

Abstract
Cancer survivors often carry disseminated tumour cells (DTCs), yet owing to DTC dormancy they do not relapse from treatment. Understanding how the local microenvironment regulates the transition of DTCs from a quiescent state to active proliferation could suggest new therapeutic strategies to prevent or delay the formation of metastases. Here, we show that implantable biomaterial microenvironments incorporating human stromal cells, immune cells and cancer cells can be used to examine the post-dissemination phase of the evolution of the tumour microenvironment. After subdermal implantation in mice, porous hydrogel scaffolds seeded with human bone marrow stromal cells form a vascularized niche and recruit human circulating tumour cells released from an orthotopic prostate tumour xenograft. Systemic injection of human peripheral blood mononuclear cells slowed the evolution of the active metastatic niches but did not change the rate of overt metastases, as the ensuing inflammation promoted the formation of DTC colonies. Implantable pre-metastatic niches might enable the study of DTC colonization and proliferation, and facilitate the development of effective anti-metastatic therapies.
Tumour-cell dissemination and subsequent metastatic relapse is the leading cause of death from nearly all cancers. This insidious event has often already occurred when a patient is first diagnosed with a tumour. However, not all disseminated tumour cells (DTCs) develop lethal metastases within the lifetime of the patient because the formation of aggressive secondary tumours is inefficient and lengthy 1. Only a few circulating tumour cells (CTCs) disseminate successfully to vital organs, and the majority of these DTCs undergo apoptosis or clearance by immune cells 2. Often, CTCs that survive extravasation do not immediately proliferate, but instead lay dormant for months to decades until the surrounding milieu becomes favorable for regrowth 3, 4. Emerging evidence suggests that metastatic relapse may not be explained solely by intrinsic genetic instability of DTCs, instead bi-directional interaction with the surrounding microenvironment needs to be considered 5–7. Understanding how the local milieu surrounding DTCs prevents or aids in regaining proliferative phenotypes is imperative to developing better therapeutic strategies to prevent or delay lethal metastasis.
CTCs theoretically spread to a wide range of distant organs, but metastasis limitedly occurs in a subset of target organs including the lung, bone, liver, and brain. This non-random development of metastasis has been recognized as the “Seed and Soil” hypothesis 8. Recently the “Pre-metastatic Niche” hypothesis further posits that CTCs are actively attracted to transiently formed pro-inflammatory microenvironments, driven by signaling from the primary tumour, in these distant organs that better support the survival and growth of DTCs 9. The key microenvironmental signatures of the pre-metastatic niche include (i) a vascular network 10, 11 and associated oxygen tension (i.e. hypoxia) 12, (ii) altered local deposition of extracellular matrix 13–15, (iii) recruitment of bone marrow-derived cells 9, 16, and (iv) pro-inflammatory immune cell activity 17–20. These niche factors are believed to attract CTCs and subsequently direct the fate of DTCs to remain in a dormant state or proliferate 21. However, the detailed mechanisms through which dormant DTCs regain their aggressive phenotype while interacting with the local microenvironment have remained uncertain due to the lack of relevant experimental models that can faithfully simulate the post-dissemination phase of a dormant-to-active transition of DTCs with high analytical power.
Mouse models have been widely used to understand various aspects of cancer biology. For example, spontaneous and experimental metastasis models simulate invasion, circulation and dissemination of cells from solid tumours in a physiologically relevant manner 22–24. The development of immunodeficient NOD-scid IL2Rgnull (NSG) mice has improved the ability to study the biology of human cancer cells in the context of living systems 25. This has greatly advanced knowledge about early stage events in human tumour metastasis and the functional interplay between human DTCs and the local stromal microenvironment 26. However, there are major limitations in current models to study metastatic relapse of dormant human DTCs. First, experimental metastasis models always induce both active and dormant DTCs simultaneously, restricting the study of late stage metastatic tumour recurrence. Second, rare dormant DTCs are impractical to detect. Metastatic relapse becomes evident only after reactivated DTCs establish a clinically detectable tumour mass. Third, immunodeficient mice cannot capture the role of the immune system in microenvironmental regulation of DTC survival and outgrowth. Fourth, most of these mouse models rely on xenografts wherein human tumour cells interact with mouse stromal cells, which does not fully recapitulate human disease. Lastly, these systems offer little opportunity to manipulate individual properties of the niche, constraining the ability to distinguish the role of individual factors of the tumour microenvironment in regulating DTC biology.
Recently, tissue engineering strategies have been applied to address the fundamental limitations of conventional murine metastasis models 27–29. These efforts can be divided into two different aspects based on the target tissues. Among the four major metastatic prone tissues, bone has been the most successful in tissue engineering. A combination of bone inducing biomaterial selection, porous scaffold design, and osteogenic growth factors have successfully been applied to generate ectopic bone and marrow, mostly beneath the skin 30, 31. The addition of human bone marrow stromal cell (hBMSC)-derived osteogenic cells and subsequently introduced human tumour cells recapitulate humanized bone-specific metastasis 32–36. The second approach exploits the transient foreign body response triggered by biomaterials that attract immune cells along with CTCs to the site of implantation 26, 37–40. These approaches have been utilized to distinguish common biological processes that support aggressive metastatic tumour formation 26, 39. Engineered tumour cell attracting niches have also demonstrated translational potential as they have been shown to significantly reduce metastatic burden in vital organs and resultantly extend the lifespan of tumour bearing mice 41–43. However, tissue engineered microenvironments have not been applied for detailed investigation of the post-dissemination phase of cancer biology.
In this report, we introduce an experimental metastasis model to study the evolution of early humanized DTC niches in immunodeficient NSG mice. Functional pre-metastatic niches can arise in multiple tissue sites and share pro-inflammatory features observed during wound healing and tissue remodeling processes 44. A recent study demonstrated that repeated injection of tumour-derived exosomes induced a pre-metastatic niche in the liver and lung of mice 45, indicating that a pre-metastatic niche can be generated by exogenously delivered factors. Based on this premise, we have developed a tissue engineered pre-metastatic niche by subdermally implanting inverted colloidal crystal (ICC) hydrogel scaffolds that induce vascularized and pro-inflammatory tissue microenvironments. This niche was humanized with hBMSCs preseeded on the scaffolds, followed by recruitment of CTCs released from an orthotopic human tumour xenograft. The established humanized pre-metastatic niche was applied to determine the long-term impact of inflammation triggered by human peripheral blood mononuclear cells (hPBMCs) in directing the fate of DTCs. Many solid tumour patients suffering metastasized lesions receive chemotherapy and/or radiation therapy and subsequent bone marrow transplantation. In these patients, graft-versus-tumour effects have been clinically observed, whereby grafted cells reduce tumour burden of the recipient 46. Therefore, we hypothesized that the introduction of hPBMCs would reduce long-term metastatic relapse by decreasing DTCs. Serial transplantation of early humanized DTC niches into syngeneic naive mice permitted prolonged evolution of the tumour microenvironments. Over a 10-week period, the kinetics of metastatic relapse were quantitatively monitored under whole body bioluminescent imaging. At the end of the study, we identified early, intermediate, and overt stages of the metastatic cascade. Deep multiplex imaging and quantitative analysis captured the ability of hPBMCs to reduce the number of single DTCs but did not affect the incidence of overt metastatic tumour development. Additionally, the importance of vasculature and innate immune cells in regulating the stable-to-active transition of human DTCs was substantiated. The presented tissue engineered, humanized, and analytical niche model may serve as an enabling tool for the study of microenvironmental regulation of the post-dissemination phase of cancer biology and development of targets for anti-metastatic therapies.
Results
hBMSC-seeded inverted colloidal crystal hydrogel scaffolds induce a vascularized tissue microenvironment in vivo
We hypothesized that engineered implantable biomaterials could create pre-metastatic niche-mimicking tissue microenvironments by directing the foreign body response to promote angiogenesis and maintain a pro-inflammatory milieu. An isotropic porous ICC structure fabricated by a simple and tunable procedure was chosen to systematically investigate the role of biomaterial design in directing host tissue response by orthogonally manipulating its physical and chemical properties 47–50. ICC geometry consists of fully interconnected spherical pore arrays and provides maximal surface area in the given volume, increasing the biomaterial-tissue interface to direct the foreign body response 51. Cavity diameter and mechanical integrity of the hydrogel matrix were recognized as important parameters to direct angiogenesis and subsequent systemic recruitment of cells including immune and bone marrow cells. ICC scaffolds made with 5–50 wt% polyacrylamide (PAA) hydrogel were subdermally implanted into immunodeficient NSG mice and retrieved 4 weeks later. There was no significant difference in tissue development from 30–50 wt%, however scaffolds made with 5–15 wt% PAA mechanically collapsed after subdermal implantation and inter-scaffold tissue development was incomplete (Supplementary Fig. 1). Next, 30 wt% PAA was examined in scaffolds with varying pore diameters from 75–500 µm. Blood vessel diameters increased with increasing pore sizes, but larger pores (400–500 µm) developed a more diverse range of vessels. Smaller pore sizes (75–150 µm) induced relatively homogenous vascular development but restricted stromal and immune cell migration (Supplementary Fig. 2). Ultimately, we determined that a 6.5 mm diameter, 1 mm thick cylindrical ICC scaffold consisting of 300 ± 15 µm pores with 30 wt% synthetic PAA hydrogel was optimal to generate a controlled humanized vascularized tissue at the subcutaneous site (Fig. 1a).
Fig. 1. Subdermally implanted hBMSC-seeded ICC hydrogel scaffolds develop vascularized humanized niches in immunodeficient NSG mice.

a, Schematic of microsphere template-based fabrication of ICC geometry polyacrylamide hydrogel scaffolds. Bright-field image shows ICC structure through transparent hydrogel matrix (top), and SEM images show interconnecting junctions and surface immobilized type I collagen (bottom). b, Confocal images of tiled top and cross-sectioned scaffolds demonstrate homogeneously seeded fluorescently labeled hBMSCs into 3D ICC hydrogel scaffolds with typical mesenchymal morphology after adhesion. c, Representative gross and histological images of subdermally implanted hBMSC seeded scaffold after 4 weeks in NSG mice. H&E (top) and trichrome staining (bottom) shows recruitment of stromal and immune cells, and organized collagen architecture, respectively. d, Immunohistostaining (IHS) of hVimentin and mCD31 for analyzing spatial characteristics of hBMSCs and vasculature within the scaffold. e, f, Quantitative analysis of blood vessels (e), and hBMSC localization (f) (n=3, independent scaffolds). g, Comparison of key human cytokine secretion from hBMSC-scaffolds before and after implantation for 4 and 12 weeks (n=3, independent scaffolds). Data are mean ± standard deviation (s.d.). *P < 0.05 via two-sided student t-test.
A humanized stromal niche was created by seeding 5×105 hBMSCs onto an ICC hydrogel scaffold coated with type I collagen. hBMSCs are adherent fibroblastic cells isolated from the human bone marrow that carry multilineage capabilities and high secretory activity (Supplementary Fig. 3) 52. Confocal imaging confirmed deep and homogeneous distribution of hBMSCs, displaying a spindle-like cellular morphology (Fig. 1b). We previously demonstrated that subdermally implanted ICC scaffolds preseeded with hBMSCs significantly enhanced vascularization and hematopoietic cell recruitment 37. Four hBMSC-scaffolds were implanted in the dorsal subcutaneous space apart from each other in a NSG mice. ICC hydrogel scaffolds facilitated rapid and deep migration of mouse immune cells, which was in turn followed by near tissue cell migration and inter-scaffold angiogenesis. After 4 weeks, complete inter-scaffold tissue development was confirmed under scanning electron microscopy (Supplementary Fig. 4). Histological analysis revealed that pores were filled with organized dense collagen extracellular matrix and populated with various types of hematopoietic cells on the pore surface (Fig. 1c). Immunohistostaining (IHS) for mouse endothelial cell marker, mCD31, and human mesenchymal marker, hVimentin, revealed a richly vascularized tissue throughout the scaffold with hBMSCs (Fig. 1d). There was no overlap between mCD31 and hVimentin, indicating that blood vessels were mouse tissue derived and hBMSCs did not incorporate with mouse vessels. Interscaffold angiogenesis was directed by the biomaterial geometry as vasculature took on branched morphology between the scaffold cavities (Supplementary Fig. 5). Quantitative analysis revealed that most blood vessels and hBMSCs resided at the outer regions of the scaffold, gradually decreasing in number toward the center (Fig. 1e,f). This indicates that the survival of hBMSCs was dependent on the rapid formation of inter-scaffold blood vessels. To determine whether engrafted hBMSCs remained functional, we examined ex vivo secretion of cytokines from hBMSCs remaining in implanted scaffolds 4 and 12 weeks post implantation. Explanted scaffolds were cultured ex vivo for 36 hours before conditioned media was collected and analyzed for a subset of human cytokines (VEGF, IL-6, and IL-8) that have implicated importance in angiogenesis and inflammation 9, 53–55. Human cytokine secretion gradually diminished overtime in vivo but was still detectable by ELISA (>35 pg/mL) 12 weeks after implantation (Fig. 1g). The directed foreign body response and hBMSC secretome in ICC hydrogel scaffolds reproducibly form an analytical tissue microenvironment that recapitulates three key features including (i) vascular networks, (ii) bone marrow derived cells, and (iii) inflammatory immune cells.
Humanized stromal niches attract human CTCs and permit colonization
We employed an orthotopic xenograft tumour-based experimental metastasis model with PC3 human prostate tumour cells that has demonstrated robust engraftment and subsequent metastasis 56–58. After 3 weeks hBMSC-scaffold implantation, we performed an intra-prostate injection of 2×106 PC3 human prostate tumour cells expressing green fluorescent protein (GFP) and luciferase (Luc). Engraftment and metastatic spread of human tumour cells was non-invasively monitored via whole-body bioluminescent imaging (BLI). The efficiency of successful orthotopic engraftment based on BLI was above 90%. Unsuccessfully engrafted mice were excluded from experiments. BLI+ mice were sacrificed at 8 weeks because mice became moribund due to continuous growth of the primary tumour and aggressive metastasis in vital organs (Fig. 2a). Previous characterization demonstrated that by this time point, primary tumours grew in weight to about 400 mg and generated roughly 10–50 CTCs per mL of blood 26. At the end of the study, gross examination of internal organs confirmed overt metastatic tumour nodules in the liver (Fig. 2b). IHS of explanted tissues confirmed the formation of micro-metastases in the liver and lung whereas DTCs remained individually in the bone marrow and spleen (Fig. 2c). IHS of humanized stromal niches revealed CTCs had successfully disseminated. DTCs were mostly observed in a single cell state but there were few colonized DTCs (Fig. 2d). These results verified that the implanted humanized stromal niches recapitulated the microenvironmental complexity, tumour cell attracting, and supportive functions of the pre-metastatic niche.
Fig. 2. Humanized implantable microenvironments recapitulate tumor cell receptive and supportive functions of the pre-metastatic niche.

a, Experimental schematic describing establishment of an orthotopic xenograft tumor and subsequent metastasis. b, Gross images of the orthotopic xenograft PC-3 human prostate tumor and liver metastasis. Arrows indicate overt metastatic nodules. c, IHS images of dissemination and colonization of DTCs in vital organs including the liver, lung, bone marrow and spleen. Micrometastatses commonly appeared in the liver and lung while DTCs remained mostly in an individual state in the bone marrow and spleen. d, IHS of implantable pre-metastatic niche showed both single and colonized DTCs. Green arrow indicates single DTC. Similar patterns of primary tumor xenograft and tissue and scaffold metastasis were confirmed from 3 independent experiments.
Intravenously delivered hPBMCs increased human immune cell complexity
Immune cells, both innate and adaptive, are important components of the pre-metastatic niche 9, 59. To add human immune cell complexity, we intravenously injected 1 × 107 hPBMCs isolated from healthy adult donors into mice bearing hBMSC-scaffolds 4 weeks after implantation (Fig. 3a). Initial distribution of hPBMCs was characterized 24 hours after injection. As expected, hPBMCs were primarily localized to the spleen and lung. A relatively low and comparable level of hPBMCs was confirmed across hBMSC-scaffolds, bone marrow, and liver (Fig. 3b and Supplementary Fig. 6). At the same time point, we also determined whether systemically migrated immune cells initiated local tissue inflammation. After 36 hours of ex vivo culture, conditioned media for human cytokines representative of inflammation (TNF-α, IL-6, and IL-10) were compared to non-hPBMC injected controls. Increased inflammatory cytokine secretion was observed in the lung, liver, and spleen but not in the bone marrow. hBMSC-scaffolds showed significantly increased pro-inflammatory IL-6 but decreased anti-inflammatory IL-10 compared to hPBMC-free controls (Fig. 3c). We then examined the long-term effect of human immune cells on tissues and hBMSC-scaffolds by IHS for the pan-leukocyte marker, hCD45, 6 weeks after injection of hPBMCs. hCD45+ cells displayed a single cell morphology and were observed widespread across the scaffold (Fig. 3d and Supplementary Fig. 7). Sublineage analysis of human immune cells confirmed both hCD4 and hCD8 T-lymphocytes were present in the scaffold (Supplementary Fig. 8). Diffuse mCD31 endothelial staining in hPBMC-injected mice compared to controls indicated leaky and permeable vasculature, which is the representative vascular morphology in inflamed tissues 60. Further quantitative image analysis confirmed a significant increase in vascular density in the scaffold tissue with hPBMCs compared to controls (Fig. 3e). Taken together, implantable humanized niches gained enhanced inflammatory characteristics after the introduction of hPBMCs, however the host animals developed symptoms of systemic graft-versus-host-disease and became moribund 10–12 weeks post hPBMC injection 61, 62.
Fig. 3. Implantable humanized stromal niches attract systemic hPBMCs.
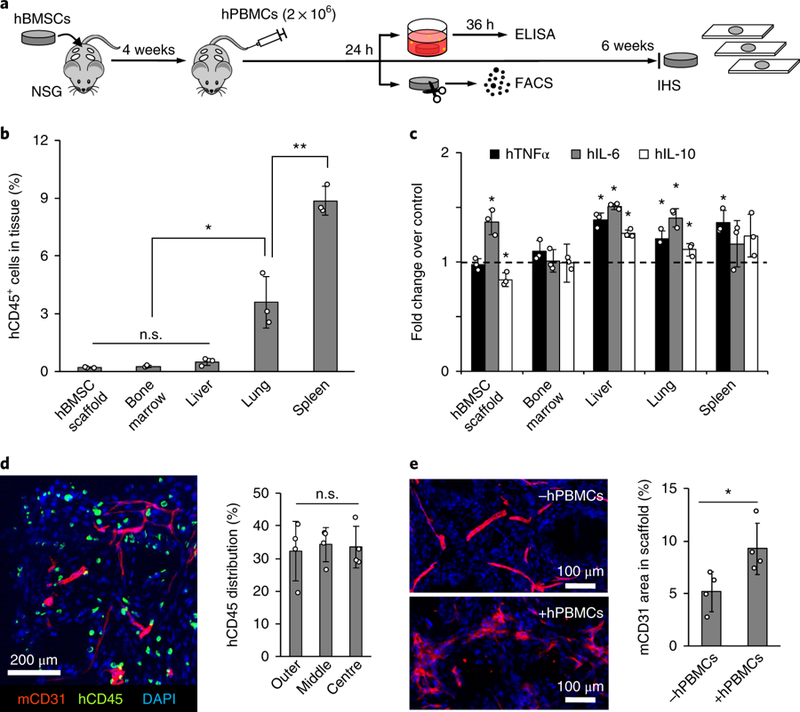
a, Experimental schematic describing systemic introduction of hPBMCs followed by functional and immunohistological characterization. b, Flow cytometry analysis of hPBMC distribution 24 hours after intravenous injection (n=3, mice) Data are mean ± s.d. *P<0.05 and **P<0.005 via two-sided student-t test. c, Analysis of inflammatory human cytokine secretion from ex vivo cultured tissues and scaffolds retrieved 24 hours after injection. Dotted line indicates cytokine levels from control tissue and scaffolds without hPBMCs (n=3, mice). Data are mean ± s.d. *P<0.05 via two-sided student-t test. d, IHS of hCD45 and mCD31 in scaffolds 6 weeks after hPBMC injection with quantitative characterization of hCD45+ cell distribution within the scaffolds (n=4, independent scaffolds). Data are mean ± s.d. Not significant, P>0.05 via two-sided student t-test. e, IHS of mCD31 6 weeks after hPBMC injection shows leaky vascular morphology when compared to non-injected control and quantitative analysis confirms significantly increased vascular density following hPBMC injection (n=4, independent scaffolds). Data are mean ± s.d. *P<0.05 via two-sided student t-test.
Systemic delivery of hPBMCs instigates early stage humanized DTC niches
Next, we challenged humanized early DTC niches with hPBMCs under the hypothesis that mature, functional human immune cells would decrease metastasis. After subdermal implantation hBMSC-scaffolds for 3 weeks, 2×106 eGFP-Luc PC3 cells were injected into the mouse prostate to form an orthotopic human prostate tumour xenograft. The primary tumour grew for 6 weeks before intravenous injection of 1×107 hPBMCs to introduce human immune cells to the implanted microenvironments. By this point, the host animals carried substantial primary and metastatic tumour burden. Thus, 5 days after hPBMC injection, we euthanized mice and characterized the establishment of the early humanized metastatic niche in the scaffolds (Fig. 4a). IHS revealed coexistence of human stromal (hVimentin), human immune (hCD45), and human tumour (hCytokeratin) cells residing in the same microenvironment (Fig. 4b). BLI analysis of explanted scaffolds revealed 8% of scaffolds produced positive signal from both control (3/36) and hPBMC (4/44) injected mice. There was no significant difference in overall bioluminescent signal intensity between hPBMC-injected and control mice (Fig. 4c). We expected hPBMC-induced inflammation of humanized DTC niches altered DTC fate, but these results indicated that a longer period of implantation was necessary to substantiate the functional consequence.
Fig. 4. Instigation of humanized pre-metastatic niches with hPBMCs and long-term monitoring of DTC niche evolution via serial transplantation.

a, Experimental schematic describing the establishment of early stage humanized DTC niches and subsequent instigation by systemic introduction of hPBMCs. b, Representative IHS of humanized early DTC niche from 3 independent experiments including hCytokeratin (tumor), hCD45 (immune), hVimentin (stroma) and mCD31 (vessel). Arrows indicate human immune (white) and tumor (green) cells. c, Representative ex vivo BLI of explanted scaffolds from primary mice and quantified bioluminescent signal with and without hPBMCs. The dotted line represents a threshold used to define BLI+ and BLI-. Data are mean ± s.d. Not significant, P > 0.05 via two-sided student t-test. d, Experimental schematic describing a strategy to monitor the long-term evolution of humanized DTC niches via serial transplantation of intact early metastatic niches to naïve secondary syngeneic mice. e, Representative whole body BLI of primary and secondary mice at different time points from 2 independent experiments. Gross image of explanted scaffolds from secondary mice showing an overt metastasis. f, Quantitative analysis of human DTC growth in scaffolds after transplantation into secondary mice via weekly BLI. Scaffolds were classified as BLI-, BLI+, or overt based on their bioluminescent intensity. Presented N values are representative of independent scaffolds.
Serial transplantation of humanized DTC niches allowed for long-term monitoring of metastatic evolution
We hypothesized that evolution of the early humanized DTC niches could be continued by intact transplantation of the microenvironment to the same anatomical site of syngeneic mice. We followed the aforementioned experimental schedule to generate humanized DTC niches with and without hPBMCs. Bioluminescent signal from scaffolds retrieved from primary mice was checked ex vivo, and BLI negative scaffolds were transplanted immediately to naïve NSG mice 6–12 weeks of age. Subsequent metastatic relapse was non-invasively monitored on a weekly basis via BLI starting 4 weeks after transplantation and continued up to 10 weeks (Fig. 4d). We did not observe noticeable necrotic tissue areas in histological analysis of serially transplanted scaffolds (data not shown). Gross observation 3 and 7 days after transplantation showed large blood vessels surrounding the scaffolds (Supplementary Fig. 9). By the end of the study, serially transplanted microenvironments captured a broad range of the metastatic cascade (Fig. 4e). Based on bioluminescent intensity and outgrowth beyond the scaffold, we categorized scaffold microenvironments into three groups: (i) BLI-, (ii) BLI+ and (iii) Overt. BLI+ microenvironments were defined by signal at least 3 times greater than the background signal, and the rest were defined as BLI- microenvironments. Background BLI signal was determined from mice without injection of luciferin. Overt growth, a subcategory of BLI+, described aggressive metastases where the tumour expanded outside of the boundary of the scaffold (Fig. 4f). hPBMC-free mice displayed a gradual increase in the number of BLI+ scaffolds over the 10-week period whereas in mice with hPBMCs the number of BLI+ scaffolds stabilized 8 weeks after transplantation. By the end of the study, 53% (17/32) and 23% (9/40) of scaffolds became BLI+ in hPBMC-free and hPBMC-injected mice, respectively. There was no significant difference in the percentage of scaffolds with overt metastasis between the two groups; 6.3% (2/32) in hPBMC-free and 10% (4/40) in hPBMC-injected mice. Our results signify the ability of hPBMCs to suppress human DTC growth but did not affect the incidence of aggressively growing metastases.
Complete optical sectioning of BLI- scaffolds captured the impact of hPBMCs on early DTCs
We hypothesized that BLI- scaffolds still attracted CTCs, but the number of DTCs were low and the cells entered a dormant state. Conventional IHS and flow cytometry may not effectively detect rare DTCs due to the technical difficulties of probing entire tissue volumes and representative cell recovery after tissue digestion, respectively. To overcome these challenges, we adapted the recently introduced CLARITY technique through which optically cleared fixed tissues permit microscopic detection of rare cell populations while retaining spatial resolution in the tissue microenvironment 63 (Fig. 5a). A PAA hydrogel meshwork (4 wt%) between fixed cellular proteins was formed before removal of light scattering lipids, achieved by passive washing with sodium dodecyl sulfate and boric acid at 37 °C. Transparency dramatically improved in the scaffold, but light scattering was still present, possibly due to ICC geometry, or mismatched hydrogel density between the pore cavities and scaffolds (Fig. 5b). We cut the scaffolds to a thickness of 250 µm using a cryostat, which allowed complete optical imaging (Fig. 5c). Sliced scaffolds were stained with antibodies for mouse α-smooth muscle actin and human cytokeratin to visualize vasculature and tumour cells, respectively. Analysis of the images revealed abundant (80–400) single DTCs as well as few (5–30) colonized DTCs throughout the BLI- scaffolds (Fig. 5d and Supplementary Video 1). DTCs were detected in all characterized BLI- (6/6) scaffolds regardless of hPBMC injection. Complete counts of single and colonized DTCs did not show a significant difference between hPBMC-injected and hPBMC-free mice (Fig. 5e). Further spatial analysis revealed widespread distribution of DTCs across inner and outer regions of the scaffolds (Supplementary Fig. 10). The frequency of colonized DTCs in total tumour counts, was two times higher in implants with hPBMCs compared to hPBMC-free implants (Fig. 5f). There was no significant difference in DTC colony size among BLI- scaffolds with an average size of 3,700 μm2 (Supplementary Fig. 11). These results suggest that the implantable pre-metastatic niches effectively attract CTCs and subsequently maintain the long-term viability of DTCs. hPBMC injection disrupted DTC survival but did not decrease colonization events.
Fig. 5. Detection of rare dormant DTCs via whole scaffold tissue clearing and optical sectioning.

a, Schematic of tissue clearing process describing hydrogel embedding, tissue clearing, thick sectioning (250 µm), and antibody staining of explanted scaffolds b, Gross images of whole scaffold before and after tissue clearing. c, Tiled confocal z-stack images of an entire scaffold sectioned into 4 slices. d, Representative tissue cleared scaffold images of DTCs as singular and micro-colonies from 3 independently characterized scaffolds. e, Comparison of complete single and colonized DTC counts between BLI- scaffolds with and without hPBMCs (n=3, independent scaffolds per group). f, Comparison of normalized frequency of DTC colony per 100 DTCs in BLI- scaffolds with and without hPBMCs. Data are mean ± s.d. *P<0.05 via two-sided student t-test. g, Representative HIS of Ki67+ tumor colony (left) and quantitative comparison of Ki67+ of single and colonized DTCs between scaffolds with and without hPBMCs (right). White arrows identify Ki67+ cells (n= 3, independent scaffolds from mice without and with hPBMCs). Data are mean ± s.d.
We then extended cytological profiling of the DTCs to determine intrinsic tumour cell properties and proliferative state using standard immunohistostaining. First, we examined CD44 expression in DTCs to identify potential cancer stem cells 64, 65. About 29% of primary tumour cells (N=3 primary tumours) co-expressed hCD44 and hCytokeratin whereas all observed DTCs (97 DTCs from 4 independent scaffolds) were double positive regardless of being in single or colonized states (Supplementary Fig. 12). To confirm cellular dormancy, we examined the functional status of the DTCs. Several studies have reported a subset of markers that are expressed while DTCs are in a dormant state, however, these markers have been shown to be dependent on the microenvironment 66–69. For example, NR2F1 was shown to promote dormancy in the lung and spleen, however did not play the same role in DTCs found in the bone marrow 70. Additionally, the expression of these markers is transient and relative, therefore not necessarily informative to distinguish the functional state of dormancy 71–73. Thus, we focused on expression of the cell proliferation marker Ki67. All singular DTCs found in the scaffold microenvironments (N=107) were Ki67-, confirming that these cells were not in a proliferative state. In DTCs that formed colonies, only 8.3% (24/252) contained at least one Ki67+ cell. We did not observe any colonies in which all cells expressed Ki67. These results suggest that the majority of initial proliferating DTCs stop growing and enter dormancy indicating that intrinsic properties of early DTCs may be important to initiate metastatic colonization but not sufficient to lead to continuous growth. Comparison between scaffolds with and without hPBMCs revealed no significant differences in the number of Ki67+ DTCs (Fig. 5g).
Comparative analysis of niche factors identifies key microenvironmental regulation of DTCs in single-to-colony transition
Next, we characterized how early DTC niches evolved by probing the local tumour microenvironments in single and colonized DTCs. By leveraging the uniform pore geometry of the scaffold, we generated a standardized imaging analysis of the microenvironment (Fig. 6a). Antibodies for IHS were chosen to determine the role of blood vessels (mCD31), human stromal cells (hVimentin), human immune cells (hCD45), and innate mouse immune cells in the single-to-colony transition of DTCs. Snapshots were collected in the tumour microenvironment of single cell and tumour colonies from at least 4 independent scaffolds (Fig. 6b). We first determined the proximity of human immune cells to single and colonized DTCs. hCD45+ cells located significantly closer to colonized DTCs. About 25% of tumour colonies were infiltrated with hCD45+ cells. However, colocalization of single tumour cell and human immune cells was rare (Fig. 6c). Next, we analyzed the correlation between single and colonized DTCs with respect to the distance from blood vessels. In general blood vessels were found within 50 µm of a tumour cell. Without hPBMCs, over 50% of tumour colonies had at least one infiltrating blood vessels, but this phenomenon was observed in less than 35% of tumour colonies in scaffolds with hPBMCs (Fig. 6d). As expected, the average colony size infiltrated with blood vessels was significantly larger than those without, but there existed no significant correlation between tumour colony size and hPBMCs (Fig. 6e). Functional characterization between Ki67+ tumour colonies and blood vessel availability revealed that nearly all (8/9) proliferative colonies had infiltrating vasculature. However infiltrating vasculature did not guarantee proliferation, only accounting for 26% (9/35) instances. These observations were not dependent on the presence hPBMCs (Supplementary Fig. 13).
Fig. 6. Quantitative comparison of vascular, stromal and immune niches between single and colonized DTCs in single pore microenvironment.

a-b, (a) Representative images of dormant human DTC bearing scaffold and schematics of pore imaging targets and mapping interpore cellular distribution. (b) Representative IHS of microenvironments surrounding single and colonized DTCs with vascular and cellular niche components including hCD45, hBMSCs, mLy6G, and mF4/80 from 2 independent experiments. Scale bars 100 µm. c, Comparison of distribution profiles of neighboring hCD45+ cells to hTumor cells between single and colonized DTCs (top) and frequency mapping of hCD45+ cells (bottom). d, Comparison of distribution profiles of nearest mCD31+ cells to hTumor cells with and without hPBMC injection (top) and frequency mapping of mCD31+ cells (bottom). e, Comparison of tumor colony size with and without infiltrating mVessel. f, Comparison of distribution profiles of mCD31+ cells to hTumor with and without neighboring hBMSCs (top) and frequency mapping of mCD31+ cells (bottom). g, Comparison of distribution profiles of neighboring mLy6G+ cells to hTumor cells with and without hPBMCs (top) and frequency mapping of mLy6G+ cells (bottom). Representative values were plotted, selected by ordering datasets and averaging every 10 points, when N>100. Data collected from 5 biologically independent scaffolds from each group. Data are mean ± s.d. *P<0.05, **P<0.005 and ***P<0.0005 via two-sided student t-test.
We extended quantitative analysis of IHS images with other microenvironmental components. First, the distance between tumour cells and blood vessels was considered when in the proximity of hBMSCs. Although the existence of DTCs and hBMSCs in the same pore was rare, as cellularity of hBMSCs decreased over the implantation period, tumour colonies that formed near hBMSCs were more likely to be infiltrated with a blood vessel than those not closely associated with human stroma (Fig. 6f). Next, we characterized the recruitment and distribution of endogenous mLy6G+, mLy6C+, and mF4/80+ cells to determine the role of innate immune cells. Abundant mLy6C+ and mF4/80+ cells were observed at the pore periphery in nearly every pore regardless of experimental condition indicating that their response was dominantly triggered by the biomaterial rather than tumour or human immune cell activities (Supplementary Fig. 14). Both mLy6C+ and mF4/80+ cells are known critical players in tissue remodeling, secreting protease enzymes including matrix metallopeptidase 9 (MMP-9) 74. IHS of MMP-9 confirmed localized tissue remodeling activity at the pore surface (Supplementary Fig. 15). Interestingly, mLy6G+ cells were preferentially localized within tumour colonies, compared to single DTCs (Fig. 6g). The distribution of mLy6G+ cells in tumour colonies was independent of their proliferative status confirmed by Ki67 staining (Supplementary Fig. 16). Taken together these results indicate that increasing vascular connection and recruitment mLy6G+ cells are common and could be important changes in DTC niches during single-to-colony transition.
Multiplex IHS of overt metastasis substantiates the importance of innate immune cell influx in continuous outgrowth of DTCs
To understand how the early DTC microenvironment evolved during metastatic relapse, we characterized tumours that had progressed beyond the initial awakening events. A panel of 9 different antibodies, hCytokeratin, hVimentin, mCD31, hCD45, Ki67, hCD4, hCD8, mLy6G, and mF4/80, and the nucleus stain DAPI were used to visualize and quantify cellular signatures in the tumour microenvironment (Supplementary Table 1). Eight consecutive histological sections, 20 µm thick, were stained with sets of 3–5 antibodies to generate a multiplexed and comprehensive view of the metastatic tumour heterogeneity (Supplementary Fig. 17). Using ImageJ, individual image channels were centered and overlaid. From these cumulative images, eight zones were manually identified that highlighted regions of diversity (Fig. 7a).
Fig. 7. Multiplex IHS imaging-based characterization of heterogeneity in overt metastatic microenvironments.

a, Overlaid IHS images from eight consecutive 20 µm thick slices and manually defined zones representing distinct microenvironment profiles with a list of antibodies and their associated targets. b, Representative IHS images of each zone with quantitative cellular complexity. Bar graphs display intra-zone composition of quantified antibody staining. Radial plots represent inter-zone comparison of cellular markers normalized to the zone with the highest signal. Top panel highlights human stromal, tumor, and immune cell localization. Bottom panel highlights human and mouse immune cell subset. Three independently formed overt metastatic scaffolds were characterized by multiplex IHS. c, Five functional zones capturing different states of metastatic tumors: (1) necrotic tumor, (2) active tumor, (3) host tissue, (4) dormant tumor, and (5) intermediate tumor. d, IHS images of hCD45 cells found within early DTC colonies (n=5, independent scaffolds) and overt scaffold metastases (n=3, independent scaffolds) with quantitative representation of overlapping hCD45+ cell coverage. Arrows show infiltrating hCD45+ cells (white). e, IHS image of mLy6G+ cell localization at the boundary of an actively growing overt metastasis and quantitative comparison of Ki67 stain overlap with human tumor and mLy6G+ cells (n=3, independent scaffolds per group). f, Quantitative comparison of overlapping mLy6G+ cells in early DTCs and overt scaffold metastases (n=3, independent scaffolds per group). Data are mean ± s.d. * P<0.05 via two-sided student t-test.
To quantitatively determine cellular and functional signatures of each zone, we developed an imaging analysis pipeline. Two different characterizations were considered for quantifying the selected regions (Supplementary Fig. 18). The first analysis compared the absolute pixel number of each stain within each zone to highlight the makeup of each region, which was displayed as a bar graph. The second was a comparative analysis between zones, which was plotted on a radial chart broken into two panels; (i) a general overview of cellularity, and (ii) the diversity of immune cells (Fig. 7b). Comparison of the different zones revealed similar phenotypes that were categorized into five separable regions. Zone 1 highlighted a typical necrotic tumour region consisting largely of weak human tumour and immune cell markers. Mouse macrophages were present but relatively low compared to other regions. A pocket of mouse tissue, zone 4, was characterized by a low signal in human cell markers and weak Ki67 signal. Zones 5 and 6 exhibited similar tissue microenvironments to a dormant DTC niche; a high number of human stromal and immune cells with relatively few mLy6G+ cells were observed, and overall Ki67 signal was weak. On the periphery of the scaffold, zones 7 and 8, an increase in vessels and immune cells was observed (Fig. 7c). In contrast to our observation of micro-metastases, the frequency of human immune cells in overt tumour regions was significantly lower (Fig. 7d). This result was possibly caused by reduction in human immune cells, but IHS of spleens revealed the presence of hCD45+ cells suggesting that a subset of human immune cells systemically migrate and proliferate in the secondary host. The frequency of hCD45+ cells in the spleen was not significantly different between secondary mice with and without overt tumours (Supplementary Fig. 19). Zone 2 and 3 contained actively growing tumours indicated by high DAPI and Ki67 staining. Zone 2 had higher hCytokeratin signal than zone 3 but only half the relative Ki67. High hVimentin in zone 3 suggests a more mesenchymal tumour phenotype, which was linked with higher proliferation compared to the more epithelial tumour phenotype in zone 2. Colocalization analysis confirmed that Ki67 staining was mostly derived from tumour cells and not mLy6G+ cells that were also observed in these actively growing tumour zones (Fig. 7e). mLy6G+ cells limitedly infiltrated overt tumour regions and remained on the tumour periphery, opposite to what was observed during the early stages of colonization (Fig 7f.). Considering the relatively short lifespan of mLy6G+ cells, about 1 day 75, this result indicates that mLy6G+ cells continuously migrated to actively growing tumours. This phenomenon was repeatedly observed in three independent overt metastatic tumours (Supplementary Fig. 20). Together these results demonstrate that the tumor microenvironment evolves during progression to an overt tumour, including phenotypic changes of tumour cells to a more mesenchymal phenotype, and continual recruitment of innate immune cells, especially mLy6G+ cells.
Discussion
Accumulating evidence suggests that systemic spread of DTCs may be unavoidable 76, but metastatic relapse can be preventable. For example, low-dosage aspirin 77, canakinumab 78, metformin 79, and cabozantinib 80 have demonstrated significantly reduced risk for metastasis. It is imperative to develop detailed understanding of how these molecules suppress revitalization of dormant DTCs in distant tissue sites. The lack of relevant experimental models that can faithfully determine the long-term functional consequence of therapeutic intervention in the context of humanized tissue microenvironments has been a critical challenge. The presented tissue engineered pre-metastatic niche model represents a new opportunity to study the post-dissemination phase of human cancer biology from three distinct aspects. First, the use of biomaterials to create a pre-metastatic niche allowed controlled and analytical experimentation to quantitatively distinguish key features of microenvironmental regulation in dormant-to-active transition of DTCs. The standardized structure of ICC scaffolds generated reproducible tissue microenvironments and streamlined multiplex imaging and comparative analysis of DTC niches. Additionally, optical transparency of PAA hydrogel permitted whole tissue clearing and complete optical sectioning of tumour microenvironments to detect rare DTCs while retaining spatial resolution. Implantation of biomaterials in a subcutaneous pocket enables easy accessibility to various functional characterizations including direct measurement of tumour growth, BLI, and possible intravital imaging via surgical engraftment of a skinfold window chamber 37, 81. The tunable design of ICC hydrogel scaffolds permits easy manipulation of physical and geometrical parameters. Recently there has been a substantial progress in understanding the interaction between implantable biomaterials and host tissue and immune cells 82. For example, poly-lactic and glycolic acid (PLGA) scaffolds 83 and methacrylated alginate cryogels 84 manipulate dendritic cell homing and immune response; scaffolds composed of extracellular matrix proteins have modulated pro-regenerative systemic immune environments 85; physical dimensions of biomaterials influence foreign body response 86. These efforts can further empower an implantable pre-metastatic niche model to study human tumour metastasis.
Second, in vitro seeding of stromal cells provided additional capability to create defined and functional implantable pre-metastatic niches. Here hBMSCs were introduced into the scaffold to create humanized stromal niche and modulate foreign body response and interscaffold angiogenesis. hBMSCs are known to secrete immunomodulatory and proangiogenic molecules, which resultantly attenuate foreign body response 87 and enhanced angiogenic activity 37 in previous studies. Similarly, different types and combinations of stromal cells can be introduced into biomaterials to induce defined functionality of microenvironments. For example, genetically engineered stromal cells that secrete specific human cytokines were used to create defined humanized soluble microenvironments 88.
Finally, the transplantation strategy separated early metastatic niches from advanced tumours and prolonged the microenvironment evolution to substantiate functional mechanisms behind metastatic relapse. Serial transplantation has been commonly conducted to maintain intrinsic characteristics of human blood cancer cells and solid tumours (e.g. patient derived xenograft tumours) in the context of living system for extended periods 89, 90. Applying this concept to metastatic tumour microenvironments overcame the fundamental limitation of mouse model for long-term evolution of the DTC niche beyond the lifespan of primary host mice, which enabled observation of the full spectrum of post-disseminated tumour progression. Here we exploited this feature to determine the functional consequence of acute inflammation triggered by hPBMC injection. Even though the experiment was halted 10 weeks after transplantation, dormant DTC microenvironment studies could continue for longer experimental duration. This capability will facilitate observation of the long-term dormant DTC niche and the screening of drugs for metastasis prevention.
Intravenous injection of hPBMCs instigated early humanized DTCs niches and subsequent metastatic relapse became evident 6 weeks after serial transplantation. The incidence of metastasis was significantly lower and stabilized when compared to hPBMC-free scaffolds. This phenomenon is reminiscent of a graft-versus-tumour response 46, 91, 92. Comparative analysis of single and colonized DTCs revealed a correlation between human immune cells and colonized DTCs, whereas single DTCs did not directly interact with human immune cells (Fig. 6c, d). Given the observation of reduced DTCs in mice that received hPBMCs, direct contact between human immune and tumour cells could be a potential mechanism in the elimination of DTCs. Indeed, a previous study showed that systemic delivery of hPBMCs prior to the subcutaneous injection of human PC3 tumour cells in NSG mice significantly decreased ectopic tumour development with increased primary tumour-infiltration of T-lymphocytes 93. Additional studies reported that T-lymphocytes within patient-derived xenografts continually proliferated and systemically spread to the host NSG mouse 94. Similarly, we confirmed the presence of hCD45+ cells in the spleens of secondary mice, indicating that a subset of human immune cells migrated out from the scaffolds and potentially continued growth. This could be the mechanism that leads to long-term stabilization of DTC growth in secondary hosts. Although hPBMCs reduced overall metastatic relapse, the incidence to develop overt metastasis was not changed. Additionally, complete optical sectioning of BLI- scaffolds revealed an increased frequency of colonized DTCs in total DTC counts in mice that received hPBMCs (Fig. 5f). These results suggest that although hPBMCs decreased DTC burden, during this process they may also increase the chance to initiate colonization of DTCs possibly due to local tissue inflammation. However, detailed mechanisms by which human immune cells promoted colonization of DTCs remains an open question.
Previous studies have highlighted vasculature and immune cell interaction as critical for reactivation of dormant DTCs 10, 95–99. Our results provide additional evidence to support these findings and quantitatively capture the changes in the local DTC microenvironments from single, to colonized to overt metastasis. The vasculature is initially used by tumour cells to migrate to distant tissues, however during the period of dormancy, DTCs remain distant from the vasculature. Quantitative analysis of the blood vessels surrounding DTCs revealed that tumour cells begin to proliferate without immediate contact with a blood vessel, but their growth could not exceed a cross-sectional area of roughly 4,000 µm2 without the connection to a blood vessel. It remains unclear if DTCs migrate toward a blood vessel, a vessel is recruited by DTCs, or DTCs and vessels meets opportunistically. Recent studies suggest the source of angiogenic signaling may be derived from local immune and stromal cells that release proinflammatory or proangiogenic factors that initiate endothelial tip cell sprouting 10, 100, 101, induce phenotypic switching of perivascular cells to a less differentiated state 102, or transiently enhance vascular remodeling activity.
Additionally, our results substantiate the important role of continuously recruited innate immune cells in forming a supportive microenvironment for awakening dormant DTCs. mLy6G+ cells consist of several myeloid cells including monocytes, granulocytes, and neutrophils. Recent studies have shown that neutrophils are associated with playing a key role in initiating metastatic outgrowth 95–99. Deep optical characterization of the metastatic niche demonstrated their presence within awakening DTC colonies during initial tumour colonization, however it is unclear how the host neutrophils seek out early stage, reactivated DTCs and their role in promoting or inhibiting growth. Once a tumour colony became an advanced large metastasis, mLy6G+ cells no longer infiltrated but remained on the periphery where they were continually recruited. The lack of infiltrating neutrophils within overt tumours indicates that the tumour may produce a repulsive microenvironment once it has formed a large enough mass. The varying activities of mLy6G+ cells within the different stages of tumour development demonstrate the need for deeper understanding of myeloid cell biology such as neutrophils in the tumour microenvironment, which may lead to more effective anti-metastatic strategies 103. It is possible, and likely that many budding metastases fail to evade immune response at the initial stages of outgrowth and become terminated. Tumours that can evade microenvironmental control will continue to proliferate and gain control of the microenvironment and dictate bi-directional interactions with surrounding cells. From our data, we demonstrated that overt tumour cell proliferation occurs together with vascularization and exclusion of innate and adaptive immune cells, while selectively attracting a subset of innate immune cells to the actively proliferative boundary, allowing for continuous growth (Fig. 8).
Fig. 8.
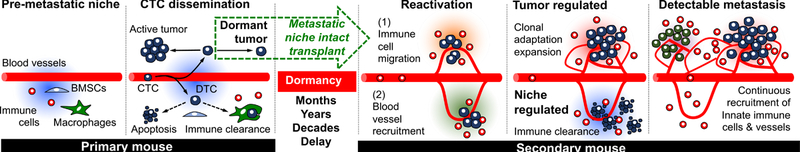
Proposed microenvironmental regulation of DTCs during initial dormancy, reactivation, and overt metastasis along with the experimental transplantation strategy from primary to secondary mice for long-term observation.
The presented implantable pre-metastatic niche model has demonstrated enabling features for detailed observation of awakening dormant DTCs in vivo, yet there is still room to improve. First, since the established humanized microenvironments are comprised of three different donors, human immune interactions are allogeneic, which limits the study of adaptive immune system mediated tumour microenvironment regulation. Creating donor-matched microenvironments is possible by obtaining different tissue cells from the same patient but this involves clinical and ethical considerations 104. One alternative solution is to use induced pluripotent stem cells that can differentiate into stromal, hematopoietic, and tumourigenic cells 105. While the current study has focused on humanized tumour microenvironments keeping in mind translational opportunity, for mechanistic studies the experimental system could be adapted to an entirely mouse-based model. Second, hPBMC injection delivers fully functional human immune cells but the innate immune cell effect is restricted to a short-term period due to their limited lifespan; the average lifespan of monocytes and neutrophils are about 1 week and 24 hours, respectively 75, 106. Instead, repeated injection of human granulocytes into the humanized mice could replenish short-lived immune cells. Alternatively, the transplantation of human hematopoietic stem cells could stably provide human innate immune cells, but differentiated human immune cells in the mouse bone marrow may not carry full functionality 107. Emerging data including our own has demonstrated that innate immune cells play a critical role in regulating the tumour microenvironment 108–110. Innate immune cell activity does not strictly rely on the highly specific binding required for adaptive immune cells, and thus human tumour and mouse innate immune cell interaction may be clinically relevant. Third, the prolonged local foreign body unnaturally elevated macrophage activity and may not accurately reflect physiological situations. Recent studies have shown that locally implanted biomaterials modulate systemic immune response and host regenerative potential 85, 111, 112. Additionally, biomaterial-induced myeloid cell responses were shown to be tunable by incorporating macrophage selective drugs e.g. clodronate for macrophage depletion 81. Finally, transplantation of the intact microenvironments possibly caused brief temporal oxygen and nutrient deprivation. Aside from intrinsic changes to DTCs, their fate can be critically influenced by the secondary host cells, especially immune cells. We minimized this potential effect by using syngeneic mice and implanted the microenvironments to the same anatomical locations. However, a similar clinical scenario has been reported in the field of organ transplantation where dormant DTCs in the donor organ developed metastasis in the recipient 113, 114. Indeed, this feature may provide a new opportunity to investigate the role of local and systemic tissue microenvironments by altering the recipient host animal in terms of age, immune competence, and implantation site.
Mouse models play a critical role in advancing our understanding of cancer metastasis, however their capability to capture the long-term evolution of the metastatic niche and their relevance to humans remains questionable. Tissue-engineered humanized metastasis models can reduce the preclinical gap and potentially uncover new biological interactions between human tumours and the surrounding microenvironment. Studies can be readily extended to characterize the extracellular matrix, and molecular and genetic profiles. One promising future direction is to combine comparative analysis based on endpoint IHS characterization with temporal observation of cellular processes via intravital or ex vivo imaging, which will enable deeper mechanistic understanding of the DTC niche evolution. The established metastasis model can also be applied to screen the effectiveness of drugs targeting DTCs and secondary outgrowths. Taken together, we envision that tissue engineered strategies will provide an invaluable tool to answer provocative hypotheses emerging in the cancer field and advance clinical translation of anti-metastatic therapeutic strategies.
Methods
All chemicals and materials were purchased from Sigma Aldrich or Fisher Scientific unless specified. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts-Amherst. Experiments and handling of mice were conducted under federal, state and local guidelines.
Type I collagen coated inverted colloidal crystal hydrogel scaffold fabrication:
Inverted colloidal crystal hydrogel scaffolds were fabricated following the previously reported methods (33). Soda lime glass beads were sorted using an Advantech Sonic Sifter for each range with ~8% deviation. Beads dispersed in deionized (DI) water were gradually loaded into a glass vial (8 × 35 mm) to a height of 2–2.5 mm and were mechanically packed into a lattice structure in an ultrasonic water bath. Orderly packed glass beads were dried in a 60 °C oven and then thermally annealed between 650 and 680 °C depending on the bead size for 4 hours in a furnace. A hydrogel precursor solution composed of 5, 15, 30, and 50 wt% acrylamide monomer, 1.5 wt% bis-acrylamide crosslinker, 0.2 vol% N,N,N’,N’-tetramethylethylenediamine accelerator, and 0.2 vol% 2-hydroxy-2-methylpropiophenone photoinitiator in nitrogen purged DI water was prepared immediately before use. 150 µL of precursor solution was infiltrated into the glass bead template and centrifuged in a microcentrifuge at 9,000 RPM for 15 min and subsequently polymerized under a 15 W ultraviolet light source for 15 minutes. Polyacrylamide hydrogel-glass templates were removed from the glass vials the next day to ensure complete polymerization. Excess hydrogel was removed by scraping the glass bead template with a razor blade on all surfaces. Glass beads were selectively dissolved in alternating washes of an acid solution containing a 1:5 dilution of hydrofluoric acid in 1.2 M hydrochloric acid and 2.4 M hydrochloric acid (Caution: These chemicals are corrosive and must be used in a fume hood with proper protective gear). Washes were completed on a shake plate and solutions were changed every four hours until the beads were removed. Scaffolds were thoroughly washed with DI water to remove residual acid and lyophilized. Following lyophilization, scaffolds were resuspended in CryomatrixTM embedding resin and cut to 1 mm thickness on a CryoStar NX70. After thorough washing in DI water, scaffolds were sterilized with 70% ethanol and stored at 4 °C in sterile phosphate buffered saline solution (PBS). The final pore dimension of the optimized scaffolds used for the remainder of the study was 300 ± 16 µm. To support stromal cell adhesion on polyacrylamide hydrogel scaffolds, type I collagen extracted from rat tails was covalently immobilized on the pore surface using Sulfo-SANPAH conjugate chemistry (Supplementary Method 1).
Human BMSC isolation and culture expansion:
50 mL of bone marrow aspirate obtained from a healthy donor was purchased from Lonza. Mononuclear cells were isolated via density gradient based centrifugation using Ficoll Paque (GE) at 1500 RPM for 30 minutes using minimum acceleration and no brake. Mononuclear cells were plated on T-225 flasks with Minimum Essential Medium Eagle, alpha modified (αMEM) supplemented with 20% FBS, 2% penicillin-streptomycin, 0.2% gentamicin, and 1 µg/L recombinant human fibroblast growth factor. After 1 week, colonies of adherent cells were harvested, cryogenically frozen in media containing 10% DMSO, and stored in a cryotank. The multilineage differentiation potential of hBMSCs were tested using standard protocols (Supplementary Method 2).
Human BMSC seeding and culture on a 3D hydrogel scaffold:
Collagen coated ICC hydrogel scaffolds were partially dehydrated by removing excess PBS and drying in a biosafety cabinet. 5 × 105 hBMSCs suspended in 20 µL of growth media were dropped on top of a partially dehydrated scaffold. The cell suspension was drawn into the dried scaffold, facilitating deep and homogenous cell seeding in the 3D scaffolds. Cell loaded scaffolds were incubated for 20 minutes in a CO2 incubator to allow initial cell adhesion. Cell-scaffolds were transferred to a 48-well plate containing 500 µL of media and allowed to grow for up to one week prior to use. Media was changed every 3 days.
Subdermal implantation of hBMSC-scaffolds in NSG mice:
A breeding pair of NOD-scid IL2Rgnull mice (005557) was initially obtained from the Jackson Laboratories. NSG mice were housed in sterile conditions with unrestricted access to food and water. In this study 6–13 week age mice were used for implantation. Mice were anesthetized with 1.5% isoflurane and dorsal hair was removed with electric clippers and Nair. The skin was sterilized using 70% isopropyl alcohol prep wipes. Prior to surgery, 2 mg meloxicam/kg mouse weight was subcutaneously injected. 2 mm horizontal incisions were made in the upper and lower dorsal spaces. A subcutaneous pocket was formed by inserting surgical scissors into the incision and expanding. One scaffold per subcutaneous pocket was implanted and the incisions were closed with 2 Reflex 7 mm wound clips. Four scaffolds were implanted per mouse. Wound clips were removed after 1 week.
Characterization of cytokine secretion of implanted hBMSC-scaffolds:
Implanted hBMSC-scaffolds were retrieved at 4 and 12 weeks from NSG mice. Explanted scaffolds were cut into 1–2mm pieces using surgical scissors and cultured in 500 µL of αMEM media supplemented with 1% mouse serum and 1% penicillin-streptomycin for 36 hours. For control samples, 1 week in vitro cultured hBMSC-scaffolds were used. Conditioned media was collected and used immediately for characterization human VEGF, IL-6 and, IL-8 secretion by DuoSet® ELISA kits (R&D Systems).
Tail vein injection of human PBMCs:
Whole blood was obtained from 3 healthy male donors age 20–38 in BD Vacutainer K2 EDTA blood collection tubes. Mononuclear cells were isolated via density gradient-based centrifugation using Ficoll Paque at 1500 RPM for 30 minutes using minimum acceleration and no brake. From roughly 30 mL of whole blood we extracted 25–30 million PBMCs. Isolated mononuclear cells were counted and re-suspended in PBS to a density of 10×106 or 20×106 cells per 100 µL PBS. Scaffold bearing or scaffold and tumour bearing mice were warmed under a heat lamp to dilate the tail vein, placed in a restrainer, and intravenously delivered by tail vein injection using a 27 Gauge needle.
Human PBMC distribution and cytokine secretion:
Mice 6–13 weeks of age were implanted with BMSC seeded scaffolds. 20×106 PBMCs suspended in 100 µL of PBS were intravenously delivered 5.5 weeks after scaffold implantation. Mice were euthanized 24 hours after PBMC injection. Lung, liver, spleen, bone, and scaffolds were retrieved. Tissue samples were aliquoted for analysis via flow cytometry or ELISA. For flow cytometry experiments, freshly harvested tissue samples were minced with scissors in a 40 µm cell strainer and further digested with Collagenase, Type II. Stock solution (100 U/µL) was diluted in PBS to 200 U/mL and added to the tissue for 15 minutes. Tissue pieces were pushed through the 40 µm cell strainer using the back side of a 1 mL syringe plunger. DMEM media containing 10% FBS was added to quench the enzymatic reaction before spinning down in a centrifuge at 1500 RPM. Cell pellets were resuspended in FACS buffer (PBS containing 1% bovine serum albumin (BSA)) and counted. Cells were spun down again and resuspended to a final concentration of 107 cells/mL in FACS buffer. 100 µL of the cell suspension was added to a U-bottom 96 well plate. 100 µL of FACS buffer containing 2.5 µg of Fc Block was added to each well and incubated for 10 minutes at room temperature. The plate was spun down and supernatant removed before adding mouse anti-human CD45-PE diluted in FACS buffer 1:100. Samples were incubated for 30 minutes at room temperature in the dark. Samples were washed 3 times with FACS buffer before being transferred to FACS tubes and analyzed with a BD LSRFortessa. BD FACSDiva software was used for both collection and analysis. For human cytokine secretion, retrieved tissues were kept relatively equal in size and cut into 1–2 mm pieces using surgical scissors. Conditioned media was prepared 36 hours after plating in 500 µL of αMEM media supplemented with 1% mouse serum and 1% penicillin and streptomycin. Conditioned media was used immediately for characterization using DuoSet® ELISA kits for human TNF-α, IL-6 and IL-10 (R&D Systems)
Orthotopic xenoengraftment of human prostate tumour:
Luciferase (Luc) and green fluorescent protein (GFP) transduced PC-3 Human Prostate Cancer cells were obtained from the Center for Engineering in Medicine, Massachusetts General Hospital 26. Luc-GFP PC3 cells were cultured with DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Scaffold bearing mice were anesthetized with 2% isoflurane followed by ventral hair removal with Nair. Skin was sterilized with 70% isopropyl alcohol prep wipes. Prior to surgery 2 mg meloxicam/kg mouse weight was subcutaneously injected. A 10 mm vertical incision was made in the lower abdomen of the mouse. The bladder was externalized through the opening to expose the prostate. 2 × 106 Luc-GFP PC-3 cells suspended in 50 µL of matrigel were injected to the prostate using a 27 G needle. The bladder was returned to the intraperitoneal cavity, the muscle layer was sutured using polyglycolic acid sutures (Henry Schein), and the skin was clamped with Reflex 7 mm wound clips. Clips were removed after 1 week.
Monitoring human tumour engraftment and metastasis by bioluminescent imaging:
Two weeks after orthotopic injection of Luc-GFP PC3 tumour cells, whole body bioluminescent imaging was performed to confirm engraftment and metastasis to distant organs using an IVIS SpectrumCT (Perkin Elmer). Luciferin was prepared by dilution in PBS to a concentration of 15 mg/mL in PBS. Mice were anesthetized with 2% isoflurane and injected intraperitoneally with 10 µL luciferin solution/g of body weight before being placed in the imaging chamber. Images were taken every 5 minutes until peak values were achieved. 6 weeks after orthotopic tumour injection, mice were euthanized, and scaffolds were retrieved and placed in a 48-well plate. Luciferin stock solution was diluted 1:100 in PBS and 300 µL were loaded into each well. Bioluminescent imaging was performed every 2 minutes until reaching peak values.
Non-invasive long-term monitoring of metastasis in serially transplanted scaffolds:
Metastatic tumour microenvironments primed in primary mice were retrieved and immediately transplanted to the same anatomical site in naïve male NSG mice 6–12 weeks of age following the same method as initial scaffold implantation. Wound clips were removed after 1 week. Whole body bioluminescent imaging was started 4 weeks after serial transplantation and continued for an additional 6 weeks with measurements taken weekly. Images were taken every 5 minutes until peak values were achieved.
Scanning electron microscopy imaging:
Microfabricated scaffolds coated with type I collagen were fixed with 2% glutaraldehyde, serially dehydrated with 50, 70, 90, and 100% ethanol and lyophilized overnight. The prepared dehydrated samples were deposited with a thin gold film using a sputter-coating machine (208HR, Cressington) and observed using a Zeiss Ultra55 Field Emission Scanning Electron Microscope.
Immunohistological staining and imaging:
At the end of in vivo experimentation, tissue and scaffold samples were frozen by embedding tissue in CryomatrixTM and snap-frozen in a metal beaker containing 2-methylbutane cooled by dry ice. Frozen samples were sectioned using a Cryostat (NX70). Scaffolds were cut to 20–25 µm, while native tissue were cut to 10 µm thickness and attached to ColorMarkTM Plus glass slides. Remaining frozen tissue blocks and sectioned slides were stored at −80 °C. Frozen tissue was briefly fixed in pre-chilled acetone for 10 minutes. Following 3 wash cycles in 0.05% Tween-20 in PBS (PBST), samples were outlined with a hydrophobic marker and blocked with 10% goat serum and 1% BSA in PBST for 2 hours at room temperature. Primary antibodies were added at a dilution ratio of 1:200 in blocking solution and left overnight in 4 °C. Samples were washed 3 times with 0.05% PBST followed by secondary antibodies at a dilution ratio of 1:200 in blocking solution and left for 2 hours at room temperature. Samples were washed 3 times with PBST prior to addition of 1 µg/mL DAPI solution and a cover slide. When required, streptavidin conjugated Qdot was added to the samples and left for 1 hour prior to the final washing cycle. 10 µL of 10 ng/µL DAPI solution was added to samples before imaging. Images were obtained using a Zeiss Cell Observer SD with 10x and 20x objectives. Tiling was performed with 10% overlap. Human antibodies were validated against relevant mouse tissue to rule out cross-reactivity (Supplemental Fig. 21). For hematoxylin & eosin and trichrome staining, frozen tissue sections were fixed with 10% neutral buffered formalin for 10 minutes. After washing with DI water, sides were stained with dye solutions following the protocol provided by the (American Master Tech) (Supplementary Method 3).
Optical tissue clearing and complete optical sectioning of the scaffolds:
Mice were transcardially perfused with 20 mL of ice cold PBS followed by 20 mL of ice cold monomer solution containing 4% acrylamide, 0.05% bis-acrylamide, 0.25% VA-044 (thermal initiator) and 4% paraformaldehyde in PBS. Tissue samples were collected and incubated in fresh monomer solution at 4 °C for 48 hours. Air in the tubes was replaced with nitrogen gas prior to incubation at 37 °C for 3–4 hours to polymerize the monomer solution. Excess polymer was removed and samples were left in either PBS or tissue clearing solution (4% SDS, 200 mM boric acid, pH 8.5). Scaffold samples required thick sectioning to achieve whole tissue imaging. Samples were frozen in CryomatrixTM and cut to a thickness of 250 µm with a cryostat for a total of 4 slices per scaffold. Sections were washed thoroughly in 0.1 vol% Triton X-100 in PBS prior to antibody staining. All of the staining procedures were performed at room temperature. Samples were blocked with 10% normal goat serum (NGS) and 1% BSA in PBST overnight. Samples were incubated for 36 hours in primary antibody solution containing chicken anti-human cytokeratin and rabbit anti-mouse smooth muscle actin diluted 1:200 in blocking solution. Samples were washed thoroughly with Triton X-100 PBS for 24 hours. Samples were incubated in secondary antibody solution containing goat anti-chicken Alexa Fluor 647 and goat anti-rabbit Alexa Fluor 568 diluted 1:200 in blocking solution for 36 hours. Samples were washed thoroughly with Triton X-100 PBS before imaging. Refractive index matching solutions did not enhance scaffold imaging depth, so samples were imaged in PBS. Samples were transferred to a glass slide with silicone isolators and covered with a cover slip. Whole tissue Z-stack images were taken with a Zeiss Cell Observer SD with a 10x objective at a 10 µm z-step. Optical slices were observed and compiled in ImageJ. Single cells and tumour colonies were manually counted and concentric circles were drawn as previously described. Nikon Elements AR v4.5 was used for 3D rendering and creation of videos. The videos were annotated in Adobe Premiere Pro CC 2014 and encoded in Adobe Media Encoder CC 2014.
Image analysis:
For characterization of mCD31 and hVimentin distribution in 4-week in vivo scaffolds, concentric circles were drawn in ImageJ. Three circles were drawn with 1/3 R-Total (Total Radius), 2/3 R-Total and R-Total. Background signal of each individual channel were removed before measuring the pixel density. Values were normalized by the zone area and the signal measured from the entire scaffold to generate a relative distribution across the scaffold. The same analysis was performed for hCD45 distribution in hPBMC injected mice 6 weeks after intravenous injection. Percent coverage analysis (mCD31, hCD44, hCD45, Ki67, and mLy6G) was calculated by measuring the overlap between positive signal and the total area of the appropriate tissue (entire scaffold, hCytokeratin, or mLy6G+).
For in depth characterization of the local tumour microenvironment, single cell and tumour colonies were manually identified by examining tiled images of stained tissue sections. Nearest vessel, vessel diameter, tumour colony diameter, and nearest cell distance were measured in ImageJ for all of the identified tumour cells. Data was binned to generate histograms.
Multiple-antibody staining of overt metastatic tumour bearing scaffold:
8 consecutive tissue slices, each 20 µm thick, from a single scaffold were stained with multiple combinations of antibodies, for a total of 11 different stains. All of the images were aligned in ImageJ. Images were then divided into a grid of 24×24 images, thresholded and pixel density was measured for each channel. Total tissue area was determined by setting a low threshold to include background signal. Values from each individual channel were normalized by total tissue area within each and plotted in a 24×24 heatmap. 10 color images were generated by combining images from different staining combinations. From these composite images, regions of interest were manually determined to highlight heterogeneity. Regions of interest were translated to the 24×24 heatmaps and pixel density was recalculated for the 8 zones. Pixel densities from each of the zones of interest were normalized to the maximum value from the 8 zones. Channels that had more than one replicate were averaged together before plotting on a radar chart. Cellular composition within each zone was achieved by normalizing the absolute pixel count of each channel by the total pixel count of the zone.
Statistics:
Unpaired Student’s t tests were performed for comparison of the mean values between two groups. Statistical significance was determined if p<0.05 for two-tailed analysis. All quantitative data represent mean and standard deviation.
Supplementary Material
Acknowledgments
We thank the University of Massachusetts Amherst Animal Care Services. Amy Burnside for assistance with animal imaging and flow cytometry. Joseph Bergan for assistance with tissue clearing and imaging analysis. James Chambers and the University of Massachusetts Amherst Light Microscope Facility and Nikon Center of Excellence for assistance with Nikon software and workstations. Lisa Minter for discussing human immune cell activity in NSG mice. R.C. was supported by a National Science Foundation Research Traineeship (1545399). This work was supported by the National Cancer Institute (R00 CA163671) and the Institute for Applied Life Sciences to J. L.
Footnotes
Data Availability: The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information.
Competing interests
The authors declare no competing interests.
References
- 1.Luzzi KJ et al. Multistep nature of metastatic inefficiency - Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153, 865–873 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chambers AF, Groom AC & MacDonald IC Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2, 563–572 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Aguirre-Ghiso JA Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7, 834–846 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng S et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 10, 8152–8162 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Sosa MS, Bragado P & Aguirre-Ghiso JA Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14, 611–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He F et al. Multiscale characterization of the mineral phase at skeletal sites of breast cancer metastasis. P Natl Acad Sci USA 114, 10542–10547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paget S The distribution of secondary growths in cancer of the breast. Lancet 1, 571–573 (1889). [PubMed] [Google Scholar]
- 9.Kaplan RN et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kienast Y et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16, 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Wong CC et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci U S A 108, 16369–16374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oskarsson T et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17, 867–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barkan D et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res 70, 5706–5716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levental KR et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erler JT et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyles J et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 120, 2030–2039 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koebel CM et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Muller-Hermelink N et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 13, 507–518 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Hanna RN et al. Patrolling monocytes control tumor metastasis to the lung. Science 350, 985–990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambert AW, Pattabiraman DR & Weinberg RA Emerging Biological Principles of Metastasis. Cell 168, 670–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang Y & Pantel K Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francia G, Cruz-Munoz W, Man S, Xu P & Kerbel RS Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer 11, 135–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bos PD, Nguyen DX & Massague J Modeling metastasis in the mouse. Curr Opin Pharmacol 10, 571–577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shultz LD, Ishikawa F & Greiner DL Humanized mice in translational biomedical research. Nat Rev Immunol 7, 118–130 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Bersani F et al. Bioengineered Implantable Scaffolds as a Tool to Study Stromal-Derived Factors in Metastatic Cancer Models. Cancer Res 74, 7229–7238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villasante A & Vunjak-Novakovic G Tissue-engineered models of human tumors for cancer research. Expert Opin Drug Discov 10, 257–268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutmacher DW et al. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol 28, 125–133 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Cuddihy MJ & Kotov NA Three-dimensional cell culture matrices: state of the art. Tissue Eng Part B Rev 14, 61–86 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Seib FP, Berry JE, Shiozawa Y, Taichman RS & Kaplan DL Tissue engineering a surrogate niche for metastatic cancer cells. Biomaterials 51, 313–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stiers P-J, van Gastel N, Moermans K, Stockmans I & Carmeliet G An Ectopic Imaging Window for Intravital Imaging of Engineered Bone Tissue. JBMR Plus 2, 92–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holzapfel BM et al. Species-specific homing mechanisms of human prostate cancer metastasis in tissue engineered bone. Biomaterials 35, 4108–4115 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Moreau JE et al. Tissue-engineered bone serves as a target for metastasis of human breast cancer in a mouse model. Cancer Res 67, 10304–10308 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Schuster J, Zhang J & Longo M A novel human osteoblast-derived severe combined immunodeficiency mouse model of bone metastasis. J Neurosurg Spine 4, 388–391 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Thibaudeau L et al. A tissue-engineered humanized xenograft model of human breast cancer metastasis to bone. Dis Model Mech 7, 299–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinisch A et al. A humanized bone marrow ossicle xenotransplantation model enables improved engraftment of healthy and leukemic human hematopoietic cells. Nat Med 22, 812–821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J et al. Implantable microenvironments to attract hematopoietic stem/cancer cells. P Natl Acad Sci USA 109, 19638–19643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ko CY et al. The use of chemokine-releasing tissue engineering scaffolds in a model of inflammatory response-mediated melanoma cancer metastasis. Biomaterials 33, 876–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguado BA et al. Secretome identification of immune cell factors mediating metastatic cell homing. Sci Rep 5, 17566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaiselbuh SR, Edelman M, Lipton JM & Liu JM Ectopic human mesenchymal stem cell-coated scaffolds in NOD/SCID mice: an in vivo model of the leukemia niche. Tissue Eng Part C Methods 16, 1523–1531 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Aguado BA, Bushnell GG, Rao SS, Jeruss JS & Shea LD Engineering the pre-metastatic niche. Nature Biomedical Engineering 1, 0077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Azarin SM et al. In vivo capture and label-free detection of early metastatic cells. Nat Commun 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao SS et al. Enhanced Survival with Implantable Scaffolds That Capture Metastatic Breast Cancer Cells In Vivo. Cancer Res 76, 5209–5218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dvorak HF Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315, 1650–1659 (1986). [DOI] [PubMed] [Google Scholar]
- 45.Costa-Silva B et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17, 816–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue M et al. Graft-versus-tumor effect in a patient with advanced neuroblastoma who received HLA haplo-identical bone marrow transplantation. Bone Marrow Transplant 32, 103–106 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Lee J, Shanbhag S & Kotov NA Inverted colloidal crystals as three-dimensional microenvironments for cellular co-cultures. Journal of Materials Chemistry 16, 3558 (2006). [Google Scholar]
- 48.Bryers JD, Giachelli CM & Ratner BD Engineering biomaterials to integrate and heal: the biocompatibility paradigm shifts. Biotechnol Bioeng 109, 1898–1911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stachowiak AN, Bershteyn A, Tzatzalos E & Irvine DJ Bioactive hydrogels with an ordered cellular structure combine interconnected macroporosity and robust mechanical properties. Adv Mater 17, 399-+ (2005). [Google Scholar]
- 50.Kotov NA et al. Inverted colloidal crystals as three-dimensional cell scaffolds. Langmuir 20, 7887–7892 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Joao CF, Vasconcelos JM, Silva JC & Borges JP An overview of inverted colloidal crystal systems for tissue engineering. Tissue Eng Part B Rev 20, 437–454 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Parekkadan B & Milwid JM Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 12, 87–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rutkowski MR et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell 27, 27–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li A, Dubey S, Varney ML, Dave BJ & Singh RK IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. The Journal of Immunology 170, 3369–3376 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Lee E et al. Breast cancer cells condition lymphatic endothelial cells within pre-metastatic niches to promote metastasis. Nat Commun 5, 4715 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Havens AM et al. An In Vivo Mouse Model for Human Prostate Cancer Metastasis. Neoplasia 10, 371–IN374 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Magnon C et al. Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Tai S et al. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 71, 1668–1679 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kitamura T, Qian BZ & Pollard JW Immune cell promotion of metastasis. Nat Rev Immunol 15, 73–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakahara T, Norberg SM, Shalinsky DR, Hu-Lowe DD & McDonald DM Effect of inhibition of vascular endothelial growth factor signaling on distribution of extravasated antibodies in tumors. Cancer Res 66, 1434–1445 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Levine JE Implications of TNF-alpha in the pathogenesis and management of GVHD. Int J Hematol 93, 571–577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeiser R & Blazar BR Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med 377, 2167–2179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung K et al. Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Visvader JE & Lindeman GJ Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8, 755–768 (2008). [DOI] [PubMed] [Google Scholar]
- 65.McFarlane S et al. CD44 increases the efficiency of distant metastasis of breast cancer. Oncotarget 6, 11465–11476 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taichman RS et al. GAS6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS One 8, e61873 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kobayashi A et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. The Journal of Experimental Medicine 209, 639–639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao H et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bragado P et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol 15, 1351–1361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sosa MS et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun 6, 6170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aguirre-Ghiso JA, Estrada Y, Liu D & Ossowski L ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res 63, 1684–1695 (2003). [PubMed] [Google Scholar]
- 72.Yumoto K et al. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep 6, 36520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ruppender N et al. Cellular Adhesion Promotes Prostate Cancer Cells Escape from Dormancy. PLoS One 10, e0130565 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Novak ML & Koh TJ Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol 183, 1352–1363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCracken JM & Allen LA Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death 7, 15–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hosseini H et al. Early dissemination seeds metastasis in breast cancer. Nature (2016). [DOI] [PMC free article] [PubMed]
- 77.Rothwell PM et al. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. The Lancet 379, 1591–1601 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Ridker PM et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. The Lancet 390, 1833–1842 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Jacob L, Kostev K, Rathmann W & Kalder M Impact of metformin on metastases in patients with breast cancer and type 2 diabetes. J Diabetes Complications 30, 1056–1059 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Yakes FM et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther 10, 2298–2308 (2011). [DOI] [PubMed] [Google Scholar]
- 81.Dondossola E et al. Examination of the foreign body response to biomaterials by nonlinear intravital microscopy. Nature Biomedical Engineering 1, 0007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nair A & Tang L Influence of scaffold design on host immune and stem cell responses. Semin Immunol 29, 62–71 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Ali OA, Huebsch N, Cao L, Dranoff G & Mooney DJ Infection-mimicking materials to program dendritic cells in situ. Nat Mater 8, 151–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bencherif SA et al. Injectable cryogel-based whole-cell cancer vaccines. Nat Commun 6, 7556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sadtler K et al. Developing a pro-regenerative biomaterial scaffold microenvironment requires T helper 2 cells. Science 352, 366–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Veiseh O et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat Mater 14, 643–651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Swartzlander MD et al. Immunomodulation by mesenchymal stem cells combats the foreign body response to cell-laden synthetic hydrogels. Biomaterials 41, 79–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee J, Heckl D & Parekkadan B Multiple genetically engineered humanized microenvironments in a single mouse. Biomater Res 20, 19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tentler JJ et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 9, 338–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.White L, Meyer PR & Benedict WF Establishment and characterization of a human T-cell leukemia line (LALW-2) in nude mice. J Natl Cancer Inst 72, 1029–1038 (1984). [PubMed] [Google Scholar]
- 91.Baron F et al. Graft-versus-tumor effects after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. J Clin Oncol 23, 1993–2003 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Korngold R, Marini JC, de Baca ME, Murphy GF & Giles-Komar J Role of tumor necrosis factor-α in graft-versus-host disease and graft-versus-leukemia responses. Biology of Blood and Marrow Transplantation 9, 292–303 (2003). [DOI] [PubMed] [Google Scholar]
- 93.Roth MD & Harui A Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J Immunother Cancer 3, 12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simpson-Abelson MR et al. Long-Term Engraftment and Expansion of Tumor-Derived Memory T Cells Following the Implantation of Non-Disrupted Pieces of Human Lung Tumor into NOD-scid IL2R null Mice. The Journal of Immunology 180, 7009–7018 (2008). [DOI] [PubMed] [Google Scholar]
- 95.El Rayes T et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. P Natl Acad Sci USA 112, 16000–16005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Cock JM et al. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res 76, 6778–6784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park J et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wculek SK & Malanchi I Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Engblom C et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rahbari NN et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci Transl Med 8, 360ra135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sainson RC et al. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 111, 4997–5007 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murgai M et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med 23, 1176–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Coffelt SB, Wellenstein MD & de Visser KE Neutrophils in cancer: neutral no more. Nat Rev Cancer 16, 431–446 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Jespersen H et al. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat Commun 8, 707 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Marin Navarro A, Susanto E, Falk A & Wilhelm M Modeling cancer using patient-derived induced pluripotent stem cells to understand development of childhood malignancies. Cell Death Discov 4, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Patel AA et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 214, 1913–1923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Drake AC, Chen Q & Chen J Engineering humanized mice for improved hematopoietic reconstitution. Cell Mol Immunol 9, 215–224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Powell DR & Huttenlocher A Neutrophils in the Tumor Microenvironment. Trends Immunol 37, 41–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reichman H, Karo-Atar D & Munitz A Emerging Roles for Eosinophils in the Tumor Microenvironment. Trends Cancer 2, 664–675 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Qian BZ & Pollard JW Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Green JJ & Elisseeff JH Mimicking biological functionality with polymers for biomedical applications. Nature 540, 386–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aguado BA et al. Biomaterial Scaffolds as Pre-metastatic Niche Mimics Systemically Alter the Primary Tumor and Tumor Microenvironment. Adv Healthc Mater (2018). [DOI] [PMC free article] [PubMed]
- 113.MacKie RM, Reid R & Junor B Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med 348, 567–568 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Strauss DC & Thomas JM Transmission of donor melanoma by organ transplantation. The Lancet Oncology 11, 790–796 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.