

SUMMARY
We explored the clinical and pathological impact of epidermal growth factor receptor (EGFR) extracellular domain missense mutations. Retrospective assessment of 260 de novo glioblastoma patients revealed a significant reduction in overall survival of patients having tumors with EGFR mutations at alanine 289 (EGFRA289D/T/V). Quantitative multi-parametric magnetic resonance imaging analyses indicated increased tumor invasion for EGFRA289D/T/V mutants, corroborated in mice bearing intracranial tumors expressing EGFRA289V and dependent on ERK-mediated expression of matrix metalloproteinase-1. EGFRA289V tumor growth was attenuated with an antibody against a cryptic epitope, based on in silico simulation. The findings of this study indicate a highly invasive phenotype associated with the EGFRA289V mutation in glioblastoma, postulating EGFRA289V as a molecular marker for responsiveness to therapy with EGFR-targeting antibodies.
In Brief
Binder et al. show that glioblastoma (GBM) expressing EGFR A289 mutants exhibit invasive features and are associated with shorter survival in patients and mice. GBM cells expressing EGFRA289V increase ERK-dependent MMP1 expression but are sensitive to an EGFR monoclonal antibody being clinically developed.
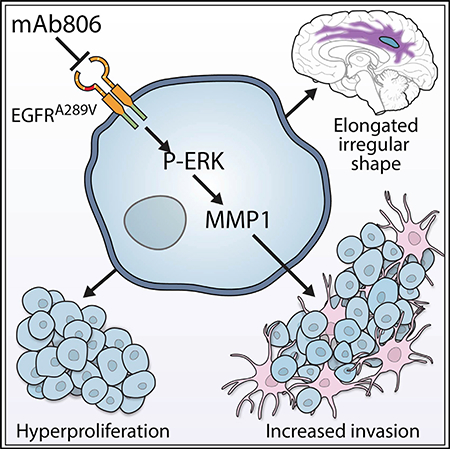
Graphical Abstract
INTRODUCTION
Glioblastoma (GBM) is the most common adult, primary, malignant brain tumor with an overall incidence rate of 3.2 per 100,000 in the US (Ostrom et al., 2015). With a median overall survival (OS) of 14.6 months following standard-of-care (SOC), patients diagnosed with GBM have a strikingly poor prognosis (Stupp et al., 2005). Much insight has been gleaned following studies describing the somatic genomic alterations in GBM, with the intended goal of defining core biological pathways to facilitate the discovery of actionable targets for diagnostic and therapeutic purposes (Brennan et al., 2013; Parsons et al., 2008). The 2016 CNS tumor classification of the World Health Organization incorporated molecular parameters into traditionally microscopy-based histological classification (Louis et al., 2016). However, regardless of classification, GBM remains a poorly margined, diffusely infiltrating necrotic mass that is difficult to treat, in part due to its highly invasive phenotype.
Intra-tumoral heterogeneity is a key factor in the poor therapeutic success for GBM. A seminal study found that 57% of GBM specimens contain a mutation, rearrangement, splicing alteration, and/or amplification of the epidermal growth factor receptor (EGFR). While the most common EGFR variant is a deletion from exons 2–7, EGFRvIII, extracellular domain (ECD) missense mutations comprise 10%–15% of transcripts and often co-occur with focal EGFR amplification (Brennan et al., 2013). The contribution of cellular crosstalk between EGFR variants to GBM pathogenicity sheds light on the importance of dissecting the GBM tumor into its single components, rather than focusing on individual mutations (Inda et al., 2010; Zanca et al., 2017). While the transforming capacity of several ECD EGFR missense mutants has been described, sufficient analysis of their downstream signaling pathways, their phenotypic impact on the tumor, clinical impact, and potential to specifically target these mutants has yet to be elucidated (Lee et al., 2006).
Multimodal MRI depicts the tumor core as consisting of enhancing (ET), non-enhancing, and necrotic sub-regions, distinct from the peritumoral edematous/invaded region (ED), which in combination with the tumor core describe the complete tumor extent (CTE) (Bakas et al., 2016). Boosted Glioma Image Segmentation and Registration (GLISTRboost) is a computer-aided method, utilizing machine-learning algorithms to accurately partition gliomas into its various distinct sub-regions in multimodal MRI scans (Bakas et al., 2017b). This partitioning enables quantitative feature analysis of precise sub-regions from which the clinician may gain valuable insight into the rational selection of targeted agents.
In this study, we examined if EGFR ECD missense mutations had distinguishable clinical effects on GBM patients and investigated the impact of specific targeting of these mutations.
RESULTS
Demographics of the GBM Patient Population from the University of Pennsylvania Resemble the Cancer Genome Atlas Data
To investigate the relationship of EGFR ECD missense mutations with GBM patient OS, patient records from the University of Pennsylvania (UPenn) from 2013 to 2016 demonstrating a confirmed diagnosis of GBM were included for analysis. Of 411 GBM cases, 260 were IDH1 wild-type (WT), de novo GBMs with accompanying next-generation sequencing (NGS) data from the UPenn Center for Personalized Diagnostics. Male to female ratio was 1.5:1 with a median age of 61 at the time of diagnosis (Table S1). The gene encoding the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) was methylated in 49% of cases. EGFR amplification was identified in 38% of cases and EGFRvIII was found in 25% of cases. The most common missense mutations were ECD mutations A289D/T/V, R108G/K, and G598V, found in 6%, 3%, and 2% of cases, respectively (Figure 1A). Comparison of the frequency and location of EGFR missense mutations in the UPenn cohort with data from the Cancer Genome Atlas (TCGA) demonstrated significant overlap (Figure 1B). Analysis of these missense mutations revealed no significant association with gender, age, or EGFRvIII status. However, each was found to co-occur with EGFR amplification (Table 1), while R108G/K was found more frequently in patients with MGMT methylation. The majority of patients received SOC, including surgical resection, concurrent radiotherapy and temozolomide (TMZ), and maintenance TMZ. There were no treatment differences in the missense mutation cohorts.
Figure 1. EGFRA289D/T/V Missense Mutations Are Associated with Inferior Survival in GBM.

(A and B) 2D representation of EGFR protein with functional domains indicated by colored segments and summary of missense mutations identified in the UPenn cohort (n = 260) (A) and the TCGA cohort (n = 591) (B). The location of mutated amino acids is indicated by a bar with a green circle. The height of the bar shows the number of patients in each cohort with the specific mutation.
(C–F) Kaplan-Meier (KM) survival curves for the UPenn cohort, comparing EGFRA289D/T/V with EGFRA289 (C), EGFRR108G/K to EGFRR108 (D), EGFRG598V to EGFRG598 (E), and EGFR amplified to non-amplified (F).
See also Figure S1 and Table S1.
Table 1.
Demographics of EGFR Extracellular Domain Missense Mutations
| Mutational Status at ECD | Fisher’s Exact Test | ||||
|---|---|---|---|---|---|
| Wild-Type | Mutated | ||||
| Gender | A289D/T/V | female | 95(92%) | 8 (8%) | 0.286 |
| male | 150 (96%) | 7 (4%) | |||
| R108G/K | female | 98 (95%) | 5 (5%) | 0.325 | |
| male | 153 (97%) | 4 (3%) | |||
| G598V | female | 102 (99%) | 1 (1%) | 0.408 | |
| male | 152 (97%) | 5 (3%) | |||
| Age (years) | A289D/T/V | 0–29 | 2 (100%) | 0 (0%) | 0.607 |
| 30–49 | 26 (100%) | 0 (0%) | |||
| 50–69 | 147(94%) | 10(6%) | |||
| 70+ | 70 (93%) | 5 (7%) | |||
| R108G/K | 0–29 | 2 (100%) | 0 (0%) | 0.795 | |
| 30–49 | 26 (100%) | 0 (0%) | |||
| 50–69 | 151 (96%) | 6 (4%) | |||
| 70+ | 72 (96%) | 3 (4%) | |||
| G598V | 0–29 | 2 (100%) | 0 (0%) | 0.695 | |
| 30–49 | 25 (96%) | 1 (4%) | |||
| 50–69 | 153 (97%) | 4 (3%) | |||
| 70+ | 74 (99%) | 1 (1%) | |||
| MGMT methylation status | A289D/T/V | unmethylated | 118 (97%) | 4 (3%) | 0.175 |
| methylated | 81 (93%) | 6 (7%) | |||
| R108G/K | unmethylated | 120(98%) | 2 (2%) | 0.036 | |
| methylated | 80(92%) | 7 (8%) | |||
| G598V | unmethylated | 118 (97%) | 4 (3%) | 0.404 | |
| methylated | 86 (99%) | 1 (%) | |||
| EGFR amplification | A289D/T/V | unamplified | 161 (99%) | 1 (1%) | <0.001 |
| amplified | 84 (86%) | 14(14%) | |||
| R108G/K | unamplified | 160 (99%) | 2 (1%) | 0.029 | |
| amplified | 91 (93%) | 7 (7%) | |||
| G598V | unamplified | 161 (99%) | 1 (1%) | 0.030 | |
| amplified | 93 (95%) | 5 (5%) | |||
| EGFRvIII status | A289D/T/V | negative | 144 (94%) | 10(6%) | 0.932 |
| positive | 48 (96%) | 2 (4%) | |||
| R108G/K | negative | 150 (97%) | 4 (3%) | 0.103 | |
| positive | 46(92%) | 4 (8%) | |||
| G598V | negative | 149 (97%) | 5 (3%) | 1.000 | |
| positive | 49 (98%) | 1 (1%) | |||
| Received standard-of-care therapy | A289D/T/V | yes | 158 (95%) | 9 (5%) | 0.582 |
| no | 78 (93%) | 6 (7%) | |||
| R108G/K | yes | 161 (96%) | 6 (4%) | 1.000 | |
| no | 81 (96%) | 3 (4%) | |||
| G598V | yes | 163 (98%) | 4 (2%) | 1.000 | |
| no | 82 (98%) | 2 (2%) | |||
EGFRA289D/T/V Missense Mutations Confer a Negative Survival Effect in GBM Patients
Median OS in the complete cohort was 15 months, with a 2-year survival rate of 21%, and a 5-year survival rate of 10% (Figure S1A). The median OS, 2-year survival rate, and 5-year survival rate of patients with A289D/T/V mutations was 6 months, 12%, and 12%, respectively (Figure 1C). Patients containing a WT EGFRA289 had a median OS of 15 months, a 2-year OS rate of 22%, and a 5-year OS rate of 11%, demonstrating a significantly shorter median OS for patients harboring the A289D/T/V mutations compared with WT at that same position (p = 0.028). Patients with R108G/K mutations had a median OS, a 2-year survival rate, and a 5-year survival rate of 17 months, 19%, and 19%, respectively, and patients with WT EGFRR108 had a median OS of 14 months, a 2-year survival rate of 22%, and a 5-year survival rate of 10%, indicating no significant difference (p = 0.77, Figure 1D). Finally, patients with a G598V mutation had a median OS, a 2-year survival rate, and a 5-year survival rate of 17 months, 33%, and 33%, respectively, and patients with WT EGFRG598 had a median OS of 15 months, a 2-year survival rate of 21%, and a 5-year survival rate of 10%, also indicating no significant difference (p = 0.54, Figure 1E). The discovered EGFRA289D/T/V-negative survival association was validated using two independent cohorts: an additional 111 patients from UPenn (Figure S1B) and 116 patients from Hôpital Pitié-Salpêtrière (Idbaih et al., 2009) (Figure S1C).
Given the association of missense mutation with EGFR amplification, we looked at the survival characteristics of patients with amplified EGFR versus those without (Figure 1F). The median OS for amplified patients was 16 months, compared with 14 months for non-amplified patients and the 2- and 5-year survival rates were 17% and 5% for the amplified EGFR population compared with 25% and 14% for the WT EGFR population, indicating no significant difference between these two populations (p = 0.72).
As EGFRR108G/K and MGMT methylation were associated, we looked at the survival characteristics of MGMT methylated versus unmethylated patients (Figure S1D). As expected, the median OS of patients with MGMT methylation was significantly longer than patients without MGMT methylation, at 24 versus 15 months, respectively, and the 2- and 5-year survival rates for MGMT methylated versus MGMT unmethylated were 50% and 18% versus 14% and 12%, respectively. Collectively, the patient survival data from the UPenn cohort revealed an oncogenic driving force behind the EGFRA289DT/V mutants, which is distinct from EGFR amplification and MGMT methylation status.
Patient-Specific MRI Signatures of EGFR Missense Mutants Suggest an Invasive and Proliferative Phenotype in GBM
The initial comprehensive set of 2,104 quantitative imaging phenomic (QIP) features was pruned into 299 statistically significant (p < 0.05) features (Table S2) using a multivariate classification framework (Gaonkar and Davatzikos, 2013). QIP features synergistically represented altered imaging signals to formulate a descriptive signature of each EGFR variant (Figures 2A and S2). Grouped together, comparison of WT EGFR with the missense mutants revealed relatively few features that demonstrated statistical significance (Figure S3A; Table S2). However, when we looked at the individual component, each mutation had a unique set of radiographically interpretable features associated with it, rather than a unifying radiophenotype (Table S3). While EGFRR108G/K and EGFRG598V did have unique features, the overall picture presented by the EGFRA289D/T/V mutations highlighted possible biological mechanisms that could result in the poor patient OS associated with this mutation; thus, we focused our attention on this specific mutant.
Figure 2. MRI Signatures of EGFR Missense Mutants Suggest an Invasive and Proliferative Phenotype.

(A) Examples of the four basic/structural MRI modalities used to segment all brain scans into healthy and tumor labels, along with the major axis, minor axis, and the color legend for each label. WM, white matter; GM, gray matter; CSF, cerebrospinal fluid.
(B) Selected quantitative imaging phenotypes (QIP) features found in the ET region presenting a picture of increased rCE due to decreased T1 signal in EGFRA289D/T/V mutant tumors.
(C) QIP features found in advanced imaging in the ET region.
(D) Morphological features found in the CTE region.
(E)Basic and advanced imaging features found in the ED region.
See also Figures S2 and S3, Tables S2 and S3.
The relative contrast enhancement (rCE) in the ET tissue, defined as the subtraction of the native T1-weighted signal (T1) from the post-contrast (gadolinium) T1 signal (T1Gd), demonstrated a higher value in the presence of an EGFRA289D/T/V mutation (Figure 2B). To investigate the cause of the increased rCE, we assessed the mean value of the T1 signal in the ET. This revealed lower values in the presence of the EGFRA289D/T/V mutation, indicating that the higher contrast value was not solely due to the presence of contrast, but also a lower native T1 signal. Examining the T2 value in the ET, we found that EGFRA289D/T/V mutant tumors had higher values. When combined with the T1 and rCE data, these values supported a radiographic phenotype of higher water content in the tissue. To elucidate a possible cause for this, we examined relative cerebral blood volume (rCBV) and peak height (PH) in the ET (Figure 2C), where both parameters showed higher values in the EGFRA289D/T/V population.
When looking at the CTE, we found the ratio of major to minor axes associated with EGFRA289D/T/V significantly different (Figure 2D). The major axis was defined as the largest 2D distance and the minor axis is perpendicular to the major axis. Additional morphological characteristics, while not demonstrating statistical significance, displayed relationships further emphasizing the more irregular shape of the EGFRA289D/T/V mutant tumors compared with that of the WT EGFR (Figure S3B). Furthermore, the quantitative assessment of the ED region revealed decreased homogeneity of fractional anisotropy (FA) for cases with EGFRA289D/T/V mutations (Figure 2E), indicating decreased tissue organization, and increased rCE signal. Comparison of EGFRA289D/T/V mutants with WT EGFR and EGFR-amplified cases confirmed the association of the imaging features with the A289D/T/V mutation, with the exception of FA homogeneity in the ED (Figures S3C and S3D). The ED was previously reported as having a unifying MRI signature across various EGFR pathway activating alterations (including EGFRvIII, EGFR amplification, EGFRA289D/T/V, EGFRR108K, and EGFRG598V) in GBM, compared with WT EGFR (Bakas et al., 2017c).
Mice Bearing Intracranial EGFRA289V Tumors Have Attenuated Survival and an Invasive Phenotype
Based on patient imaging analysis, we hypothesized that EGFRA289 missense mutations conveyed enhanced tumor growth and invasion distinguishable from amplified WT EGFR. To test this, we engineered U87 glioma cells and HK281 GBM-spheres to express either WT EGFR or EGFRA289V at levels associated with amplified EGFR found in GBM (Nishikawa et al., 1994). Corroborating our findings in the GBM patient population, mice bearing intracranial tumors harboring the EGFRA289V mutation had a significantly worse survival rate compared with those engrafted with WT EGFR-expressing tumors (Figures 3A and 3B). Histological examination of these tumors revealed a striking increase in invasive fronts as well as increased Ki67 staining (Figures 3C, 3D, S4A, and S4B).
Figure 3. Mice Bearing Intracranial EGFRA289V Tumors Have Attenuated Survival and an Invasive Phenotype.

(A and B) KM survival curves comparing mice implanted with U87 (A) or HK281 (B) tumors expressing either WT EGFR or EGFRA289V, n = 6 per group.
(C and D) Representative H&E and Ki67 stained sections of U87 (C) and HK281 (D) tumors expressing WT EGFR or EGFRA289V harvested at time of sacrifice from mice in (A and B) Scale bars, 100 μm.
(E and F) T2 weighted MRI (left) and 3D volume segmentation map (right) of mouse brains and tumors for WT EGFR (E) and EGFRA289V U87 (F) tumors at 20 days after orthotopically implantation. Whole brain is in red and tumor is in green.
See also Figure S4.
Comparison of EGFRA289V with WT EGFR tumors by T2-weighted MRI at day 14 further demonstrated a striking difference between the two conditions (Figures 3E and 3F), and corroborated our radiographic findings in GBM patients. The scans of WT EGFR tumors showed little if any disruption of the normal brain; however, the EGFRA289V images demonstrated large, necrotic tumors with poorly demarcated borders, suggesting an invasive phenotype. While limited modalities of animal imaging do not allow for a quantitative analysis, increased T2 signal throughout the EGFRA289V tumors and in the peritumoral region showed a similar pattern to the patient data. Follow-up histological examination of the imaged brains revealed highly cellular, poorly demarcated, and invasive neoplasms mainly developing unilaterally within the injected neuroparenchyma, confirming the invasive edge of the U87 EGFRA289V tumors (Figures S4C and S4D).
EGFRA289V Missense Mutation Induces ERK Activation and Increased MMP1 Expression
The results above indicated an aggressive tumor growth phenotype that was imparted by the EGFRA289V mutation in both patients and animal models. To characterize the molecular underpinnings of this mutation, we first examined the expression of matrix metalloproteinases (MMP) 2 and 9, known to convey enhanced glioma cell invasion (Das et al., 2011; Nakada et al., 2003). Surprisingly, we did not find any effect of EGFRA289V on MMP2 or MMP9 expression in U87 cells (Figure S5A). In addition, invasion through gelatinous substrates was unaffected by EGFRA289V expression (Figure S5B). Next, we examined a panel of MMPs known to be expressed in glioma cells (Hagemann et al., 2012). Here we found a significant increase in MMP1 expression in U87 cells expressing EGFRA289V compared with WT EGFR, EGFRR108K, and EGFRG598V, which was also induced in HK281 GBM-spheres expressing EGFRA289V compared with WT EGFR (Figures S5A, 4A, and 4B).
Figure 4. EGFRA289V Missense Mutation Induces ERK Activation and Increased MMP1 Expression.

(A) RT-PCR analysis of MMP1 expression in U87 glioma cells expressing WT EGFR, EGFRvIII (V3), EGFRR108K, EGFRA289V, or EGFRG598V.
(B) RT-PCR analysis of MMP1 in HK281 GBM-spheres expressing WT EGFR or EGFRA289V.
(C) Western blotting analysis of the indicated proteins in serum starved U87 glioma cells expressing WT EGFR, EGFRvIII, EGFRR108K, EGFRA289V, or EGFRG598V in the presence or absence of 100 ng/mL EGF for 10 min at 37°C.
(D) Densitometric quantification of (C).
(E) Western blot analysis of the indicated proteins in U87 glioma cells expressing either WT EGFR or EGFRA289V following treatment with gefitinib (Gef) or lapatinib (Lap) (4 μM, 24 hr).
(F) Densitometric quantification of (E).
(G) Western blot analysis of the indicated proteins in U87 glioma cells expressing either WT EGFR or EGFRA289V following treatment with U0126 or PD98059 (10 μM, 24 hr).
(H) Densitometric quantification of (G).
(I) RT-PCR analysis of MMP1 expression in U87 glioma cells expressing EGFRA289V following treatment with gefitinib or lapatinib (4 μM, 24 hr).
(J) RT-PCR analysis of MMP1 expression in U87 glioma cells expressing EGFRA289V following treatment with U0126 or PD98059 (10 μM, 24 hr).
(K) Western blot analysis of the indicated proteins in HK281 GBM-spheres expressing either WT EGFR or EGFRA289V following treatment with gefitinib (4 μM), lapatinib (4 μM), or U0126 (10 μM) for 24 hr.
(L) Densitometric quantification of (K).
(M) RT-PCR analysis of MMP1 expression in HK281 GBM-spheres expressing EGFRA289V following treatment with gefitinib (4 μM), lapatinib (4 μM), or U0126 (10 μM) for 24 hr. RT-PCR data shown are fold-change gene expression relative to GAPDH. Error bars are SEM of at least three replicates and represent at least three independent experiments.
ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S5.
The EGFRA289V missense mutation has been shown previously to be constitutively activated (Lee et al., 2006). To examine the downstream signaling pathways that may have contributed to increased MMP1 expression, we interrogated known signaling effectors by immunoblot analysis of U87 glioma cells harboring EGFR mutants. Our results confirm that the EGFRA289V mutation leads to constitutive EGFR activation (Figures 4C and 4D). Furthermore, analysis of downstream MAPK, AKT, and STAT3 signaling pathways revealed a striking constitutive activation in the signaling molecule p42/44 MAPK (ERK) in cells expressing the EGFRA289V mutant. Importantly, when we inhibited this signaling pathway using two different MEK inhibitors, U0126 and PD98059, or the tyrosine kinase inhibitors, gefitinib and lapatinib, there was a significant reduction in MMP1 gene expression (Figures 4E and 4J). We confirmed this EGFRA289V-driven EGFR/ERK/MMP1 signaling pathway in the HK281 GBM-spheres (Figures 4K–4M).
EGFRA289V Constitutive EGFR/ERK/MMP1 Signaling Results in Increased Invasion and Proliferation In Vitro
To further delineate the effect of EGFRA289V on EGFR/ERK/MMP1 signaling, we used modified Boyden Transwells coated with collagen, in the presence or absence of MEK inhibitors (Figures S6A and S6B). Corroborating our in vivo findings, both U87 cells and HK281 GBM-spheres expressing EGFRA289V showed increased invasion compared with their WT counterparts. When we blocked the ERK signaling pathway by U0126, we reduced the effect in EGFRA289V cells to the level of WT (Figures 5A and 5B). To test the role of MMP1 in this assay, we transduced the EGFRA289V-expressing cells with either a small hairpin RNA (shRNA) targeting MMP1 or a control shRNA (Figures S6C and S6D), and showed that in both in vitro glioma models, invasion was attenuated upon MMP1 knockdown (Figure 5D).
Figure 5. Constitutive EGFRA289V/ERK/MMP1 Signaling Results in Increased Invasion and Proliferation In Vitro.

(A and B) Quantification of U87 glioma cells (A) and HK281 GBM-spheres (B) expressing WT EGFR or EGFRA289V cell invasion following treatment with U0126 (10 μM) or control for 24 hr.
(C and D) Quantification of invaded EGFRA289V U87 glioma cells (C) and HK281 GBM-spheres (D) expressing an shMMP1 vector compared with control shRNA.
(E and F) Quantification of % bromodeoxyuridine (BrdU)-positive U87 (E) or HK281 (F) cells treated with DMSO control or U0126 (10 μM) 24 hr prior to BrdU incorporation.
(G and H) H&E staining of intracranial U87 (G) and HK281 (H) tumors without (shControl) or with MMP1 knockdown (shMMP1). Scale bars, 100 μm. Error bars are SEM of at least three replicates and represent at least three independent experiments. ns, not significant; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
See also Figure S6.
Next, we tested if increased Ki67 staining in vivo was due specifically to the expression of EGFRA289V (Figures 3B and 3D) by quantifying bromodeoxyuridine incorporation (Figure S6E). We found that EGFRA289V-expressing cells had a significantly higher percentage of active proliferation than WT cells in both U87 and HK281 models. Treatment with U0126 again reversed the phenotypic difference, bringing the proliferation percentage in the EGFRA289V cells to a level comparable with the WT EGFR cells (Figures 5E and 5F). Finally, MMP1 knockdown in vivo nullified the invasive phenotype attributed to the EGFRA289V mutation (Figures 5G and 5H). The results from these experiments indicated that the increased invasion and proliferation in EGFRA289V-expressing mutant cells was due to a constitutively active EGFR/ERK/MMP1 signaling pathway that can be rescued through pharmacological inhibition.
mAb806 Is a Potential Therapeutic Option for Patients Expressing EGFRA289V
The antibody-drug conjugate ABT-414, which specifically recognizes EGFRvIII and WT EGFR when expressed at amplified levels, has shown promise in phase I/II clinical trials for GBM patients (Gan et al., 2017; Phillips et al., 2016; Reardon et al., 2017). Previously, we have proposed that certain mutations such as EGFRA289V could bind and respond to mAb806 based on simulations (Orellana et al., 2014). To examine the potential efficacy of this therapy for patients with GBM expressing EGFRA289V, we used flow cytometry to confirm the ability of mAb806 to bind EGFRA289V. We found that mAb806 bound EGFRA289V significantly better than WT EGFR in both U87 and HK281 models (Figures 6A and 6B). We next assessed the efficacy of this drug in vivo using subcutaneous and intracranial tumor models. To directly test the ability for mAb806 to reduce tumor growth, we engrafted mice subcutaneously with U87 glioma cells expressing WT EGFR, EGFRA289V, or EGFRvIII, and treated mice with either mAb806 or vehicle. Using a low dose of mAb806 (0.1 mg/mouse) previously shown to have no effect on WT EGFR-expressing cells (Mishima et al., 2001), we verified these results and also show that there is a similar reduction in tumor growth when cells express the EGFRA289V mutation compared with the EGFRvIII deletion mutation (Figure 6C).
Figure 6. mAb806 as a Therapeutic Option for Patients Expressing EGFRA289V.

(A and B) Fluorescent-activated cell sorter analysis of U87 glioma cells (A) and HK281 GBM-spheres (B) expressing WT EGFR, EGFRvIII, or EGFRA289V. Serum-starved cells were incubated with either mAb806 or mAb528 (1 μg/1 × 106 cells) followed by secondary staining with a fluorescein isothiocyanate-conjugated antibody. Results are shown as mAb806 staining normalized to mAb528 (total EGFR).
(C) Mice bearing subcutaneous U87 tumors expressing WT EGFR, EGFRvIII (V3), or EGFRA289V were treated with PBS (100 μL/mouse) or mAb806 (0.1 mg/100 μL/mouse) intraperitoneally (i.p.) 3 times per week for 2 weeks once tumors reached an average of 100 mm3. Mean tumor growth after treatment is shown as a function of time.
(D) Representative images by fluorescence molecular topography (FMT) at day 23 of mice implanted intracranially with iRFP720 expressing U87 glioma cells expressing WT EGFR, EGFRvIII, or EGFRA289V and treated with PBS (100 μL) or mAb806 (1 mg/100 μL/mouse) i.p. every other day from days 0 to 14.
(E) Quantification of FMT signal intensity on day 23 post-implantation for each region of interest of mice in (D).
(F) KM survival curve of mice in (D and E).
(G) KM survival curve of mice bearing HK281 intracranial tumors as described in (D). n = 5 for each animal group.
Error bars are SEM. ns, not significant; *p < 0.05, **p < 0.01. See also Figure S7.
To test if mAb806 recognition of the EGFRA289V mutation would lead to an increase in survival, iRFP720-labeled U87 cells (Figure S7A) were engrafted orthotopically in mice, followed by treatment with mAb806 or vehicle and fluorescence molecular tomography (FMT) imaging. By day 16, FMT imaging indicated a strong reduction in fluorescence intensity in mAb806-treated mice bearing tumors expressing EGFRA289V or EGFRvIII, and this difference further intensified over the next week (Figures 6D and 6E, day 23). It became clear that the difference in FMT intensity, a direct indicator of intracranial tumor volume, correlated with survival (Figure 6F). In addition to the EGFRvIII model (p = 0.0025), mAb806 therapy significantly enhanced animal survival in mice bearing EGFRA289V-expressing tumors (p = 0.0015), with only a mild effect on mice bearing WT EGFR-expressing tumors (p = 0.06). This result was reproduced in the HK281 GBM-sphere model (Figure 6G: WT EGFR-expressing tumors p = 0.085, EGFRA289V-expressing tumors p = 0.0048, Figures S7B–S7D). This study shows that an EGFR-directed therapy, mAb806, is beneficial to tumors expressing EGFRA289V, confirming our in silico predictions. These results suggest that the EGFRA289V mutation, in addition to EGFRvIII, may be considered relevant for patients undergoing clinical trials with ABT-414. Collectively, this data justifies the further development of EGFR ECD targeting reagents for GBM therapy.
DISCUSSION
In this study, we have demonstrated that the UPenn cohort of GBM patient specimens matches the literature, in terms of demographics, survival, and EGFR mutational frequency and location (Cerami et al., 2012; Stupp et al., 2005). Median OS of patients with GBM following SOC treatment, in the literature, is 14.8 months, closely matching the 15 months OS of our cohort (Stupp et al., 2005). Median age at diagnosis and male:female ratio also closely matched the population data (Dubrow and Darefsky, 2011), as did MGMT promoter methylation status, the only prognostic biomarker in GBM (Hegi et al., 2005). EGFRvIII expression and EGFR amplification were lower in the UPenn cohort than in the literature (Heimberger et al., 2005), potentially because our assessment of EGFR amplification was based on the read depth from NGS, compared with fluorescent in situ hybridization (FISH), which is often used in the literature. While NGS offers multiple advantages over FISH, including high-throughput nature and objective quantification, it also averages the reads across the tumor sample. In contrast, FISH assesses individual cells, avoiding the possibility of sample dilution via non-neoplastic sources such as stroma and microglia.
A deeper look at EGFR alterations revealed a trio of ECD missense mutations second to EGFRvIII in frequency. EGFRR108G/K, EGFRA289D/T/V, and EGFRG598V are the most common missense mutations in both the UPenn cohort and the TCGA population (Cerami et al., 2012). Prior work has demonstrated that these missense mutations have tumorigenic potential (Lee et al., 2006). In addition, structural work on these mutations has pointed toward increased ligand affinity (Bessman et al., 2014). Thus, the overall impact of these mutations could be anticipated to be over-activation of EGFR-driven pathways, leading to increased cell proliferation and invasion (Talasila et al., 2013; Xing et al., 2013). In addition, we have shown that certain mutations, such as EGFRA289V, can expose the mAb806 binding epitope, which is accessible in EGFRvIII (Orellana et al., 2014).
Turning toward the clinical outcomes from our patient cohort, EGFRA289D/T/V was associated with a worse OS when compared with WT EGFR at the A289 position. In contrast, EGFRG598V was not associated with survival and EGFRR108G/K presented a complex picture: while the OS trend was neither positive nor negative, EGFRR108G/K mutations were shown to occur in the presence of MGMT methylation. As MGMT methylation is known to confer sensitivity to TMZ treatment and result in increased OS (Stupp et al., 2005), the true survival impact of the EGFRR108G/K mutation could not be elucidated.
There is increasing evidence demonstrating the validity of advanced computational analysis of QIP features that has shown promise in predicting clinical outcome and molecular characteristics (Aerts, 2016; Bakas et al., 2017a, 2017c; Ellingson et al., 2017; Gevaert et al., 2017; Gill et al., 2014; Gutman et al., 2013; Itakura et al., 2015; Macyszyn et al., 2016; Zhang et al., 2017). In this study, examination of QIP features associated with the EGFR missense mutations helped to generate hypotheses for the negative survival impact of EGFRA289D/T/V. The water content picture presented in the ET could have been due to either increased “leakiness” of existing vessels or increased total blood content as a result of increased neovascularization (Aronen et al., 1994). Together with the relationship of PH values and rCBV values between EGFRA289D/T/V and WT EGFR, this presents a picture of increased proliferation in the ET, and we hypothesized that there was increased total neovascularization. The morphological characteristics of the CTE presented a picture of a highly invasive tumor, penetrating further from the tumor in an asymmetrical pattern. The ED, considered a mix of tumor and normal brain, showed a loss of normal tissue structure accompanied by neovascularization at a level indiscernible to the naked eye, as supported by the decreased homogeneity in the FA signal. A shifting of the composition toward tumor cells, due to increased invasion, would result in a more chaotic picture as the existing brain architecture is disrupted by the tumor cells. Tumor cells would also stimulate neovascularization, leading to the increased gadolinium presence (i.e., increased rCE) we discovered in the EGFRA289D/T/V mutants. Taken together, the major:minor axes ratio, the FA, and the rCE signals suggest increased invasion in the peritumoral ED region of the EGFRA289D/T/V mutant tumors. Although some of the features discussed here, and shown in Figures 2B–2E, were not statistically significant, they were included in our analysis and discussion as they supported the overall evaluated radiographic phenotype of the EGFRA289D/T/V mutants.
While imaging characteristics can suggest underlying biological processes, verification using animal studies is more conclusive. U87 often displays bulky tumors with little presentation of invasion in orthotopic implantations (Miura et al., 2010). The invasive picture presented by the EGFRA289V mutation represents a significant alteration to both the baseline of U87 and U87-expressing WT EGFR. Importantly, we recapitulated this result in the patient-derived HK281 GBM-sphere model, indicating invasion is an attribute of the EGFRA289V mutant in a more pathologically appropriate model.
EGFR-mediated signaling acts through two main pathways, RAS/RAK/ERK and PIK3CA/AKT, resulting in increased nuclear transcription of genes involved in cellular proliferation and tumor invasion. To explore the mechanism behind the in vivo phenotypes we found, we examined the activation statuses of these two pathways and performed inhibition studies. In both U87 cells and HK281 GBM-spheres, EGFRA289V expression resulted in constitutive phosphorylation of EGFR, as seen previously (Lee et al., 2006). Further analyses revealed that A289V-mediated EGFR activation resulted in constitutive cellular signaling through phosphorylated ERK, ultimately enhancing the expression of MMP1. Interestingly, while WT EGFR signals mainly through the STAT3 and MAPK pathways, EGFRvIII preferentially acts through the PI3K/AKT pathway (Thorne et al., 2016); EGFRA289V acting primarily through the MAPK pathway thus indicates a divergence in oncogenic signaling activation between EGFR ECD missense and deletion mutants.
A role for MMPs in cancer progression and invasion has been widely characterized. Specifically, MMP2 and MMP9 have been extensively implicated in GBM progression. It has been shown that, while MMP1 is not typically expressed in the normal brain, it is elevated in gliomas, correlating with tumor grade and survival (Stojic et al., 2008). Here, we reveal a link between the EGFR ECD missense mutant A289V and MMP1 expression, which results in a pro-invasive phenotype. A previous study showed that MMP1 is induced by EGF stimulation in glioma cells (Anand et al., 2011), indicating the likelihood of an exacerbated effect of the EGFRA289V-mediated invasive phenotype following stimulation. Interestingly, this study also indicated EGFR/MAPK signaling for its effect, and therefore it is likely that EGFRA289V would mediate its pro-invasive phenotype through this pathway regardless of ligand stimulation.
Directly targeting MMPs in cancer has proven to be a challenge due to structural homology between members of the MMP family and a bilateral role in many cancers (Levin et al., 2017). To date, there have been few clinical trials testing MMP inhibitors for GBM. Most notably, a phase II trial combining the broad spectrum MMP inhibitor marimastat with TMZ resulted in a progression-free survival at 6 months that significantly exceeded the literature target. However, a separate randomized, double-blind, placebo-controlled trial indicated that the inhibitor on its own had no effect on patient survival (Groves et al., 2002; Levin et al., 2006) and further studies examining MMP inhibitors for GBM were not conducted.
Along these lines, a significant contributor to the overwhelmingly poor outcome for GBM patients is the relative dearth of active treatments. There are four US Food and Drug Administration-approved therapies for GBM: TMZ, carmustine implants, bevacizumab, and NovoTTF-100A (Brem et al., 1995; Friedman et al., 2009; Stupp et al., 2005, 2012). More effective therapies are needed to improve patient outcomes. Following promising results indicating safety, efficacy studies are currently underway for ABT-414, which specifically targets tumor cells expressing aberrant EGFR (Reardon et al., 2017). Here we examined the prospects of using ABT-414 against GBMs containing the EGFRA289V missense mutant by assessing the efficacy of its non-conjugated precursor, mAb806, in subcutaneous and orthotopic animal models. We found that mAb806 therapy significantly reduced tumor burden and prolonged animal survival in mice bearing EGFRA289V-positive tumors to a similar degree as to what has been previously published in mice bearing EGFRvIII-positive tumors (Mishima et al., 2001). Significantly, our data suggest that mAb806 might be a viable therapeutic option for tumors harboring EGFR alterations other than EGFRvIII or amplification of WT EGFR. Our study indicates that when deciding measurement outcomes for clinical trials with EGFR-targeted therapies, we should not ignore the smaller EGFR ECD missense mutation populations, as they may provide valuable insight for patient stratification. Importantly, with a baseline poor OS in the EGFRA289D/T/V mutation population, survival benefits in this population may be overshadowed by a lack of significant survival improvement in the WT EGFR cohort.
We have demonstrated the clinical significance of the EGFRA289D/T/V missense mutations in the context of WT IDH1, de novo GBMs. This negative survival impact was reinforced by quantitative imaging analysis suggesting hyperproliferation and increased invasion in patients. Decreased OS, increased proliferation, and increased invasion were demonstrated using modified cell lines in vivo. Mechanistic exploration revealed increased MMP1 expression driven by ERK activation leading to both the increased proliferation and invasion. Finally, the tumor driver status of EGFRA289V was demonstrated by in vivo targeting via mAb806, increasing animal survival and inhibiting tumor growth. These results serve to highlight the complexity of the EGFR signaling cascade and pathway nuances of ECD mutations in the context of cancer.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank B. Furnari (ffurnari@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Cohort
All UPenn patient data was obtained retrospectively under a protocol approved by the University of Pennsylvania’s Institutional Review Board, with a waiver for patient consent. For the initial UPenn cohort, patients who had undergone surgical resection for a cranial malignancy between December 2009 and June 2016 were identified through the UPenn CPD. GBM diagnoses were confirmed via medical records containing neuropathological assessment. Exclusion criteria were IDH1 mutation as determined by NGS, 1p19q co-deletion, recurrent resection, and prior diagnosis of a lower grade glioma. There were a total of 260 cases that fit all the criteria. Full patient demographics are presented in Table S1.
For the validation UPenn cohort, patients seen at UPenn between June 2016 and June 2017 with tissue submitted to the UPenn CPD were included. All patients were IDH1 wild-type, de novo GBMs. The Hôpital Pitié-Salpêtrière cohort was obtained prior to 2009 and did not have testing for IDH1 mutational status. Patient data from the Hôpital Pitié-Salpêtrière was obtained with written patient consent under a protocol approved by the Comité de Protection des Personnes-Ile de France VI (Idbaih et al., 2009).
Animals
All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee at either the University of California, San Diego or the University of Pennsylvania, in accordance with NIH and institutional guidelines. Four to six week old female Athymic nu/nu mice were used for the subcutaneous tumor studies and six to eight week old female Athymic nu/nu mice were used for the intracranial tumor studies (Charles River Laboratories, Frederick, MD; The Jackson Laboratory, Bar Harbor, ME). For subcutaneous studies, mice were injected into the rear right flank with 2.5×105 U87 glioma cells expressing WT EGFR, EGFRvIII, or EGFRA289V. When tumors reached an average size of 100 mm3 mice were injected intraperitoneally with either PBS control or mAb806 antibody (0.1 mg/mouse in 100 μL PBS) 3×/week, for two weeks. Tumor width (a) and length (b) were obtained using calipers and tumor volumes were determined using the formula V = ½ × a2 × b, where b ≤ a. Mice were euthanized when tumor volumes exceeded 1,500 mm3. For intracranial studies, mice were anesthetized and fixed in a stereotactic apparatus, and a burr hole was drilled at 2 mm right or left lateral to bregma. U87 glioma cells or HK281 GBM-spheres expressing WT EGFR, EGFRvIII, or EGFRA289V (1 × 105 cells) were implanted at a depth of 3 mm. Every other day, from day 0 – 14, mice were injected intraperitoneally with mAb806 or PBS control (1 mg/mouse in 100 μL PBS). Animals were observed daily and were euthanized when they showed signs of morbidity.
Cell Lines
U87 glioma cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS and 1% penicillin/streptomycin. HK281 GBM-spheres [a kind gift from Harley Kornblum, UCLA, (Visnyei et al., 2011)] were cultured in DMEM/F12 medium supplemented with 1× B27 (GIBCO/Life Technologies), 1% penicillin/streptomycin, human recombinant EGF (20 ng/mL), bFGF (20 ng/mL) and 2 mg/mL heparin (StemCell Technologies). All cells were maintained at 37°C, 5% CO2, and 100% relative humidity for the duration of the experiment. Reagents used in this study were obtained from the following sources: Antibodies: EGFR (BD Biosciences), pan-phospho-Tyrosine-HRP (R&D Systems), p42/44 MAPK, phospho-p42/44 MAPK, STAT3, phospho-STAT3 y705, AKT, phospho-AKT s473 (Cell Signaling Technology), Ki67 (Santa Cruz Biotechnology), and β-Actin (Sigma). Inhibitors: U0126 and PD98059 (LC Laboratories). The monoclonal antibody 806 was produced in the Biological Production Facility at the Olivia-Newton John Cancer Research Institute (Melbourne, Australia).
METHOD DETAILS
EGFR Mutational Status
EGFR mutation status (single nucleotide variants, indels) was obtained from the CPD through an NGS assay. Briefly, formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks were selected by a neuropathologist and sent to the CPD for processing. DNA was extracted from the tissue following manufacturer’s instructions (Qiagen, Valencia, CA). After extraction, DNA quality and concentration were assessed (Agilent, Santa Clara, CA; Life Technologies, Waltham, MA). For NGS, between 10 and 200 ng of DNA was used to prepare the library. Samples were multiplexed and read on a MiSeq (Illumina, San Diego, CA) with an average read depth of 2500×. NGS data was processed through an in-house bioinformatics pipeline, identifying variants and amplifications of EGFR by standard methods.
EGFRvIII Expression
EGFRvIII determination was made by NGS sequencing through the CPD. A total of 204 of the 260 cases were assessed for EGFRvIII expression. Briefly, using primers designed to capture both EGFRvIII and WT EGFR, NGS library preparations were amplified and sequenced using MiSeq (Illumina, San Diego, CA). The resulting data was processed through an in-house bioinformatics pipeline that quantified EGFRvIII as a fraction of total EGFR. Samples with an EGFRvIII fraction greater than 5% were considered to be positive.
MGMT Methylation
A total of 209 cases were examined for MGMT promoter methylation. The initial 91 samples were sent out to an outside laboratory (ARUP, Salt Lake City, UT). The remaining 118 samples were processed at UPenn. Briefly, DNA was extracted from FFPE blocks following manufacturer’s instructions (Qiagen, Valencia, CA). Bisulfite conversion was carried out according to manufacturer’s instructions (Zymo Research, Irvine, CA) and DNA was then amplified using PCR targeting 4 CpG islands in exon 1 of MGMT. The PCR results were then pyrosequenced to assess the presence of methylation at each CpG island (Qiagen, Valencia, CA).
Magnetic Resonance Imaging Acquisition
The assessed patients were pre-operatively scanned, as part of the UPenn standard care protocol of patients with brain tumors, using an advanced MRI acquisition protocol of 6 different modalities, comprising native (T1) and contrast-enhanced (T1Gd) T1-weighted, T2-weighted (T2), T2 Fluid-Attenuated Inversion Recovery (T2-FLAIR), Diffusion Tensor Imaging (DTI), and Dynamic Susceptibility Contrast (DSC) MRI volumes. The GBM patients with available advanced MRI brain scans included 13 WT EGFR, 11 EGFR-amplified, 11 EGFRA289D/T/V, 4 EGFRR108G/K, and 4 EGFRG598V. The protocol was approved by the Institutional Review Board at the Hospital of the University of Pennsylvania (HUP), and informed consent was obtained from all subjects. No randomization method was used for allocating samples to experimental groups.
All MRI scans were acquired in the axial plane using 3-Tesla Siemens Magnetom Trio A Tim clinical MRI systems (Erlangen, Germany), according to the standardized advanced acquisition protocol followed at the HUP. MRI specifics are available in Table S4. The contrast material used in the scans included in this study was either gadodiamide (Omniscan, GE Healthcare, Mickleton, NJ), or gadobenate dimeglumine (MultiHance, Bracco SpA, Milan), and administered intravenously (IV). The total dose of contract material was divided into two IV injections to help minimize errors due to potential contrast leakage out of intravascular space. The initial loading dose described the 25% of the total injected contrast material and the second bolus injection the remaining 75%, with a delay of 5min. The exact contrast material dosage was dependent on patient weight and given on a relative proportion of 0.3 mL/kg. The T2 volumes were acquired prior to any contrast administration. The T2-FLAIR volumes were acquired between the initial IV injection and the DSC acquisition. The DSC acquisition was performed during the second IV injection of the contrast material. DTI scans (Axial 2D) were acquired using a single-shot spin echo planar imaging sequence (Variant: segmented k-space\spoiled, Options: Partial Fourier-Phase\Fat Saturation), with 95 phase encoding steps. Following acquisition at b=0 s/mm2 (repeated 3 times), diffusion weighted images were acquired (b=1000 s/mm2) with diffusion gradients applied in 30 directions.
Image Pre-processing
All acquired volumes were converted from DICOM to NIfTI and oriented to the RAI coordinate system convention, as part of the requirements of the segmentation algorithm we used (Bakas et al., 2017b). All patient scans were co-registered to a single T1Gd template using an affine registration, through the Oxford center for Functional MRI of the Brain (FMRIB) Linear Image Registration Tool (FLIRT) (Jenkinson et al., 2002) of the FMRIB Software Library (FSL) (Jenkinson et al., 2012). All brain scans were then skull-stripped using a template library of 216 MRI brain scans and their corresponding masks. This library was used for target specific template selection and subsequent registrations MUlti-atlas Segmentation utilizing Ensembles (MUSE) (Doshi et al., 2016). A final step based on region-growing guided by the T2 signal was applied to obtain a brain mask that includes the intra-cranial CSF. High frequency intensity variations in regions of uniform intensity profile, while preserving the underlying tissue structure was then applied using the Smallest Univalue Segment Assimilating Nucleus (SUSAN) approach (Jenkinson et al., 2002). Non-uniformities of the image intensity caused by the inhomogeneity of the magnetic field during acquisition were removed using a non-parametric, non-uniform intensity normalization algorithm (Sled et al., 1998), and the intensity histograms of all modalities of all patients were then matched to the corresponding modality of a single reference patient. A set of DTI measurements were extracted from the DTI volumes for comprehensive analysis, namely the tensor’s fractional anisotropy DTI(FA), radial diffusivity DTI(RAD), axial diffusivity DTI(AX), and apparent diffusion coefficient DTI(ADC). Furthermore, parametric maps of clinical parameters from the temporal perfusion dynamic volumes (i.e., DSC-MRI) were extracted, comprising the relative cerebral blood volume (rCBV), peak height (PH) and percentage signal recovery (PSR). Both the DTI and the DSC extracted maps were treated as individual image modalities.
Tumor Segmentation
The method used to segment each tumor into its various histologically distinct sub-regions is named GLISTRboost (Bakas et al., 2017b) and it is based on a hybrid generative-discriminative model. The generative part (Gooya et al., 2012) incorporates a glioma growth model (based on a reaction-diffusion-advection model) (Hogea et al., 2007), and follows an Expectation-Maximization (EM) framework to segment the brain scans into tumor (i.e., ET, NET and ED), as well as healthy tissue labels (i.e., WM, GM, CSF, vessels and cerebellum), and register a healthy population probabilistic atlas to glioma patients’ brain scans using the tumor growth model to account for mass effects. The discriminative part is based on a gradient boosting multi-class classification scheme and used to refine the tumor sub-region labels based on information from multiple patients. Lastly, a Bayesian strategy (Bakas et al., 2017d) is employed to further refine and finalize the tumor segmentation based on patient-specific intensity statistics from the multimodal MRI scans available (Figure 2A).
GLISTRboost is designed to tackle cases with both solitary and multifocal masses of complex shapes with heterogeneous texture. It requires manual input of tissue seed-points for each segmentation label, to capture the intensity variation and model the intensity distribution (i.e., mean and variance) across all modalities. However, the coordinate position of these input seed-points is not taken into consideration by the segmentation method, but only the corresponding intensity value. Therefore, any potential variation in the coordinates of seed-points initialized during independent initialization attempts should not affect the output segmentation labels, given that the modelled intensity distributions during these attempts are the same. In addition to the seed-points for each tissue class, a single seed-point and a radius are also required to approximate the bulk volume of each apparent tumor by a spherical parametric model, which is then used as a prior to the tumor growth model (Hogea et al., 2007). This growth model deforms a healthy population probabilistic atlas into a glioma patient brain scan matching the input scan, while approximating the brain tissue deformations occurred due to the mass effect of the tumors. A random-walk-based generative model estimates a tumor shape prior, initialized by the spherical parametric model. This prior is incorporated in the generative part of GLISTRboost via an empirical Bayes model (Gooya et al., 2012) and produces a probability map for each tissue label, leading to an integrative non-overlapping segmentation label map. This label map is then refined by a voxel-level multi-label classification through a gradient boosting ensemble model of decision trees. We trained decision trees of maximum depth 3 in a subset of the training data to introduce randomness and using a cross-validation framework to avoid overfitting. Sampling rate of 0.6 was used, while additional randomness was introduced by sampling stochastically a subset (square root of the total number of features) of imaging features at each node. The exact features use for training this discriminative part of GLISTRboost, consisted of i) intensity, ii) image derivatives, ii) geodesic distance transform, iv) texture parameters and the tissue probability maps. The intensity parameters comprise the raw intensity voxel value at each MRI modality, as well as their differences across all four modalities. The image derivatives are summarized by the Laplacian of Gaussians and the image gradient magnitude. These sets of parameters are extracted after performing an intensity normalization based on the median intensity of the current cerebrospinal fluid segmentation label. The geodesic distance transform was estimated for each voxel from the initialized tumor center seed-point (Gaonkar et al., 2015). Texture features were based on a gray-level co-occurrence matrix (GLCM) and extracted following discretization of the input volumes into 64 gray levels and using a bounding box of 125 voxels (5×5×5) for each voxel of each image. The GLCM accounted for intensity values within a radius of 2 voxels and for 26 3D directions to extract the energy, entropy, dissimilarity, homogeneity (i.e., inverse difference moment of order 2), and inverse difference moment of order 1. The tumor segmentation labels of each patient were then further refined based on individual patient intensities (Bakas et al., 2017d). First, the intensity distributions of the WM, ED, NET and ET were populated, considering the corresponding voxels of tissue probability equal to 1. The histograms of the 3 pair-wise distributions considered (i.e., ED vs WM in T2-FLAIR, ET vs ED in T1-Gd, and ET vs NET in T1-Gd) were then normalized. The maximum likelihood estimation was then used to model the class-conditional probability densities (Pr(I(vi)|Class) of each class by a distinct Gaussian model for each class. The voxels of each class in a close proximity (4 voxels) to the voxels of the paired class, were then iteratively evaluated by assessing their intensity I(vi) and comparing the Pr(I(vi)|Class1) with Pr(I(vi)|Class2). The voxel vi was then classified into the class with the larger conditional probability, which is equivalent to a classification based on Bayes’ Theorem with equal priors for the two classes, i.e., Pr(Class1) = Pr(Class2) = 0.5.
Extraction of Quantitative Imaging Phenomic Features
Accurate delineation of the three histologically distinct tumor sub-regions (i.e., ET, NET, ED) enabled us to extract QIP features (n=2104), accurately corresponding to each of the sub-regions. Specifically, these QIP features comprise: 21 volumetric measurements, 11 parameters describing the spatial configuration and distribution of the tumor, 1650 radiomic features (150 for each modality assessed), 330 histogram-based intensity parameters (30 for each modality), 66 first order statistics of image intensities (the mean and standard deviation of each sub-region in each modality), 21 parameters descriptive of the tumor’s shape, and 5 glioma diffusion properties extracted from tumor biophysical models using reaction-diffusion-advection equations.
Multivariate Machine Learning Analysis
An analytic estimation of statistical significance based on multivariate analysis (Gaonkar and Davatzikos, 2013) was employed to calculate the relative contribution of each QIP feature extracted on distinguishing the EGFR missense mutants from WT EGFR by assigning a corresponding p value. The multivariate approach was based on a support vector machine formulation for classification using a linear kernel function. The parameter for the soft margin cost function (c) was optimized on the training data based on a five-fold cross validated grid search; c=2α, where αε[−5,5]. This parameter controls the influence of each individual support vector that involves trading error penalty for stability. This analytic estimation assesses the relative individual contribution of each feature, while considering the synergistic interactions with other features, and assigns a conservative p value to each feature for evaluating/quantifying the statistical significance of each. Feature selection was conducted by selecting those features with a p value above a certain threshold (p > 0.05). Next, we analytically estimated the statistical significance (i.e., p values) of all extracted radiomic features distinguishing the EGFR missense mutants from WT EGFR, as well as between the EGFR mutants, based on a multivariate classification framework (Gaonkar and Davatzikos, 2013). Note that WT EGFR here described only those tumors that had confirmed wild-type EGFR at normal expression levels, thereby excluding EGFRvIII, amplification, and mutations at A289, R108, and G598 residues. This multivariate analysis enabled us to prune the extensively comprehensive set including 2,104 QIP features into 299 statistically significant (p<0.05) features (Table S2). Subsequent selection of features based on radiographic interpretability, according to a board-certified Neuroradiologist (M.B.), further reduced these features to a more manageable set of 17 QIP features (Table S3).
Typically, p value corrections for multiple comparisons are considered for assignment of statistical significance. However, statistical significance was not relevant to the scope of our imaging analysis, since the latter was used to generate hypotheses that were then tested both in silico and in vivo. Even if the outcome findings of the imaging analysis were considered trends, the conclusions of this study, after their evaluation in silico and in vivo, remained unchanged. Furthermore, traditional correction methods used (e.g. false discovery rate, and/or Bonferroni correction) were designed for multiple independent univariate tests. In the multivariate method we used, the significance of a feature was not calculated through univariate tests that use each feature independently. In contrast, our feature selection considered all features together and found the optimally discriminative combination of all features jointly by a multivariate classifier (Gaonkar and Davatzikos, 2013). Specifically, the p value for a feature indicated the significance of the weight assigned to this specific feature by the multivariate classifier. Accordingly, in the permutation tests, the weights of each feature depended on the weights of all other features. Considering the assumption of traditional correction methods for feature independence did not hold true, their application was not appropriate.
Reagents
For U87 transduction, pLRNL retroviral plasmids containing the full-length wild-type EGFR and the truncated EGFRvIII constructs were previously reported (Zanca et al., 2017). The EGFR ECD missense mutations were generated from the WT EGFR construct by site-directed mutagenesis (Agilent Technology), according to manufacturer’s instructions. Primers used were as follows: EGFRA289V forward 5’-caaatacagctttggtgtcacctgcgtgaagaagt-3’ and reverse 5’-acttcttcacgcaggtgacaccaaagctgtatttg-3’, EGFRR108K forward 5’-aaacctgcagatcatcaaaggaaatatgtactac-3’ and reverse 5’-gtagtacatatttcctttgatgatctgcaggttt-3’, EGFRG598V forward 5’-caagacctgcccggcagtagtcatgggagaaaaca-3’ and reverse 5’-tgttttctcccatgactcatgccgggcaggtcttg-3’. For HK281 transduction, the pLRNL constructs were used to subclone WT EGFR and EGFRA289V into the pLV-EF1a-MCS-IRES-Hyg plasmid (Biosettia). The near-infrared fluorescent protein iRFP720 cDNA construct was described previously (Zanca et al., 2017). The MMP1 Mission shRNA pLKO.1 plasmid was obtained from Sigma (clone NM_002421.3–986s21c1) and the pLKO.1 shGFP2 construct was previously reported (Fenton et al., 2012).
Retro- and Lentivirus Production and Transduction
To produce retrovirus, HEK 293T cells were co-transfected with pLRNL vectors and the pCL10A1 packaging construct using Lipo-fectamine 2000 reagent (Life Sciences). Viral supernatants were collected and filtered at 48 and 72 hr following transfection and used to infect cells overnight in the presence of 10 μg/mL polybrene. Cells were then selected and maintained with neomycin (400 μg/mL). To produce lentivirus, HEK 293T cells were co-transfected with pLV or pLKO.1 vectors, along with pRev, pMDL, and VSVg packaging constructs using Lipofectamine 2000. Supernatants were collected and filtered at 48 and 72 hr after transfection. Viral supernatant was concentrated by centrifugation at 23,000 × g for 2 hr at 4°C and used to infect cells overnight in the presence of 10 μg/mL polybrene. Cells were selected and maintained with hygromycin (200 μg/mL, pLV-EF1a-MCS-IRES-Hyg), puromycin (2 μg/ml, shMMP1), or blastocidin (5 μg/ml, iRFP720), depending on vector. Gene expression was verified by western blot, real time PCR, and FACS analysis.
FACS (Fluorescent-Activated Cell Sorter) Sorting and Analysis
For the generation of stable U87 cell lines and HK281 GBM-spheres expressing equal levels of EGFR, cells were collected at 1 × 106 cells in 100 μL FACS buffer (PBS + 1% FBS) and exposed to 1 μg of the EGFR monoclonal antibody 528, which recognizes WT and mutant EGFR, for 1 hr. Cells were then exposed to FITC-conjugated goat anti-mouse IgG antibody for 1 hr. Stained cells were analyzed by SH800 Cell Sorter (Sony Biotechnology) and sorted to within 10% of the mean FITC intensity of control cells expressing EGFRvIII. Sorted cells were cultured and reanalyzed to confirm stability of receptor levels. To measure binding affinity to the mAb806 antibody, the cells were processed as above but each cell line was stained separately with 1 μg of either antibody 528 or mAb806, followed by a secondary staining with a FITC-conjugated antibody. Mean FITC intensity following mAb806 staining was normalized to the corresponding mean FITC intensity for antibody 528.
Western Blotting
Cells were seeded in 10% FBS overnight to allow for adherence and then serum starved for 24 hr and treated with or without EGF at 100 ng/mL for 10 min at 37°C. Cell lysates were collected on ice with RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS, 5 mM EDTA) supplemented with protease (Roche) and phosphatase (Sigma) inhibitors. Protein concentration was determined using a standard DC assay (Bio-Rad) and equal amounts were loaded and separated by SDS-PAGE and then transferred to PVDF membranes. The membranes were blocked with buffer containing 5% BSA in tris-buffered saline with 0.1% Tween-20 (TTBS) for 1 h and incubated overnight with primary antibodies at 4°C. Membranes were washed with TTBS, and then incubated with HRP-conjugated secondary antibodies (Sigma-Aldrich) and developed with an enhanced chemiluminescence detection kit (Pierce). ImageJ software was used to quantify band signal intensity normalized to total protein.
FMT Imaging
For the intracranial in vivo experiments, tumor growth was monitored via fluorescence molecular tomography using the FMT 2500 Fluorescence Tomography System (PerkinElmer). Signal intensity was calculated based on ROI and analyzed using Perkin Elmer software.
Immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) tissue sections were prepared by the Histology Core Facility at UCSD Moores Cancer Center. Immunohistochemistry was performed according to standard procedures using the Scytek SensiTek HRP (AEC) Staining System. Antigen was retrieved by boiling slides in 0.01 M of sodium citrate (pH 6.0) in an oven for 30 min. Sections were incubated with primary antibody at 4°C overnight, followed by incubation with biotinylated secondary antibodies at room temperature for 20 min. Nine representative images from each immunostained section were taken with a Keyence BZ-X700 microscope and analyzed with BZ-X Analyzer Keyence software.
For pathological examination, the entire head was collected and fixed for approximately 5 days in 4% PFA at 4°C. After fixation, brain was removed from the skull and serially sliced using the coronal brain matrix system (Zivic Instruments BSMYS001–1). Five slices were obtained from each brain using the following anatomical landmarks as references: paraflocculi, infundibulum/median eminence, optic chiasm, cranial border of olfactory tubercles. Sliced brains were then processed for paraffin embedding (Thermo Scientific Excelsior™ AS Tissue Processor and HistoStar™ Embedding Workstation) and serial sagittal sections of 5 μm were obtained (Reichert Jung 2030 microtome). Sections were mounted on Superfrost microscope slides (Fisherbrand 12–550-14) and stained with hematoxylin and eosin (H&E) using the Gemini AS automated slide stainer (Thermo Fisher Scientific). H&E-stained slides were finally evaluated by a board certified veterinary pathologist (ER) and representative pictures were captured using an Olympus BX53 microscope coupled with an Olympus DP25 camera.
Animal MRI Studies
All MRI studies were performed in the Small Animal Imaging Facility in the Department of Radiology at the University of Pennsylvania, on a 9.4T horizontal bore scanner (Agilent Inc, Palo Alto, CA) equipped with a 12 cm ID, 40 gauss/cm gradient tube. Animals were prepared for study by induction of general anesthesia using 1–2% inhaled isoflurane through a nose cone. A respiration pillow and rectal temperature probe were placed on the animal and attached to a MR compatible vital signs monitoring system (Small Animal Instrument Inc, Stonybrook, NY). The animals were then mounted in a 20 mm ID quadrature birdcage RF coil (M2M Imaging, Cleveland, OH) and positioned in the magnet. The vital signs monitoring system was equipped with a regulated warm air source which was directed over the animal during the study to maintain the core body temperature at 37±1°C. Respiration and core body temperature were monitored throughout the study and the isoflurane level was adjusted as needed to maintain a deep anesthesia. Following optimization of RF transmitter power and generation of scout images, a contiguous series of T2-weighted, fast spin echo, axial slices spanning the brain (cerebral cortex or cerebellum) was generated with the following parameters: TR = 3800 msec; ETL = 8; ESP = min (10.96); kzero = 5; Effective TE = 54.80 msec; averages = 8; dummy scans = 2; FOV = 20 × 20; matrix = 256 × 256; slice thickness = 0.5 mm; 30 slices total. Images were converted to the Meta file format for 3D volumetric analysis using the ITK-SNAP program. The total brain volume and volume of the T2 abnormality were determined (Yushkevich et al., 2006). The overlay maps were generated using ParaView (Hansen and Johnson, 2005).
Cell Invasion Assay
In vitro cell invasion was evaluated using a 24-well chemotaxis chamber equipped with a polycarbonate filter with 8 μm pores (Costar, Corning, NY). Prior to plating, transwells were coated with Pure Col, Bovine Collagen Solution, Type I collagen for 30 min at 37°C (1:30 in 0.1% FBS, DMEM for U87 cells, 1:30 in F12 DMEM for HK281 GBM-spheres). Serum starved cells were incubated with the indicated inhibitor for 24 hr prior to plating. Cells were then counted and plated in the upper chamber and left to migrate for 6 hr towards 10% FBS, DMEM. Following incubation, cells that traversed the membrane were fixed and stained with crystal violet and non-migrated cells were removed with a cotton-tipped applicator. Cells were quantified by averaging the cell count of 5 different view fields at 20× magnification from at least three independent transwells.
BrdU Assay
For U87 glioma cells, BrdU assessment was carried out as follows: cells were plated on coverslips (10,000 cells/well in a 24 well plate), cultured for 7 days, and then incubated in the presence of 10 μM BrdU for 4–6 hr. The cells were fixed in 4% PFA (Sigma), stained with anti-BrdU antibody (BD), and counterstained with Hoechst 33258 (Sigma). The number of BrdU-positive cells was scored as a percentage of the total number of cells counterstained with Hoechst 33258. All pictures were captured using Nikon DS-Ri1 microscope. The assay was performed in triplicate for all conditions. For HK281 GBM-spheres, cells were plated in 6-well dishes (200,000 cells/well), cultured for 24 hr and then incubated in the presence of 10 μM BrdU for 6 hr. Cells were washed once with PBS, and 50,000 were plated on coverslips in a 24-well dish. The plate was centrifuged at 1500 rpm for 5 min to allow cells to adhere. Cells were then fixed with 4% PFA and stained with anti-BrdU antibody as described above.
Real Time-PCR
Cells were harvested with 0.5% trypsin-EDTA, centrifuged for 5 min at 2,000 rpm, and cell pellets frozen. Cell pellets were homogenized using a QIAshredder (Qiagen, Valencia, CA) and RNA was isolated using an RNeasy Mini Kit (Qiagen, Valencia, CA). Real time continuous detection of PCR product was achieved using Sybr Green (Applied Biosystems, Carlsbad, CA). GAPDH was used as an internal control with relative quantification being expressed as a ratio of the difference in the number of cycles needed for expression of a gene. Primers were designed using the Primer3 version 4.0.0. (Table S5).
QUANTIFICATION AND STATISTICAL ANALYSIS
All population analyses were conducted using Stat 14.2 (Stata Corporation, College Station, TX). Time to event data were analyzed using Kaplan-Meier methods, and group differences were tested using the log-rank test. Other associations among binary or categorical variables were analyzed using contingency table methods, and tested using the chi-square statistic. Results from experiments are presented as mean values ± standard error of the mean (SEM). Statistical analyses were carried out by unpaired Student’s t-test using GraphPad Prism® 5.01 software. P values < 0.05 were considered statistically significant. For the animal survival analysis, Kaplan–Meier curves were plotted and compared using the log rank test. Statistical details can be found in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The software tool used for skull-stripping (i.e., MUSE (Doshi et al., 2016)) is available in www.med.upenn.edu/sbia/muse.html. The software tool used for co-registration (i.e., FLIRT (Jenkinson et al., 2002)) is available in: fsl.fmrib.ox.ac.uk. We developed the Cancer Imaging Phenomics Toolkit (CaPTk) (Davatzikos et al., 2018) to facilitate clinical translation of complex computational algorithms, without requiring computational background (e.g., identification of genetic mutation imaging signatures (Bakas et al., 2017a)). Specifically for this study, CaPTk was used for 1) initialization of seed-points required by GLISTRboost, 2) image smoothing, as well as 3) extracting the quantitative imaging features. The code source, as well as executable installers, of CaPTk are available in: www.med.upenn.edu/cbica/captk. Finally, our segmentation approach, GLISTRboost (Bakas et al., 2017b), is available for public use through CBICA’s Image Processing Portal (IPP - ipp.cbica.upenn.edu), which allows users to perform data analysis using integrated algorithms, without any software installation, whilst using CBICA’s High Performance Computing resources.
Animal MRIs were converted to the Meta file format for 3D volumetric analysis using the ITK-SNAP (www.itksnap.org) program. The total brain volume and volume of the T2 abnormality were determined (Yushkevich et al., 2006). The overlay maps were generated using ParaView (Hansen and Johnson, 2005).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGFR antibody | BD Biosciences | 61007 |
| Pan-phospho-Tyrosine HRP antibody | R&D Systems | HAM1676 |
| p42/44 MAPK antibody | Cell Signaling Technology | 9102 |
| Phospho-p42/44 MAPK antibody | Cell Signaling Technology | 9101 |
| STAT3 antibody | Cell Signaling Technology | 4904 |
| Phospho-STAT3 y705 antibody | Cell Signaling Technology | 9145 |
| AKT antibody | Cell Signaling Technology | 9272 |
| Phospho-AKT s473 | Cell Signaling Technology | 4060 |
| Ki67 | Santa Cruz Biotechnology | Sc-15402 |
| β-Actin | Sigma | A 3854 |
| mAb806 | Biological Production Facility | N/A |
| BrdU (clone B44) antibody | BD Biosciences | BDB347580 |
| Hoechst 33258 | Sigma-Aldrich | B2883 |
| Bacterial and Virus Strains | ||
| pLRNL vector | Nishikawa et al., 1994 | N/A |
| pLV-EF1a-MCS-IRES-Hyg | Biosettia | cDNA-pLV02 |
| iRFP720 cDNA | Zanca et al., 2017 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| U0126 | LC Laboratories | U-6770 |
| U0126 | Cell Signaling Technology | 9903S |
| PD98059 | LC Laboratories | P-4313 |
| Experimental Models: Cell Lines | ||
| Human: U87 cells | ATCC | HTB-14 |
| Human: HK281 GBM-spheres | Laboratory of Harley Kornblum, UCLA | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Female NCI Ath/Nu | Charles River Labs | Strain 563 |
| Mouse: Female J:NU Homozygous | The Jackson Laboratory | 007850 |
| Oligonucleotides | ||
| shRNA targeting sequence: shMMP1: CCGGGC TAACCTTTGATGCTATAACCTCGAGGTTATAGC ATCAAAGGTTAGCTTTTTG | Sigma | TRCN0000372933 |
| shControl: pLKO.1shGFP2 | Fenton et al., 2012 | N/A |
| Primers for Real Time PCR, see Table S2 | This paper | N/A |
| Software and Algorithms | ||
| MUSE | Doshi et al., 2016 | www.med.upenn.edu/sbia/muse.html |
| FLIRT | Jenkinson et al., 2002 | fsl.fmrib.ox.ac.uk |
| Cancer Imaging Phenomics Toolkit (CaPTk) | Davatzikos et al., (2018) | www.med.upenn.edu/sbia/captk.html |
| GLISTRboost | Bakas et al., 2016, Bakas et al., 2017b | ipp.cbica.upenn.edu http://www.med.upenn.edu/sbia/glistrboost.html |
| ITK-SNAP | Yushkevich et al., 2006 | www.itksnap.org |
| ParaView | Hansen and Johnson, 2005 | www.paraview.org |
Significance.
Epidermal growth factor receptor (EGFR) extracellular domain (ECD) missense mutations at A289 confer increased invasiveness, increased proliferation, and decreased patient survival in glioblastoma (GBM). Importantly, when designing clinical trials for EGFR-targeted therapies in GBM, EGFR ECD missense mutations may give valuable insight into responsive patient populations.
Highlights.
EGFRA289D/T/V-mutated GBM tumors have distinct in vivo imaging characteristics
EGFRA289D/T/V-associated decrease in OS is driven by tumor proliferation and invasion
EGFRA289V leads to phosphorylation of Erk followed by increased MMP1 secretion
Targeting of EGFRA289V via mAb806 can reduce tumor growth and increase survival
ACKNOWLEDGMENTS
We would like to thank the following organizations for their support: Templeton Family Initiative in Neuro-Oncology (to Z.A.B.); NCI: 2T32CA009523–29A1 (to A.H.T.); Ligue National Contre le Cancer (to A.I.); Lawski Fund for Biomedical Research (to L.O.); NINDS: R01NS042645 (to C.D. and D.M.O.), NCI: U24CA189523, NCATS: UL1TR001878, ITMAT of the University of Pennsylvania (to S.B., C.D., and D.M.O.); NBTS: Defeat GBM Research Collaborative, NIH: RO1NS080939 (to F.F). We would also like to thank Kent Osborn, D.V.M., Ph.D., Associate Director for the Animal Care Program at UCSD, and Enrico Radaelli, M.S., V.M.D., D.E.C.V.P., Technical Director for the Comparative Pathology Core in the Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, for their assistance with histology.
DECLARATION OF INTERESTS
Spyridon Bakas has a patent application related to the detection of EGFRvIII mutation by imaging methods that are different from the ones used herein. David Reardon has received support from Acerta Pharmaceuticals, Agenus, Celldex, EMD Serono, Incyte, Midatech, Omniox, and Tragara. He is a consultant for and has received honoraria from Abbvie, Agenus, Bristol-Myers Squibb, Celldex, EMD Serono, Genentech/Roche, Inovio, Merck, Merck KGaA, Monteris, Novocure, Oncorus, Oxigene, Regeneron, Stemline, Taiho Oncology. Ahmed Idbaih has received support from Carthera, BMS, Hoffmann-La Roche, and Cipla. Michael Weller has received research grants from Abbvie, Acceleron, Actelion, Bayer, Merck, Sharp & Dohme (MSD), Merck (EMD), Novocure, OGD2, Piqur, Roche, and Tragara, and honoraria for lectures or advisory board participation or consulting from Abbvie, BMS, Celgene, Celldex, Merck, Sharp & Dohme (MSD), Merck (EMD), Novocure, Orbus, Pfizer, Progenics, Roche, Teva, and Tocagen. Jennifer Morrissette is a consultant for Novartis. Andrew Scott is an inventor on patents related to mAb806 and has received research support from Abbvie, National Health and Medical Research Council, and Cancer Council Victoria. He is also a consultant with Life Science Pharmaceuticals. Christos Davatzikos has a patent application related to the detection of EGFRvIII mutation by imaging methods that are different from the ones used herein. Frank Furnari is an inventor on patent 9,023,356 on a treatment method using EGFR antibodies and src inhibitors in related formulations. Donald O’Rourke is an inventor on patents 7,625,558 and 6,417,168 held by the University of Pennsylvania on methods of treating cancer cells with erbB inhibition and cytotoxic therapy. In addition, he has a patent application related to the detection of EGFRvIII mutation by imaging methods that are different from the ones used herein.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.06.006.
REFERENCES
- Aerts HJ (2016). The Potential of radiomic-based phenotyping in precision medicine: a review. JAMA Oncol. 2, 1636–1642. [DOI] [PubMed] [Google Scholar]
- Anand M, Van Meter TE, and Fillmore HL (2011). Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J. Neurooncol 104, 679–687. [DOI] [PubMed] [Google Scholar]
- Aronen HJ, Gazit IE, Louis DN, Buchbinder BR, Pardo FS, Weisskoff RM, Harsh GR, Cosgrove GR, Halpern EF, Hochberg FH, et al. (1994). Cerebral blood volume maps of gliomas: comparison with tumor grade and histologic findings. Radiology 191, 41–51. [DOI] [PubMed] [Google Scholar]
- Bakas S, Akbari H, Pisapia J, Martinez-Lage M, Rozycki M, Rathore S, Dahmane N, O’Rourke DM, and Davatzikos C (2017a). In vivo detection of EGFRvIII in glioblastoma via perfusion magnetic resonance imaging signature consistent with deep peritumoral infiltration: the phi-index. Clin. Cancer Res 23, 4724–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakas S, Akbari H, Sotiras A, Bilello M, Rozycki M, Kirby JS, Freymann JB, Farahani K, and Davatzikos C (2017b). Advancing the Cancer Genome Atlas glioma MRI collections with expert segmentation labels and radiomic features. Sci. Data 4, 170117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakas S, Binder ZA, Morrissette JJD, Akbari H, O’Rourke D, and Davatzikos C (2017c). NIMG-07. Unifying magnetic resonance imaging signature of EGFR pathway activation in glioblastoma consistent with uniformly aggressively infiltration. Neuro Oncol. 19, vi143. [Google Scholar]
- Bakas S, Chatzimichail K, Hunter G, Labbé B, Sidhu PS, and Makris D (2017d). Fast semi-automatic segmentation of focal liver lesions in contrast-enhanced ultrasound, based on a probabilistic model. Comput. Methods Biomech. Biomed. Eng. Imaging Vis 5, 329–338. [Google Scholar]
- Bakas S, Zeng K, Sotiras A, Rathore S, Akbari H, Gaonkar B, Rozycki M, Pati S, and Davatzikos C (2016). GLISTRboost: combining multimodal MRI segmentation, registration, and biophysical tumor growth modeling with gradient boosting machines for glioma segmentation. Brainlesion (2015) 9556, 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessman NJ, Bagchi A, Ferguson KM, and Lemmon MA (2014). Complex relationship between ligand binding and dimerization in the epidermal growth factor receptor. Cell Rep. 9, 1306–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G, et al. (1995). Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The polymer-brain tumor treatment group. Lancet 345, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das G, Shiras A, Shanmuganandam K, and Shastry P (2011). Rictor regulates MMP-9 activity and invasion through Raf-1-MEK-ERK signaling pathway in glioma cells. Mol. Carcinog 50, 412–423. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Rathore S, Bakas S, Pati S, Bergman M, Kalarot R, Sridharan P, Gastounioti A, Jahani N, Cohen E, et al. (2018). Cancer imaging phenomics toolkit: quantitative imaging analytics for precision diagnostics and predictive modeling of clinical outcome. J. Med. Imaging (Bellingham) 5, 011018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi J, Erus G, Ou Y, Resnick SM, Gur RC, Gur RE, Satterthwaite TD, Furth S, Davatzikos C, and Alzheimer’s Neuroimaging, I. (2016). MUSE: MUlti-atlas region Segmentation utilizing Ensembles of registration algorithms and parameters, and locally optimal atlas selection. Neuroimage 127, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrow R, and Darefsky AS (2011). Demographic variation in incidence of adult glioma by subtype, United States, 1992–2007. BMC Cancer 11, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingson BM, Gerstner ER, Smits M, Huang RY, Colen R, Abrey LE, Aftab DT, Schwab GM, Hessel C, Harris RJ, et al. (2017). Diffusion MRI phenotypes predict overall survival benefit from anti-VEGF monotherapy in recurrent glioblastoma: converging evidence from phase II trials. Clin. Cancer Res 23, 5745–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton TR, Nathanson D, Ponte de Albuquerque C, Kuga D, Iwanami A, Dang J, Yang H, Tanaka K, Oba-Shinjo SM, Uno M, et al. (2012). Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240. Proc. Natl. Acad. Sci. USA 109, 14164–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R, et al. (2009). Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol 27, 4733–4740. [DOI] [PubMed] [Google Scholar]
- Gan HK, van den Bent M, Lassman AB, Reardon DA, and Scott AM (2017). Antibody-drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat. Rev. Clin. Oncol 14, 695–707. [DOI] [PubMed] [Google Scholar]
- Gaonkar B, and Davatzikos C (2013). Analytic estimation of statistical significance maps for support vector machine based multi-variate image analysis and classification. Neuroimage 78, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaonkar B, Macyszyn L, Bilello M, Sadaghiani MS, Akbari H, Atthiah MA, Ali ZS, Da X, Zhan Y, O’Rourke D, et al. (2015). Automated tumor volumetry using computer-aided image segmentation. Acad. Radiol 22, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevaert O, Echegaray S, Khuong A, Hoang CD, Shrager JB, Jensen KC, Berry GJ, Guo HH, Lau C, Plevritis SK, et al. (2017). Predictive radiogenomics modeling of EGFR mutation status in lung cancer. Sci. Rep 7, 41674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A, Yun J, Samanamud J, Sims JS, Banu M, et al. (2014). MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 111, 12550–12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooya A, Pohl KM, Bilello M, Cirillo L, Biros G, Melhem ER, and Davatzikos C (2012). GLISTR: glioma image segmentation and registration. IEEE Trans. Med. Imaging 31, 1941–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves MD, Puduvalli VK, Hess KR, Jaeckle KA, Peterson P, Yung WK, and Levin VA (2002). Phase II trial of temozolomide plus the matrix metalloproteinase inhibitor, marimastat, in recurrent and progressive glioblastoma multiforme. J. Clin. Oncol 20, 1383–1388. [DOI] [PubMed] [Google Scholar]
- Gutman DA, Cooper LA, Hwang SN, Holder CA, Gao J, Aurora TD, Dunn WD Jr., Scarpace L, Mikkelsen T, Jain R, et al. (2013). MR imaging predictors of molecular profile and survival: multi-institutional study of the TCGA glioblastoma data set. Radiology 267, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann C, Anacker J, Ernestus RI, and Vince GH (2012). A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol 3, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CD, and Johnson CR (2005). The Visualization Handbook (Elsevier-Butterworth Heinemann; ). [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med 352, 997–1003. [DOI] [PubMed] [Google Scholar]
- Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, Sawaya R, and Aldape K (2005). Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res 11, 1462–1466. [DOI] [PubMed] [Google Scholar]
- Hogea C, Biros G, Abraham F, and Davatzikos C (2007). A robust framework for soft tissue simulations with application to modeling brain tumor mass effect in 3D MR images. Phys. Med. Biol 52, 6893–6908. [DOI] [PubMed] [Google Scholar]
- Idbaih A, Aimard J, Boisselier B, Marie Y, Paris S, Criniere E, Carvalho Silva R, Laigle-Donadey F, Rousseau A, Mokhtari K, et al. (2009). Epidermal growth factor receptor extracellular domain mutations in primary glioblastoma. Neuropathol. Appl. Neurobiol 35, 208–213. [DOI] [PubMed] [Google Scholar]
- Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, et al. (2010). Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 24, 1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura H, Achrol AS, Mitchell LA, Loya JJ, Liu T, Westbroek EM, Feroze AH, Rodriguez S, Echegaray S, Azad TD, et al. (2015). Magnetic resonance image features identify glioblastoma phenotypic subtypes with distinct molecular pathway activities. Sci. Transl. Med 7, 303ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M, Bannister P, Brady M, and Smith S (2002). Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17, 825–841. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens TE, Woolrich MW, and Smith SM (2012). Fsl. Neuroimage 62, 782–790. [DOI] [PubMed] [Google Scholar]
- Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, Yoshimoto K, King JC, Nghiemphu P, Yuza Y, et al. (2006). Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 3, e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M, Udi Y, Solomonov I, and Sagi I (2017). Next generation matrix metalloproteinase inhibitors - novel strategies bring new prospects. Biochim. Biophys. Acta 1864 (11 Pt A), 1927–1939. [DOI] [PubMed] [Google Scholar]
- Levin VA, Phuphanich S, Yung WK, Forsyth PA, Maestro RD, Perry JR, Fuller GN, and Baillet M (2006). Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J. Neurooncol 78, 295–302. [DOI] [PubMed] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, and Ellison DW (2016). The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. [DOI] [PubMed] [Google Scholar]
- Macyszyn L, Akbari H, Pisapia JM, Da X, Attiah M, Pigrish V, Bi Y, Pal S, Davuluri RV, Roccograndi L, et al. (2016). Imaging patterns predict patient survival and molecular subtype in glioblastoma via machine learning techniques. Neuro Oncol. 18, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima K, Johns TG, Luwor RB, Scott AM, Stockert E, Jungbluth AA, Ji XD, Suvarna P, Voland JR, Old LJ, et al. (2001). Growth suppression of intracranial xenografted glioblastomas overexpressing mutant epidermal growth factor receptors by systemic administration of monoclonal antibody (mAb) 806, a novel monoclonal antibody directed to the receptor. Cancer Res. 61, 5349–5354. [PubMed] [Google Scholar]
- Miura FK, Alves MJ, Rocha MC, da Silva R, Oba-Shinjo SM, and Marie SK (2010). Xenograft transplantation of human malignant astrocytoma cells into immunodeficient rats: an experimental model of glioblastoma. Clinics (Sao Paulo) 65, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada M, Okada Y, and Yamashita J (2003). The role of matrix metalloproteinases in glioma invasion. Front. Biosci 8, e261–e269. [DOI] [PubMed] [Google Scholar]
- Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, and Huang HJ (1994). A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 91, 7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana L, Hospital A, and Orozco M (2014). Oncogenic mutations of the EGF-receptor ectodomain reveal an unexpected mechanism for ligand-independent activation. bioRxiv. 10.1101/009068. [DOI] [Google Scholar]
- Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, and Barnholtz-Sloan JS (2015). CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 17 (Suppl 4), iv1–iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AC, Boghaert ER, Vaidya KS, Mitten MJ, Norvell S, Falls HD, DeVries PJ, Cheng D, Meulbroek JA, Buchanan FG, et al. (2016). ABT-414, an antibody-drug conjugate targeting a tumor-selective EGFR epitope. Mol. Cancer Ther 15, 661–669. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, Fichtel L, Sulman EP, Gomez E, Fischer J, et al. (2017). Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol. 19, 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sled JG, Zijdenbos AP, and Evans AC (1998). A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans. Med. Imaging 17, 87–97. [DOI] [PubMed] [Google Scholar]
- Stojic J, Hagemann C, Haas S, Herbold C, Kuhnel S, Gerngras S, Roggendorf W, Roosen K, and Vince GH (2008). Expression of matrix metalloproteinases MMP-1, MMP-11 and MMP-19 is correlated with the WHO-grading of human malignant gliomas. Neurosci. Res 60, 40–49. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Stupp R, Wong ET, Kanner AA, Steinberg D, Engelhard H, Heidecke V, Kirson ED, Taillibert S, Liebermann F, Dbaly V, et al. (2012). NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur. J. Cancer 48, 2192–2202. [DOI] [PubMed] [Google Scholar]
- Talasila KM, Soentgerath A, Euskirchen P, Rosland GV, Wang J, Huszthy PC, Prestegarden L, Skaftnesmo KO, Sakariassen PO, Eskilsson E, et al. (2013). EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 125, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne AH, Zanca C, and Furnari F (2016). Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro Oncol. 18, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visnyei K, Onodera H, Damoiseaux R, Saigusa K, Petrosyan S, De Vries D, Ferrari D, Saxe J, Panosyan EH, Masterman-Smith M, et al. (2011). A molecular screening approach to identify and characterize inhibitors of glioblastoma stem cells. Mol. Cancer Ther 10, 1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing WJ, Zou Y, Han QL, Dong YC, Deng ZL, Lv XH, Jiang T, and Ren H (2013). Effects of epidermal growth factor receptor and phosphatase and tensin homologue gene expression on the inhibition of U87MG glioblastoma cell proliferation induced by protein kinase inhibitors. Clin. Exp. Pharmacol. Physiol 40, 13–21. [DOI] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, and Gerig G (2006). User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 31, 1116–1128. [DOI] [PubMed] [Google Scholar]
- Zanca C, Villa GR, Benitez JA, Thorne AH, Koga T, D’Antonio M, Ikegami S, Ma J, Boyer AD, Banisadr A, et al. (2017). Glioblastoma cellular cross-talk converges on NF-kappaB to attenuate EGFR inhibitor sensitivity. Genes Dev. 31, 1212–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chang K, Ramkissoon S, Tanguturi S, Bi WL, Reardon DA, Ligon KL, Alexander BM, Wen PY, and Huang RY (2017). Multimodal MRI features predict isocitrate dehydrogenase genotype in high-grade gliomas. Neuro Oncol. 19, 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The software tool used for skull-stripping (i.e., MUSE (Doshi et al., 2016)) is available in www.med.upenn.edu/sbia/muse.html. The software tool used for co-registration (i.e., FLIRT (Jenkinson et al., 2002)) is available in: fsl.fmrib.ox.ac.uk. We developed the Cancer Imaging Phenomics Toolkit (CaPTk) (Davatzikos et al., 2018) to facilitate clinical translation of complex computational algorithms, without requiring computational background (e.g., identification of genetic mutation imaging signatures (Bakas et al., 2017a)). Specifically for this study, CaPTk was used for 1) initialization of seed-points required by GLISTRboost, 2) image smoothing, as well as 3) extracting the quantitative imaging features. The code source, as well as executable installers, of CaPTk are available in: www.med.upenn.edu/cbica/captk. Finally, our segmentation approach, GLISTRboost (Bakas et al., 2017b), is available for public use through CBICA’s Image Processing Portal (IPP - ipp.cbica.upenn.edu), which allows users to perform data analysis using integrated algorithms, without any software installation, whilst using CBICA’s High Performance Computing resources.
Animal MRIs were converted to the Meta file format for 3D volumetric analysis using the ITK-SNAP (www.itksnap.org) program. The total brain volume and volume of the T2 abnormality were determined (Yushkevich et al., 2006). The overlay maps were generated using ParaView (Hansen and Johnson, 2005).