

Abstract
Sex chromosome gene dosage compensation is required to ensure equivalent levels of X-linked gene expression between males (46, XY) and females (46, XX). To achieve similar expression, X-chromosome inactivation (XCI) is initiated in female cells during early stages of embryogenesis. Within each cell, either the maternal or paternal X chromosome is selected for whole chromosome transcriptional silencing, which is initiated and maintained by epigenetic and chromatin conformation mechanisms. With the emergence of small-molecule epigenetic inhibitors for the treatment of disease, such as cancer, the epigenetic mechanism underlying XCI may be inadvertently targeted. Here, we test 2 small-molecule epigenetic inhibitors being used clinically, GSK126 (a histone H3 lysine 27 methyltransferase inhibitor) and suberoylanilide hydroxamic acid (a histone deacetylase inhibitor), on their effects of the inactive X (Xi) in healthy human female fibroblasts. The combination of these modifiers, at subcancer therapeutic levels, leads to the inability to detect the repressive H3K27me3 modification characteristic of XCI in the majority of the cells. Importantly, genes positioned near the X-inactivation center (Xic), where inactivation is initiated, exhibit robust expression with treatment of the inhibitors, while genes located near the distal ends of the X chromosome intriguingly exhibit significant downregulation. These results demonstrate that small-molecule epigenetic inhibitors can have profound consequences on XCI in human cells, and they underscore the importance of considering gender when developing and clinically testing small-molecule epigenetic inhibitors, in particular those that target the well-characterized mechanisms of X inactivation.
Keywords: epigenetic inhibitors, gene dosage compensation, X-chromosome inactivation, X-linked gene expression, X-linked gene regulation, gender-specific medicine, sex-specific genetic architecture, environment and epigenetics, biological sex and gene expression
Introduction
In early human embryogenesis, X-chromosome inactivation (XCI) is initiated in female cells (46, XX) to establish equivalent levels of X-linked gene expression with male cells (46, XY).1,2 To achieve dosage compensation, either the maternal- or paternal-derived X chromosome in female cells is randomly selected for whole chromosome transcriptional silencing through epigenetic and chromatin conformation mechanisms.3 Xist, a 19-kb long noncoding RNA, is expressed from the X-inactivation center (Xic) of the chromosome selected for inactivation.4,5 Xist spreads in cis to transcriptionally active regions, initiating conformational changes and creating newly accessible regions for silencing.6 Shortly after Xist RNA accumulates, histone modifications associated with gene expression, including H3K4me2/3, H3K36 methylation, and H3 and H4 acetylation, are lost.7–12 Next, Xist recruits repressive epigenetic protein complexes PRC2, HBiX1, and SMCHD1, resulting in repressive epigenetic modifications on histone H3 including, K27me3 and K9me3.13–15 The inactive X (Xi) undergoes whole chromosome heterochromatinization facilitated by heterochromatin protein 1 forming the transcriptionally inactive Barr body.16 Although a subset of X-linked genes (approximately 15%) are known to escape inactivation, the Xi is maintained during all subsequent cell divisions.17–19 Thus, maintenance of the epigenetic environment of the Xi is critical for dosage compensation in human female cells.
Epigenetic protein complexes are increasingly targeted in cancer treatment and currently comprise a significant number of ongoing clinical trials in drug therapy pipelines.20 Currently, approximately 100 clinical trials testing epigenetic inhibitors are listed on clinicaltrials.gov, including classes that target DNA methyltransferases (DNMTs), histone deacetylases (HDACs), and histone methyltransferases.20–23 In addition, combinatorial strategies targeting more than 1 class of epigenetic enzyme are also being developed.20,24 However, many of the epigenetic inhibitors currently in clinical trials target key chromatin modifications necessary for maintaining X chromosome dosage compensation in human female cells, adding an important caveat to consider when designing epigenetic inhibitor cocktails.21,22,24 Due to the nonspecific gene regulatory nature of these epigenetic modifiers, their use may result in unintended consequences for maintenance of the Xi, possibly leading to additional deleterious side effects from these drugs, specifically in female patients.
Here, we have treated healthy human female fibroblasts with an EZH2 inhibitor, GSK126 (GSK2816126), which targets the enzymatic activity of the PRC2 complex, and a Food and Drug Administration (FDA)-approved HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA; also known as vorinostat and Zolinza). We chose to target EZH2 due to the tight association of its repressive epigenetic mark, H3K27me3, with Xist localization on the Xi and because EZH2 is one of the most commonly upregulated epigenetic regulators in human cancers.25,26 The SAHA was chosen as HDAC activity is integral to Xi gene silencing and the increasing popularity of epigenetic combinatorial treatments including at least 1 HDAC inhibitor. We observe a significant drop in H3K27me3 association with the Xi in all human female cells after treatment with GSK126 and SAHA. In addition, X-linked genes are significantly upregulated and downregulated after drug incubation, with gene regulatory impact dependent on chromosome X location. Our results indicate the importance of considering gender when developing and testing small-molecule epigenetic modulators to be used clinically, specifically those that target the well-characterized marks of XCI.
Material and Methods
Cell Culture and Epigenetic Inhibitor Incubations
Primary human lung fibroblasts (IMR-90; ATCC, Manassas, Virginia) were cultured in Dulbecco modified Eagle medium α supplemented with 15% fetal bovine serum, 1% penicillin/streptomycin, and 1% L-glutamine. Cells were incubated with GSK126 (APExBIO, Houston, Texas) for 4 days and HDAC inhibitor, SAHA (Sigma-Aldrich, St Louis, Missouri) for 24 hours, or an equivalent volume of dimethyl sulfoxide as a control.
Immunoblot
IMR-90s were harvested, washed, and resuspended in 100 μL of harsh lysis buffer (50 mM Tris, 300 mM NaCl, 50 mM NaF, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM DTT, and 1X protease inhibitors; Sigma-Aldrich) for 30 minutes on ice. Lysates were sonicated for 30 seconds (amplitude 35%, 1-second pulses) and centrifuged for 10 minutes at 4°C at maximum speed. Supernatants were collected and protein concentrations quantified. Equivalent protein was loaded and run for each sample on 4% to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels for gel electrophoresis, transfer, and immunoblotting. Super-Signal West Pico (Thermo Fisher Scientific, Waltham, MA) was used for detection and imaging with the luminescent image analyzer (LAS-4000; Fujifilm, Tokyo, Japan).
Antibodies/Reagents
Rabbit polyclonal antibody against H3K27me3 (Millipore Sigma, St Louis, Missouri) was diluted 1:1000 or 1:50 for Western blot or immuno-DNA fluorescence in situ hybridization (FISH), respectively. Rabbit polyclonal antibody against acetyl-histone H3 (Millipore Sigma) was diluted 1:2000 and rabbit polyclonal antibody against histone H3 (Abcam, Cambridge, United Kingdom) was diluted 1:4000 for Western blot. Antirabbit secondary antibody (Abcam) conjugated with horseradish peroxidase was diluted 1:5000. For immuno-DNA FISH, biotin-conjugated antirabbit antibody (Thermo Fisher Scientific) was diluted 1:250 and secondary antibody antimouse biotin conjugated with fluorescein (Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania) was diluted 1:250. A total of 8 μL of human chromosome X paint (MetaSystems, Heidelberg, Germany) was hybridized per 22 mm × 22 mm coverslip.
Three-Dimensional Interphase Immuno-DNA FISH
Standard 3-dimensional (3D) interphase immuno-DNA FISH protocol was performed based on the protocol of Cremer et al27 of sections 3.2.6, 3.2.4, and 3.2.5 with minimal modifications. Minimal modifications include 2-hour 20% glycerol incubation step, no liquid nitrogen freezing steps, and chromosome X paints were incubated overnight. Slides were imaged on an A1R confocal microscope (Nikon, Tokyo, Japan) with 0.2 μm z-stacks and 3D images were analyzed with Image-Pro Plus software (Media Cybernetics, Rockville, Maryland).
RNA Isolation, Complementary DNA, and Quantitative Polymerase Chain Reaction
IMR-90s were harvested, washed, and pelleted, followed by total RNA isolation with TRIzol (Thermo Fisher Scientific). Complementary DNA was synthesized from RNA using the SuperScript IV First-Strand Synthesis System (Thermo Fisher Scientific) for quantitative polymerase chain reaction performed with the 480 SYBR Green 1 master reagent (Roche, Basel, Switzerland) on the LightCycler 480 (Roche). Primer bank IDs are listed in Supplemental Table 1.
Results
GSK126 and SAHA Treatment Decreases Total H3K27me3 Levels and Increases Total Histone H3 Acetylation Levels in Human Female Fibroblasts
To identify the most effective drug combination, we first tested a range of concentrations of GSK126 (0-2 μM) and SAHA (0-15 μM) independently and in combination on total H3K27me3 and histone H3 acetylation levels in normal human female fibroblasts (IMR-90). Concentration ranges for both drugs did not exceed concentrations tested as cancer therapeutics.22–24 GSK126 treatment revealed lowered levels of total H3K27me3 with increasing GSK126 concentrations, as previously shown, while total H3 acetylation levels were unaffected (Figure 1A, left panels).23,28 The concentration of GSK126 that exhibited the greatest level of H3K27me3 depletion was 2 μM, with a 0.7-fold reduction in H3K27me3 signal. The SAHA increased total H3K27me3 up to 1.5-fold and H3 acetylation levels up to 8-fold with increasing drug concentrations (Figure 1A, center panels). Next, we tested the same range of SAHA concentrations in combination with 2 μM GSK126 (Figure 1A, right panels). In all conditions, H3K27me3 levels slightly decreased by approximately 1-fold. In addition, the level in which total H3 acetylation levels begin to increase starts at 5 μM of SAHA as opposed to 1 μM on its own (Figure 1A, center panels), while H3 acetylation levels only increase by approximately 2.5-fold in drug conditions 3, 4, and 5. From this analysis, we concluded the optimal drug combination to be 2 μM GSK126 + 5 μM SAHA (drug combination 3), the lowest concentration of drugs which resulted in the largest decrease in H3K27me3 levels and the highest H3 acetylation levels. These drug concentrations are below the dose used for cancer intervention.22–24
Figure 1.
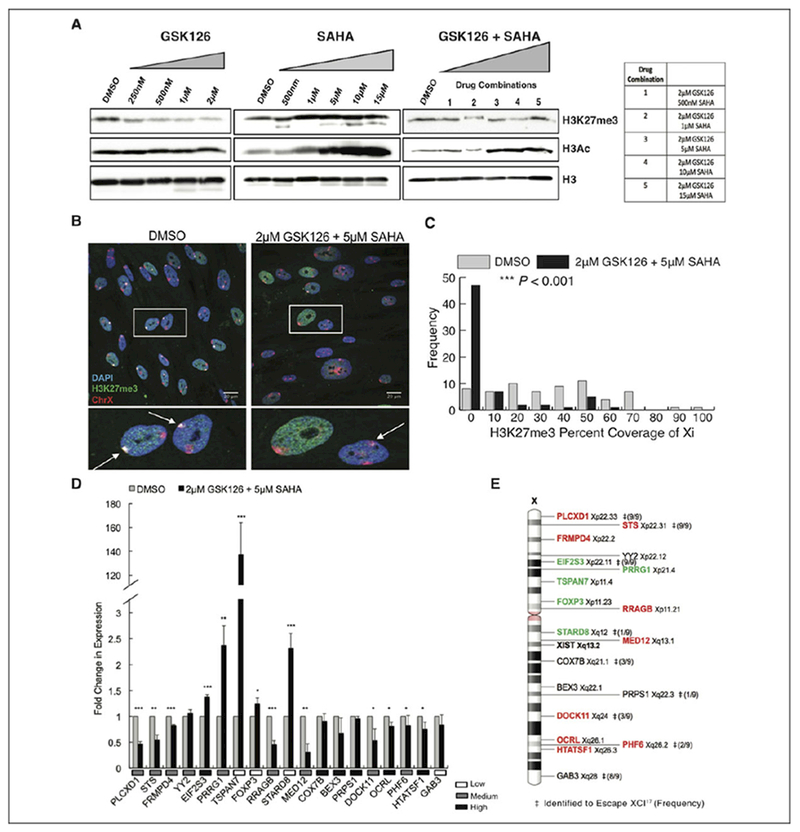
GSK126 and SAHA disrupt chromosome X gene dosage compensation in human female cells. A, Total H3K27me3, H3 acetylation, and histone H3 protein levels of IMR-90s treated with various concentrations and combinations of epigenetic inhibitors, GSK126 and SAHA, or DMSO alone as the control. Total histone H3 was used as a loading control. B, Maximum intensity projections of H3K27me3 (green) and chromosome X (red) localization in IMR-90 nuclei (blue). Insets show enlarged nuclei, arrows highlight location of the Xi. C, Distribution display of 3-dimensional volumetric measurements of H3K27me3 signal over total Xi signal for either DMSO or 2 μM GSK126 + 5 μM SAHA treatment (n = 130 nuclei, 65 nuclei per condition). *P < .05, **P < .01, ***P < .001, Student t test. D, Quantitative PCR analysis of X-linked genes with either DMSO treatment or 2 μM GSK126 + 5 μM SAHA treatment. Values presented as mean (standard deviation) of 3 technical replicates. *P < .05, **P < .01, ***P < .001, Student t test. Relative expression levels of genes in DMSO-treated cells indicated under graph. E, Schematic diagram of chromosome X, with corresponding positions of X-linked genes analyzed in (D) as well as the Xist gene locus. Significantly upregulated genes (green) and significantly downregulated genes (red). ‡Genes identified to escape XCI in human hybrid fibroblasts,17 and frequency of escape listed in parenthesis. DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; SAHA, suberoylanilide hydroxamic acid; XCI, X-chromosome inactivation; Xi, inactive X.
GSK126 and SAHA Disrupt the Epigenetic Environment of the Xi Chromosome
We next examined the effects of the epigenetic inhibitor cocktail specifically on the epigenetic environment of the Xi. The Xi can be visualized by the overlap of the epigenetic repressive mark H3K27me3 and the X chromosome by 3D interphase immuno-DNA FISH. Control human fibroblasts showed standard frequencies of H3K27me3 coverage of the Xi using this technique (Figure 1B, left panels and 1C). However, in fibroblasts incubated with 2 μM GSK126 and 5 μM SAHA, the majority of cells exhibit no evidence of H3K27me3 labeling on either X chromosome, and the remaining cells exhibit significantly decreased levels of H3K27me3 coverage of the Xi (Figure 1B, right panels and 1C). To determine changes in X-linked gene expression after drug incubation, quantitative PCR was performed. X-linked genes located in the most central portion of the X chromosome and proximal to the Xic demonstrate the highest levels of upregulation (Figure 1D and E). These genes include EIF2S3, PRRG1, TSPAN7, FOXP3, and STARD8. Interestingly, genes located near the distal ends of the X chromosome are downregulated, even though they are typically highly expressed (Figure 1D). Genes previously identified to escape XCI in human hybrid fibroblasts17 show no correlation between escapee status and a particular pattern of gene regulation disruption from the epigenetic inhibitor cocktail (Figure 1E). These results indicate that GSK126 in combination with the FDA-approved HDAC inhibitor SAHA are capable of severely disrupting the epigenetic environment required for gene dosage compensation in human female cells resulting in misregulation of X-linked genes.
Discussion
In this study, we have demonstrated that the combination of popular epigenetic inhibitors, GSK126 and SAHA, severely disrupt the repressive epigenetic environment of the Xi in human female cells. Moreover, this treatment results in significant upregulation and downregulation of X-linked genes, leading to a loss of proper X-linked gene dosage compensation. Misregulation of X-linked genes leads to serious physiological consequences, such as increased propensity for autoimmunity in women and hematological cancer in mice.29,30 Surprisingly, disruption of the epigenetic repressive mark H3K27me3 of the Xi in human cells only requires targeting EZH2-mediated H3K27me3 and histone H3 acetylation. These results may be due to the effectiveness of the epigenetic inhibitors utilized. GSK126 is the most highly potent and selective EZH2 inhibitor developed thus far (0.3 nM, 150-fold selectivity against EZH1).23 Several other EZH2 inhibitors have been generated with various levels of potency and selectivity including but not limited to DZNep, GSK343, El1, EPZ005687, EPZ-6438 (tazemetostat), and CP1-169.31 The SAHA targets class I HDACs at various levels of potency, ranging from 21 to 1524 nM.22 Other class I HDAC inhibitors developed for clinical interventions include but are not limited to TSA (trichostatin A), MS-275 (entinostat), MGCD0103 (mocetinostat), and FK228 (romidepsin).22 The development of a myriad of epigenetic inhibitors creates useful tools for determining the significance and role of epigenetic mechanisms underlying initiation and maintenance of XCI.
Our results also indicate the importance of considering gender when developing and testing small-molecule epigenetic modifiers, specifically those that target well-characterized marks of X inactivation. Currently, singular administration of EZH2 inhibitors is in either stage 1 or 2 of clinical trials (http://ClinicalTrials.gov). The SAHA (vorinostat, Zolinza) is approved by the FDA for the treatment of cutaneous T-cell lymphoma32 and currently enrolled in 80 clinical trials for combinatorial treatment with other established chemotherapies (ClinicalTrials.gov). Testing of combinatorial epigenetic inhibitor strategies in vitro and in vivo has started to increase in popularity. The particular combination of an EZH2 inhibitor and HDAC inhibitor has only been tested thus far in vitro on human breast cancer cell lines.24 A large proportion of studies are focused on combinations of HDAC and DNMT inhibitors.33–35 With the large increase in novel epigenetic inhibitor development for treatment of disease, effects on XCI maintenance in human female cells should be analyzed. Also, during early stages of clinical trials, epigenetic inhibitor side effects should be delineated by gender. Finally, our work suggests that developing gender-specific interventions when utilizing epigenetic therapeutics in the treatment of disease is warranted.
Supplementary Material
Acknowledgments
Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. The authors also thank the tremendous technical imaging assistance of Dina Arvanitis of the Center for Advanced Microscopy.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by research grants from the National Institutes of Health, National Institute of General Medical Sciences, New Innovator Award (DP2 OD008717), awarded to S.T.K. as well as the National Institutes of Health, National Institute of General Medical Sciences, National Research Service Postdoctoral Award (F32 GM115046) awarded to A.I.L.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplementary material for this article is available online.
References
- 1.Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–373. [DOI] [PubMed] [Google Scholar]
- 2.Moreira de Mello JC, Fernandes GR, Vibranovski MD, Pereira LV. Early X chromosome inactivation during human preimplantation development revealed by single-cell RNA-sequencing. Sci Rep. 2017;7(1):10794. doi: 10.1038/s41598-017-11044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avner P, Heard E. X-chromosome inactivation: counting, choice and initiation. Nat Rev Genet. 2001;2(1):59–67. doi: 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- 4.Bacher CP, Guggiari M, Brors B, et al. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat Cell Biol. 2006;8(3):293–299. doi: 10.1038/ncb1365. [DOI] [PubMed] [Google Scholar]
- 5.Xu N, Tsai CL, Lee JT. Transient homologous chromosome pairing marks the onset of X inactivation. Science. 2006;311(5764):1149–1152. doi: 10.1126/science.1122984. [DOI] [PubMed] [Google Scholar]
- 6.Engreitz JM, Pandya-Jones A, McDonel P, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341(6147):. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boggs BA, Cheung P, Heard E, et al. Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat Genet. 2002;30(1):73–76. doi: 10.1038/ng787. [DOI] [PubMed] [Google Scholar]
- 8.Chaumeil J, Okamoto I, Guggiari M, Heard E. Integrated kinetics of X chromosome inactivation in differentiating embryonic stem cells. Cytogenet Genome Res. 2002;99(1-4):75–84. [DOI] [PubMed] [Google Scholar]
- 9.Jeppesen P, Turner BM. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell. 1993;74(2):281–289. [DOI] [PubMed] [Google Scholar]
- 10.Keohane AM, O’neill LP, Belyaev ND, Lavender JS, Turner BM. X-inactivation and histone H4 acetylation in embryonic stem cells. Dev Biol. 1996;180(2):618–630. doi: 10.1006/dbio.1996.0333. [DOI] [PubMed] [Google Scholar]
- 11.Mermoud JE, Popova B, Peters AH, Jenuwein T, Brockdorff N. Histone H3 lysine 9 methylation occurs rapidly at the onset of random X chromosome inactivation. Curr Biol. 2002;12(3):247–251. [DOI] [PubMed] [Google Scholar]
- 12.O’Neill LP, Spotswood HT, Fernando M, Turner BM. Differential loss of histone H3 isoforms mono-, di- and tri-methylated at lysine 4 during X-inactivation in female embryonic stem cells. Biol Chem. 2008;389(4):365–370. doi: 10.1515/BC.2008.046. [DOI] [PubMed] [Google Scholar]
- 13.Nozawa RS, Nagao K, Igami KT, et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol. 2013;20(5):566–573. doi: 10.1038/nsmb.2532. [DOI] [PubMed] [Google Scholar]
- 14.Plath K, Fang J, Mlynarczyk-Evans SK, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300(5616):131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 15.Silva J, Mak W, Zvetkova I, et al. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell. 2003;4(4):481–495. [DOI] [PubMed] [Google Scholar]
- 16.Chan KM, Zhang H, Malureanu L, van Deursen J, Zhang Z. Diverse factors are involved in maintaining X chromosome inactivation. Proc Nati Acad Sci U S A. 2011;108(40):16699–16704. doi: 10.1073/pnas.1107616108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434(7031):400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 18.Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet. 2011;130(2):237–245. doi: 10.1007/s00439-011-1011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Castillo-Morales A, Jiang M, et al. Genes that escape x-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol. 2013;30(12):2588–2601. doi: 10.1093/molbev/mst148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma M, Banerjee HN. Epigenetic inhibitors. Methods Mol Biol. 2015;1238:469–485. doi: 10.1007/978-1-4939-1804-1_24. [DOI] [PubMed] [Google Scholar]
- 21.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hull EE, Montgomery MR, Leyva KJ. HDAC inhibitors as epigenetic regulators of the immune system: Impacts on cancer therapy and inflammatory diseases. BioMed Res Int. 2016;2016. doi: 10.1155/2016/8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 24.Sun F, Chan E, Wu Z, et al. Combinatorial pharmacologic approaches target EZH2-mediated gene repression in breast cancer cells. Mol Cancer Ther. 2009;8(12):3191–3202. doi: 10.1158/1535-7163.MCT-09-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mut Res. 2008;647(1-2):21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17(9):2613–2618. doi: 10.1158/1078-0432.CCR-10-2156. [DOI] [PubMed] [Google Scholar]
- 27.Cremer M, Grasser F, Lanctot C, et al. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods in Mol Biol. 2008;463:205–239. doi: 10.1007/978-1-59745-406-3_15. [DOI] [PubMed] [Google Scholar]
- 28.Rao RA, Dhele N, Cheemadan S, et al. Ezh2 mediated H3K27me3 activity facilitates somatic transition during human pluripotent reprogramming. Sci Rep. 2015;5:8229. doi: 10.1038/srep08229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Syrett CM, Kramer MC, et al. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc Natl Acad Sci U S A. 2016;113(14):E2029–E2038. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yildirim E, Kirby JE, Brown DE, et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152(4):727–742. doi: 10.1016/j.cell.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics. 2016;8:57. doi: 10.1186/s13148-016-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The Oncologist. 2007;12(10):1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 33.Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pera B, Tang T, Marullo R, et al. Combinatorial epigenetic therapy in diffuse large B cell lymphoma pre-clinical models and patients. Clin Epigenetics. 2016;8:79. doi: 10.1186/s13148-016-0245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thangavel J, Malik AB, Elias HK, et al. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am J Pathol. 2014;184(8):2237–2249. doi: 10.1016/j.ajpath.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.