

The vast majority of bacteria, including human pathogens and microbiome species, lack genetic tools needed to systematically associate genes with phenotypes. This is the major impediment to understanding the fundamental contributions of genes and gene networks to bacterial physiology and human health. CRISPRi, a versatile method of blocking gene expression using a catalytically inactive Cas9 protein (dCas9) and programmable single guide RNAs (sgRNAs), has emerged as a powerful genetic tool to dissect the functions of essential and non-essential genes in species ranging from bacteria to humans1–6. However, the difficulty of establishing effective CRISPRi systems across bacteria is a major barrier to its widespread use to dissect bacterial gene function. Here, we establish “Mobile-CRISPRi”, a suite of CRISPRi systems that combine modularity, stable genomic integration and ease of transfer to diverse bacteria by conjugation. Focusing predominantly on human pathogens associated with antibiotic resistance, we demonstrate the efficacy of Mobile-CRISPRi in γ-Proteobacteria and Bacillales Firmicutes at the individual gene scale by examining drug-gene synergies and at the library scale by systematically phenotyping conditionally essential genes involved in amino acid biosynthesis. Mobile-CRISPRi enables genetic dissection of non-model bacteria, facilitating analyses of microbiome function, antibiotic resistances and sensitivities, and comprehensive screens for host-microbe interactions.
CRISPRi (Clustered Regularly Interspaced Short Palindromic Repeats interference) is a programmable method for controlling gene expression that has enabled systematic interrogation of gene phenotypes in diverse organisms1–6. In bacterial CRISPRi, an sgRNA-dCas9 complex binds to a target gene by base-pairing and reduces gene expression by sterically blocking transcription elongation (Fig. 1a)1,4. New CRISPRi targets are easily programmed by substituting the first 20 nt of the sgRNA sequence (spacer) to match the non-template strand of the target gene, making design and construction of CRISPRi libraries that target specific sets of genes or the entire genome straightforward4,7,8. Genetic screens using CRISPRi libraries have contributed new insights into fundamental biology and molecular medicine including identifying functions for uncharacterized essential genes4,7 and drug modes of action4,9.
Fig. 1 |. Mobile-CRISPRi overview.
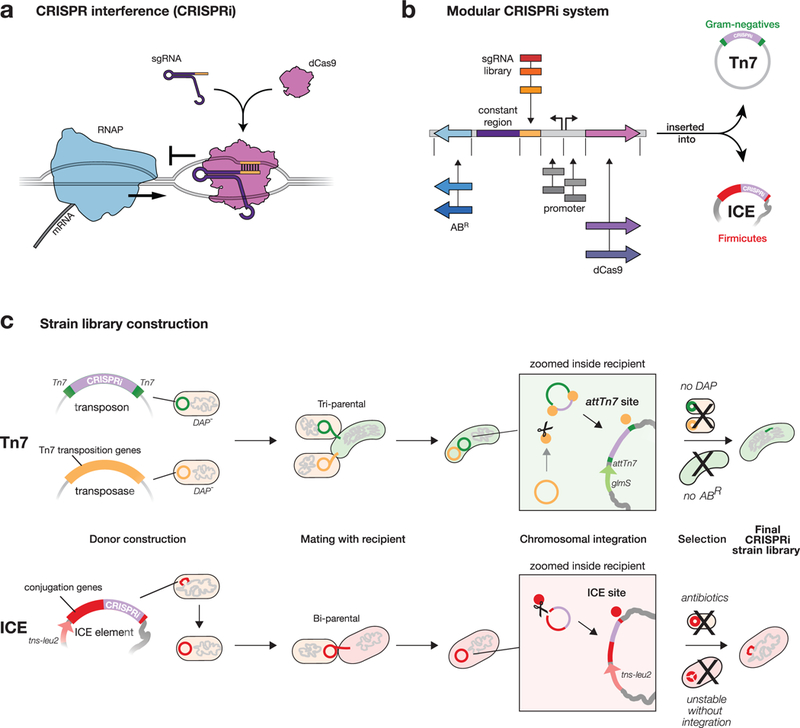
a, Mechanism of CRISPRi repression. A dCas9-sgRNA complex binds to DNA by base-pairing and sterically blocks progression of RNA polymerase (RNAP), reducing gene expression. b, Mobile-CRISPRi modularity. Individual modules are flanked by unique restriction sites, which can be used for cloning sgRNA libraries or exchanging other components (e.g. antibiotic resistance marker (ABR), or dcas9 promoter). c, Strain construction using Mobile-CRISPRi. Top: a Tn7 transposon carrying CRISPRi components (shown in b) and a plasmid containing Tn7 transposition genes are transferred to recipient bacteria by tri-parental mating. Donor cells contain a chromosomal copy of the RP4 transfer machinery used to mobilize the Tn7 plasmids. Once inside the recipient cell, Tn7 transposition proteins integrate the CRISPRi DNA (purple) flanked by left and right Tn7 end sequences (green) into the recipient genome downstream of the glmS gene. Selection on antibiotic plates lacking DAP eliminates the E. coli donors and retains recipients with an integrated CRISPRi system. Bottom: An ICE element carrying CRISPRi components is transferred to recipient bacteria by bi-parental mating. Once inside the recipient cell, the ICE integrase inserts ICE into trnS-leu2. Double antibiotic plates that select for ICE and for the intrinsic resistance of the recipient strain (Streptomycin-resistance in this work) are used to identify recipients with an integrated CRISPRi system.
CRISPRi provides several advantages over other methods for genetic manipulation in bacteria. CRISPRi knockdowns can be induced1,3–6 and titrated/tuned4,10, enabling depletion of essential gene products without complex strain construction strategies that remove genes from their native regulation. Dissecting genetic redundancy via multiplexed CRISPRi targeting of several genes in the same cell4,11 requires markedly less effort than construction of multiple-deletion strains. At the genome scale, CRISPRi expands on prior transposon-based gene perturbation methods such as Tn-seq12 by allowing all genes—including essential genes that cannot be studied through deletion—to be systematically targeted so that a relatively small strain library provides comprehensive coverage of the genome. Moreover, the DNA sequences encoding sgRNAs serve as unique barcodes to differentiate CRISPRi strains mixed in a pool, allowing for competitive fitness measurements using next generation sequencing8. CRISPRi blocks expression of downstream genes in operons1,4, but this property can be used to further simplify libraries by targeting operons instead of genes.
Despite these advantages, CRISPRi has been used in only a few bacterial species both because CRISPRi has been transferred using species-specific4 or narrow host-range1,3,5,6,13 strategies, and because components need to be optimized for function in different species. To overcome this barrier, we developed “Mobile-CRISPRi”—a suite of modular and transferable CRISPRi components that can stably integrate into the genomes of diverse bacteria. The modularity of every component of Mobile-CRISPRi makes it straightforward to clone in organism-specific sgRNA libraries and other components (e.g. promoters, Fig. 1b, Supplementary Fig. 1 and Supplementary Fig. 2). Mobile-CRISPRi achieves transfer and genomic integration by distinct mechanisms for γ-Proteobacteria and Firmicutes. For γ-Proteobacteria , Mobile-CRISPRi is transferred from Escherichia coli using the broad host range RP4 plasmid conjugation machinery, and is integrated into the recipient genome downstream of the highly conserved glmS gene using the extensively characterized Tn7 transposition system (Fig. 1c, top)14,15. Consistent with previous reports15, this strategy was unsuccessful in Bacillales Firmicutes (Supplementary Fig. 3), leading us to transfer CRISPRi using the ICEBs1 conjugation and integration machinery. Here, Mobile-CRISPRi is transferred from B. subtilis to other Bacillales Firmicutes (e.g., Staphylococcus aureus), and integrated into trnS-leu216 (Fig. 1c, bottom). Notably, ICEBs1 host range is likely broader than previously thought17. Critically, Mobile-CRISPRi integrations either downstream of glmS (Tn7) or into trnS-leu2 (ICEBs1) do not disrupt those gene functions14,16, occur in a specified orientation, and are stable (Fig. 2a) and functional (Supplementary Fig. 4) in the absence of selection for ≥ 50 generations, enabling studies of gene and antibiotic function in which maintaining selection is problematic or impossible.
Fig. 2 |. Mobile-CRISPRi stability, transfer, and knockdown efficiency.

a, Mobile-CRISPRi stability after 50 generations of growth in the absence of antibiotic selection. Stability was calculated as the plating efficiency on kanamycin (the marker associated with Mobile-CRISPRi) vs no antibiotic n=4 for B. subtilis and E. coli and n=3 for E. cloacea and K. pneumoniae. b, Mobile-CRISPRi transfer and integration efficiency. ICE or Tn7 containing CRISPRi was transferred to the species listed. Efficiency was calculated as: %ABR/total recipients n=3 for all strains except V. casei for which n=1. c, Efficiency of Mobile-CRISPRi knockdown in various species. Knockdown was tested using a Mobile-CRISPRi variant containing a constitutively expressed rfp reporter and an sgRNA targeting rfp. RFP expression was normalized to a strain lacking either dcas9 (for P. aeruginosa) or an sgRNA (all others; no sgRNA controls are recommended for future experiments). n=4 for all strains except E. faecalis, P. mirabilis, and V. casei for which n=3 Data are represented as mean ± s.d. The full species names for the strains used in panels b and c and their corresponding rfp fold knockdowns are: Bacillus subtilis (182-fold), Staphylococcus aureus (150-fold), Listeria monocytogenes (105-fold), Enterococcus faecalis (45-fold), Escherichia coli (65-fold), Enterobacter cloacae (32-fold), Enterobacter aerogenes (40-fold), Pseudomonas aeruginosa (8-fold), Klebsiella pneumoniae (34-fold), Vibrio casei (8-fold), Acinetobacter baumannii (10-fold), Salmonella enterica (54-fold), and Proteus mirabilis (35-fold).
We assessed the efficacy of Mobile-CRISPRi in multiple bacterial species, focusing on those involved in human disease. We first measured CRISPRi transfer by quantifying the number of recipient colonies (i.e., transconjugants) on selective agar plates as a fraction of total recipients (from non-selective plates; Fig. 2b). Most species showed transfer efficiencies sufficient for genome-scale sgRNA library construction (e.g., Enterobacter sp. ~10−2-10−3, and L. monocytogenes ~10−2), but some were more suited for single gene knockdown approaches (e.g., Acinetobacter baumanii ~10−6). Transfer efficiencies can also vary significantly at the strain level (Supplemental Fig. 5).
We then assessed CRISPRi knockdown efficacy with a “test” version of Mobile-CRISPRi consisting of rfp (encoding Red Fluorescent Protein, or RFP) and either an sgRNA targeting rfp (to measure knockdown) or lacking an sgRNA (a control to normalize rfp expression). Quantification of rfp knockdown in single cells using flow cytometry indicated that knockdown efficiency ranged from ~8-fold in Pseudomonas aeruginosa (possibly due to dCas9 degradation, Supplementary Fig. 7) to ~150-fold in S. aureus, with a median knockdown of ~40-fold across all measured species (Fig. 2c and Supplementary Fig. 6). Additionally, knockdown was titratable (Supplementary Fig. 8). We confirmed that CRISPRi was also functional against native genes by targeting P. aeruginosa pyocyanin production (Supplementary Fig. 9). In this visual assay, the culture appears yellow because production of the blue pigment pycocyanin has decreased.
Our initial assessment of Mobile-CRISPRi used pathogenic strains possessing at least rudimentary tools for perturbing gene function. To determine whether Mobile-CRISPRi functions in an environmental isolate with no existing genetic system, we tested transfer and knockdown in Vibrio casei, a γ-Proteobacterium originally isolated from French wash-rind cheeses and broadly associated with cheese microbiomes18. We found that Mobile-CRISPRi transferred to V. casei with library scale efficiency (~10−3, Fig. 2a), and a modest, but useful knockdown efficiency (~8-fold, Fig. 2b). The modular nature of Mobile-CRISPRi allows for further optimization of knockdown efficiency; for instance, by using Vibrio-specific promoters for dcas9 and sgRNA expression. We conclude that Mobile-CRISPRi is an effective genetic tool for gene knockdowns in diverse, non-model bacteria.
The emergence of multi-drug resistant pathogenic bacteria is an urgent threat to human health that requires both new antibiotics and a better understanding how existing antibiotics function. Knowledge of the mechanisms by which antibiotics kill bacteria—the mode of action (MOA)—is critical to advance new antibiotics from the laboratory to the clinic19. Because the full complement of genes in a bacterial genome (i.e., genetic background) can affect antibiotic function19, the MOA should ideally be determined directly in clinically relevant strains. However, most pathogenic bacterial lack genetic tools to systematically perturb the functions of essential genes that typically encode antibiotic targets. Importantly, we previously showed that the ability to titrate the knockdown level enables the systematic study of essential genes in B subtilis. A low (~3-fold) level of knockdown allowed sufficient growth to determine the MOA of an uncharacterized antibiotic by virtue of its synergistic effects on growth4.
We tested whether Mobile-CRISPRi could be used to explore MOA in pathogenic γ-Proteobacteria associated with antibiotic resistance (i.e., Gram-negative rods). We expected that strains with a small reduction in expression of the direct target of an antibiotic would be sensitized to low concentrations of that antibiotic (i.e., drug-gene interaction or synergy). Partial knockdown of the essential gene, folA, which encodes the trimethoprim target dihydrofolate reductase20, increased sensitivity to trimethoprim, shifting the minimal inhibitory concentration (MIC) by 2–4-fold, depending on the species (Fig. 3 and Supplementary Table 7), indicating synergy. Even though CRISPRi knockdown in P. aeruginosa was at a lower efficiency compared to other strains (Fig. 2b), there was still a clear shift toward sensitivity (Fig. 3). Moreover, concentrations of trimethoprim below the MIC for the WT, completely inhibited growth of the folA knockdown strains, clearly demonstrating synergy (Supplementary Fig. 10). As expected for an essential gene, fully induced CRISPRi targeting folA was lethal in Enterobacter aerogenes, Klebsiella pneumoniae, and P. aeruginosa (Supplemental Fig. 10), We conclude that Mobile-CRISPRi targeting essential genes can be used to generate sensitized strains for antibiotic MOA studies.
Fig. 3 |. CRISPRi knockdown of folA increases sensitivity to trimethoprim in multiple species.

MIC assays for trimethoprim sensitivity in P. aeruginosa and E. aerogenes with or without Mobile-CRISPRi targeting folA (n=3). Data are represented as mean ± s.d.
A compelling feature of CRISPRi is the ease of pooled knockdown library construction, either for defined gene sets or at the genome scale8. As a proof of principle, we construct a 40-member library of selected Enterobacter cloacae genes (Supplementary Tables 1 and 2). In the pooled context, each sgRNA functions as a barcode, enabling quantification of each knockdown strain in the pool. We first evaluated strain representation in our pipeline by performing two different pooled experiments. In the first, all steps from initial cloning to analysis were performed in a pool (Fig. 4a). This revealed that all sgRNA strains were present and had reasonable representation in the pool (31/40 sgRNA counts were within one standard deviation of the median, with a maximum 50-fold difference in representation). In the second, each sgRNA plasmid was constructed individually, and an equimolar mixture of plasmids was used to transform E. coli and perform downstream steps (Fig. 4b). This assessed the variability of all steps downstream of cloning and revealed a maximum 2-fold difference in representation. Thus, Mobile-CRISPRi transfer and integration is highly uniform, with essentially all variability derived from the initial cloning step. Further optimization or alternative cloning strategies8 may decrease the variability in sgRNA representation.
Fig. 4 |. A Mobile-CRISPRi library targeting auxotrophic genes in E. cloacae.
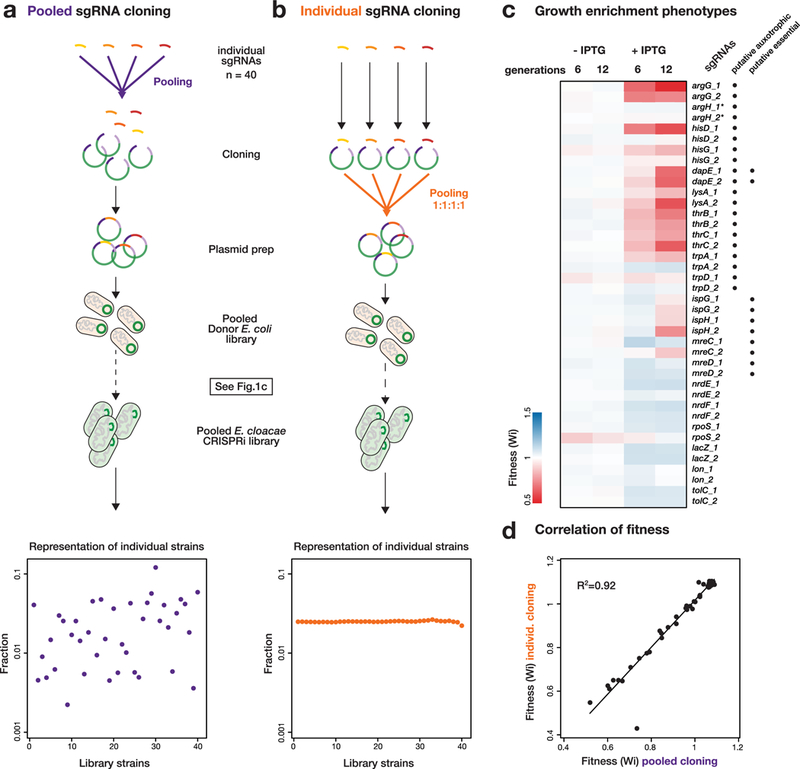
a, Tn7 Mobile-CRISPRi library construction. sgRNAs were cloned as a pool, transformed, and mated into E. cloacae, or b, sgRNAs were cloned individually, mixed as a pool with equal representation, and mated into E. cloacae as a pool. Representation of individual CRISPRi strains was determined by Illumina sequencing. c, Fitness of CRISPRi strains in glucose minimal media after 6 or 12 doublings with or without CRISPRi induction by IPTG, determined from the library constructed by pooled cloning (n=2). Asterisks indicate strains had no fitness change in pooled screen but decreased fitness in the arrayed screen. Putative auxotrophic or essential gene knockdown strains are indicated next to sgRNA names. d, Correlation between fitness measurements in libraries constructed by pooled cloning or individual cloning (n=2); a linear fit is shown.
We next evaluated fitness of our strain library which includes knockdowns of 10 amino acid biosynthesis genes, 4 putative essential genes and 6 well-characterized genes, each targeted by 2 sgRNAs (Supplementary Table 1). We grew our library in glucose minimal medium in competition with a 100-fold excess of wild-type E. cloacae and then measured the relative frequency of each strain in the library after 6 and 12 generations with or without CRISPRi induction to initiate knockdown. Using the fitness calculation of van Opijnen et al.12, we found that fitness of strains with sgRNAs targeting amino acid biosynthesis and those targeting some putative essential genes decreased, whereas representation of non-essential genes that are unrelated to amino acid biosynthesis remained constant (Fig. 4c; Supplementary Table 2). Fitness for affected strains was more pronounced at 12 doublings than at 6 doublings, suggesting that a larger number of generations was required to dilute out existing protein products. Additionally, both guides generally decreased the fitness of the essential and auxotrophic genes, but with more variability than previously observed4. Finally, the fitness measurements from the completely pooled construction (Fig 4a), and those in the equal representation library (Fig. 4b, Supplementary Table 2) were highly correlated (R2=0.92) indicating that the initial frequency of the strain in the pooled library did not affect the measurement of the fitness (Fig. 4d).
We also screened an arrayed library of each individual knockdown strain. Importantly, we confirmed the auxotrophy of amino acid biosynthesis gene knockdown strains, finding that their poor growth in minimal medium was suppressed by relevant amino acids (Supplementary Fig. 11 and Supplementary Table 3). Thus, knockdown effects are specific to the targeted gene and do not represent off-target effects of CRISPRi. We found that knockdown of some putative essential genes (e.g., mreD) showed no apparent phenotype, possibly because of limited growth after induction or low sgRNA efficacy. We conclude that Mobile-CRISPRi enables both pooled and arrayed library construction and straightforward assaying of phenotypes.
We anticipate that Mobile-CRISPRi will be a transformative technology for non-model bacteria lacking genetic tools and will facilitate cross-species genetic analysis, complementing existing TnSeq technology. The advantages of CRISPRi over other technologies is its ability to interrogate essential genes and double mutant combinations, as well as construction of parsimonious genome-scale knock down libraries. Mobile-CRISPRi transfer and knockdown systems brings this technology to a broad array of organisms, and its modularity makes it straightforward to expand host range (e.g., combining different transfer and integration functions, anti-restriction proteins21) and increase knockdown efficacy (e.g., use of “alternative” dcas9 genes5,6). The stability of Mobile-CRISPRi in the absence of selection suggests that it could be a valuable tool for dissecting the genetics of host-microbe interactions in both pathogenic and microbiome contexts, and aid in MOA studies in relevant human pathogens. Interestingly, our approach for transferring CRISPRi mirrors natural transfer of CRISPR systems by transposons related to Tn722. We will continue to look to nature for future approaches to explore the vast landscape of bacterial genetics.
Methods
Construction of Mobile-CRISPRi vectors
A complete list of Mobile-CRISPRi vectors can be found in Supplementary Table 4. All plasmids were constructed by restriction enzyme digestion of vector DNA followed by either ligation or NEBuilder HiFi DNA Assembly with insert DNA (all enzymes were purchased from NEB). To generate the Mobile-CRISPRi vectors, the pUC origin of replication in the Tn7 transposon plasmid pTJ1 was replaced with the R6K γ origin that requires the π protein (encoded by the pir gene) for replication, generating pJMP1050, and ensuring that Mobile-CRISPRi vectors cannot replicate in recipient cells. Mobile-CRISPRi “backbone” DNA containing unique restriction sites that flank the cloning modules was synthesized as a gBlock (IDT), and inserted into a pJMP1050 derivative (pJMP1054) that lacked those restriction sites, generating pJMP1055. pJMP1055 served as a base for all Tn7-based Mobile-CRISPRi derivatives. Derivatives were constructed by inserting components into the following modules/restriction sites: antibiotic markers/XhoI, reporter genes (e.g., rfp)/PmeI, sgRNA promoters and sgRNAs/EcoRI, sgRNA spacers (for creating sgRNA libraries)/BsaI, regulatory genes (e.g., lacI)/SmaI, dcas9 promoters and ribosome binding sites/SpeI, and dcas9/SpeI-AscI. To create a Mobile-CRISPRi plasmid that integrates into the ICEBs1 element, two ~1kb DNA fragments flanking the rapI gene were amplified from B. subtilis 168 gDNA and used to replace the Tn7 transposon ends in a pJMP1055 derivative (pJMP1106), generating pJMP1290. pJMP1290 served as a base for all ICE-based Mobile-CRISPRi derivatives and has the same unique restriction sites listed for the modules above. sgRNAs were cloned into the BsaI sites of Mobile-CRISPRi plasmids by ligating annealed oligos8. Oligos were designed to include overlaps that were complementary to the sticky ends generated by BsaI. Oligos were added to 1X NEB buffer 4 at 5 μM concentration, denatured for 5 min at 95 °C, and then annealed by transferring the reactions to room temperature. Annealed oligos were then diluted 1:20, 2μl of the dilution was ligated to 100ng of BsaI-digested vector for 1hr at room temperature. sgRNAs were designed as previously described4.
Construction of Mobile-CRISPRi strains and mating assays
A complete list of strains used in the study can be found in Supplementary Table 5. Tn7-based Mobile-CRISPRi strains were constructed by tri- or quad-parental mating as previously described in Choi et al.15,23, with several modifications. All Tn7 matings used MFDpir24 (a pir+ strain that is dependent on DAP for growth and contains the RP4 transfer machinery) transformed with either a Tn7 transposase plasmid (pJMP1039—a derivative of pTNS325 with a spontaneous small deletion upstream of the Pc promoter) or transposon plasmid (various pJMP1055 derivatives) as mating donors. Matings with Acinetobacter baumannii ATCC19606 required the presence of a third donor strain containing the self-mobilizing RP4 transfer plasmid pRK201315 for unknown reasons. Cultures of the two E. coli donor strains (transposon and transposase donors) were grown overnight (~16 hrs) at 37 °C in Lysogeny Broth (LB) + 300 μM DAP (Alfa Aesar B22391) + 100 μg/ml ampicillin. Recipient strains assayed here also grew to saturation in LB after incubation at 37 °C for ~16 hrs. 100 μl of each donor and recipient strain was added to 700 μl of LB and mixed by pipetting. Mixes of donor and recipient strains were pelleted for 2 min at 7000 × g, washed twice with 1 ml of LB, resuspended in 30 μl of LB after the final wash, pipetted onto a cellulose filter (MF-Millipore HAWG01300) placed on a pre-warmed LB + 300 μM DAP plate, and incubated at 37 °C for 6 hrs. Filters were then transferred to microcentrifuge tubes containing 200 μl of PBS and vortexed to liberate the cells. Cells were spread onto on media that selects for the Mobile-CRISPRi plasmid and recipient (e.g., LB + kanamycin) without DAP (the absence of DAP will select against donor E. coli). Antibiotic concentrations used for selection were: 30 μg/ml kanamycin, and 30 μg/ml gentamicin (for P. aeruginosa).
ICE-based Mobile-CRISPRi strains were constructed by bi-parental mating as previously described26,27 with modifications. ICE donor strains were generated by transformation of B. subtilis with Mobile-CRISPRi integration plasmids using natural competence as previously described4. Expression of the ICE anti-repressor, RapI, induces conjugation genes found on the ICE element and promotes excision26. ICE excision and the large insert size of Mobile-CRISPRi plasmids resulted in very few transformants. To produce a strain with a stable ICE element in the presence of an IPTG-inducible rapI gene that transformed at high efficiency, a dcas9 gene linked to a chloramphenicol-resistance marker was integrated into ICE—selection for the chloramphenicol marker and the extra homology present in the dcas9 gene improved transformation efficiency. For mating, one 3 ml LB culture of each donor and recipient strain was grown from single colonies to exponential phase (~2 hrs at 37 °C); donors were grown in LB + 3.25 μg/ml kanamycin to select for ICE retention. Exponential phase cultures were then back diluted to an OD600 of 0.02 and grown until OD600 0.2 before inducing rapI expression with 1mM IPTG for 1 hr. 2.5 ml of donor and recipient cells adjusted to an OD600 of 0.9 were mixed with 5 ml of 1X Spizizen salts and vacuum filtered using an analytical CN filter (Nalgene 145–0020). Filters were transferred to Spizizen agar plates and incubated for 3 hrs at 37 °C. Transconjugants were selected for plating on kanamycin + streptomycin plates as all recipient strains were streptomycin resistant. The ICE mating procedure used was the same for all Bacillales Firmicutes in this study. Antibiotic concentrations used for selection were: 6 μg/ml (B. subtilis) chloramphenicol, 7.5/50/1000 μg/ml kanamycin (B. subtilis/S. aureus and L. monocytogenes/E. faecalis), and 100 μg/ml streptomycin.
Transfer efficiency assays
Tn7 or ICE mating experiments were carried out in triplicate (n = 3 matings). Transfer efficiency was calculated by taking the ratio of transconjugants (antibiotic-resistant Dap+ colonies for Tn7 matings, and KanR/StrR colonies for ICE matings) to viable cells (LB colonies for Tn7 matings, and StrR colonies for ICE matings). For Tn7 transfer to the B. subtilis attTn7 site in E. coli (Supplementary Fig. 1a), the native attTn7 site in E. coli K-12 DH10B was occupied by an unmarked Tn7 to prevent chromosomal transposition, while test attTn7 sites were cloned onto a chloramphenicol resistant plasmid.
Mobile-CRISPRi stability assays
Four independently generated isolates (n = 4 isolates) of E. coli K-12 BW25113 and B. subtilis 168 containing Mobile-CRISPRi systems targeting rfp were grown to saturation overnight at 37 °C in LB + kanamycin (30 μg/ml for E. coli, E. cloacae, and K. pneumoniae and 7.5 μg/ml for B. subtilis) to select for retention of the of the Tn7 or ICE element containing CRISPRi. One ml of each culture was centrifuged at 6000 × g for 3 min and washed twice with LB to remove any residual kanamycin. The washed cells were diluted 1:1000 in LB and grown to saturation. The procedure of dilution and growth to saturation was repeated a total of 5 times for ~50 generations of growth. Cells were then serially diluted and plated on selective (LB + kanamycin) and non-selective plates (LB). The ratio between colony counts on LB and LB + kanamycin was used to determine the fraction of cells that retained the Tn7 or ICE element.
RFP knockdown assays
RFP knockdown was measured using flow cytometry or a plate reader (for A. baumannii and V. casei; n = 4 independently constructed isolates for all strains except P. mirabilis [n = 3 isolates] and V. casei [n = 3 isolates]). Flow cytometry was performed by diluting overnight cultures of Mobile-CRISPRi rfp knockdown strains 1:10,000 into fresh media (LB for all γ-Proteobacteria and B. subtilis, Brain Heart Infusion broth for S. aureus) containing CRISPRi inducer (1mM IPTG for all γ-Proteobacteria except P. aeruginosa, 1% arabinose for P. aeruginosa, and 0.1 μg/ml anhydrotetracycline for Firmicutes) and incubating cultures at 37 °C with rotation until the cultures reached mid-log phase (OD600 0.3–0.6). Cultures were then cross-linked with 1% formaldehyde [final] for 10 min, followed by quenching for 10 min with 0.5 M glycine [final]. Cross-linked cells were then diluted 1:10 in phosphate buffered saline and flowed on a BD LSRII using 610/20 BP filter (PE-Texas-Red fluorochrome). Data for at least 10,000 cells was collected for four independently constructed strain isolates. In all cases, data for 100% of the cells collected were used in the analysis. For V. casei, overnight cultures were normalized to 2.0 OD600 and then diluted 1:200 in LB with or without 0.5 mM IPTG. After 6 hours growth post-induction the strains were normalized to 0.2 OD600 and washed once in 1X PBS. The samples were then transferred to a 96well plate (200ul in each well) in triplicate and measured for ds-Red fluorescence (Ex 557nm Em 592nm) using a bottom-read plate reader (Tecan). For A. baumannii, overnight cultures were diluted 1:10,000 into fresh LB with or without 0.1 mM IPTG. Cells were grown in a 96 well plate with measurements of OD600 and RFP every 10 min. The values reported reflect the RFP knockdown at mid-log growth. The values reported are mean values and error bars reflect the standard deviation from the mean.
Pyocyanin knockdown assays
Strains were grown overnight in Kings Medium A Base (HiMedia M1543) to induce pyocyanin and pyorubin production and 1% arabinose to fully induce dcas9 expression. Growth was repeated 3 times (n = 3); representative results are shown.
Antibiotic sensitivity assays
MIC assays were performed using the broth microdilution method as previously described28, except that 0.1% arabinose (for P. aeruginosa) or 100 μM IPTG (for E. aerogenes) was added to induce dcas9 expression, and K. pneumoniae cultures were shaken to reduce clumping. Three biological replicates were grown for MIC assays (Supplementary Table 7). Growth curves shown in Fig. 3 were set up in exactly the same manner as the MIC assays, except that cultures were grown with agitation in a plate reader (BioTek) for ~16 hrs. The data for two biological replicates is plotted on the same graph in Supplementary Fig. 10. P-values for comparing MICs between control sgRNA and folA sgRNA strains obtained using an unpaired two-tailed t-test with Welch’s correction to account for unequal variances between samples and calculated using GraphPad Prism 7.0e (Supplementary Table 7).
Construction of Mobile-CRISPRi strains and mating assays
Pooled Tn7-based Mobile-CRISPRi libraries for E. cloacae were constructed by following the procedure for single gene CRISPRi strain construction with several modifications. Equal concentration of annealed oligonucleotides for each sgRNA (Supplementary Table 1) were pooled and ligated into a BsaI digested plasmid. Ligation product was transformed into an E. coli pir+ strain. Colonies on selection plates (LB + 100 μg/ml ampicillin) were collected and resuspended in LB and plasmids were purified from of pooled transformants. Purified pooled plasmids were transformed into donor strain, MFDpir. Transformants were collected and resuspended in LB + 300 μM DAP + 100 μg/ml ampicillin + 12.5% glycerol and stored at −80 °C. For comparison, the other donor was prepared by transformation of a pool of individually cloned plasmids with equal concentration. Tri-parental mating and selection were performed as described above and selected colonies of E. cloacae CRISPRi strains were collected and resuspended in MOPS salts solution29 + 12.5% glycerol and stored at −80 °C after measurement of OD450 of stock. In order to prepare inoculum of library to screen fitness of library in minimal media, frozen stock was diluted in glucose minimal medium to OD450 of 5 and incubated for recovery for 1 hr. Recovered cell culture was mixed with a 100-fold excess wild-type E. cloaca, then diluted to an OD450 of 0.01 in 30 ml glucose minimal media with or without IPTG, then grown in 125 ml flasks at 30 °C with shaking (250 rpm). When the culture reached OD450 of 0.64, 1 ml of culture was collected for preparation of sequencing library of 6 doubling sample. For 12 doubling sample, this culture was diluted to an OD450 of 0.01 in 30 ml and was grown until the culture reached OD450 of 0.64. in order to prepare the Illumina sequencing library, genomic DNA was purified using the Qiagen DNeasy Blood & Tissue kit and sequencing region was amplified by PCR using the primers harboring indices for different sampling time and growth conditions. Differentially indexed PCR products were purified by agarose gel electrophoresis prior Illumina sequencing. Sequences of primers used for preparation of sequencing libraries are listed in (Supplementary Table 6). Frequencies of strains in each sample were calculated by dividing the number of reads of sgRNA encoding sequence from each strain by the number of total read and used for calculation of fitness. Fitness was calculated as described in van Opijnen et al.12 Wi = ln(Ni(t2)Xd/Ni(ti))/ln((1-Ni(t2))Xd/(1-Ni(t1))), N(t) is frequency of the mutant in the population at the time points, and d represents the growth of the bacterial population during library selection. We calculated d using OD450 change. Average fitness from two biological replicates (n = 2) is presented in Supplementary Table 2. Pearson’s r was calculated from a linear fit of the data using Excel.
Ordered Tn7-based Mobile-CRISPRi libraries for E. cloacae were constructed by following the procedure for single gene CRISPRi strain construction with modifications for automation (Fig. S4). Each donor Tn7::CRISPRi strains were prepared by transformation of individually cloned plasmids into MFDpir strain and arrayed in 96 well plate. Equal amount of transposase strain was added to each well and pinned to LB + 300 μM DAP + 2% agar plate using a Singer ROTOR robot. Wild-type E. cloacae cells arrayed in 96 colony format were pinned to the same plate, which was incubated for 6 hrs. Kanamycin resistant E. cloacae CRISPRi strains were selected on LB supplemented with kanamycin two times and stored at −80 °C as a glycerol stock. To screen growth phenotype of each strain, cells were pinned from glycerol stocks onto rectangular LB agar plates in 384-format using a Singer ROTOR robot (four technical replicates on one plate in this screen). For each screen, exponentially growing cells in 384-format were then pinned to defined media plates and incubated for 16 hrs at room temperature to avoid mucoid colony formation. Plates were imaged using a Powershot G10 camera (Canon) when at a time point at which fitness differences were apparent but growth had not saturated. The calculation of RF was carried out as described in Koo et al.29 with minor modifications. Relative fitness (RF) was measured by the colony opacity of each mutant determined with Iris colony sizing software30. The RF of each mutant was calculated as: RF = (average colony opacity of CRISPRi strain)/(average colony opacity of CRISPRi with no sgRNA strain); knockdown strains were grown in quadruplicate (n = 4) . The average RF calculated from two same media plates is presented in Supplementary Table 3.
dCas9 Western Blot
Cultures of P. aeruginosa were diluted back from stationary phase and grown to saturation in the presence/absence of 1% arabinose at 37°C and then 1ml of culture was added to 0.25ml 5X SDS-PAGE sample buffer and boiled at 100C for 10min before storage. Samples were boiled at 90C for 2min before running on a Bolt 10% Bis-Tris Plus gel (ThermoFisher) alongside PagerRuler Plus Protein Ladder (ThermoFisher) at 150V for 1hr. Proteins were transferred to a nitrocellulose membrane (Bio-Rad, 0.45um) at 100V for 2hr at 4C using the Mini Trans-Blot Cell system (Bio-rad). Protein amounts were checked by Ponceau staining (0.1% Ponceau S, 5% (v/v) acetic acid) for 25min at room temperature, followed by washing in H2O to destain. Membranes were blocked in Odyssey Blocking Buffer (Licor) at 4C overnight. Each primary antibody (Anti-CRISPR-Cas9 (AbCam 191468) and c-Myc (Santa Cruz Biotechnology 9E10)) were used at 1:1000 in PBS + 0.05% Tween80 + 3% BSA at room temperature for 2hr. Secondary antibody (IRDye 680RD Goat anti-Mouse IgG) was used at 1:10000 in Odyssey Blocking Buffer (Licor) at room temperature for 1hr. All membrane washes were performed using PBS + 0.05% Tween80. Blots were images on a Licor Odyssey Aerius at 700nm.
Supplementary Material
Acknowledgements
We thank Joanna Goldberg and Herbert Schweizer for Tn7 plasmids, Lei (Stanley) Qi for a plasmid encoding Human codon optimized dCas9, the American Type Culture Collection, Harry Mobley, Bill DeGrado, KC Huang, Amy Banta, and Paula Welander for strains, Joan Garbarino and Marco Jost for help with flow cytometry, and the Carol Gross and Oren Rosenberg labs for helpful comments. This work was supported by NIH F32 GM108222 (to J.M.P.), US Department of Agriculture National Institute of Food and Agriculture Hatch Project NYC-189438 (to J.E.P.), and NIH R01 GM102790 (to C.A.G.)
Footnotes
Data collection
Flow cytometry data was collected using BD FACSDIVA v8.0.1.
Data analysis
Data analysis was performed in Galaxy v18.01 (pooled sequencing data), FlowJo v10.4.2 and FCS Express 6 Plus (flow cytometry data), GraphPad Prism 7.0e (graphing and statistical analysis) and Excel v16.12. Plasmid sequence maps were created using SnapGene v3.1.4.
Code availability
sgRNA design code is available from Github (https://github.com/traeki/sgrna_design).
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
Competing interests
The authors declare no competing interests.
References
- 1.Qi LS et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mimee M, Tucker AC, Voigt CA & Lu TK Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli in the Murine Gut Microbiota. Cell Syst. 1, 62–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters JM et al. A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rock JM et al. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat. Microbiol. 2, 16274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan SZ, Reisch CR & Prather KLJ A Robust CRISPR Interference Gene Repression System in Pseudomonas. J. Bacteriol. 200, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X et al. High‐throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 13, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert LA et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jost M et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 68, 210–223.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vigouroux A, Oldewurtel E, Cui L, Bikard D & Teeffelen S van. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 14, e7899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H et al. Depletion of Undecaprenyl Pyrophosphate Phosphatases Disrupts Cell Envelope Biogenesis in Bacillus subtilis. J. Bacteriol. 198, 2925–2935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Opijnen T, Bodi KL & Camilli A Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji W et al. Specific gene repression by CRISPRi system transferred through bacterial conjugation. ACS Synth. Biol. 3, 929–931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peters JE Tn7. Microbiol. Spectr. 2, (2014). [DOI] [PubMed] [Google Scholar]
- 15.Choi K-H et al. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2, 443–448 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Johnson CM & Grossman AD Integrative and Conjugative Elements (ICEs): What They Do and How They Work. Annu. Rev. Genet. 49, 577–601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brophy JAN et al. Engineered integrative and conjugative elements for efficient and inducible DNA transfer to undomesticated bacteria. Nat. Microbiol. 3, 1043–1053 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Bokulich NA & Mills DA Facility-specific ‘house’ microbiome drives microbial landscapes of artisan cheesemaking plants. Appl. Environ. Microbiol. 79, 5214–5223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardona ST, Selin C & Gislason AS Genomic tools to profile antibiotic mode of action. Crit. Rev. Microbiol. 41, 465–472 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Baccanari D, Phillips A, Smith S, Sinski D & Burchall J Purification and properties of Escherichia coli dihydrofolate reductase. Biochemistry 14, 5267–5273 (1975). [DOI] [PubMed] [Google Scholar]
- 21.McMahon SA et al. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 37, 4887–4897 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters JE, Makarova KS, Shmakov S & Koonin EV Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc. Natl. Acad. Sci. 114, E7358–E7366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi K-H & Schweizer HP mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 1, 153–161 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Ferrières L et al. Silent Mischief: Bacteriophage Mu Insertions Contaminate Products of Escherichia coli Random Mutagenesis Performed Using Suicidal Transposon Delivery Plasmids Mobilized by Broad-Host-Range RP4 Conjugative Machinery. J. Bacteriol. 192, 6418–6427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi K-H et al. Genetic Tools for Select-Agent-Compliant Manipulation of Burkholderia pseudomallei. Appl. Environ. Microbiol. 74, 1064–1075 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auchtung JM, Lee CA, Monson RE, Lehman AP & Grossman AD Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc. Natl. Acad. Sci. U. S. A. 102, 12554–12559 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auchtung JM, Lee CA, Garrison KL & Grossman AD Identification and characterization of the immunity repressor (ImmR) that controls the mobile genetic element ICEBs1 of Bacillus subtilis. Mol. Microbiol. 64, 1515–1528 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiegand I, Hilpert K & Hancock REW Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Koo B-M et al. Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Syst. 4, 291–305.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kritikos G et al. A tool named Iris for versatile high-throughput phenotyping in microorganisms. Nat. Microbiol. 2, 17014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.