

Abstract
A significant proportion of neonatal and childhood seizures are poorly controlled by existing anti-seizure drugs (ASDs), likely due to prominent differences in ionic homeostasis and network connectivity between the immature and mature brain. In addition to the poor efficacy of current ASDs, many induce apoptosis, impair synaptic development, and produce behavioral deficits when given during early postnatal development. There is growing interest in new targets, such as cannabidiol (CBD) and its propyl analog cannabidivarin (CBDV) for early life indications. While CBD was recently approved for treatment of refractory childhood epilepsies, little is known about the efficacy or safety of CBDV. Here, we addressed this gap through a systematic evaluation of CBDV against multiple seizure models in postnatal day (P) 10 and 20 animals. We also evaluated the impact of CBDV on acute neurotoxicity in immature rats. CBDV (50–200 mg/kg) displayed an age and model-specific profile of anticonvulsant action. In P10 rats, CBDV suppressed seizures only in the pentylenetetrazole model. In P20 rats, CBDV suppressed seizures in the pentylenetetrazole, DMCM, and maximal electroshock models. Between P10 and P20, we identified significant increases in mRNA expression of TRPV1 in multiple brain regions; when CBDV was tested in P20 TRPV1 knockout mice, anticonvulsant effects were attenuated. Finally, CBDV treatment generally avoided induction of neuronal degeneration in immature rats. Together, the efficacy and safety profile of CBDV suggest it may have therapeutic value for early life seizures.
Keywords: seizure, cannabinoid, neonatal, cannabidivarin, cell death
1. INTRODUCTION
Seizure disorders of infancy and childhood are common, significant causes of morbidity (Glass et al., 2018), and respond poorly to current first-line anti-seizure drugs (ASDs) (Glass et al., 2012; Painter et al., 1999). Distinct characteristics of the developing brain, including immature network connectivity and divergent ionic homeostasis, likely contribute to the pharmacoresistance of early life seizures (Glykys et al., 2009). In addition to inferior efficacy, early life pharmacotherapy also carries a significant risk to the developing brain: early life exposure to many ASDs including phenobarbital, phenytoin, and valproate are associated with the induction of apoptosis in both grey and white matter structures (Bittigau et al., 2002; Forcelli et al., 2011; Kaushal et al., 2016). Several ASDs likewise produce lasting disruptions in synaptic development after even a single exposure during early postnatal development (Al-Muhtasib et al., 2018; Forcelli et al., 2012a). Moreover, clinical evidence also suggests that gestational and early life exposure to ASDs may produce lasting deficits in cognitive function (Farwell et al., 1990; Meador et al., 2012).
The lack of efficacy, as well as concerns about safety of existing ASDs have prompted the search for new therapies. The FDA approval of Epidiolex (cannabidiol, CBD) for the treatment of Lennox-Gastaut and Dravet syndromes (FDA, 2018) underscores the growing interest in phytocannabinoids for refractory epilepsies. In a recent clinical trial, CBD displayed efficacy in children with a favorable side effect profile.(Devinsky et al., 2017) Along with CBD, cannabidivarin (CBDV), a propyl analog of CBD, is currently being evaluated in clinical trials for a range of indications. These compounds avoid the intoxicating effects of other phytocannabinoids (e.g., Δ9-tetrahydrocannabinol [THC]) likely due to their lack of activity at the orthosteric binding site of CB receptors (Morales et al., 2017). Both CBD and CBDV have multiple proposed mechanisms of action including allosteric modulation of the cannabinoid 1 receptor (CB1R), antagonism of G protein-coupled receptor 55 (GPR55), and activation of the transient receptor potential vanilloid (TRPV) family of receptors (Morales et al., 2017). Both CBD and CBDV demonstrate significant anti-seizure effects in preclinical models of adult epilepsy, but only two studies has evaluated CBD in the context of seizures during early development (Alvarez et al., 2008; Friedman and Wongvravit, 2018); CBDV has not been similarly examined. Thus, the anticonvulsant efficacy of CBDV treatment in neonates/infants is unknown. Moreover, the neuroprotective effects of CBD (Alvarez et al., 2008) suggest this class of compounds may avoid the acute developmental neurotoxicity seen with many other ASDs. To fill this gap, we sought to determine the preclinical efficacy and safety profiles of CBDV.
2. METHODS
Animals were maintained in temperature-controlled (21°C) rooms with a 12:12 h light:dark cycle with food (Lab Diet #5001) and water available ad libitum. Experimental manipulations occurred during the light phase. Procedures were performed in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care standards and were approved by the Georgetown University Animal Care and Use Committee. Treatments were counterbalanced within and across litters, and all experiments and analyses were conducted by individuals blinded to treatment status.
2.1. Animal Testing Paradigm
Female Sprague-Dawley rats with male pups were obtained from Envigo (Frederick, MD) when the litters were P7 (except for NMDA-evoked spasms). For experiments in P20 mice, heterozygous TRPV1-null mice (Trpv1tm1Jul, Jackson Laboratories, Stock #003770, Bar Harbor, ME) (Caterina et al., 2000) were bred and both male and female wildtype and TRPV1-null littermate mice were used for seizure testing. Two age groups were used for seizure testing: P10 and P20; these ages were selected model infancy and pre-adolescence in humans, respectively (Dobbing and Sands, 1979). At P10 or P20, pups were weighed, numbered, and randomly assigned to a treatment group. The pups were pretreated with CBDV (50, 100, or 200 mg/kg) or vehicle and returned to their dam for 60 min before seizure testing. During the observation period, pups were not subjected to any passive or active heating.
Synthetic CBDV (Cayman Chemical, Ann Arbor, MI) was prepared in a 2:1:17 ratio of ethanol:Kolliphor:0.9% saline (Sigma-Aldrich, St. Louis, MO), which also served as the corresponding vehicle solution. Drug was dissolved at concentrations of 5, 10, or 20 mg/mL and administered intraperitoneally (ip) at a volume of 10 mL/kg of body weight to deliver 50, 100 or 200 mg/kg of drug. These doses of CBDV are effective in adult models of epilepsy (Hill et al., 2012, 2013).
2.2. Seizure Models
We evaluated CBDV against a battery of ASD screening models: pentylenetetrazole (PTZ), methyl-6,7-dimethoxy-4-ethyl-beta-carboline-3-carboxylate (DMCM), kainic acid (kainate, KA), and maximal electroshock (MES) screening models were used in both P10 and P20 rats. Graded global hypoxia and N-Methyl-D-Aspartate (NMDA)-evoked betamethasone primed infantile spasms were also used in P10 rats. This combination of models was selected to provide broad characterization of the range of CBDV effects modeling infancy (P10) and preadolescence (P20).
2.2.1. PTZ evoked tonic-clonic seizures
PTZ evoked seizures in developing rats are characterized by tonic-clonic behavioral manifestations (Kubová and Mares, 1993; Mares and Schickerová, 1980). We selected the PTZ seizure model as it is one of the most common models for preclinical ASD screening; a wide range of ASDs display efficacy in this model in immature rats (Forcelli et al., 2012b; Huizenga et al., 2017; Kubova and Mares, 1991; Kubová and Mares, 1993).
For both P10 and P20 rats, PTZ (Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% saline at a concentration of 10 mg/mL and administered via subcutaneous (sc) injection at a dose of 100 mg/kg. This dose is consistent with that used in our prior reports assessing anticonvulsant drug efficacy in developing rats (Forcelli et al., 2013, 2012b; Huizenga et al., 2017).
Following PTZ pretreatment, behavioral manifestations were observed and recorded for 20 min. Seizure behavior was scored using a 5-point rating system as previously described (Forcelli et al., 2012b; Huizenga et al., 2017; Kubova and Mares, 1991): 0 = no change in behavior; 0.5 = scratching, chewing, tremors; 1 = 0.5 + myoclonic jerks; 2 = unilateral clonus and back/shuffling; 3 = bilateral facial and forelimb clonus; 4 = clonic seizure with loss of righting; 4.5 = clonic seizure with running/bouncing or swimming like behavior; 5 = tonic extension (both forelimb and hindlimb). A score of 0.5 – 3 was considered a clonic seizure, while a score of 4 or above was considered a tonic-clonic seizure.
2.2.2. DMCM evoked clonic seizures
DMCM can be used to preferentially and reliably evoke clonic seizure manifestations in developing rats (C. Kulick et al., 2014). DMCM (Tocris, Bristol, UK) was dissolved in a small amount of 1 N hydrochloric acid, then diluted in 0.9% saline to a concentration of 0.1 mg/mL (pH 2.0). DMCM was administered (ip) at a dose of 600 ug/kg for P10 rats and 900 ug/kg for P20 rats, based on our prior reports (C. Kulick et al., 2014; C. V. Kulick et al., 2014). Following DMCM pretreatment, behavioral manifestations were observed and recorded for 20 min. Seizure behavior was scored using the same 5-point rating system for PTZ seizures.
2.2.3. Kainic acid evoked seizures
KA (Cayman Chemical, Ann Arbor, MI) was dissolved in 0.9% saline to a concentration of 0.1 mg/mL and administered (ip) at a dose of 4 mg/kg in P10 rats and 7 mg/kg in P20 rats, based on prior reports (Velíšek et al., 1992).
Following KA administration, behavioral manifestations were observed and recorded for 40 min. Recorded behavioral seizure manifestations were assigned a score on a 3-point rating system: 0 = no behavior; 1 = automatisms; 2 = clonic activity; 3 = clonic-tonic activity. Acute KA seizures in immature rats are suppressed by a range of ASDs (Velíšek et al., 1992).
2.2.4. Maximal electroshock evoked tonic seizures
MES is a prevalent and rapid ASD screening method (Löscher, 2011). The primary endpoint of MES is tonic limb extension, modeling tonic seizures in humans.
Rats and mice received 0.5% tetracaine HCl eyedrops 15–45 min before stimulation and 0.9% saline eyedrops immediately before transorbital (P10) or transcorneal (P20) stimulation. Stimulation occurred 60 min following VEH or CBDV (200 mg/kg in rat; 100 mg/kg in mouse) pretreatment and seizure responses were scored for the presence and duration of tonic extension. We used a lower dose for mice, as compared to rats, as the higher dose of CBDV produced severe sedation in P20 mice.
P10 rats received a 60Hz sinusoidal train of pulses at 110mA for 200ms (Wahlquist stimulator, Salt Lake City, UT). P20 rats received a 200Hz train of 0.9ms-wide square pulses at 50mA for 300ms; P20 mice received the same pulse sequence at 17mA for 900ms (Ugo Basile #57800 stimulator, Stoelting Co., Wood Dale, IL). Stimulation thresholds were based on those previously described (London and Buterbaugh, 1978). P10 pups predominately display tonic forelimb extension as the endpoint for MES, whereas P20 pups displayed tonic hindlimb extension as the maximal seizure response (London and Buterbaugh, 1978).
2.2.5. Hypoxia-evoked generalized seizures
Hypoxia (or hypoxic-ischemia) is the most common cause of neonatal seizures, and thus a model of particular interest. In immature rodents, hypoxia produces both clonic and tonic-clonic seizures, which display sensitivity to topiramate (Koh and Jensen, 2001).
P10 rat pups were placed in individual observation boxes within a chamber regulated by an OxyCycler Oxygen Profile Controller (Biosphereix, Redfield, NY). Hypoxia was induced as previously described (7% O2 for 8 min, 5% for 6 min, and 4% for 1 min) (Huizenga et al., 2017). Behavioral seizure manifestations were scored using a 3-point rating system: 0 = no behavior, 1 = automatisms, 2 = clonic activity; 3 = tonic-clonic activity.
2.2.6. Infantile spasms
Timed pregnant rat dams were injected on gestational day 15 with two doses of betamethasone (0.4 mg/kg, ip in saline, at 07:00 and 17:00 hours) (Chachua et al., 2011; Velíšek et al., 2010). On P10, flexion spasms were induced in the primed rat pups with a single injection of NMDA (7.5 mg/kg, ip, in saline) (Chachua et al., 2011; Velíšek et al., 2010). The number and latency of flexion spasms were observed and recorded for 75 min. Spasms evoked in this model mimic primary motor flexion spasms and are responsive to pharmacotherapy (Chachua et al., 2011; Velíšek et al., 2010).
2.3. mRNA Expression Level of CBDV Targets
RNA was extracted from punches through frontal cortex, hippocampus, amygdala, and thalamus from P10 and P20 rat pups using the Direct-zol RNA Miniprep Kit (Zymo, Irvine, CA). iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) was used to generate cDNA. Reverse transcription reactions were performed under the recommended reaction protocol.
mRNA expression levels of targets were determined using real time quantitative PCR (RT-qPCR). The following All-in-One qPCR primers were used to quantify target expression: TRPA1 (NM_207608.1), TRPV1 (NM_031982.1), TRPV2 (NM_017207.2), TRPV3 (NM_001025757.1), and GAPDH (NM_017008.3) as a reference gene (GeneCopoeia, Rockville, MD). Primer sequences for GPR55 were created as previously described(AlSuleimani and Hiley, 2015) (Eurofins Genomics, Louisville, KY). RT-qPCR was performed in an a CFX Real-Time PCR Detection System (Bio-Rad, Hercules, CA) with PowerUp SYBR Green Master Mix (Applied Biosystems, Foster City, CA). Reactions were incubated at 95°C for 10 min followed by 45 cycles (95°C for 10 s, 60°C for 20 s, and 72°C for 15 s). Threshold cycle numbers (CT) were determined using CFX software.
2.4. Drug induced neurotoxicity
Female Sprague-Dawley rats with male pups were obtained from Envigo (Frederick, MD) when the litters were P5; rats pups were treated at P7. P7 was selected because it is the time point of peak sensitivity to ASD induced neuronal apoptosis (Bittigau et al., 2002).
Phenobarbital (PB; 75 mg/kg, Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% saline and a concentration of 7.5 mg/mL was administered (ip) at a volume of 10 mL/kg. PB was used a positive control because it consistently induces a significant increase in developmental cell death at this dose (Bittigau et al., 2002; Forcelli et al., 2011; Kaushal et al., 2016). Pups received a single dose of CBDV (100 or 200 mg/kg), vehicle solution, PB (75 mg/kg in saline), or saline.
24 h after drug treatment, pups were perfused transcardially with phosphate buffered saline (pH 7.4, 10mL) followed by 4% paraformaldehyde (10mL). This survival interval was based on prior reports examining drug-related apoptosis in the developing brain (Bittigau et al., 2002; Kim et al., 2007). Brains were removed and post-fixed in paraformaldehyde for 24 h, then transferred to 30% sucrose before cryosectioning.
40 μm coronal sections were stained using Fluoro-Jade B (EMD Millipore, Burlington, MA) to fluorescently label degenerating neurons as previously described (Schmued and Hopkins, 2000). Fluorescent photomicrographs were collected on a Nikon 80i microscope with a QImaging QIClick camera. Images at 10x magnification (1.18 mm2 region) of three sequential sections at 200 μm intervals for each brain region were taken. Regions were defined using the neonatal rat brain atlas of Ramachandra and Subramanian (Ramachandra and Subramanian, 2011) and selected based on prior reports of anti-seizure drug induced enhancements in cell death from our lab (Brown et al., 2016; Forcelli et al., 2011) and others (Bittigau et al., 2002). Fluoro-Jade B positive cells were counted manually to quantify neurodegeneration as previously described (Huizenga and Forcelli, 2018).
2.5. Statistics
Statistical analyses were performed using GraphPad Prism 7.0c (GraphPad Software; La Jolla, CA). Behavioral seizure scores were analyzed using Kruskal-Wallis test followed by Dunn’s multiple comparisons test. Latencies to seizure onset were analyzed using analysis of variance (ANOVA) followed by Dunnett’s test. The number of spasms produced by NMDA were not normally distributed and thus analyzed using Mann Whitney U test; the latency to spasm onset was analyzed with an unpaired t-test. The incidence of tonic MES seizures was analyzed using Chi-Square followed by Fisher’s exact test (Holm-Sidak corrected). mRNA expression was analyzed using the ΔΔCt method, with GAPDH as a reference. Data were log-normally distributed and the log-transformed values were analyzed using a one-tailed, one-sample t-test. For TRPV3 (non-normally distributed), data were analyzed using a one-sample Wilcoxon Sign Rank test. In all cases, P values were adjusted for multiple comparisons (Holm-Sidak) within each family of comparisons. Probability values <0.05 were considered statistically significant.
3. RESULTS
3.1. Effect of CBDV on DMCM and KA evoked seizures in P10 and P20 rats.
To evaluate the anti-seizure efficacy of CBDV in models of clonic seizures, we examined the effect of CBDV on DMCM evoked seizures in both P10 and P20 rats. The DMCM model reliably evokes seizures characterized by clonic behavior and is sensitive to traditional (e.g. phenobarbital) and newer anti-seizure drugs (e.g. levetiracetam and tiagabine).(C. V. Kulick et al., 2014) In vehicle treated P10 rats, DMCM administration produced a median seizure score of 3. CBDV administration was without effect on seizure severity in this age group, as revealed by Kruskal-Wallis test (H=0.85, p=0.84; Fig. 1A). As another measure of anti-seizure efficacy, we also analyzed the latency to first clonic seizure behavior (characterized as a score of 0.5–3; Fig. 1B). As with seizure severity, latency to seizure onset did not differ as a function of CBDV treated groups (F3,28=0.24, p=0.86, ANOVA).
Figure 1. CBDV attenuates the severity of DMCM-evoked seizures in postnatal day 20, but not postnatal day ten old rats.

Mean (+standard error of the mean [SEM]) seizure score as a function of drug treatment and dose in P10 (A) and P20 rats (C). Mean (+SEM) latency (in seconds) to first behavioral seizure manifestation in P10 (B) and P20 (D) rats. Asterisks indicate significant difference from vehicle control group at *p<0.05, ^p=0.085, Dunn’s tests for multiple comparisons.
By contrast, CBDV treatment in P20 rats resulted in a significant decrease in behavioral seizure severity (Fig. 1C). In vehicle treated rats, the median seizure score was 3. Kruskal-Wallis test revealed a significant difference between groups (H=11.62, P=0.0088, followed by Dunn’s test). Rats treated with 200 mg/kg CBDV displayed significantly reduced seizure severity (p=0.038) to a median score of 2. The 100 mg/kg dose of CBDV trended toward, but did not reach the level of statistical significance (p=0.085). The mean latency to clonic seizure activity in vehicle and CBDV treated rats did not differ (F3,34=0.40, p=0.76, ANOVA, Fig. 1D).
We next assessed the anti-seizure efficacy of CBDV against KA evoked seizures in P10 and P20 rats. Under control conditions, vehicle treated P10 rats exhibited a median seizure score of 2.5. Kruskal-Wallis test revealed no significant difference in seizure score between treatment groups (H=5.24, p=0.15; Fig. 2A) in P10 animals. Likewise, the latency to first seizure manifestation did not differ between groups (F3,28=1.91, p=0.16, ANOVA; Fig. 2B).
Figure 2. CBDV does not alter KA-evoked seizures in immature rats.

Mean (+SEM) seizure score as a function of drug treatment and dose in P10 (A) and P20 (C) rats. Mean (+SEM) latency to first behavioral seizure manifestation in P10 (B) and P20 (D) rats.
In P20 rats, the vehicle treated group exhibited a median seizure score of 2. As in P10 rats, neither seizure severity (H=0.68, p=0.89, Kruskal-Wallis; Fig. 2C) nor latency to seizure onset (F3,36=0.70, p=0.10, ANOVA, Fig. 2D) differed between groups.
In both P10 and P20 rats, latency to seizure onset was numerically (but not statistically) increased by CBDV; in both age groups this resulted from a population of “responders” and “non-responders” with the data bimodally distributed.
3.2. Effect of CBDV on PTZ and MES evoked tonic seizures in P10 and P20 animals.
We next tested CBDV for efficacy in two models of tonic seizures: those evoked by PTZ or MES. In the PTZ model, vehicle treated P10 rats displayed a median seizure score of 4.5. Kruskal-Wallis test revealed a significant difference in seizure score between groups (H=12.03, p=0.0073; Fig. 3A), driven by a significant decrease in seizure severity in rats treated with the highest two doses of CBDV. When treated with 100 or 200 mg/kg CBDV median seizure score decreased to 4 (p=0.0093, Dunn’s test). Neither the latency to exhibit the first minimal (score 0.5–3) nor the first maximal (score 4–5) seizure behavior differed across groups (F3,32=1.35, p=0.27, ANOVA; Fig. 3B; F3,32=2.17, p=0.11, Fig. 3C, respectively).
Figure 3. CBDV attenuates the severity of PTZ-evoked seizures in immature rats.

Mean (+SEM) seizure score as a function of drug treatment and dose in P10 (A) and P20 (D) rats. Asterisks indicate significant difference compared to vehicle control, *p<0.05, **p<0.01, Dunn’s test for multiple comparisons. Mean (+SEM) latency (in seconds) to first behavioral clonic seizure manifestation in P10 (B) and P20 (E) rats. Mean (+SEM) latency (in seconds) to first behavioral tonic or tonic-clonic seizure manifestation in P10 (C) and P20 (F) and rats.
Vehicle treated P20 rats displayed a median seizure score of 5. There was a near-significant effect of drug treatment (Kruskal-Wallis test, H=7.47, p=0.059, Fig. 3D), driven by a significant decrease in seizure score in rats treated with 200 mg/kg CBDV, as compared to vehicle (median seizure score of 3 and 5, respectively; p=0.024). The average latency to minimal seizure behavior did not differ as a function of treatment (F3,34=0.28, p=0.84, ANOVA), however, latency to onset of maximal seizure behavior did differ between groups (F3,34=4.30, p=0.011, ANOVA; Fig. 3E). This effect was driven by increased latency in the 200 mg/kg CBDV group (p=0.0045).
We further assessed the effect of CBDV against tonic seizures using the MES model. The endpoint for MES seizures are tonic forelimb or hindlimb extension in P10 and P20 rats, respectively.(London and Buterbaugh, 1978) CBDV did not protect against maximal electroshock seizures in P10 rat pups (p=1.000, Fisher’s exact test, Fig. 4A). By contrast, in P20 rats, CBDV completely protected against tonic hindlimb extension (p<0.0001, Fisher’s exact test; Fig. 4B).
Figure 4. CBDV protects against MES-evoked tonic seizures in postnatal day 20 rats.

Percent of P10 animals (A) and P20 (B) exhibiting tonic extension as a function of drug treatment. Asterisks indicate significant reduction in tonic seizures in P20 rats compared to vehicle control, ****p<0.0001, Fisher’s exact test.
3.3. Effect of CBDV on hypoxia and NMDA evoked infantile seizures in P10 animals.
We next sought to determine if CBDV would display efficacy in two etiologically relevant models in P10 rats: hypoxia evoked generalized seizures and NMDA evoked flexion spasms. In the hypoxia model, vehicle treated rats exhibited a median seizure score of 3. Kruskal-Wallis test revealed no significant difference in seizure score between treatment groups (H=6.24, p=0.10; Fig. 5A).
Figure 5. CBDV does not protect against hypoxia-induced seizures or NMDA evoked spasms in postnatal day 10 rats.
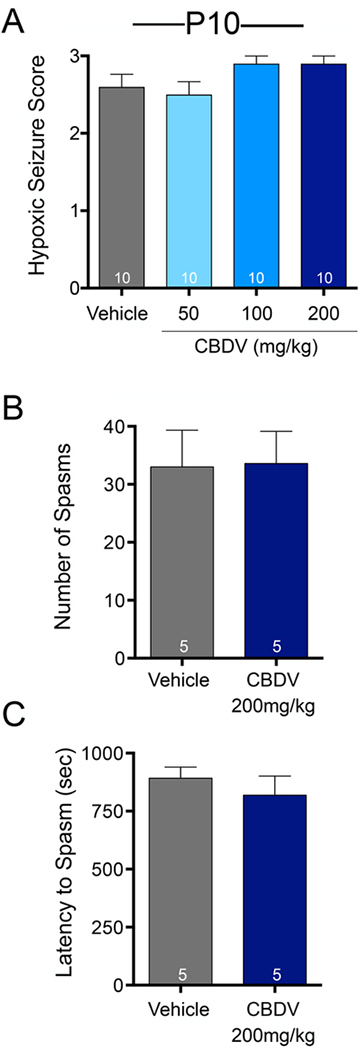
(A) Mean (+SEM) hypoxic seizure score as a function of drug treatment and dose. (B) Mean (+SEM) number of spasms (C) and latency (in seconds) to first flexion spasm as a function of drug treatment.
In the NMDA evoked model of infantile spasms, the average number of spasms did not differ between vehicle and CBDV treated rats (U=12.0, p=0.94, Mann-Whitney; Fig. 5B). The latency to the first spasm also did not differ between vehicle and CBDV treated rats (t(8)=0.76, p=0.46, t-test; Fig. 5C).
3.4. Age-specific mRNA expression of CBDV targets between P10 and P20 animals.
CBDV displayed more striking anticonvulsant effects in older as compared to younger rats. To determine if developmental increases in receptor expression between P10 and P20 could contribute to this, we compared the relative mRNA transcript levels of several CBDV targets. We evaluated TRPV1, TRPV2, TRPV3, TRPA1, and GPR55 transcript levels in the frontal cortex, hippocampus, amygdala, and thalamus (Fig. 6). Data are expressed as fold change in P20 rats as compared to P10 rats. TRPV1 was increased in the amygdala and hippocampus (p=0.052 and p=0.021 Holm-Sidak corrected one-sample t-test), TRPV3 was significantly increased in thalamus (p=0.025, Holm-Sidak corrected one-sample t-test), and GPR55 was significantly increased in the amygdala (p=0.01, Holm-Sidak corrected one sample t-test). We did not statistically evaluate decreases in P20 as compared to P10 rats, as this was not relevant to our a priori hypothesis.
Figure 6. mRNA expression levels of TRPV1, TRPV3, and GPR55 increase as a factor of age from postnatal day 10 to postnatal day 20 in rats.

Violin plot illustrating the fold change in TRPV1 (A), TRPV2 (B), TRPV3 (C), TRPA1 (D), and GPR55 (E) mRNA expression in P20 rats over P10 expression. *p<0.05, ^p=0.052 over P10 levels, one-sample t-test, Holm-Sidak corrected.
3.5. TRPV1 knockout mice show reduced anticonvulsant effect of CBDV in the MES model
TRPV1 was the one transcript to show clear increases in multiple brain regions implicated in seizure generation, as a function of postnatal day. Additionally, the strongest mechanism-based evidence for CBDV’s neuroactive site of action remains TRPV1 (Hill et al., 2013; Iannotti et al., 2014). Therefore, we hypothesized that one mechanism responsible for the anticonvulsant effects of CBDV in P20, but not P10 animals, could be the developmental increase of TRPV1 expression. To test this, we pretreated P20 wildtype and TRPV1 knockout mice with either CBDV (100mg/kg) or vehicle prior to MES seizure testing. The MES model was selected as it displayed the strongest anti-seizure protection in P20 rats. Loss of the TRPV1 receptor attenuated CBDV protection against maximal tonic extensor seizures in P20 mice (Fig. 7). In the vehicle control condition, 100% (8/8) wildtype and TRPV1 knockout mice exhibited tonic hindlimb extension. Pretreatment with CBDV protected the majority of wildtype mice (χ2=19.69, df=3, p=0.0002). Fisher’s exact test (Holm-Sidak corrected) revealed a significant protection in CBDV treated wildtype mice (p=0.018) with only 37.5% (3/8) exhibiting tonic extension compared to 100% (10/10) of the TRPV1 knockout mice.
Figure 7. TRPV1 expression mediates anticonvulsant effects of CBDV against MES seizures in postnatal day 20 mice.

Proportion of tonic seizures as a function of drug treatment and genotype. 100% of vehicle treated wild-type (8 of 8) and TRPV1 knockout (9 of 9) mice displayed tonic hindlimb extension. 100% of CBDV treated TRPV1 knockout (10/10) mice displayed tonic hindlimb extension. 37.5% of CBDV treated wild-type (3/8) mice displayed tonic hindlimb extension. The difference between CBDV-treated wild-type and TRPV1 mice differed significantly; *p<0.05, Chi-Square followed by Fisher’s exact test, Holm-Sidak corrected for multiple comparisons.
3.6. Effect of CBDV on developmental cell death in P7 animals.
The developing rodent brain is uniquely susceptible to drug induced enhancement in apoptosis, an effect that is most pronounced at P7. Drug induced increases in developmental apoptosis are a widely characterized effect of many ASDs, including phenobarbital which we used here as a positive control. As expected, PB treatment resulted in significant increases in the number of Fluoro-Jade B positive cells in all brain regions surveyed (Fig. 8). Accordingly, there was a significant effect of drug treatment in cingulate cortex (F4,31=9.87, p<0.0001; Fig. 8A), motor cortex (F4,29=19.67, p<0.0001; Fig. 8B), somatosensory cortex (F4,29=29.7, p<0.0001; Fig. 8C), striatum (F4,31=20.62, p<0.0001; Fig. 8D), lateral septum (F4,31=14.28, p<0.0001; Fig. 8E), and lateral thalamus (F4,29=125.6, p<0.0001, ANOVA; Fig. 8F). In all regions, this was driven by significant differences between PB and saline treated groups (Ps<0.001, Holm-Sidak corrected). In addition, in the lateral thalamus, we observed a small but significant increase in cell death in rats treated with the 200mg/kg dose of CBDV (P=0.007), but the magnitude was smaller than that observed with PB (2-fold vs. 11-fold increases, respectively).
Figure 8. CBDV increases cell death in the lateral thalamus, but not other regions in the postnatal day 7 rat brain.

Quantification of cell death as indicated by Fluoro-Jade B positive cells in: (A) cingulate cortex, (B) motor cortex, (C) somatosensory cortex, (D) striatum, (E) lateral septum, and (F) lateral thalamus from P7 rat pups treated with SAL, PB 75 mg/kg, VEH, or CBDV (100 or 200 mg/kg). Values are expressed as mean (+SEM) of 3 sequential 40 μm-thick, 1.18 mm2 tissue samples through each region of interest, averaged per animal. *p<0.05, ***p=0.0001, ****p<0.0001; significantly different from SAL or VEH control group. Scale bar = 200 μm. Red boxes indicate the area of interest imaged for each brain region.
4. DISCUSSION
Despite the success of CBD as a novel, FDA approved anti-seizure medication for early life refractory seizures, little is known about the potential anti-seizure effects of the CBD analog, CBDV. Ongoing clinical trials of CBDV for indications ranging from epilepsy to autism, underscore the need for further characterization of this compound. Historically, ASDs “migrate” in use from initial approval in adult patients with refractory epilepsy, to first-line therapies in adults, and eventually to child, infant, and neonatal populations (Chapman et al., 2012). However, the efficacy and safety of ASDs differs dramatically across the lifespan. The poverty of information regarding CBDV effects in the immature brain was the primary motivator for this study.
We found that CBDV displayed anticonvulsant efficacy against clonic and tonic seizures, with effects substantially more striking in P20, as compared to P10 rats. This anticonvulsant effect paralleled a developmental increase in the expression of a CBDV target, the TRPV1 receptor. Moreover, genetic deletion of the TRPV1 receptor reduced the efficacy of CBDV in mice. Beyond efficacy, CBDV displayed a generally benign profile in an established assay of ASD-induced neurotoxicity, although modest cell death was detected in one of six regions surveyed.
The anticonvulsant effects we observed were both model- and age-specific. In P10 rats, which approximate the developmental stage of a human neonate, we only observed an anti-seizure effect of CBDV against PTZ induced seizures. This is consistent with a prior report in adult rats, (Hill et al., 2012) with preferential suppression of tonic seizure components following CBDV pretreatment. Surprisingly, however, the tonic forelimb extension following MES was unaltered at this same age. This differs both from our findings in P20 rats and mice, and with prior reports of CBDV efficacy against MES seizures in adult animals (Hill et al., 2012). However, P10 animals display an immature motor response to electroshock, characterized primarily by tonic extension of the forelimbs, rather than the hindlimbs. This phenotype changes progressively with age into the characteristic tonic hindlimb extension observed in P20 and adult animals (London and Buterbaugh, 1978), and this difference in seizure type may underlie the apparent discrepancy in CBDV action. Second, this may more broadly reflect the general decreased efficacy of CBDV in younger animals, which may be secondary to differences in TRP receptor expression.
CBDV was also without effect against hypoxia induced seizures or in the infantile spasm model, which represent two common seizure types in infancy. While clinical trials of CBD reported a significant seizure reduction in refractory seizures, none of the participants enrolled exhibited seizures as a result of hypoxic-ischemic events or presented with infantile spasms. (Devinsky et al., 2017, 2016) Our current data suggest that these seizure types may not be amenable to CBDV therapy, and may provide guidance for patient selection in future clinical studies.
In contrast, we found a more robust response in P20 animals. The third postnatal week in rodents corresponds to late childhood in humans, and thus more closely mirrors the age of patients enrolled in prior clinical trials. In the PTZ, DMCM, and MES models, CBDV decreased the severity of evoked seizures. These effects were primarily evident against tonic seizures in the PTZ and MES models. The effects against MES were particularly striking with 100% of animals protected. This effect mirrors what has been reported in adult animals (Hill et al., 2012) for PTZ and MES.
CBDV also displays efficacy against clonic seizures evoked by DMCM. CBDV has not been previously evaluated against DMCM evoked seizures in adult animals, however, it is without effect against pilocarpine-evoked limbic seizures in adult animals (Hill et al., 2012). By contrast, CBD does suppress pilocarpine-evoked seizures (Jones et al., 2012). Pilocarpine, like DMCM and KA is primarily associated with limbic (e.g., temporal lobe) seizures, and not tonic (e.g., brainstem-mediated) responses. Interestingly, in P20 rats, there was a trend toward increased latency to onset of KA evoked seizures following the higher doses of CBDV. 80% of vehicle treated rats displayed seizure activity within five minutes of KA administration, whereas latencies in CBDV treated rats ranged from 10–20 min. Interestingly, in a recent study, CBD was reported to significantly suppress KA-evoked seizures (Friedman and Wongvravit, 2018); this treatment was more effective when administered intrahippocampally as compared to intraperitoneally, but significantly attenuated seizure activity when administered by either route. Our findings here, together with these other reports, suggest that the profile of CBDV action against complex-partial like seizures may differ from that of CBD.
Abundant preclinical evidence demonstrates that CB1R agonists decrease seizure incidence and severity in both developing and adult animal models (Huizenga et al., 2017; Rosenberg et al., 2017). However, evidence suggests that while CBD and CBDV share a mechanism or target pathway that is CB-receptor independent (Hill et al., 2013). Several other targets have been suggested for CBD/CBDV action, including activity at the TRP family of receptors and at GPR55, a putative “CB3” receptor with pharmacology distinct from that of CB1 and CB2 (Morales et al., 2017). Both CBD and CBDV are partial agonists at TRPV1, 2, and 3 and TRPA1 channels (De Petrocellis et al., 2012, 2011; Morales et al., 2017), and both TRPV1 agonists and antagonists have been reported to display anticonvulsant properties (Gonzalez-Reyes et al., 2013; Socała et al., 2015). Consistent with this, TRPV1 selective antagonists attenuate the anticonvulsant effect of CBD and CBDV in several models (Iannotti et al., 2014; Vilela et al., 2013).
While the transcript levels of all these targets was low across brain regions surveyed, we identified transcripts that increased between P10 and P20. TRPV1, in particular, was increased in the hippocampus and amygdala of P20 rats, key regions involved in seizure generation and propogation (Vismer et al., 2015). Additionally, we found a substantial increase in the mRNA expression of TRPV2 in the hippocampus of some P20 rats. In support of our findings, TRPV1 and TRPV2 transcripts have been shown to be expressed in rat hippocampal tissues and both receptors are activated by CBDV (Huang et al., 2014; Morales et al., 2017). We observed a striking, but incomplete, decrease in the anticonvulsant effect of CBDV in TRPV1 knockout mice, suggesting that increased TRPV1 expression may contribute to the developmental change in sensitivity to CBDV, and that CBDV actions may be partially TRPV1 independent.
There is a well-documented effect of current ASDs increasing programmed cell death during the first week of postnatal development in rodents (Bittigau et al., 2002; Forcelli et al., 2011). Given the migration in use of ASDs from adult to neonatal populations, it is essential to assess the safety of putative therapies in the developing brain. Both the cannabinoids THC and WIN 55,212–2 avoid inducing apoptosis in developing animals, but display a complex spectrum of effects including exacerbating injury caused by other drugs (Hansen et al., 2008) and in some cases, neuroprotection (Huizenga and Forcelli, 2018). Here we found that CBDV produced a small but significant increase in the number of Fluoro-Jade B positive cells in the lateral thalamus of P7 rats. The thalamus was the only region with CBDV increases in cell death, and interestingly, this region displays the most prominent cell death following exposure to other drugs (Bittigau et al., 2002; Kim et al., 2007). However, the CBDV induced increase in cell death in the thalamus remains significantly lower than with PB and many other ASDs. We did not evaluate co-treatment with other compounds in the present study, and notably, several ASDs that avoid toxicity when administered as part of a monotherapy regimen significantly exacerbate toxicity as part of polytherapy (Bittigau et al., 2002; Kim et al., 2007). Whether this holds true for CBDV remains to be determined.
5. Conclusions
Together, the low efficacy we observed in P10 rats suggests that CBDV may hold less promise for seizures in infancy, although examining other models (e.g., Dravet syndrome) may reveal effects not detected in our screen. However, the prominent anti-seizure effects in P20 rats and mice and relatively safe profile with respect to neuronal apoptosis suggests therapeutic value of CBDV as a novel treatment for childhood epilepsy.
Cannabidiol (CBD) was recently approved for early life refractory seizures
The efficacy and safety profile of the CBD analog, cannabidivarin (CBDV) is unkown
CBDV displayed age- and model-specific efficacy of in developing animals.
CBDV effects are mediated in part by TRPV1 expression
CBDV has a favorable safety profile for use during development
Acknowledgments
Funding: MNH was supported by TL1TR001431; PAF was supported by R01NS097762 and KL2TR001432; ASR was supported by CONACyT (Mexican Council on Science and Technology) Scholarship #381291. Supported by R01HD091994 to PAF
Abbreviations:
- ASD
anti-seizure drug
- CBD
cannabidiol
- CBDV
cannabidivarin
- P
postnatal day
- PTZ
pentylenetetrazole
- KA
kainic acid
- MES
maximal electroshock
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Al-Muhtasib N, Sepulveda-Rodriguez A, Vicini S, Forcelli PA, 2018. Neonatal phenobarbital exposure disrupts GABAergic synaptic maturation in rat CA1 neurons. Epilepsia 59, 333–344. 10.1111/epi.13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlSuleimani YM, Hiley CR, 2015. The GPR55 agonist lysophosphatidylinositol relaxes rat mesenteric resistance artery and induces Ca2+ release in rat mesenteric artery endothelial cells. Br J Pharmacol 172, 3043–3057. 10.1111/bph.13107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Lafuente H, Rey-Santano MC, Mielgo VE, Gastiasoro E, Rueda M, Pertwee RG, Castillo AI, Romero J, Martínez-Orgado J, 2008. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr. Res 64, 653–658. 10.1203/PDR.0b013e318186e5dd [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, Dzietko M, Pesditschek S, Mai I, Dikranian K, Olney JW, Ikonomidou C, 2002. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proceedings of the National Academy of Sciences 99, 15089–15094. 10.1073/pnas.222550499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L, Gutherz S, Kulick C, Soper C, Kondratyev A, Forcelli PA, 2016. Profile of retigabine-induced neuronal apoptosis in the developing rat brain. Epilepsia 57, 660–670. 10.1111/epi.13335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D, 2000. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313. [DOI] [PubMed] [Google Scholar]
- Chachua T, Yum M-S, Velíšková J, Velíšek L, 2011. Validation of the rat model of cryptogenic infantile spasms. Epilepsia 52, 1666–1677. 10.1111/j.1528-1167.2011.03220.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KE, Raol YH, Brooks‐Kayal A, 2012. Neonatal seizures: controversies and challenges in translating new therapies from the lab to the isolette. European Journal of Neuroscience 35, 1857–1865. 10.1111/j.1460-9568.2012.08140.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, Stott CG, Di Marzo V, 2011. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol 163, 1479–1494. 10.1111/j.1476-5381.2010.01166.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Orlando P, Moriello AS, Aviello G, Stott C, Izzo AA, Di Marzo V, 2012. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol (Oxf) 204, 255–266. 10.1111/j.1748-1716.2011.02338.x [DOI] [PubMed] [Google Scholar]
- Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, Scheffer IE, Thiele EA, Wright S, 2017. Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome. New England Journal of Medicine 376, 2011–2020. 10.1056/NEJMoa1611618 [DOI] [PubMed] [Google Scholar]
- Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, Miller I, Flamini R, Wilfong A, Filloux F, Wong M, Tilton N, Bruno P, Bluvstein J, Hedlund J, Kamens R, Maclean J, Nangia S, Singhal NS, Wilson CA, Patel A, Cilio MR, 2016. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. The Lancet Neurology 15, 270–278. 10.1016/S1474-4422(15)00379-8 [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J, 1979. Comparative aspects of the brain growth spurt. Early Hum Dev 3, 79–83. [DOI] [PubMed] [Google Scholar]
- Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB, 1990. Phenobarbital for febrile seizures--effects on intelligence and on seizure recurrence. N. Engl. J. Med 322, 364–369. 10.1056/NEJM199002083220604 [DOI] [PubMed] [Google Scholar]
- Forcelli PA, Janssen MJ, Vicini S, Gale K, 2012a. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann. Neurol 72, 363–372. 10.1002/ana.23600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Kim J, Kondratyev A, Gale K, 2011. Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia 52, e207–211. 10.1111/j.1528-1167.2011.03297.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Soper C, Duckles A, Gale K, Kondratyev A, 2013. Melatonin potentiates the anticonvulsant action of phenobarbital in neonatal rats. Epilepsy Research 107, 217–223. 10.1016/j.eplepsyres.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcelli PA, Soper C, Lakhkar A, Gale K, Kondratyev A, 2012b. Anticonvulsant effect of retigabine during postnatal development in rats. Epilepsy Research 101, 135–140. 10.1016/j.eplepsyres.2012.03.006 [DOI] [PubMed] [Google Scholar]
- Friedman LK, Wongvravit JP, 2018. Anticonvulsant and Neuroprotective Effects of Cannabidiol During the Juvenile Period. J. Neuropathol. Exp. Neurol 10.1093/jnen/nly069 [DOI] [PubMed] [Google Scholar]
- Glass HC, Grinspan ZM, Shellhaas RA, 2018. Outcomes after acute symptomatic seizures in neonates. Semin Fetal Neonatal Med 23, 218–222. 10.1016/j.siny.2018.02.001 [DOI] [PubMed] [Google Scholar]
- Glass HC, Kan J, Bonifacio SL, Ferriero DM, 2012. Neonatal seizures: treatment practices among term and preterm infants. Pediatr. Neurol 46, 111–115. 10.1016/j.pediatrneurol.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Dzhala VI, Kuchibhotla KV, Feng G, Kuner T, Augustine G, Bacskai BJ, Staley KJ, 2009. Differences in cortical versus subcortical GABAergic signaling: a candidate mechanism of electroclinical uncoupling of neonatal seizures. Neuron 63, 657–672. 10.1016/j.neuron.2009.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reyes LE, Ladas TP, Chiang C-C, Durand DM, 2013. TRPV1 antagonist capsazepine suppresses 4-AP-induced epileptiform activity in vitro and electrographic seizures in vivo. Exp. Neurol 250, 321–332. 10.1016/j.expneurol.2013.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HH, Krutz B, Sifringer M, Stefovska V, Bittigau P, Pragst F, Marsicano G, Lutz B, Ikonomidou C, 2008. Cannabinoids enhance susceptibility of immature brain to ethanol neurotoxicity. Ann. Neurol 64, 42–52. 10.1002/ana.21287 [DOI] [PubMed] [Google Scholar]
- Hill A, Mercier M, Hill T, Glyn S, Jones N, Yamasaki Y, Futamura T, Duncan M, Stott C, Stephens G, Williams C, Whalley B, 2012. Cannabidivarin is anticonvulsant in mouse and rat. Br J Pharmacol 167, 1629–1642. 10.1111/j.1476-5381.2012.02207.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TDM, Cascio M-G, Romano B, Duncan M, Pertwee RG, Williams CM, Whalley BJ, Hill AJ, 2013. Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br J Pharmacol 170, 679–692. 10.1111/bph.12321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W-X, Min J-W, Liu Y-Q, He X-H, Peng B-W, 2014. Expression of TRPV1 in the C57BL/6 mice brain hippocampus and cortex during development. Neuroreport 25, 379–385. 10.1097/WNR.0000000000000105 [DOI] [PubMed] [Google Scholar]
- Huizenga MN, Forcelli PA, 2018. Neuroprotective action of the CB1/2 receptor agonist, WIN 55,212–2, against DMSO but not phenobarbital-induced neurotoxicity in immature rats. Neurotox Res. 10.1007/s12640-018-9944-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizenga MN, Wicker E, Beck VC, Forcelli PA, 2017. Anticonvulsant effect of cannabinoid receptor agonists in models of seizures in developing rats. Epilepsia 58, 1593–1602. 10.1111/epi.13842 [DOI] [PubMed] [Google Scholar]
- Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, Russo E, Whalley BJ, Di Marzo V, Stephens GJ, 2014. Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability. ACS Chem. Neurosci 5, 1131–1141. 10.1021/cn5000524 [DOI] [PubMed] [Google Scholar]
- Jones NA, Glyn SE, Akiyama S, Hill TDM, Hill AJ, Weston SE, Burnett MDA, Yamasaki Y, Stephens GJ, Whalley BJ, Williams CM, 2012. Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure 21, 344–352. 10.1016/j.seizure.2012.03.001 [DOI] [PubMed] [Google Scholar]
- Kaushal S, Tamer Z, Opoku F, Forcelli PA, 2016. Anticonvulsant drug-induced cell death in the developing white matter of the rodent brain. Epilepsia 57, 727–734. 10.1111/epi.13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kondratyev A, Gale K, 2007. Antiepileptic Drug-Induced Neuronal Cell Death in the Immature Brain: Effects of Carbamazepine, Topiramate, and Levetiracetam as Monotherapy versus Polytherapy. J Pharmacol Exp Ther 323, 165–173. 10.1124/jpet.107.126250 [DOI] [PubMed] [Google Scholar]
- Koh S, Jensen FE, 2001. Topiramate blocks perinatal hypoxia-induced seizures in rat pups. Ann. Neurol 50, 366–372. [DOI] [PubMed] [Google Scholar]
- Kubová H, Mares P, 1993. Anticonvulsant action of oxcarbazepine, hydroxycarbamazepine, and carbamazepine against metrazol-induced motor seizures in developing rats. Epilepsia 34, 188–192. [DOI] [PubMed] [Google Scholar]
- Kubova H, Mares P, 1991. Anticonvulsant effects of phenobarbital and primidone during ontogenesis in rats. Epilepsy Res. 10, 148–155. [DOI] [PubMed] [Google Scholar]
- Kulick C, Gutherz S, Kondratyev A, Forcelli PA, 2014. Ontogenic profile of seizures evoked by the beta-carboline DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) in rats. European Journal of Pharmacology 740, 662–668. 10.1016/j.ejphar.2014.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulick CV, Gutherz SB, Beck VC, Medvedeva N, Soper C, Forcelli PA, 2014. Profile of anticonvulsant action of levetiracetam, tiagabine and phenobarbital against seizures evoked by DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) in neonatal rats. European Journal of Pharmacology 743, 63–68. 10.1016/j.ejphar.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ED, Buterbaugh GG, 1978. Modification of electroshock convulsive responses and thresholds in neonatal rats after brain monoamine reduction. J Pharmacol Exp Ther 206, 81–90. [PubMed] [Google Scholar]
- Löscher W, 2011. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 20, 359–368. 10.1016/j.seizure.2011.01.003 [DOI] [PubMed] [Google Scholar]
- Mares P, Schickerová R, 1980. Seizures elicited by subcutaneous injection of metrazol during ontogenesis in rats. Act Nerv Super (Praha) 22, 264–268. [PubMed] [Google Scholar]
- Meador KJ, Baker GA, Browning N, Cohen MJ, Bromley RL, Clayton-Smith J, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW, NEAD Study Group, 2012. Effects of fetal antiepileptic drug exposure: outcomes at age 4.5 years. Neurology 78, 1207–1214. 10.1212/WNL.0b013e318250d824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Hurst DP, Reggio PH, 2017. Molecular Targets of the Phytocannabinoids-A Complex Picture. Prog Chem Org Nat Prod 103, 103–131. 10.1007/978-3-319-45541-9_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MJ, Scher MS, Stein AD, Armatti S, Wang Z, Gardiner JC, Paneth N, Minnigh B, Alvin J, 1999. Phenobarbital Compared with Phenytoin for the Treatment of Neonatal Seizures. New England Journal of Medicine 341, 485–489. 10.1056/NEJM199908123410704 [DOI] [PubMed] [Google Scholar]
- Press Announcements - FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy [WWW Document], 2018. . FDA U.S. Food & Drug; URL https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm611046.htm (accessed 7.7.18). [Google Scholar]
- Ramachandra R, Subramanian T, 2011. Atlas of the Neonatal Rat Brain. CRC Press, Boca Raton. [Google Scholar]
- Rosenberg EC, Patra PH, Whalley BJ, 2017. Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis, and epilepsy-related neuroprotection. Epilepsy Behav 70, 319–327. 10.1016/j.yebeh.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ, 2000. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874, 123–30. [DOI] [PubMed] [Google Scholar]
- Socała K, Nieoczym D, Pieróg M, Wlaź P, 2015. α-Spinasterol, a TRPV1 receptor antagonist, elevates the seizure threshold in three acute seizure tests in mice. J Neural Transm (Vienna) 122, 1239–1247. 10.1007/s00702-015-1391-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšek L, Chachua T, Yum M-S, Poon K-L, Velíšková J, 2010. Model of cryptogenic infantile spasms after prenatal corticosteroid priming. Epilepsia 51, 145–149. 10.1111/j.1528-1167.2010.02630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšek L, Kubová H, Velíšková J, Mareš P, Ortová M, 1992. Action of Antiepileptic Drugs Against Kainic Acid-Induced Seizures and Automatisms During Ontogenesis in Rats. Epilepsia 33, 987–993. 10.1111/j.1528-1157.1992.tb01748.x [DOI] [PubMed] [Google Scholar]
- Vilela LR, Medeiros DC, Rezende GHS, de Oliveira ACP, Moraes MFD, Moreira FA, 2013. Effects of cannabinoids and endocannabinoid hydrolysis inhibition on pentylenetetrazole-induced seizure and electroencephalographic activity in rats. Epilepsy Res. 104, 195–202. 10.1016/j.eplepsyres.2012.11.006 [DOI] [PubMed] [Google Scholar]
- Vismer MS, Forcelli PA, Skopin MD, Gale K, Koubeissi MZ, 2015. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 9 10.3389/fncir.2015.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]