reiter, 1977). A 14 μm section from the center of the murine temporal bone (Figure S1) contains only one visible IHC, and by counting nuclei in representative sections (apical section, 459 nuclei; middle section, 565 nuclei; basal section, 605 nuclei) and multiplying by the number of such sections (255 sections in each region), we estimated the number of cells in the murine membranous labyrinth to be ~415,395 (Figures S1A–S1E) (Ehret and Frankenreiter, 1977). IHCs and OHCs represent 0.19% and 0.59% of this cell count (Figure S1F).
Cell Isolation
To enrich for rare cell types of interest and facilitate full-length cDNA library preparation, we isolated cells using a pulled glass micropipette technique, as reported by Liu et al. (2014). IHCs, OHCs, and DCs were identified using an inverted microscope with a 20× differential interference contrast (DIC) objective and harvested at p15, p30, p70, and p228 to confirm our ability to isolate HCs at different time points. These cells have distinct morphology (Figures 1C and 1D), and although we also collected small numbers of other cell types with less distinct morphological characteristics, we focused in the present study on the IHC (n = 42), OHC (n = 127), and DC (n = 39) datasets (Figure S2). To increase sample size, cells from both genders, apical and basal tonotopic positions, and multiple time points were collected. These variables were recorded for each cell. For clarity and to eliminate confounding variables from our analysis, the main figures presented in this study include cells exclusively from the p15 time point, leveraging the largest number of available single-cell samples (n = 132 after filtering). Smaller numbers of cells from the p30 (n = 6), p70 (n = 54), and p228 (n = 6) time points confirm the applicability of this technique across a broad range of mature time points and facilitated temporal and tono-topic differential expression analysis. The single-cell nature of this dataset permits re-filtering to complete additional analyses beyond those presented here.
Quality Control
Prior to clustering, quality control filters were applied to the gene expression matrix, eliminating cells expressing fewer than 2,000 genes at a threshold >0 counts. Next, principal-component analysis and t-distributed stochastic neighbor embedding (tSNE) clustering were applied to refine the dataset. Seven cells were eliminated because of discordant expression levels of the top 100 most highly expressed genes across all cell types. Three cells were eliminated because of discordant cluster assignments in an effort to achieve 100% concordance between morphologically defined and bioinformatically assigned cell type identities. The remaining set of high-quality cells contained low percentages of mitochondrial transcripts among the total read counts per cell transcriptome, which is noteworthy, as an increased percentage of mitochondrial reads per cell is an indication of increased cell death. Sequencing performance and quality metrics are provided in Figure S3.
Unbiased Clustering
Seurat, an R package for single-cell analysis, was used to identify differentially expressed genes, perform clustering, and complete additional statistical analyses (Satija et al., 2015). Unbiased clustering on the basis of principal components 1 and 2 was performed using Seurat (Figure 1E). The resultant tSNE clusters showed that IHCs, OHCs, and DCs segregated cleanly to form unbiased cell-specific groups in agreement with their morphological assignments (Figure 1F). To identify the characteristic expression pattern of each cell type, we extracted the top 100 cluster-defining genes for each cell type ranked by Seurat’s area under the receiver-operating characteristic curve (AUC) classifier (Figures 1G and S4).
Expression Profiles
To compare the expression profile among cell types, we calculated mean expression levels for each gene across all cells within a cluster. Comparison of mean expression profiles by Venn diagrams showed that each cell type expressed about 700 unique genes (OHCs, 753; IHCs, 655; DCs, 713) above a cutoff of 10 Seurat scaled counts. Seven hundred seventy-one genes were expressed by both IHCs and OHCs, although we found that IHCs and DCs also had a surprisingly high level of overlap (750 genes compared with 564 genes shared by OHCs and DCs) (Figure 2A). To more rigorously test the transcriptional similarity of each cell type, we performed Pearson correlation and R2 regression analysis using all genes. We found a strong correlation in all three comparisons, with the highest correlation, as expected, between OHCs and IHCs, and the lowest correlation between OHCs and DCs (a Pearson correlation of 1 indicates a perfect linear relationship, and a score of 0 reflects no linear correlation). That the OHC and DC profiles were the least similar may reflect a high level of OHC specialization (Figure 2B).
Figure 2. Characterizing the Transcription Profile of Each Cell Type.

To assess the variability in expression profiles of p15 IHCs (n = 33), OHCs (n = 61), and DCs (n = 37), we performed several transcriptome-level analyses.
(A) Venn diagram shows the number of genes with shared and unique expression among the three cell types. Genes included meet an average minimum expression threshold of 10 Seurat scaled counts.
(B) To compare similarities and differences in global expression profiles by cell type and to assess similarity, we performed correlation and regression analysis. For visualization, mean expression levels were log2 transformed.
(C, E, and G) To assess the gene expression profile of each cell type, we calculated mean expression level from Seurat scaled counts values. We then rank-ordered genes by mean expression level: (C) IHCs, green; (E) OHCs, blue; and (G) DCs, red. The 50 most highly expressed genes, by mean expression level, are listed beneath bar plots showing log mean expression levels by gene and cell type.
(D, F, and H) The top 10 cluster-defining genes for each cell type ranked by receiver-operating characteristic (ROC) area under the curve (AUC): (D) IHCs, (F) OHCs, and (H) DCs. *Genes that are in the top 50 genes ranked by log expression level and the top 10 cluster-defining genes ranked by the AUC classifier. Violin plots for any gene of interest may be generated using our online violin plot tool at morlscrnaseq.org.
To define the characteristic expression profile of each cell type, we rank-ordered all genes by normalized expression level across individual cells within a cluster. General profiles of each cell type were characterized by a small number of genes with high expression (reaching log mean expression levels between 2 and 7) and a large number of genes with low expression (log mean expression levels of <2) (Figures 2C, 2E, and 2G). Among the top 50 most highly expressed genes by cell type, some genes, such as Fbxo2 and Skpa1, were highly expressed in all three cell types, whereas others, such as Ocm and Fgfr3, were expressed primarily in only one cell type. Using AUC rankings (Figure S4), we extracted cluster-defining genes for each group to identify defining cell type-specific markers (Figures 2D, 2F, and 2H; Tables S2, S3, and S4). Violin plots of the top 10 cluster-defining genes revealed well-known marker genes for OHCs (Ocm and Slc26a5), IHCs (Otof and Atp2a3), and DCs (Bace2 and Ceacam16). Violin plots for the complete list of genes are accessible using our scRNA-Seq Browser Tool (Web Resources: morlscrnaseq.org).
Differential Expression Analysis
We found high differential expression of known cell type markers, such as prestin (Slc26a5), which is robustly and differentially expressed in OHCs, and after confirming high concordance between our data and published OHC and IHC transcriptomes (Liu et al., 2014; Li et al., 2018), we searched for genes with discordant rankings, hypothesizing that ranking genes by AUC rather than expression level would identify cell type-defining genes across a wider range of expression levels (Figure S4). When ranked by AUC classification, two calcium-associated genes, Ocm and Sri, topped the list of OHC-defining genes, outranking even Slc26a5, which placed third (Figure 2F). Expression of Ocm has been previously reported in OHCs. The encoded protein, oncomodulin, is a calcium-buffering protein that localizes to the lateral cell membrane, where it is required for cochlear amplification (Simmons et al., 2010; Tong et al., 2016). Sri encodes sorcin, a protein expressed in cardiac myocytes that is essential for calcium-mediated excitation-contraction coupling. It carries out calcium-dependent inhibition of calcium-induced calcium release (CICR) through inhibition of Ryr channels (Ca2+ release channels/ryanodine receptors). In myocytes, Ryr channels mediate CICR (Farrell et al., 2003; Lokuta et al., 1997) enabling rapid release of Ca2+ ions from the sarcoplasmic reticulum, which creates the spatiotemporally restricted calcium sparks essential for muscle contraction (Gambardella et al., 2018).
Immunofluorescence localized sorcin to only the OHCs (Figures 3A, 3B, and S6), leading us to hypothesize that a similar mechanism may regulate prestin-based motility (Figures 3C and 3D). Consistent with this possibility, we identified expression of the essential components of a sorcin-mediated excitation-contraction pathway in OHCs, including (1) a voltage-gated calcium-release channel, (2) channels to facilitate CICR, (3) a mechanism to terminate CICR (sorcin), and (4) a calcium pump to return released Ca2+ to the Ca2+ reservoir (Figure 3E). The first component detected in our dataset and others is the deafness gene Cacna1d, a voltage-gated calcium-release channel highly expressed in OHCs and implicated in syndromic deafness with bradycardia (Liu et al., 2014; Baig et al., 2011); the second component, channels to facilitate CICR, includes Ryr1, Ryr2, and Ryr3 (Beurg et al., 2005; Grant et al., 2006). Upon induction of CICR by Ryr channels, the role of sorcin may be to rapidly terminate CICR by blocking Ryr receptors in the presence of Ca2+, a pathway previously reported in cardiac myocytes (Farrell et al., 2003; Lokuta et al., 1997). Ca2+ ATPase pumps, which are known to localize to the OHC subsurface cisternae, may remove Ca2+ from the cytosol (Schulte, 1993). Accessory proteins such as oncomodulin (Ocm), which binds and buffers Ca2+, may contribute to this process.
Figure 3. Excitation-Contraction Pathway Component Expression in OHCs.

(A) Immunofluorescence localization of sorcin protein. The images are maximum projections of the described confocal z stack images.
(B) To visualize the organ of Corti in cross section, YZ and XZ planes of the white boxed regions were extracted from the confocal z stack. Maximum projections of the entire z stack and indicated YZ and XZ regions are shown.
(C) Cartoon of the OHC lateral membrane space decorated with prestin motor particles and an actin cytoskeletal lattice in close proximity to the membranous subsurface cisternae.
(D) The hypothesized excitation-contraction pathway components occupying this space in OHCs, with Ca2+ depicted as gray dots.
(E) Violin plots of p15 DCs (n = 37), IHCs (n = 33), and OHCs (n = 61) depicting expression levels of the genes shown in (B), y axis is set to a log scale and values represent Seurat scaled counts. p values were obtained using SCDE, an R package for single-cell differential expression analysis using a Bayesian approach. Two-tailed p values were adjusted to control for multiple testing. p values < 0.05 were considered significant. NS, non-significant.
Transcript Structure Analysis
The majority of deafness-associated genes are expressed in IHCs and OHCs, and yet their isoform structure in these cells has remained poorly defined. Single-cell RNA-seq allows us to overcome both cellular heterogeneity and low abundance to define gene product structure in auditory HCs. We analyzed transcript structure by aligning Illumina scRNA-Seq reads to the mm10 genome using the STAR aligner, visualizing alternative splicing, unannotated exons, and alternative transcription start site (TSS) and transcription end site (TES) positions with the Mixture of Isoforms (MISO) Sashimi Plot feature (Integrative Genome Viewer; Broad Institute) (Katz et al., 2010; Robinson et al., 2011; Thorvaldsdóttir et al., 2013). To validate this approach, in mouse Myo15 we detected (1) a TSS at mm10 chr11:60,480,418–60,480,621, (2) a 6 bp exon at position chr11:60,486,893–60,486,898, and (3) a splice acceptor site that results in the premature stop at position chr11:60,497,434–60,497,576 (Figures S7A–S7D), three features that have been reported by Rehman et al. (2016). In addition, we detected several unannotated features, including an unreported OHC-specific alternative splice acceptor site (chr11:60,483,616–60,483,806), the inclusion of which results in a frameshift and premature stop suggesting possible function as a regulatory element similar to the reported alternative splice acceptor site located at position chr11:60,497,434–60,497,576 (Figures S7D and S7E) (Rehman et al., 2016), and a splice site at position chr11:60,527,450–60,527,710 in the 3′UTR (Figure S7F).
We next sought unannotated features in genes implicated in Mendelian deafness and identified 20 highly conserved unannotated exons in 12 hearing-loss genes (Table 1; Figures 4 and 5). The average size of the identified coding and non-UTR containing exons is 55 bp, which places these exons in the smallest decile of murine exons (33% of mouse exons are <100 bp; 6.9% are <50 bp) (Sakharkar et al., 2005).
Table 1.
Unannotated Exons in 12 Genes Associated with Genetic Hearing Loss
| Gene | Exon | Previously Detected (Mouse EST) |
Conserved | In RefSeq | In Ensembl | Mouse (mm10) Position | Human (hg19) Position | ORF | Coding | TSS | UTR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cabp2 | exon 1B | no | yes | no | no | chr19:4,081,450–4,081,607 | chr11:67,292,851–67293045 | no | no | yes | 5′ UTR |
| Cacnald | exon 1B | no | yes | no | no | chr14:30,469,793–30,469,918 | chr3:53,393,487–53,393,623 | no | no | yes | 5′ UTR |
| Cacnald | exon 22B | no | yes | no | no | chr14:30,107,653–30,107,712 | chr3:53,774,446–53,774,505 | yes | yes | no | no |
| Cacnald | exon 9B | no | yes | no | no | chr14:30,137,042–30,137,125 | chr3:53,744,421–53,744,504 | yes | yes | no | no |
| Cacnald | exon 31B | no | yes | no | no | chr14:30,085,907–30,086,890 | chr3:53,799,478–53,800,954 | yes | yes | no | 3′ UTR |
| Coch | exon 3B | no | yes | no | no | chr12:51,594,118–51,594,298 | chr14:31,344,766–31344969 | no | no | yes | 5′ UTR |
| Eps8 | exon 4B | BG081059 | yes | no | no | chr6:137,530,542–137,530,592 | chr12:15,825,388–15,825,438 | yes | yes | no | no |
| Eps8 | exon 18B | no | yes | no | no | chr6:137,498,345–137,498,380 | chr12:15,792,360–15,792,395 | yes | yes | no | no |
| Eps8 | exon 18C | no | yes | no | no | chr6:137,493,548–137,493,571 | chr12:15,787,673–15,787,696 | yes | yes | no | no |
| Myh14 | exon 1C | Cj142596, BY016326, CJ069721, BY305319, BI647808 | yes | no | yes (mm10), no (hg19) |
chr7:44,669,243–44,669,392 | chr19:50,708,659–50,708,811 | yes | no | yes | 5′ UTR |
| Myo7a | exon 2B | no | yes | no | no | chr7:98,111,310–98,111,522 | chr11:76,849,258–76,849,463 | yes | yes | yes | 5′ UTR |
| Myo7a | exon 15B | no | yes | no | no | chr7:98,090,360–98,090,413 | chr11:76877917–76878106 | yes | yes | no | no |
| Otof | exon 6B | By752695, By231086 | yes | no | no | chr5:30,407,915–30,408,063 | chr2:26,727,461–26,727,733 | yes | no | yes | 5′ UTR |
| Ptprq | exon 35B | no | no | no | no | chr10:107,544,527–107,544,607 | chr12:81041403–81041480 | no | no | no | no |
| Ptprq | exon 35C | no | yes | no | no | chr10:107,550,522–107,550,566 | chr12:81034354–81034398 | yes | yes | no | no |
| Smpx | exon 1B | AI787904 | yes | no | yes (mm10), no (hg19) |
chrX:157,702,696–157,702,737 | chrX:21,772,641–21,772,685 | yes | no | yes | 5′ UTR |
| Smpx | exon 1C | no | yes | no | no | chrX:157,702,769–157,702,848 | chrX:21,772,530–21,772,609 | yes | yes | yes | 5′ UTR |
| Tectb | exon 1B | yes many | yes | yes (mm10), no (hg19) |
yes (mm10), no (hg19) |
chr19:55,180,726–55,180,816 | chr10:114,043,164–114,043,253 | no | no | yes | 5′ UTR |
| Triobp | exon 4B | no | yes | no | no | chr15:78,957,305–78,957,465 | chr22:38,105,438–38105593 | no | no | yes | 5′ UTR |
| Ush1c | exon 24B | no | yes | no | no | chr7:46,197,846–46,197,940 | chr11:17,519,066–17,519,165 | yes | yes | no | no |
Supporting evidence for four representative regions is included in Figures 4 and 5. Each region listed may be visualized using the transcript structure browser at morlscrnaseq.org.
Figure 4. Myo7a Exon 15B Splicing Analysis.

(A) Violin plot showing gene expression levels of Myo7a in p15 DCs (n = 37), IHCs (n = 33), and OHCs (n = 61).
(B) Sashimi plot of mouse Myo7a showing exon 15B. The tracks from top to bottom represent summed reads from p15 DCs (red), IHCs (green), and OHCs (blue). The bottom two tracks display RefSeq annotation and phyloP placental mammal conservation. Detection of Myo7a exon 15B was highly reproducible and present in 17 of 44 individual p15 OHCs and 13 of 31 individual p15 IHCs from 15 different mice. Expression of this exon was detected in 100% of OHC cells with reads mapping across this position, suggesting co-expression of isoforms with and without exon 15B.
(C) The orthologous region of human MYO7A.
(D) Pairwise sequence alignment between human and mouse amino acid sequences of exon 15B.
(E) Cartoon depiction of the nucleotide and amino acid sequences show that inclusion of exon 15B preserves the reading frame in both mouse and human sequence.
(F) Modeling of the actin binding domain of canonical Myo7a (left) and Myo7a containing exon 15B (right). Exon 15 is colored green, exon 15B is colored red, and exon 16 is colored blue. Unstructured loops involved in actin binding are labeled. Exon 15B (red) forms an unstructured loop domain, the form and position of which suggest a possible role in actin binding.
Figure 5. Representative Unannotated Exons in Hearing-Loss Genes.

(A) Unannotated exons 18B and 18C in Eps8 are highlighted in red between canonical exons 18 and 19. Exons 18B and 18C are expressed in both IHCs and OHCs but not in DCs. Both exons lie in highly conserved positions. Pairwise sequence alignment between the predicted mouse and human amino acid sequences shows 100% amino acid sequence identity in for both exons 18B and C. Inclusion of these exons in the canonical Eps8 sequence does not alter the reading frame or introduction of a stop codon.
(B) Unannotated exon 6B in Otof is the product of an alternative transcription start site. This exon contains a 5′UTR sequence without the presence of a Met start codon. Translation of mRNAs originating in exon 6B would contain an open reading frame starting at a Met codon in canonical exon 7.
(C) Ptprq exon 35B has not been previously annotated. We detected expression in all three cell types. The expression of Ptprq is relatively low, but inclusion of highly conserved exon 35B was observed in all reads mapping across this position from all cell types. Exon 35B exhibits high conservation among mammals and shares 100% amino acid conservation between mouse and human reference sequences. The inclusion of exon 35B maintains the open reading frame and is predicted to be protein coding (*the unannotated exon at this position is poorly conserved and has unappreciable homology between mouse and human). These datasets can be used to visualize and investigate any of the unannotated exons reported or to query splicing of any gene of interest at morlscrnaseq.org under the “Transcript Structure Browser” tab.
Full-Length Isoform Identification
To resolve full-length isoform structure, we adapted our library preparation for Nanopore long-read RNA-seq following a protocol developed by Byrne et al. (2017) and used the Illumina short-read sequence data as a confirmatory scaffold against which to map the long-read data. Unlike Illumina reads, which require 150–800 bp fragment sizes, the Nanopore MinION sequencer does not require a fragmented input library and can generate single reads that span the length of an mRNA transcript (Figures 6A and 6B). Twelve OHCs at the p15 time point were sequenced using individual multiplexing to run all samples on four MinION R9.4 flow cells while retaining single-cell resolution (Figure 6A). This choice was made because OHCs represent our largest and most robust Illumina dataset, which facilitated cross-platform comparison to validate long reads and quantify isoforms.
Figure 6. Single-Cell Isoform Quantification.

(A) Bar plot showing the total number of genes detected in each of the 12 OHC cells sequenced using the Nanopore MinION. All 12 OHCs were obtained from male mice at the p15 time point and were isolated from the apex of the cochlea.
(B) Tracks under the heading “Isoforms Detected” depict isoform consensus sequence alignments generated by Mandalorion and aligned to the mm10 genome. Each track shows the structure of the indicated isoform; only iso-form 1 is predicted to be protein coding. Note the unannotated exon (^), which is non-coding. Also shown are annotated isoforms from RefSeq and phyloP placental mammal basewise conservation from 60 species (*artifact caused by mapping of poly-A tail remnants to a downstream position, which have been trimmed for clarity).
(C) Ten of the 12 OHCs expressed full-length reads mapping to Cabp2. Iso-forms were defined and quantified for each cell and are shown as pie charts color-coded for each isoform. The expression level, reads per gene per 10,000 reads (RPG10K), of Cabp2 in each cell is indicated.
(D) The relative per cell expression level RPG10K for each isoform across all cells.
(E) The number of cells expressing each isoform configuration.
Long-read RNA-seq data revealed striking isoform heterogeneity. For example, we detected 445 reads mapping to the deafness-associated gene Cabp2. Fourteen distinct isoforms were identified by Mandalorion, a software package for isoform quantification from Nanopore reads, but this number dropped to five when highly similar isoform configurations were combined (Figure 6B). By quantifying the abundance of each isoform, we identified the predominant isoform configuration in each individual cell and in the broader OHC group (Figures 6C–6E). The most frequently observed and abundantly expressed configuration is isoform 1. We consulted publicly available murine annotation databases (RefSeq, GenBank, GENCODE, and UCSC Genes) and datasets (mRNAs from GenBank and the UCSC Genome Browser) but found no annotation of this isoform. In addition to its absence in mouse, we were unable to identify a transcript with structure orthologous to isoform 1 in the RefSeq, GenBank, GENCODE, and UCSC human databases.
DISCUSSION
In this study, we used single-cell RNA-seq to analyze rare and difficult-to-isolate cell types from the mature organ of Corti and in so doing identify cell type-defining genes and challenge many annotation-derived assumptions about the structure of gene products expressed in auditory HCs. The technique we implemented is a simple micropipette-based single-cell isolation approach that enables precise selection of auditory HCs and supporting cells that is not achievable using microfluidics and droplet-based systems (Figure 1). The approach facilitates isolation of ultra-low-abundance cell types that can be visually identified and imaged prior to reverse transcription (Figures 1C–1G). Single-cell samples were meticulously collected and serve as high-quality biological replicates from which cell type-specific transcriptome profiles were generated (Figures 2A–2E). The resultant data afford a global view of gene expression and the ability to characterize the spectrum of isoforms in mature cochlear HCs.
To assess variability in gene expression across IHC, OHC, and DC cell types, we compared transcriptomes in several ways. Distinct differences in expression profiles were obvious on a heatmap of the top 100 cluster-defining genes in each cell type and provide refined insight in HC function (Figure 1G). For example, the top two genes that define the OHC transcriptome are associated with calcium regulation. The first, Ocm, encodes oncomodulin, a calcium-binding protein, and the second, Sri, encodes sorcin, a regulator of calcium-based excitation-contraction (Figure 2F). The third most differentially expressed gene in OHCs is the well-known motor protein prestin (Slc26a5).
Differential expression of Sri is notable because this gene has not been investigated as a marker of OHCs. Its encoded protein, sorcin, plays a prominent role in excitation-contraction coupling in cardiac tissue by increasing the speed of Ryr-mediated Ca2+ release events and reducing cytosolic free Ca2+ to facilitate the creation of rapid Ca2+ sparks. Like cardiac myocytes, which express Ryr channels in the endoplasmic reticulum (ER)-derived sarcoplasmic reticulum, OHCs express Ryr channels in a comparable pattern in the ER-derived subsur-face cisternae (Grant et al., 2006). In cardiac myocytes, it is the proximity of the Ca2+-rich sarcoplasmic reticulum to the myosin motor proteins that confers rapid and precise control of muscle contraction (Endo, 1977; Farrell et al., 2003). Similarly, the proximity of the Ca2+-rich subsurface cisternae to the prestin motors studding the lateral membrane of OHCs raises the possibility that Ca2+ may play a similar role in regulating OHC motility (Figure 3). Consistent with this hypothesis, we detected expression of the other requisite components for calcium-mediated excitation-contraction in the OHC transcriptome (Figure 3E) and found sorcin to be robustly and specifically localized throughout the body and lateral membrane of OHCs (Figures 3A and 3B). Its presence in OHCs and absence in IHCs suggests a unique requirement for tight regulation of calcium. Although we hypothesize that the role of sorcin may be related to somatomotility, other roles are also possible, making this finding an exciting opportunity for further investigation and validation.
We anticipated that deafness-associated genes may express unannotated transcripts and isoforms in auditory HCs, consistent with a specialized role in audition. To test this hypothesis, we completed a focused analysis of 12 deafness-associated genes and detected 20 unannotated, highly conserved exons (Table 1; Figures 4 and 5). The average length of these exons was 55 bp, which places them by size in the smallest 10% of murine exons (Sakharkar et al., 2005). Their identification may have important implications for the genetic diagnosis of human deafness, as this finding suggests that current comprehensive genetic tests are not complete. These exons may also be relevant to studies focused on gene therapy, as targeting these regions with knockdown-based therapeutics may confer cell type spec-ificity, thereby limiting off-target effects. Conversely, if gene replacement strategies are used, it is possible that the omission of required exons or the introduction of foreign isoforms may retard therapeutic efficacy.
Identification of unannotated exons in rare cochlear cell types is not surprising givenboth the high level of specialization throughout the cochlea and the lack of representation among tissues commonly included in annotation databases. Alternative splicing, another driver of transcript variability, was widespread in our data-set, as detected by Illuminashort-read sequencing. To resolve and quantify full-length isoform structures, we generated long-read RNA sequence data using Nanopore MinION (Figure 6). The large number of unreported spliceoforms we observed in deafness-associated genes reflects an unappreciated isoform heterogeneity in rare cochlear cell types. The precise clinical relevance of this diversity remains unclear and elucidating the nuanced roles of various isoforms of different genes will be challenging. Because pathogenicity prediction software is based on current annotations and does not capture this diversity, cell type-specific annotation databases for all cochlear cell types are needed.
There remain important challenges in quantifying isoform diversity. First, we found that SmartSeq2-based reverse transcription can generate truncated transcripts because of a mis-priming effect caused by locked nucleic acid (LNA)-modified template switching oligo (TSO) and oligo-dT30VN primers. Sequence-specific mis-priming occurs at ATGG motifs complementary to the TSO primer at the LNA-modified end and to poly A repeat sequences complementary to the oligo-dT primer. Second, long-read sequencing has a high nucleotide error rate, which we measured at 13.5% in aligned reads generated using a MinION sequencer and 1D read chemistry. These errors complicate alignment, isoform definition, and quantification and make complementary Illumina sequencing essential. Improved accuracy through platform advancement or the implementation of read correction may decrease the MinION sequencer error rate. Third, there are few bioinformatics tools for long-read RNA-seq isoform definition and quantification, which requires the adoption of imperfect solutions, extensive troubleshooting, and the construction of custom software.
In summary, we show that by leveraging single-cell RNA-seq for the analysis of three cell types from the mature organ of Corti, a multidimensional overview of IHC, OHC, and DC transcriptomes can be generated. These data provide insight into the cell type-defining genes and pathways and highlight the complex array of isoforms found in genes associated with genetic hearing loss. The technique offers the promise of advancing our knowledge of the diverse assortment of highly specialized cells in the cochlea and refining our understanding of the biology of hearing and deafness.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Smith (Richard-smith@uiowa.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All cells reported in this study were obtained from Mus musculus (C3HeB/FeJ). Animals were housed at the University of Iowa. The University of Iowa Institutional Animal Care and Use Committee (protocol 8041338) approved all relevant procedures. Single-cell sample information including serial number, cell type, mouse number, gender, age, tonotopic position, date of birth, date of death, time of death and time of cell lysis can be found in our mendeley archive doi https://doi.org/10.17632/5frjd5rv27.1.
METHOD DETAILS
Estimation of Cochlear Cell Abundance
To estimate cochlear cell abundance, temporal bones were harvested from euthanized wild-type C3HeB/FeJ mice (p15) and fixed in 4% paraformaldehyde at RT for 2 h. After decalcification in 120mM EDTA for 2 days and cryoprotection in 15% and 30% sucrose for embedding in OCT solution, midmodiolar cryosections (14 μm) were prepared for immunohistochemistry and mounted in ProLong® Diamond antifade mountant with DAPI (Thermo Fisher Scientific). A Leica TCS SP8 confocal microscope (Leica Microsystems Inc.) was used to identify nuclei, which were counted to estimate the total number of cells in the murine membranous labyrinth.
Immunohistochemistry and Fluorescence Microscopy
For immunofluorescence localization of sorcin, cochlear tissues were permeabilized with 0.3% Triton X-100 and blocked with 5% normal goat serum prior to overnight incubation with one of two different anti-sorcin primary rabbit antibodies at a 1:100 concentration in 1xDPBS (Sorcin(ab71983), Abcam; Sorcin(PA5–64975), Invitrogen). Filamentous actin was labeled with phalloidin conjugated to Alexa Fluor 488 (Invitrogen, Molecular Probes) at a 1:500 concentration for 30 min; Alexa Fluor 568 labeled anti-rabbit IgG was used as a secondary antibody. Mounting was performed using ProLong Diamond mounting medium with or without DAPI (Life Technologies). Z stack images of cochlear whole mounts and cross sections were collected at 10x-40x on a Leica SP8 confocal microscope (Leica Microsystems). Digital zoom up to 5 X was applied to assess subcellular localization. Maximum intensity and average intensity projections were generated from z stack images using Fiji.
Cochlear Dissection and Single Cell Isolation
Cochlear tissue was harvested from p15, p30, p70 and p228 male and female C3-Heb/FeJ mice (Mus musculus) following NIH guidelines for care/use of laboratory animals as approved by the Institutional Animal Care and Use Committee at the University of Iowa (Protocol #0608169). Our single-cell isolation protocol closely follows the procedure described in Liu et al. with several modifications to facilitate downstream single-cell RNA-seq (Liu et al., 2014). In brief, murine temporal bones were removed after euthanasia via CO2 and placed into a small Petri dish containing ice-cold 1xDPBS. To access the cochlear membranous labyrinth, the apical portion of the bony labyrinth was visualized under a dissecting microscope model M165FC (Leica Microsystems). Bone fragments were chipped off with #5 forceps (Fine Science Tools) and the membranous labyrinth was gently removed, placed into a 1.5mL tube containing collagenase (Sigma Aldrich) + 1xDPBS at a working concentration of 3mg/500uL and after 5min digestion at RT dissociated by gentle pipetting. The disrupted tissue was transferred to a glass microscope slide (Superfrost Plus 25 × 75 × 1.0mm; Fisher Scientific) and placed on an inverted microscope (Model DMI3000B; Leica Microsystems) equipped with 20x and 40x DIC objectives and two 3D micromanipulators (Model MN-153; Narishige) each driving a pulled glass micropipette attached to a nitrogen gas powered Pico-Injector (Model PLI-100; Harvard Apparatus), which was used to control aspiration pressure. A separate wash slide of 1mL 1xDPBS was immediately adjacent.
Candidate cells were identified by scanning the field of view. Cells of interest were aspirated into the first glass micropipette in a slow and controlled manner (Figures 1C and 1D). The cell was washed to remove extracellular debris by expelling it into the fresh 1xDPBS on the adjacent wash slide and reaspirated using a second clean glass micropipette. The washed cell was expelled into a 0.2mL tube containing 4ul of RNase inhibitor containing lysis buffer. Lysed samples were stored on ice until the ongoing batch of isolations was completed.
Reverse Transcription and Library Preparation
Reverse transcription and library preparation were performed immediately following single-cell isolations using the SmartSeq2 protocol as described by Pirelli et al., with several minor differences (Picelli et al., 2014). For reverse transcription, Superscript III Reverse Transcriptase (RT) was used rather than Superscript II RT (Invitrogen) and the number of PCR preamplification cycles was increased to 22. The number of amplification cycles for adaptor-ligated fragments was increased to 14. Tagmentation and amplification of adaptor-ligated fragments were carried out using half-volume reactions. For Illumina sequencing, barcoded single-cell libraries were pooled into batches containing between 30 and 60 cells. For Nanopore sequencing, four barcoded single-cell libraries were pooled to run on a single MinION R9.4 flowcell.
Sequencing
Data reported in this paper were obtained using either Illumina or Nanopore sequencing (as designated). For Illumina sequencing, pooled libraries were sequenced on a single lane of an Illumina HiSeq 2500 or 4000 (as designated) using 150bp paired-end read chemistry. cDNA libraries prepared with SmartSeq2 were used as the input for Nanopore Library preparation and 1D MinION sequencing, as described in Byrne et al., 2017 (Byrne et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Illumina Sequencing and Data Analysis
For transcript abundance quantification and differential expression analysis, Illumina reads were aligned to the mm10 transcriptome (GRCm38) available from Ensembl. Alignment for gene expression abundance and quantification was done using Kallisto pseudoalignment. Estimated counts values were generated for each cell by Kallisto. Estimated counts values were aggregated into a gene expression matrix using a custom python script and were input into various downstream R packages (SCDE, Seurat and Sleuth) for analysis and visualization (Kharchenko et al., 2014; Satija et al., 2015; Pimentel et al., 2017).
Prior to clustering, quality control filters were applied to the gene expression matrix eliminating cells expressing fewer than 2000 genes at a threshold of > 0 counts. PCA and TSNE clustering were used to refine the dataset. Seven cells were eliminated due to discordant expression levels of the top 100 most highly expressed genes across all cell types and three cells were eliminated due to discordant cluster assignments.
Differential expression analysis was performed using a Bayesian approach to single-cell RNA-Seq computed through the SCDE package for R (Figures 3E and S5). Two-tailed P values were adjusted to control for multiple testing. P values < 0.05 were considered significant.
Splicing analysis was completed with a different alignment strategy using STAR to align reads to the mm10 genome (GRCm38) available from Ensembl (Dobin et al., 2013). We used Samtools to sort and index .bam files for each sample (Li et al., 2009). Opening indexed .bam files using IGV enabled us to visualize splicing junctions using the Sashimi plot tool. To identify unannotated exons and isoforms and to visualize differential expression and alternative splicing across cell types (OHCs, IHCs and DCs), we merged the .bam files from each cell type at the p15 time point. Sorted, indexed and merged .bam files were viewed using IGV and Sashimi Plot (Katz et al., 2010).
Clustering and cluster analysis was performed using Seurat. Clustering was performed using Seurat FindClusters, which uses the Shared Nearest Neighbor (SNN) modularity optimization-based clustering algorithm. Cluster defining genes were extracted using Seurat FindMarkers, which employs a Area Under the Receiver Operating Characteristic curve (AUROC) classifier.
Nanopore Sequencing and Data Analysis
Individually processed single cell samples were multiplexed and pooled on a single MinION flow cell, preparing 12 OHCs this way. 1D reads were generated using three Nanopore R9.5 flowcells run on a MinION sequencer and aligned to the mm10 (GRCm38) genome using the Mandalorion pipeline described in Byrne et al., 2017 (Byrne et al., 2017). Alignments were visualized using IGV. Expression abundance at the gene and isoform levels was quantified with the Mandalorion pipeline.
Numbers of Cells and Mice Used
Cells obtained at experimental time point p15 are the focus of Figures 1, 2, 3, 4, and 5. 132 total p15 cells were used. When categorized by cell type we obtained the following numbers of single-cell samples IHC (n = 42), OHC (n = 127) and DC (n = 39). These cells originated from a total of 70 c3HeB/FeJ mice. Detailed metadata on each cell used in this study are available in our archived metadata described in the Data and Software Availibility section.
DATA AND SOFTWARE AVAILABILITY
Data availability
The accession number for the sequence data reported in this paper is Gene Expression Omnibus (GEO): GSE114157. The accession number for the single-cell counts matrix and experimental metadata used in this paper is Mendeley: https://doi.org/10.17632/5frjd5rv27.1.
Software availability
Software resources utilized for data analysis are listed along with their source and version information in the Key Resources Table. Custom scripts were written to streamline analysis and provide consistency between single-cell samples. These scripts are computing environment dependent thus requiring significant modification and setup before implementation by other groups. As such they will be distributed upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Sorcin_1 | Abcam | Cat# Ab71983; RRID:AB_1270905 |
| Rabbit anti-Sorcin_2 | Invitrogen | Cat# PA5–64975; RRID:AB_2662771 |
| Goat anti-Rabbit Alexa Fluor 568 | Invitrogen, Molecular Probes | Cat# A-11011; RRID:AB_143157 |
| Phalloidin conjugated Alexa 488 | Invitrogen, Molecular Probes | Cat# A-12379; RRID:AB_2315147 |
| Biological Samples | ||
| 132 Biological Samples | This Study / GEO | GSE114157 |
| Counts Matrix | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Experimental Metadata | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ProLong Diamond Dapi | Invitrogen | Cat#P36962 |
| Deposited Data | ||
| 507 Raw and Processed scRNA-Seq Data Files | This Study / GEO | GSE114157 |
| Counts Matrix and Metadata | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus (C3HeB/FeJ) | The Jackson Laboratory | 000658 |
| Oligonucleotides | ||
| 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′ | Exiqon | TSO (Smartseq2) |
| 5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ | IDT | Oligo-dt30VN (Smartseq2) |
| 5′-AAGCAGTGGTATCAACGCAGAGT-3′ | IDT | ISPCR oligo (Smartseq2) |
| Software and Algorithms | ||
| Fiji (Fiji Is Just) ImageJ | https://imagej.net | Fiji-macosx |
| R v3.3.2 | www.r-project.org | R |
| R Studio v1.0.136 | www.rstudio.com | N/A |
| Mandalorion v0.2 | Github | Mandalorion v0.2 |
| Samtools v1.3.1 | http://samtools.sourceforge.net/ | N/A |
| STAR v020201 | https://github.com/alexdobin/STAR | STAR |
| Kallisto v0.43.0 | https://github.com/pachterlab/ | Kallisto |
| Seurat v1.4.0.8 | https://satijalab.org/seurat/ | Seurat |
| Other | ||
| Dissecting Microscope | Leica Microsystems | M165FC |
| Inverted Microscope | Leica Microsystems | DMI3000B |
| Micromanipulators | Narishige | MN-153 |
| Nitrogen Pico-Injector | Harvard Apparatus | PLI-100 |
| Nanopore MinION | Oxford Nanopore | MinION |
ADDITIONAL RESOURCES
We have created a interactive website enabling users to query gene expression abundance and transcript structure of any gene from the single-cell data generated in this study: morlscrnaseq.org
Supplementary Material
Highlights.
Single-cell RNA-seq identifies inner and outer hair cell defining genes by AUC-ROC
Sorcin, a key component for cardiac excitation-contraction, is a top marker of OHCs
Analysis of deafness-associated genes identifies heretofore unrecognized exons
Nanopore long-read RNA-seq reveals splicing diversity and isoform abundance
ACKNOWLEDGMENTS
We would like to thank Einat Snir and Jennifer Bair from the Genomics Division of the Iowa Institute of Human Genetics for their guidance in optimizing our library preparation for Illumina sequencing, Ann Black-Ziegelbein and Alexis Divincenzo for their assistance in bioinformatics troubleshooting, and Christopher Vollmers for his comments and assistance in adapting Mandalorion for 1D read isoform definition and quantification. This research was supported in part by NIDCD RO1 grants DC003544, DC002842, and DC012049 to R.J.H.S.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.053.
DECLARATION OF INTERESTS
R.J.H.S. directs the Molecular Otolaryngology and Renal Research Laboratories, which offer genetic testing for hearing loss, and is a co-founder of Akouos, a precision genetic medicine company developing gene therapies that restore and preserve hearing.
WEB RESOURCES
MORL scRNA-Seq Browser, morlscrnaseq.org
REFERENCES
- Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nürnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N, et al. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci 14, 77–84. [DOI] [PubMed] [Google Scholar]
- Beurg M, Hafidi A, Skinner LJ, Ruel J, Nouvian R, Henaff M, Puel JL, Aran JM, and Dulon D (2005). Ryanodine receptors and BK channels act as a presynaptic depressor of neurotransmission in cochlear inner hair cells. Eur. J. Neurosci 22, 1109–1119. [DOI] [PubMed] [Google Scholar]
- Burns JC, Kelly MC, Hoa M, Morell RJ, and Kelley MW (2015). Single-cell RNA-Seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nat. Commun 6, 8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, DuBois RM, Forsberg EC, Akeson M, and Vollmers C (2017). Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun 8, 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Jen HI, Kang H, Klisch TJ, Zoghbi HY, and Groves AK (2015). Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. J. Neurosci 35, 5870–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gong TW, Stöver T, Lomax MI, and Altschuler RA (2002). Gene expression profiles of the rat cochlea, cochlear nucleus, and inferior colliculus. J. Assoc. Res. Otolaryngol 3, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Sloan KR, Kalina RE, and Hendrickson AE (1990). Human photoreceptor topography. J. Comp. Neurol 292, 497–523. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G, and Frankenreiter M (1977). Quantitative-analysis of cochlear structures in house mouse in relation to mechanisms of acoustical information-processing. J. Comp. Physiol 122, 65–85. [Google Scholar]
- Elkon R, Milon B, Morrison L, Shah M, Vijayakumar S, Racherla M, Leitch CC, Silipino L, Hadi S, Weiss-Gayet M, et al. (2015). RFX transcription factors are essential for hearing in mice. Nat. Commun 6, 8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M (1977). Calcium release from the sarcoplasmic reticulum. Physiol. Rev 57, 71–108. [DOI] [PubMed] [Google Scholar]
- Farrell EF, Antaramian A, Rueda A, Gómez AM, and Valdivia HH (2003). Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J. Biol. Chem 278, 34660–34666. [DOI] [PubMed] [Google Scholar]
- Gambardella J, Trimarco B, Iaccarino G, and Santulli G (2018). New insights in cardiac calcium handling and excitation-contraction coupling. Adv. Exp. Med. Biol 1067, 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant L, Slapnick S, Kennedy H, and Hackney C (2006). Ryanodine receptor localisation in the mammalian cochlea: an ultrastructural study. Hear. Res 219, 101–109. [DOI] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, and Burge CB (2010). Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Silberstein L, and Scadden DT (2014). Bayesian approach to single-cell differential expression analysis. Nat. Methods 11, 740–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu H, Giffen KP, Chen L, Beisel KW, and He DZZ (2018). Transcriptomes of cochlear inner and outer hair cells from adult mice. Sci. Data 5, 180199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Pecka JL, Zhang Q, Soukup GA, Beisel KW, and He DZ (2014). Characterization of transcriptomes of cochlear inner and outer hair cells. J. Neurosci 34, 11085–11095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuta AJ, Meyers MB, Sander PR, Fishman GI, and Valdivia HH (1997). Modulation of cardiac ryanodine receptors by sorcin. J. Biol. Chem 272, 25333–25338. [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Pujol R, Nouvian R, and Lenoir M (2016). Hair cells: overview, http://www.cochlea.eu/en/hair-cells.
- Rehman AU, Bird JE, Faridi R, Shahzad M, Shah S, Lee K, Khan SN, Imtiaz A, Ahmed ZM, Riazuddin S, et al. (2016). Mutational spectrum of MYO15A and the molecular mechanisms of DFNB3 human deafness. Hum. Mutat 37, 991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar MK, Perumal BS, Sakharkar KR, and Kangueane P (2005). An analysis on gene architecture in human and mouse genomes. In Silico Biol. (Gedrukt) 5, 347–365. [PubMed] [Google Scholar]
- Sarafoleanu C, Mella C, Georgescu M, and Perederco C (2009). The importance of the olfactory sense in the human behavior and evolution. J. Med. Life 2, 196–198. [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer DI, Shen J, Corey DP, and Chen ZY (2015). Gene expression by mouse inner ear hair cells during development. J. Neurosci 35, 6366–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmang T, and Maconochie M (2016). Gene expression profiling of the inner ear. J. Anat 228, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte BA (1993). Immunohistochemical localization of intracellular Ca-ATPase in outer hair cells, neurons and fibrocytes in the adult and developing inner ear. Hear. Res 65, 262–273. [DOI] [PubMed] [Google Scholar]
- Simmons DD, Tong B, Schrader AD, and Hornak AJ (2010). Oncomodulin identifies different hair cell types in the mammalian inner ear. J. Comp. Neurol 518, 3785–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong B, Hornak AJ, Maison SF, Ohlemiller KK, Liberman MC, and Simmons DD (2016). Oncomodulin, an EF-hand Ca2+ buffer, is critical for maintaining cochlear function in mice. J. Neurosci 36, 1631–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data availability
The accession number for the sequence data reported in this paper is Gene Expression Omnibus (GEO): GSE114157. The accession number for the single-cell counts matrix and experimental metadata used in this paper is Mendeley: https://doi.org/10.17632/5frjd5rv27.1.
Software availability
Software resources utilized for data analysis are listed along with their source and version information in the Key Resources Table. Custom scripts were written to streamline analysis and provide consistency between single-cell samples. These scripts are computing environment dependent thus requiring significant modification and setup before implementation by other groups. As such they will be distributed upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Sorcin_1 | Abcam | Cat# Ab71983; RRID:AB_1270905 |
| Rabbit anti-Sorcin_2 | Invitrogen | Cat# PA5–64975; RRID:AB_2662771 |
| Goat anti-Rabbit Alexa Fluor 568 | Invitrogen, Molecular Probes | Cat# A-11011; RRID:AB_143157 |
| Phalloidin conjugated Alexa 488 | Invitrogen, Molecular Probes | Cat# A-12379; RRID:AB_2315147 |
| Biological Samples | ||
| 132 Biological Samples | This Study / GEO | GSE114157 |
| Counts Matrix | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Experimental Metadata | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ProLong Diamond Dapi | Invitrogen | Cat#P36962 |
| Deposited Data | ||
| 507 Raw and Processed scRNA-Seq Data Files | This Study / GEO | GSE114157 |
| Counts Matrix and Metadata | This Study / Mendeley | https://doi.org/10.17632/5frjd5rv27.1 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus (C3HeB/FeJ) | The Jackson Laboratory | 000658 |
| Oligonucleotides | ||
| 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′ | Exiqon | TSO (Smartseq2) |
| 5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ | IDT | Oligo-dt30VN (Smartseq2) |
| 5′-AAGCAGTGGTATCAACGCAGAGT-3′ | IDT | ISPCR oligo (Smartseq2) |
| Software and Algorithms | ||
| Fiji (Fiji Is Just) ImageJ | https://imagej.net | Fiji-macosx |
| R v3.3.2 | www.r-project.org | R |
| R Studio v1.0.136 | www.rstudio.com | N/A |
| Mandalorion v0.2 | Github | Mandalorion v0.2 |
| Samtools v1.3.1 | http://samtools.sourceforge.net/ | N/A |
| STAR v020201 | https://github.com/alexdobin/STAR | STAR |
| Kallisto v0.43.0 | https://github.com/pachterlab/ | Kallisto |
| Seurat v1.4.0.8 | https://satijalab.org/seurat/ | Seurat |
| Other | ||
| Dissecting Microscope | Leica Microsystems | M165FC |
| Inverted Microscope | Leica Microsystems | DMI3000B |
| Micromanipulators | Narishige | MN-153 |
| Nitrogen Pico-Injector | Harvard Apparatus | PLI-100 |
| Nanopore MinION | Oxford Nanopore | MinION |
The accession number for the sequence data reported in this paper is Gene Expression Omnibus (GEO): GSE114157. The accession number for the single-cell counts matrix and experimental metadata used in this paper is Mendeley: https://doi.org/10.17632/5frjd5rv27.1.