

Abstract
Purpose of this review:
There has been rapid progress in defining novel causative gene variants responsible for a large spectrum of human epilepsy syndromes and subtypes. Of particular interest is the discovery that somatic mutations, e.g., non-inherited mutations occurring in neuroglial progenitor cells during embryonic brain development, are highly linked to malformations of cortical development (MCD) such as focal cortical dysplasia (FCD) type II and hemimegalencephaly (HME).
Recent Findings:
Somatic gene variants have been identified in genes encoding regulatory proteins within the mechanistic target of rapamycin (mTOR) signaling cascade and have thus comprised the group classified as mTORopathies. FCDII and HME often result from mutations in indentical genes suggesting that these are spectrum disorders. An exciting recent development has been the identification of somatic mutations causing both FCD Ia and non-lesional neocortical epilepsy.
Summary:
Defining somatic gene mutations in brain tissue specimens has shed new light on how MCD form and the mechanisms of epileptogenesis associated with MCD. Trials of mTOR inhibitors (mTORi) in tuberous sclerosis complex (TSC) have demonstrated that inhibition of mTOR activation in mTORopathies can reduce seizure frequency. New somatic mutations found for a variety of epilepsy syndromes may provide new targets for clinical therapeutics.
Keywords: mTOR, malformations of cortical development, focal cortical dysplasia, epilepsy
Introduction
Over the past decade there has been rapid progress in epilepsy genetics research in which single gene mutations have been linked to specific epilepsy syndromes, i.e., Dravet syndrome, benign familial neonatal convulsions, and epileptic encephalopathies (EE) [for review, see 1]. Most causative genes have been identified as part of large pedigree analyses to define inherited or de novo germline variants but in some cases such as EE, focused analysis of trios (proband and parents) has revealed causative genes. A major impact of gene discovery is a greater understanding of the pathogenic mechanisms causing each epilepsy subtype by positing encoded proteins into functional roles regulating, for example, signaling cascades or synaptic transmission, within neurons or glia.
Malformations of cortical development (MCD) are among the most common causes of intractable pediatric epilepsy and account for a substantial number of adult epilepsy cases as well. While germline genes mutations causing MCD such as lissencephaly, polymicrogyria, and periventricular nodular heterotopia have been identified, the molecular genetic etiology of MCD such as focal cortical dysplasia (FCD) and hemimegalencephaly (HME) has remained poorly defined until recent years [2*]. The central conundrum facing investigators has been how to explain the focal nature of FCD and HME, i.e., structural lesions confined to a brain region, surrounded by normal cerebral cortex.
Embryonic Development of the Cerebral Cortex
A brief overview of cerebral cortical development is needed to fully understand how somatic mutations can cause MCD [for review, see 3]. The cerebral cortex develops in humans at approximately gestational weeks 8-20 with the clear delineation of the telencephalic ventricular zone (VZ) within the neural tube epithelium. Within the VZ, successive rounds of mitosis of neuroglial progenitor cells (the proliferative phase) proceeds until gestational week 20 to generate the hexalaminar cortical structure seen in mature human brain. Between weeks 8-20, neuroglial progenitor cells in the VZ are directed to a neural lineage at specific birthdates guided by a complex molecular framework, and then exit the mitotic cycle, and embark on a migratory journey from the VZ to the nascent cortical plate (the migratory phase). Each cell born at a specific birthdate is destined for a specific layer between layers II-VI (layer I is populated and defined in the earliest stages of cortical development). Once the rudimentary 6 layered structure has been established, cortical neurons begin to extend dendrites and axons, and to receive numerous axonal projections from distant brain areas such as the brainstem, thalamus, and other cortical regions (organizational phase). Through a completely separate mechanism, GABAergic interneurons, derived not from the VZ but instead from the medial ganglionic eminence, will migrate into the cortex to populate the 6 cortical layers. Once the cortical cellular matrix has been assembled, further connectivity, myelination, and pruning will refine the cortical structure. The effects of germline or somatic gene mutations may alter normal neuronal or glial function throughout all phases of cortical development. Of note, somatic mutations will only occur during the mitotic (proliferative) phase.
Histopathology of MCD
The defining histopathological features of FCD have been formalized by a recent and revised ILAE Task Force [4**] to include FCD subtypes Ia, Ib, Ic, IIa, IIb, IIc, IIIa, IIIb, IIIc, and IIId; there is no formalized histopathological classification scheme for HME. Histopathological features of FCD IIa and IIb and many HME tissue specimens are similar to tubers in tuberous sclerosis complex (TSC) in which the classical findings of enlarged (cytomegalic) dysmorphic neurons and balloon cells are observed in all three disorders. The histopathological similarities between tubers, FCD, and HME suggest commonalities in pathogenesis and posed compelling questions regarding the developmental pathogenesis such as: 1) how do the distinct cell types form in FCD/HME; 2) what regulates the size and extent of each lesion; 3) what makes these lesions so highly epileptogenic?
Major breakthroughs in understanding MCD began with the identification of the TSC1 and TSC2 genes and their function within the mTOR signaling cascade [5]. The mTOR pathway is highly activated in resected tuber specimens as evidenced by selective hyperphosphorylation of the mTOR substrates p70S6 kinase, ribosomal S6 protein, and 4-EBP1 [6]. Interestingly, the profile of mTOR hyperactivation in FCD type IIa, IIb, and HME was shown to be similar, though not identical, to tubers [6-9] suggesting a common molecular pathogenesis linked to mTOR. While TSC can be inherited as an autosomal dominant disorder or can result from spontaneous germline mutations, FCD and HME were diagnosed almost exclusively as sporadic disorders with only rare examples of family pedigrees. Since FCD and HME affect restricted regions of the brain, a logical molecular mechanism to account for these lesions was somatic mutation occurring during early stages of embryogenesis.
Somatic Mutations and mTORopathies – FCDIIa, FCDIIb, and HME
Experimental strategies to identify somatic mutations relied on next-generation sequencing analysis of genomic DNA extracted from paired brain and blood samples (Fig.1). The first definitive evidence for a somatic mutational mechanism responsible for MCD demonstrated a somatic mosaic mutation in AKT3 in surgically resected HME tissues [10*]. The investigators defined trisomy of chromosome 1q in HME encompassing the AKT3 locus and then identified an activating mutation in AKT3 (c.49G>A, p.E17K); confirmation of the somatic mechanism was assured by absence of the mutation in either peripheral blood or buccal swab DNA. A subsequent report [11] using exome sequencing and mass spectrometry analysis in surgically resected HME specimens (n = 20 cases) discovered de novo somatic mutations within the PIK3CA, AKT3 and MTOR genes in 30% of affected individuals. A recurrent PIK3CA mutation (c.1633G>A, E545K) was found in four HME cases. Hyperphosphorylation of ribosomal S6 protein was identified immunohistochemically in these specimens, demonstrating mTOR hyperactivation. A number of more recent studies have identified mutations in genes encoding canonical signaling proteins within the mTOR pathway including PI3KCA, AKT3, and MTOR itself in FCDIIa, FCDIIb as well as HME [12*-19]. Two very recent studies identified somatic variants in TSC1 and TSC2 in FCD [16] and Rheb in HME [17]. Somatic MTOR variants were first identified (c.7280T>C; p.L2427P) in two subjects with FCDII [12] and subsequent deep sequencing of MTOR in resected brain tissue revealed 8 distinct somatic missense MTOR variants, all predicted to cause mTOR kinase hyperactivation, in an additional 10/73 subjects, together accounting for 15.6% of all subjects with FCDII studied. Expression of mutant MTOR constructs in mice caused altered brain architecture and seizures [12]. Four distinct somatic MTOR variants were identified in 6 of 13 (46%) individuals with FCD type IIb [13]. Somatic MTOR variants (p.L1460P mutation within the focal adhesion targeting (FAT) domain of MTOR, and the p.S2215F and p.S2215Ymutations in the mTOR kinase domain) were identified in 4 individuals with FCDIIa [14]. Somatic MTOR mutations have been identified in HME [18] within the kinase domains and in the FAT domain [19]. Histopathological evidence of hyperactive mTOR signaling was shown in these reports in resected brain tissue specimens, thus supporting the activating effect of the MTOR mutations.
Figure 1.
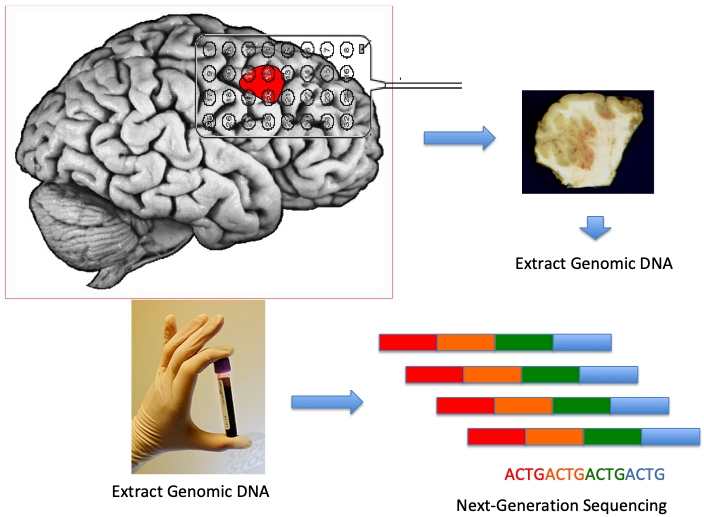
Experimental strategy to identify somatic gene mutations in epilepsy. The ictal onset zone (depicted in red), often an MCD visualized via pre-operative MRI, is defined by EEG (grid, depth or stereo-EEG) electrodes. Genomic DNA is extracted from the resected tissue and from a blood sample. Next-generation sequencing approaches are implemented to define variants within known and novel genes.
A key feature of the identified somatic variants within the reported cases was the often very low allelic frequency rate i.e., the number of cells showing mutant allele rates within the resected brain tissue. For example, in several FCD cohorts, allelic frequency varied from of ~1% to 10% [e.g., 12,13] In contrast, in the HME specimens where more extensive area of the brain is abnormal, the allelic frequencies tended to be higher, i.e., 20-40% [e.g., 11,12,18,19]. These findings suggest an interesting gene dosage effect in which a greater variant burden is associated with more extensive and often more severe cortical malformations. Thus, as more cells within an MCD contain mTOR pathway gene variants, there is a great burden of hyperactive mTOR signaling which in turn causes more disruption of normal cortical cytoarchitecture [20].
Interestingly, germline mutations in several mTOR pathway genes including PI3KCA, PTEN, AKT3, DEPDC5, NPRL3, and MTOR have been reported in association with mTORopathies including FCDIIa, FCDIIb, and HME [21,22]. As in MCD associated with somatic mutations, histopathological hyperactive mTOR signaling is detected in in resected brain tissue specimens from cases associated with germline variants. One interesting challenge in understanding the pathogenesis of MCD associated with germline variants is the problem that had previously faced investigators working on TSC i.e., how could a heterozygous germline mutation, affecting all cells in the body and not causing mTOR hyperactivation, cause a focal malformation within a restricted brain area associated with hyperactivated signaling? In TSC, this question was answered by demonstrating that a somatic “second-hit” mutation in the unaffected gene allele can be detected in cortical tubers both in DNA extracted from whole tubers and in single, microdissected cells [23, 24]. The somatic mutations in the unaffected gene allele yield a homozygous condition, fully inactivating the gene, leading to loss of function of the encoded protein and mTOR hyperactivation. In FCD associated with germline DEPDC5 mutations, somatic second hit mutations have recently been identified in DEPDC5 [25*] suggesting a mechanism like TSC. In both TSC and DEPDC5-associated FCD, allelic heterozygosity does not lead to mTOR hyperactivation nor confer an MCD phenotype, and instead the combined effects of a germline mutation plus a somatic second hit mutation, causing homozygosity is required. In attempts to model FCD, clustered regularly interspaced short palindromic repeats (CRISPR) gene editing of Depdc5 in fetal mouse brain led to histopathological changes similar to FCDIIa and spontaneous seizures [26*]. In disorders such as TSC, where somatic second hit mutations appear to account for tuber formation, multiple brain lesions may be detected likely because of the frequency of somatic events in neural progenitor cells superimposed on the ubiquitous germline mutations present in every cell. In contrast, in FCD type II and HME, there is only a single somatic event that alters the genotype of cells within the lesion and thus lesions tend to be singular in these disorders.
Mechanisms of Somatic Mutational Events
There are numerous unanswered questions regarding the pathogenesis of somatic mutational events occurring during brain development. First, by what mechanisms do these mutations occur during brain development? While the precise mechanisms causing somatic variants leading to MCD remains unknown, several mechanisms have been explored in cancer biology to explain somatic variants. Somatic mutational events occur frequently in all mitotic cells during replication and transcription, but are largely rescued or prevented by a number of protective DNA editing programs and fail-safe mechanisms [27]. However, errors in mismatch repair, base excision repair, and transcription-coupled repair are contributors to somatic variants in human cancer and likely contribute to errors occurring in neuroglial progenitors cells during cortical development. Alternatively, some variants i.e., PI3KCA E545K are recognized as mutational hotspots prone to sequence variation. The enzyme APOBEC3 can cause G>A transitions across the genome and non-homologous end joining (NHEJ) errors can cause genomic variability. Finally, intermittent retrotransposition of long interspersed elements (LINE-1) can lead to somatic alterations in DNA sequence even in normal neurons [28].
A second question is when do somatic mutational events occur? Do these occur at a critical time window during embryonic brain development or are they random events scattered throughout corticogenesis? The exact time point at which these changes in gene sequence occur is not known, but likely the mutations occur in mitotic neuroglial progenitor cells within the VZ. Third, what is the progenitor cell of origin for MCD i.e., in what cell types do somatic mutations occur? Are they the same for each MCD subtype i.e., IIa versus IIb or do these pathological subtypes reflects distinct effects of mTOR pathway variants in distinct subsets of progenitor cells? For example, data in both human [29] and mouse [20] studies suggests that MCD result from somatic mutational events in neuroglial progenitor cells within the VZ. Since neuroglial progenitors are not heterogeneous, it is possible that mutations occurring in distinct progenitor cell subsets could account for some of the phenotypic variability of MCD i.e., FCDIIa versus FCDIIb (Fig.2).
Figure 2.
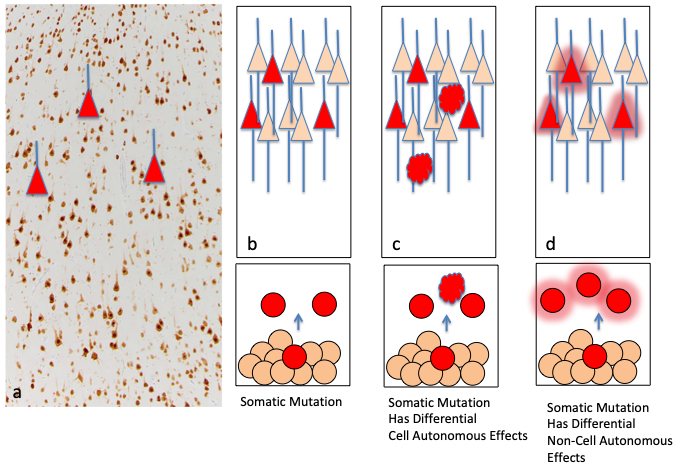
Effects of somatic mutations in epilepsy. In panel (a), a NeuN labeled brain section demonstrates cortical neurons and normal histopathology (modified from [34]). The red triangles represent neurons carrying somatic mutations scattered within the samples. Implicit in the somatic mechanism is that only a portion of cells carry the mutation and these are adjacent to cells with no genomic variants identified, thus representing a mosaic of affected and unaffected cells. (b) The developmental mechanism of somatic mutations begins with a mutation (red circle) in a mitotic progenitor cell within the VZ. The cell gives rise to daughter progeny also carrying the mutation which migrate to the cortex (red triangles as in (a)). The effects on cortical structure and network excitability may be cell autonomous i.e., from the mutation itself, (b) may reflect differential effects on different types of progenitors causing distinct cell types in MCD such as balloon cells (red ovoid shapes) in FCDII, or (c) may have non-cell autonomous effects on surrounding cells through changes in release of growth factors or neurotransmitters (red haze around circles).
Somatic Mutations, FCDIa and Non-Lesional Neocortical Epilepsy
Virtually all of the known somatic variants associated with MCD and epilepsy have been identified within mTOR pathway genes, and all have been associated with either FCDIIa or IIb histopathological subtypes. The conceptual paradigm in MCD is that somatic mutations in neuroglial progenitor cells disrupts normal programs that govern cell shape, size, and movement, yielding a focal malformation, often visualized by brain imaging, that is the pathological substrate for focal onset seizures. As such, these lesions are amenable to resective epilepsy surgery. In contrast, there a number of non-lesional neocortical focal epilepsies i.e., ADNFLE, and ADPEAF, that result from germline gene mutations, that do not cause MCD and are not amenable to epilepsy surgery. In these cases, differential patterns of gene expression dictate seizure onset zone. Understanding how seizures arise from a focal brain area without evidence of MCD, and where no germline variant can be detected has been a major research challenge. One possibility is that the normal radiological appearance belies an occult lesion e.g., MCD, glial scar, vascular malformation, that is below the limit of radiographic detection by a standard 3-Tesla MRI [see 30]. Alternatively, somatic mutations that do not alter cortical architecture and that are limited to a small number of neurons within a restricted brain area could provide an answer. Indeed, somatic mutational changes have been reported in a number of epilepsy syndromes without MCD but these have been due to somatic mosaisicm and have been detected throughout the body. For example, mixed somatic mosaicism has been reported for PCDH-19 in males with epileptic encephalopathy [31, 32]. Somatic deletions in SCN1A have been rarely reported in Dravet syndrome [33].
A recent study sought gene variants in a mixed cohort of radiographically non-lesional and MCD cases (n=58) using next-generation sequencing of DNA extracted from resected epilepsy surgery tissue samples [34**]. In this cohort, mutations in SLC35A2 were identified in 17% of the cases; in two cases, occult FCD1a pathology was found, and in 3 there was no MCD. SLC35A2 encodes a protein UGT1, that functions as a galactose transporter across the Golgi to post-translationally modify a wide array of proteins and glycolipids in neurons. Interestingly, germline SLC35A2 mutations have been reported in several case series of congenital disorders of glycosylation associated with epileptic encephalopathies [e.g., 35]. This study had several important implications for understanding epilepsy pathogenesis. First, germline mutations causing deficits in galactose transport are clearly linked to intractable epilepsy and thus it is logical that somatic SLC35A2 variants cause focal epilepsy. Second, the study identified the first FCD1a gene and may open new avenues for other genes either linked to galactose transport, or in completely distinct signaling cascades to account for FCD1a. Third, the data demonstrated that non-lesional brain MRIs often harbor very subtle MCD below the resolution of a conventional 3-Tesla magnet strength. Finally, the results show that a somatic mutation can cause focal onset epilepsy in the absence of structural brain lesion.
Mechanisms for Epileptogenesis
A pivotal unanswered question is why somatic mutations are causative in epilepsy? Clearly the disruption of cortical cytoarchitecture caused by mTOR pathway mutations leads to alterations in cortical network integrity and seizures (Fig.3). In addition, the gene mutation itself likely confers excitability in a cell autonomous mechanism to a focal cortical region. For example, spontaneous ictal discharges and seizures have been identified in a number of MCD mouse models [12,25,26], and a recent study demonstrated that V600E mutations in BRAF, a major cause of pediatric ganglioglioma, are independently linked to epileptogenesis [36].
Figure 3.
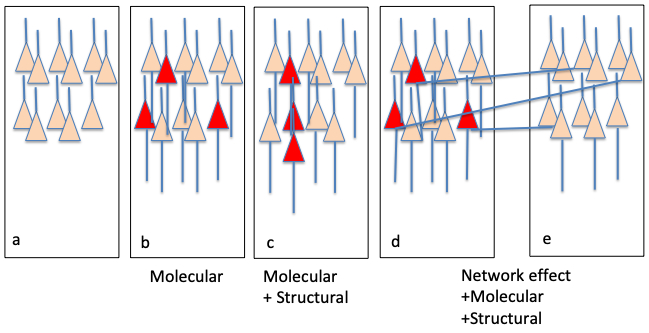
Possible functional links to epileptogenesis. There are 3 mechanisms that somatic mutations could cause seizures. The onset zone (b-d) is adjacent to normal cortex (a). The effects of variants can be purely “molecular” and causing intrinsic changes in excitability as in SLC35A2; (b), they may be a combination of “molecular” effects plus “structural” effects causing an MCD as in mTORopathies, or (c) the effects may include “molecular”, “structural”, and “network” alterations through aberrant connections to surrounding cortex (e).
Conclusion: Targeted Therapy and Precision Medicine Approaches
The identification of somatic gene variants in both lesional and non-lesional focal epilepsies provides invaluable therapeutic direction for clinical patient care. For example, the more than 10 mTORopathy genes clearly points toward clinical trial testing of mTOR inhibitors such as sirolimus or everolimus as has been shown in TSC by the EXIST-3 trial [37]. Other compounds that work downstream of mTOR or that target mTOR itself plus other kinases in the pathway i.e., PI3K or AKT3, may provide more efficacy for seizures and could even benefit cognitive and behavioral disorders associated with MCD. As other somatic mutational variants, i.e., SLC35A2, are identified, new compounds or new strategies to target the functional protein dysfunction e.g., galactose transport could yield new clues for pharmacotherapeutics.
Key Points:
Somatic mutations within the mTOR pathway cause focal MCD such as HME and FCD.
Somatic mutations may cause seizures by disrupting cortical connectivity or generating enhanced excitability in neurons.
Somatic mutations provide new potential therapeutic targets for drug development.
Acknowledgements
There are no conflicts of interest. Peter B. Crino M.D., Ph.D. is a member of the Scientific Advisory Board of Evogen, Inc. This work was supported by grants to PBC (NINDS R01NS089552-01 and NINDS R01NS094596-01A1).
Footnotes
There are no conflicts of interest.
References
- 1.Nolan D, Fink J. Genetics of epilepsy. Handb Clin Neurol 2018; 148: 467–491 [DOI] [PubMed] [Google Scholar]
- 2.Iffland PH 2nd, Crino PB. Focal Cortical Dysplasia: Gene mutations, cell signaling, and therapeutic implications Annu Rev Pathol. 2017; 12:547–571• A comprehensive review of the pathogenesis of focal cortical dysplasia and related malformations.
- 3.Casanova MF, Casanova EL. The modular organization of the cerebral cortex: Evolutionary significance and possible links to neurodevelopmental conditions. J Comp Neurol 2018. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najm IM, Sarnat HB, Blümcke I. Review: The international consensus classification of Focal Cortical Dysplasia - a critical update 2018. Neuropathol Appl Neurobiol 2018; 44: 18–31• An outstanding review of how cortical dysplasias are classified.
- 5.Peron A, Au KS, Northrup H. Genetics, genomics, and genotype-phenotype correlations of TSC: Insights for clinical practice. Am J Med Genet C Semin Med Genetic 2018; 178: 281–290 [DOI] [PubMed] [Google Scholar]
- 6.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G 2nd, Aronica E, Crino PB.mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 2004; 56: 478–87 [DOI] [PubMed] [Google Scholar]
- 7.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol 2004; 56: 510–9 [DOI] [PubMed] [Google Scholar]
- 8.Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol 2006; 60: 420–9 [DOI] [PubMed] [Google Scholar]
- 9.Samadani U, Judkins AR, Akpalu A, Aronica E, Crino PB. Differential cellular gene expression in ganglioglioma. Epilepsia 2007; 48: 646–53 [DOI] [PubMed] [Google Scholar]
- 10.Poduri A, Evrony GD, Cai X, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012; 74: 41–48•The first paper to definitively demonstrate somatic gene mutation in a malformation of cortical development.
- 11.Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet 2012; 44: 941–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med 2015; 21:395–400** The first paper to demonstrate mutations in the MTOR gene in cortical dysplasia.
- 13.Nakashima M, Saitsu H, Takei N, et al. Somatic Mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann. Neurol 2015; 78:375–386 [DOI] [PubMed] [Google Scholar]
- 14.Mirzaa GM, Campbell CD, Solovieff N, et al. Association of MTOR Mutations With Developmental Brain Disorders, Including Megalencephaly, Focal Cortical Dysplasia, and Pigmentary Mosaicism. JAMA Neurol 2016; 73:836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moller RS, Weckhuysen S, Chipaux M, et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet 2016; 2:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim JS, Gopalappa R, Kim SH, et al. Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Hum Genet 2017; 100:454–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salinas V, Vega P, Piccirilli MV et al. Identification of a somatic mutation in the RHEB gene through high depth and ultra-high depth next generation sequencing in a patient with Hemimegalencephaly and drug resistant Epilepsy. Eur J Med Genet 2018; pii: S1769-7212: 30571–8 [DOI] [PubMed] [Google Scholar]
- 18.Griffin NG, Cronin KD, Walley NM, et al. Somatic uniparental disomy of Chromosome 16p in hemimegalencephaly. Cold Spring Harb Mol Case Stud 2017; 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leventer RJ, Scerri T, Marsh AP, et al. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology 2015; 84:2029–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Gama AM Woodworth MB, Hossain AA, et al. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep. 2017; 21: 3754–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alcantara D, Timms AE, Gripp K, et al. Mutations of AKT3 are associated with a wide spectrum of developmental disorders including extreme megalencephaly. Brain 2017; 140:2610–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen LA, Mirzaa GM, Ishak GE, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015; 138:1613–1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crino PB, Aronica E, Baltuch G, et al. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010; 74:1716–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin W, Chan JA, Vinters HV, et al. Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol 2010; 20:1096–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribierre T, Deleuze C, Bacq A, et al. : Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest 2018; 128: 2452–2458• This paper shows clear evidence of somatic “second-hit” mutation in DEPDC5 in cortical dysplasia.
- 26.Hu S, Knowlton RC, Watson BO, et al. Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann Neurol 2018; 84: 140–146• An elegant mouse model analysis of the effects of DEPDC5 deletion on neuronal excitability.
- 27.McConnell MJ, Moran JV, Abyzov A, et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science. 2017; 356(6336) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evrony GD, Cai X, Lee E, et al. : Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 2012; 151: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lamparello P, Baybis M, Pollard J, et al. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 2007, 130:2267–2276. [DOI] [PubMed] [Google Scholar]
- 30.Veersema TJ, Ferrier CH, van Eijsden P, et al. : Seven tesla MRI improves detection of focal cortical dysplasia in patients with refractory focal epilepsy. Epilepsia Open 2017; 2:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiffault I, Farrow E, Smith L, et al. : PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. Am J Med Genet A 2016; 170:1585–1589. [DOI] [PubMed] [Google Scholar]
- 32.Liu A, Yang X, Yang X, et al. : Mosaicism and incomplete penetrance of PCDH19 mutations. J Med Genet 2018; pii: jmedgenet-2017-105235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama T, Ishii A, Yoshida T, et al. : Somatic mosaic deletions involving SCN1A cause Dravet syndrome. Am J Med Genet A 2018, 176:657–662. [DOI] [PubMed] [Google Scholar]
- 34.Winawer MR, Griffin NG, Samanamud J, et al. : Somatic SLC35A2 variants in the brain are associated with intractable neocortical epilepsy. Ann Neurol 2018; 83:1133–1146.**An exciting paper that demonstrates a novel gene associated with non-lesional neocortical epilepsy but also is the first gene variant found in association with focal cortical dysplasia type Ia.
- 35.Kimizu T, Takahashi Y, Oboshi T, et al. A case of early onset epileptic encephalopathy with de novo mutation in SLC35A2: Clinical features and treatment for epilepsy. Brain Dev. 2017; 39(3): 256–260 [DOI] [PubMed] [Google Scholar]
- 36.Koh HY, Kim SH, Jang J, et al. : BRAF somatic mutation contributes to intrinsic epileptogenicity in pediatric brain tumors. Nat Med 2018, 24:1662–1668. [DOI] [PubMed] [Google Scholar]
- 37.French JA, Lawson JA, Yapici Z, et al. : Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388:2153–2163. [DOI] [PubMed] [Google Scholar]