

Summary
Rapid eye movement (REM) sleep was discovered nearly 60 years ago. This stage of sleep accounts for approximately a quarter of total sleep time in healthy adults and it is mostly concentrated in the second half of the sleep period. The majority of research on REM sleep has focused on neurocognition. More recently, however, there has been a growing interest in understanding whether obstructive sleep apnea (OSA) during the two main stages of sleep (REM and non-REM sleep) lead to different cardiometabolic and neurocognitive risk. In this review we discuss the growing evidence indicating that OSA during REM sleep is a prevalent disorder that is independently associated with adverse cardiovascular, metabolic and neurocognitive outcomes. From a therapeutic standpoint, we discuss limitations of CPAP therapy given that 3 or 4 hours of CPAP use from the beginning of the sleep period would leave 75% or 60% of obstructive events during REM sleep untreated. We also review potential pharmacologic approaches to treating OSA during REM sleep. Undoubtedly further research is needed to establish best treatment strategies in order to effectively treat REM OSA. Moreover, it is critical to understand whether treatment of REM OSA will translate into better patient outcomes.
Keywords: Rapid eye movement, sleep, OSA, cardiovascular, neurocognitive, diabetes, memory, mood, treatment, pharmacologic
Introduction
Obstructive sleep apnea (OSA) occurs when the muscles and tissues surrounding the upper airway collapse partially or completely during sleep, resulting in a period where breathing stops or is significantly attenuated before the airway opens again. This phenomenon occurs repeatedly during sleep; in individuals with severe OSA, it can occur nearly every minute or more. In many people, several factors converge making OSA more severe during rapid eye movement (REM) sleep. The body naturally loses muscle tone during REM sleep, perhaps most significantly at the level of the genioglossus due to cholinergic mediated inhibition, and it becomes easier for muscles surrounding the upper airway to collapse [1]. There is also a reduction in the hypoxic and hypercapnic ventilatory drive [2, 3]. The worsening of OSA during REM can be manifested in a number of ways, including more frequent events, longer duration events, and greater oxygen desaturation associated with events [4–6]. Figure 1 highlights the mechanisms linking REM sleep to adverse health outcomes. Exactly how these transient events during sleep lead to persistent daytime consequences remains to be fully determined. In one of the first experimental demonstrations that OSA is causally linked to persistent daytime hypertension, it was determined that chronic sleep fragmentation alone was insufficient to elicit the same increases in daytime blood pressure as chronic experimentally induced OSA [7]. Additionally, in this canine model of OSA, the maximal daytime hypertension response took 4–5 weeks to develop, and when the experimentally induced OSA was discontinued, nocturnal blood pressure immediately returned to baseline levels, however, the daytime blood pressure took about 3 weeks to normalize. This dichotomy in the timing of return to baseline blood pressure levels between sleep and wake argues for a mechanism related to hypertension that is tied to state, and therefore likely mediated by plastic neural input to vessel walls.
Figure 1:
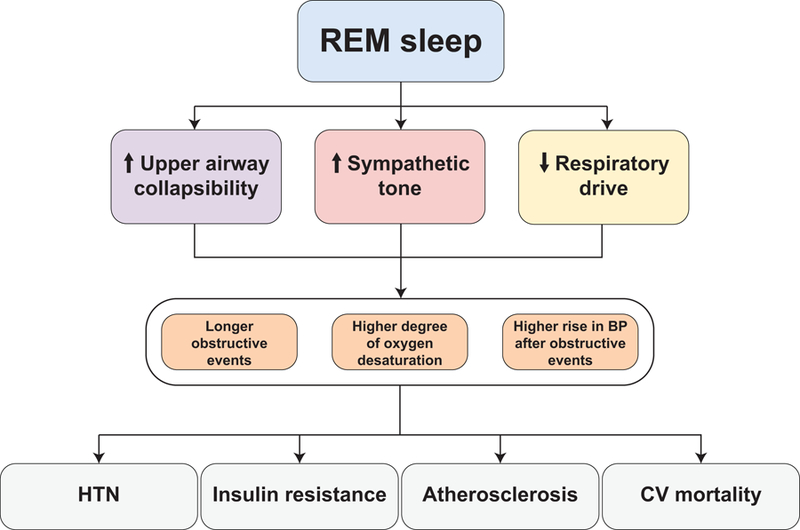
Multiple mechanisms by which REM sleep can lead to increased frequency and severity of obstructive respiratory events and disproportionately toxic consequences on health outcomes. BP – blood pressure. HTN – hypertension. CV – cardiovascular.
OSA has been clearly linked to a number of adverse cardiovascular, endocrine, and neurocognitive outcomes, but whether the risks associated with these adverse outcomes depend on the stage of sleep in which the events occur has remained a matter of debate. The concept that REM OSA can be associated with increased risks is based on at least 2 possibilities that are not mutually exclusive. The first idea is that REM OSA induces intermittently severe disease, and that severe disease, even in limited doses, is sufficient to increase risks associated with adverse outcomes. This argument would suggest that a person who spends 25% of total sleep time in REM sleep, with a non-REM apnea hypopnea index (AHI) using 4% oxygen desaturation criteria (AHI4%) of 4/hour and REM AHI4% of 24/hour with an overall AHI4% of 9/hour would experience increased risk in comparison to an individual with an overall AHI4% of 9/hour composed of REM and non-REM AHI4%’s that are each 9/hour. Of note, this conceptual framework that intermittently severe disease carries increased risks would be true for positional OSA as well, where severity of OSA in the supine position is often greater than severity in the non-supine position. The other idea is that REM sleep constitutes some state in which the brain and other end organs are particularly vulnerable, due to its physiological features including the precise brain neurochemical milieu, degree of neuronal synchrony, frequency of cortical local field potential oscillations, and autonomic tone, which summate to influence heart rate and rhythm and control blood flow to end organs.
In this review, we discuss attempts to capture the epidemiology of REM OSA before evaluating the specific adverse outcomes of REM OSA on the cardiovascular, endocrine, and neurocognitive systems and how these determinations were derived within the context of assumed comorbid risks associated with non-REM OSA. Finally, we discuss some of the particular cellular and molecular mechanisms associated with REM inhibition of muscle tone, which may lead to novel drug targets for REM OSA.
Epidemiology of REM OSA
Determining how frequently REM OSA occurs in a population depends in part on having a definition of what constitutes REM OSA. This is somewhat problematic as there is no standard consensus for what constitutes REM OSA by any AHI cutoff, and using the AHI3% and/or arousal (AHI3%a) criteria instead of the AHI4% can lower the prevalence of REM OSA [8]. Additionally, how much REM sleep needs to be captured on any given sleep study in order to feel confident that the REM AHI is likely to generalize across nights is a matter of some debate [9]. In the research setting, 30 minutes of REM sleep has been largely accepted as the minimum required to make meaningful statements about REM OSA by the REM AHI [9], although in some studies a duration as low as 10 minutes is used. Similarly, many research studies employ a ratio of REM AHI/non-REM AHI > 2 as a working definition of REM OSA. It is important to point out that there are several inherent limitations in just using a ratio of rates (i.e., events/hour in REM sleep divided by events/hour in non-REM sleep) when classifying REM OSA. First, classifying patients with OSA based on the REM AHI/non-REM AHI ratio by itself is problematic because it will undoubtedly designate some patients as having REM-related OSA when, in fact, there is also substantial disease during non-REM sleep. For example, a patient with a REM AHI of 80 events/hour and a non-REM AHI of 35 events/hour would be classified as having REM OSA when, in fact, severe OSA is also present during non-REM sleep. The REM AHI/non-REM AHI ratio does not accurately depict the occurrence of OSA predominantly during REM sleep because it can be high due to: (1) a high REM AHI, (2) a low non-REM AHI, or (3) a combination of both [9]. To overcome this limitation, some studies add additional criteria of a total AHI minimum (usually 5/hour) and non-REM AHI maximum (typically between 8 and 15/hour).
One of the earliest studies to examine REM OSA prevalence included patients with a total AHI between 5/hour and 25/hour, a non-REM AHI < 15/hour and REM AHI/non-REM AHI > 2. This occurred in 24% of 632 men and 62% of 206 women with PSG-identified OSA [10]. A similar pattern in which increased prevalence of REM OSA was observed in women was recapitulated in subsequent studies in which 21.0% of men and 40.8% of women expressed REM OSA in 2,486 OSA patients [11], or 4% of men and 35% of women among 45 severely obese patients [12]. In studies that did not segregate by sex, REM OSA was observed in 36.4% of 415 OSA patients using a sole criterion of REM AHI/non-REM AHI >2 [13], and in 14.4% of 1,540 OSA patients when stricter criteria were added [14]. Indeed, the precise effect of altering the definition of REM OSA was assessed in 931 patients with a minimum total AHI3%a of 5/hour [15]. REM OSA was observed in 36.7% when only REM AHI/non-REM AHI > 2 was used, 24.4% when the criterion of non-REM AHI3%a < 15/hour was added, and 13.5% when the criterion of non-REM AHI3%a < 8/hour was added.
While these prevalence estimates are from patients referred to a sleep center for evaluation, REM OSA prevalence estimates have also been derived from large community based epidemiological studies, although different criteria for REM OSA were typically employed. In the Wisconsin Sleep Cohort, among 2,953 sleep studies with 30 minutes of REM and a non-REM AHI4% ≤ 5 events/hour, 12% demonstrated a REM AHI ≥ 15 events/hour [16]. Furthermore, among studies where the total AHI4% was less than 15 events/hour, 22% demonstrated a REM AHI4% ≥ 15 events/hour. In the Sleep Heart Health Study, among 3,265 subjects with at least 30 minutes of REM sleep and a non-REM AHI4% < 5 events/hour, 27.7% had a REM AHI4% of 5.0–14.9 events/hour, 13.0% had a REM AHI4% of 15.0–29.9 events/hour, and 5.5% had a REM AHI > 30 events/hour [17]. Furthermore, among 4648 subjects with 30 minutes of REM sleep and all levels of OSA severity, 464 (10%) had a non-REM AHI4% < 5 events/hour and REM AHI4% > 15 events/hour [18]. In 2,044 older men >65 years of age in the MrOS study with a total AHI4% < 15 events/hour, 20% had a REM AHI4% of 15–30 events/hour and 6.5% had a REM AHI4% > 30 events/hour [19]. In the HypnoLaus study, among 2074 subjects with at least 30 minute of REM sleep, 40.8% had a REM AHI3a ≥ 20/hour [20]. In sum, although REM OSA definitions may vary, REM OSA is prevalent in both community-based and clinic population samples.
Cardiovascular Outcomes and REM OSA
OSA has been shown to be associated with several adverse cardiovascular outcomes including hypertension [15, 21], myocardial infarction [22, 23], and stroke [24, 25] in both community-based and clinic-based studies. In longitudinal studies of incident hypertension in the Wisconsin Sleep Cohort, OSA severity at baseline predicted the presence of hypertension four years later in a dose-dependent manner after controlling for several potential confounding factors [26]. In the Vitoria study, a large community-based study of middle-aged adults in Spain, OSA severity was not associated with incident hypertension over a 7-year follow-up [27], however, a subsequent reanalysis demonstrated that moderate to severe OSA was associated with the incidence of more severe forms of hypertension in men, but not women [28]. In the Sleep Heart Health Study, OSA severity was associated with incident hypertension, but this association appeared to be significantly driven by obesity, as the strength of the association was significantly weakened when BMI was included as a covariate [29]. Given this heterogeneity, it seems plausible that not all apnea influences hypertension equally, and because REM sleep is associated with greater sympathetic activity, lower vagal tone and more cardiovascular instability compared to non-REM sleep [30], obstructive events during REM sleep could disproportionately lead to hypertension and other adverse cardiovascular outcomes. It is theoretically possible that the increased sympathetic tone during REM sleep creates a ceiling effect, whereby obstructive events during this stage do not add much additional sympathetic tone. However, this is not well supported by studies examining sympathetic tone quantified by both sympathetic burst frequency and amplitude in subjects with and without OSA. In subjects without OSA, burst freuquency during wake was 24 bursts/minute and increased to 34 bursts/minute during REM sleep. Burst amplitude during REM sleep was 215% of the average value obstained during wake [31]. In individuals with OSA, wake burst frequency was already significantly elevated in comparison to subjects without OSA at 59 bursts/minute. While burst frequency was not significantly different during REM sleep in individuals with OSA, burst amplitude increased nonetheless to 141% on average during REM sleep [30].
Indeed, analyses of REM OSA in the Wisconsin Sleep Cohort [16], the Men Androgens Inflammation Lifestyle Environment and Stress (MAILES) study [32], and the HypnoLaus study [20] suggest it is significantly associated with hypertension, independent of non-REM OSA. In the analysis of the Wisconsin Sleep Cohort comprising 1,451 individuals completing 4,385 sleep studies, REM AHI4% considered as a categorical variable (AHI4% 1–4.9/hour, 5–14.9/hour, and ≥ 15/hour versus reference AHI4% < 1/hour) was associated with significantly increased risk of prevalent hypertension, after controlling for age, sex, race, body mass index, waist-to-hip ratio, smoking alcohol, and non-REM AHI4% expressed as a continuous variable. Additionally, in a subset of individuals with ambulatory blood pressure monitoring data and a non-REM AHI4% < 5 events/hour, REM AHI4% considered either categorically or continuously was significantly associated with increasing hypertension prevalence. Notably, non-REM AHI4% did not significantly predict prevalent hypertension in any models. Finally, longitudinal assessment of hypertension in 428 individuals whose hypertension status changed from absent to present (or vice versa) revealed a significant association of REM OSA severity by REM AHI4% categories and development of incident hypertension over time after controlling for non-REM AHI4% [16].
In a separate follow-up study, 269 adults enrolled in the Wisconsin Sleep Cohort Study who completed two or more 24 hour ambulatory blood pressure studies over an average of 6.6 years were evaluated for non-dipping blood pressure [33]. Blood pressure ordinarily dips 10–20% across sleep, and when this does not occur, there is increased risk for the future development of hypertension in normotensive adults, as well as increased risk for cardiac damage, including left ventricular hypertrophy, angina, myocardial infarction, and cardiovascular death [34, 35]. In subjects with at least 30 minutes of REM sleep, REM AHI4% by category was significantly associated with increased risk for incident non-dipping of both systolic and diastolic blood pressure when controlling for non-REM AHI4% and other covariates.
In the MAILES study, a community-based study of adult men in Australia, 739 men with at least 30 minutes of REM sleep on polysomnography had completed prior clinical assessments for hypertension between the years 2002 and 2010. REM OSA severity by REM AHI3%a evaluated categorically was significantly associated with both prevalent and recent-onset hypertension, particularly in those with a REM AHI3%a ≥ 20 events/hour, when controlling for a variety of potential confounders, including non-REM AHI3%a. In the subset of men with non-REM AHI3%a < 10 events/hour, hypertension was also significantly associated with REM AHI3%a by category, similar to the observations in the Wisconsin Sleep Cohort. Also similarly, hypertension was not associated with non-REM AHI3%a. In the MAILES study, mean REM oxygen desaturation (i.e. mean of the drop in oxygen saturation per obstructive event ) was significantly associated with prevalent hypertension independent of mean non-REM oxygen desaturation, and non-REM mean oxygen desaturation showed no significant associations with prevalent hypertension.
In assessing multiple polysomnographic variables that predict hypertension in the Multi-Ethnic Study of Atherosclerosis (MESA), REM OSA variables exhibited the largest absolute differences between groups when subjects were dichotomized by median systolic and diastolic blood pressures. This possibly suggests that to detect a difference between median BPs, a larger change in REM AHI vs. NREM AHI is needed [21]. That said, REM AHI4% indices did not remain associated with BP in the final multivariate regression models in this analysis, whereas overall AHI4% and arousals associated with periodic limb movement of sleep were.
HypnoLaus is a community-based study of men and women in Switzerland who are middle aged (median age 56), and largely white and non-obese. In 2074 individuals with at least 30 minutes of REM sleep, REM AHI3a, considered categorically in those with REM AHI3a ≥ 20/hour, was associated with increased risk for hypertension after controlling for age, sex, BMI, waist-to-hip ratio, total sleep time, smoking, alcohol consumption, and non-REM AHI3a [20].
Given the strong association of hypertension with other adverse cardiovascular outcomes, a logical extension of the established relationship between REM OSA and hypertension would include a potential role for REM OSA in outcomes such as angina, myocardial infarction, stroke, and heart failure. Additionally, OSA-driven sympathetic surges associated with paroxysmal arrhythmias may occur most frequently in REM sleep, due to significantly higher sympathetic tone, heart rate, and heart rate variability in REM versus non-REM sleep [36, 37]. Atrial arrhythmias in particular could lead to atrial embolus generation and stroke. The possibility of REM OSA being tied to such cardiovascular outcomes was evaluated in a study of subjects from the Sleep Heart Health Study [17]. In this study, 3,265 participants were evaluated who had at least 30 minutes of REM sleep and no significant OSA during non-REM sleep (non-REM AHI4% < 5/hour). The participants were mostly older adults (mean age 62±11 years) who were slightly overweight (BMI 28±5 kg/m2), and mostly women (63% female). Subjects were followed for 9.5 years on average, and a composite adverse cardiovascular outcome, including myocardial infarction, coronary artery revascularization, congestive heart failure, or stroke, was evaluated for prevalence and incidence. In participants with prevalent cardiovascular disease at baseline (n = 452), the hazard ratio for the composite cardiovascular endpoint was 2.56 (95% CI, 1.46–4.47) for severe REM OSA (REM AHI4% > 30 events/hour) compared to no OSA during REM sleep (REM AHI <5 events/hour) after adjusting for age, sex, race, body mass index, smoking status, prevalent hypertension and diabetes. The association was much weaker in participants without prevalent cardiovascular disease. As noted by the authors, the overall number of individuals with prevalent cardiovascular disease at baseline and REM AHI4% > 30/hour was relatively small (n = 33), and a separate study evaluating “REM OSA” as a phenotype from cluster analysis of a largely male veteran population did not observe increased cardiovascular risk in this cluster [38], so caution is warranted in interpretation of this finding. Nonetheless, these findings potentially offer an explanation for the divergent observations that OSA is significantly associated with adverse cardiovascular outcomes and yet that CPAP treatment of OSA in randomized clinical trials has yielded ambiguous or negative results [39, 40]. In such trials, the average duration of CPAP use is typically much lower than the expected total sleep duration. Because REM sleep is significantly weighted toward the second half of sleep, and especially in the minutes just prior to waking, it is likely that subjects assigned to CPAP treatment still had significant periods of untreated REM OSA. Such a hypothesis is supported by a subanalysis of the SAVE trial, where 561 patients with nightly CPAP adherence > 4 hours/night had significantly lower risk for cerebrovascular events compared to propensity score-matched subjects with usual care alone. Additional work appears to be needed to evaluate the specific effect of treatment of REM OSA on hypertension and cardiovascular outcomes.
Endocrine Outcomes and REM OSA
Sleep and endocrine function are neuroanatomically linked through the hypothalamus, and lateral hypothalamic neurotransmitters orexin and melanin concentrating hormone (MCH) have strong and reciprocal effects on both feeding behaviors [41–43] and promotion of REM sleep [44]. Disruptions to sleep, such as short sleep duration [45] and OSA [46–48], have been associated with insulin resistance, glucose intolerance, and development of diabetes (for a comprehensive review see Reutrakul and Mokhlesi [49]). A role for OSA in diabetes is confounded by the mutual strong associations of OSA and diabetes with obesity. Nonetheless, OSA has been found to be associated with diabetes development even in non-obese populations [50, 51].
A specific role for REM sleep in this process has been observed in human subjects and animal models. In non-diabetic human subjects, continuous glucose monitoring combined with polysomnography revealed steep drops in the interstitial glucose concentration across periods of REM sleep [52], and rodents experiencing acute REM sleep disruption showed reduced activity of enzymes that typically break down glucose, including hexokinase and glucose-6-phosphatase [53]. Additionally, intermittent hypoxia has been demonstrated to impair pancreatic beta cell function [54, 55], and severity of hypoxemia correlated with HbA1c levels in subjects with OSA and no previously recognized diabetes [56]. Taken together, these observations suggest that REM OSA may play a substantial role in the development of insulin resistance and diabetes, given that obstructive events during REM sleep significantly fragment REM sleep quality and are often associated with the greatest oxygen desaturations during sleep [4, 6].
Several studies lend credence to this specific hypothesis. First, continuous glucose monitoring across sleep in subjects with untreated OSA demonstrated that the occurrence of OSA in REM sleep abrogated the expected decline in interstitial glucose concentration across REM sleep, whereas OSA in non-REM sleep had no effect on interstitial glucose concentration [57]. In obese type 2 diabetics with and without OSA undergoing continuous glucose monitoring during sleep, the mean glucose level was 38% higher during REM sleep in those with OSA versus those without OSA. These smaller studies were bolstered by the observations from larger population-based studies. In the Sleep Heart Health Study consisting of middle-aged and older subjects, REM AHI4% treated as a continuous variable was significantly associated with increasing levels of insulin resistance by homeostatic model of insulin resistance (HOMA-IR), after controlling for age, sex, race, body mass index, waist circumference, sleep duration, and enrollment cite [58]. In contrast, non-REM AHI4% was significantly associated with both fasting and post-prandial glucose levels. In a prospective study of obese subjects with type 2 diabetes, increasing quartiles of REM AHI3%a were significantly associated with increasing levels of HbA1c after adjustment for age, sex, BMI, race risk, years of type 2 diabetes, insulin use, and non-REM AHI3%a [59]. The mean adjusted HbA1c increased from 6.3% in subjects with REM AHI <12.3 events/hour (lowest quartile) to 7.3% in subjects with REM AHI >47 events/hour (highest quartile), suggesting a clinically significant effect. Importantly, increasing levels of NON-REM AHI3%a quartiles were not associated with HbA1c. Overall, the development and maintenance of diabetes is likely multifactorial with potentially differential effects of OSA in individual sleep stages, but nonetheless, OSA during REM sleep appears to confer specific risks toward this end point.
Neurocognitive Outcomes and REM OSA
One of the most obvious, immediate, and mostly reversible [60] adverse neurocognitive outcomes associated with OSA is excessive daytime sleepiness (EDS). EDS can be measured subjectively with questionnaires such as the Epworth sleepiness scale and Stanford sleepiness scale and objectively with measurements such as the sleep latency of a multiple sleep latency test (MSLT) or mean reaction time and number of lapses on a psychomotor vigilance test. While sleepiness tends to correlate with overall OSA severity, most studies examining a specific role of REM OSA have found little evidence for association with EDS. In a clinic-based study of 1,146 patients undergoing both polysomnography (PSG) and MSLT, OSA severity was found to explain 11% of the MLST result variance, with non-REM OSA severity explaining 10.8% and REM OSA severity explaining just 6% [61]. In another clinic-based study of 1,821 subjects with both PSG and MSLT, REM OSA severity was not associated with daytime sleep propensity by sleep latency on the MSLT after controlling for age, gender, body mass index, and the duration of non-REM and REM sleep [62]. In contrast, non-REM OSA severity was associated with daytime sleep propensity. In the community-based Sleep Heart Health Study of 5,649 subjects, REM AHI4% was not associated with daytime sleepiness by the Epworth sleepiness score after controlling for demographics, BMI, and non-REMAHI4%, whereas non-REM AHI4% did associate with Epworth sleepiness scores after controlling for demographics, BMI, and REM AHI4% [63]. Finally, in 18 subjects with severe OSA treated chronically with therapeutic CPAP, withdrawal of CPAP exclusively during REM sleep recapitulated severe REM OSA while maintaining normal sleep breathing in non-REM sleep [64]. Following induction of severe OSA in REM sleep, there was no change in mean reaction time or number lapses on morning psychomotor vigilance testing compared to ordinarily consolidated sleep, suggesting no significant increase in sleepiness or alertness.
Studies linking REM OSA to changes in mood have been more mixed. While one recent large clinic-based study of 1,281 individuals with OSA demonstrated an association of REM OSA severity with worsened mood by Beck Depression Inventory scores in men but not women [65], assessment of older men in the MrOS cohort demonstrated no association of REM OSA severity with depression scores on the Geriatric Depression Scale-15 [19]. Similarly, in 142 predominantly male clinic patients, having REM AHI > non-REM AHI was not associated with increased Beck Depression Inventory depression scores [66]. Of note, although REM OSA does not generally appear to be associated with sleepiness, the comorbid presence of depressive symptoms predicted sleepiness assessed by the Epworth sleepiness scale in subjects with REM OSA [67].
Memory processing has long been suspected to be a significant function of sleep, and evidence is building that sleep may impart not only consolidation, but also more complex and nuanced cognitive functions such as rule learning, pattern separation, gist extraction, and even forms of creativity [68, 69]. Because REM sleep differs from non-REM sleep in several crucial features that are important for neural processing, including the neurochemical milieu, the prominent frequencies of cortical field potentials, and the degree of synchrony among cortical regions, the precise mnemonic functions subserved by REM sleep, and their vulnerability to REM OSA, may be unique from those supported by non-REM sleep. While REM sleep has been implicated in the processing of perceptual [70], procedural [71, 72], probabilistic [73], and emotional memory [74–76], effects of REM OSA on these functions has not been investigated.
A role for REM sleep in spatial navigational memory in rodents dates back to at least 1972 [77], and more recently, optogenetic suppression of hippocampal theta rhythm during REM (but not non-REM) impaired spatial object placement learning [78]. These observations raise the possibility that REM OSA in human subjects could impair the processing of spatial navigational information that ordinarily occurs during sleep. In subjects with severe OSA well treated with therapeutic CPAP, performance on a 3D virtual navigation task improved by an average of 30% across normally consolidated sleep. When REM OSA was induced in these same subjects via CPAP withdrawal exclusively during REM sleep, this benefit of sleep was abolished, and in fact subjects’ performance worsened by an average of 5% overnight [64]. Importantly, as noted above, there were no changes in psychomotor vigilance associated with apnea-induced REM sleep disruption, suggesting that any spatial navigation performance deficits were unlikely to be due to any sleepiness or inattention that may have resulted from the intervention.
Finally, it bears noting that sleep disturbances may not only impair cognitive function acutely, but also increase risk in the long term for cognitive dysfunction through neurodegenerative processes including Alzheimer’s disease [79, 80]. A recent intriguing report investigating older subjects from the Sleep Hearth Health Study (average age 67 years) followed longitudinally for an average of 12 years, demonstrated that lower REM sleep percentage and longer REM sleep latency were both associated with a higher risk of incident dementia after controlling for age and sex [81]. Notably, the effect between REM sleep percentage and dementia was reduced after excluding subjects with a high number of arousals from REM sleep due to hypopneas, suggesting a possible contributing role of REM OSA. Additional work will be needed to help clarify the potential role of REM OSA in both memory and risk for neurodegenerative disease.
Nonpharmacological and Pharmacological Intervention for REM OSA
Treatment options for OSA are varied and include variants of positive airway pressure (PAP), oral appliances providing mandibular advancement, positional therapy, hypoglossal nerve stimulator, nasal expiratory positive airway pressure (EPAP), and a variety of upper airway surgeries. PAP is generally viewed as the gold standard treatment, and appears to have the greatest efficacy, particularly in severe REM OSA; however, long-term adherence to PAP is often poor [82]. In clinical practice, 4 hours of nightly CPAP use for 70% of the nights is considered adequate adherence to therapy. This translates into an average CPAP use of 2.8 hours every night. Indeed, it is plausible that reduced CPAP adherence and the predominantly untreated OSA during REM sleep (which prevails during the latter hours of normal nocturnal sleep) may explain the negative or modest effects of CPAP therapy on blood pressure control in randomized clinical trials. Indeed, using CPAP for 3 or 4 h from the time lights are turned off will cover only 25% or 40% of REM sleep, respectively, and will leave most obstructive events during REM sleep untreated [59] (Table 1). As such, in order to effectively treat REM OSA, patients need to use CPAP during most of their sleep period. Given that many patients cannot achieve such high levels of CPAP adherence, it is imperative to explore alternative treatment strategies or even combine less effective therapeutic approaches (e.g. oral appliance plus nasal EPAP) in order to achieve clinical efficacy in lowering REM AHI.
Table 1:
Cumulative minutes of REM and non-REM sleep over 8 h of bedtime. Data are summarized as mean ± SD of cumulative REM and non-REM sleep minutes from lights off to lights on in 115 subjects with type 2 diabetes who underwent in-laboratory polysomnography with 8 hours of total recording time. The mean duration of REM and non-REM sleep in this cohort was 82 and 298 minutes, respectively. Using CPAP for 3 h or 4 h from the time lights are turned off will cover only 25% or 40% of REM sleep, respectively, and will leave most obstructive events during REM sleep untreated. In contrast, 7 h of CPAP use would treat 87% of REM sleep. Data extracted from Grimaldi et al [59].
| Time after lights turned off | Cumulative minutes of non-REM sleep (mean±SD) | Cumulative percentage of non-REM sleep | Cumulative minutes of REM sleep (mean±SD) | Cumulative percentage of REM sleep |
|---|---|---|---|---|
| 1 hour | 38±14 | 13% | 2±6 | 2% |
| 2 hours | 81±18 | 27% | 11±11 | 13% |
| 3 hours | 123±23 | 41% | 20±16 | 25% |
| 4 hours | 161±27 | 54% | 33±20 | 40% |
| 5 hours | 201±31 | 67% | 44±23 | 53% |
| 6 hours | 236±36 | 79% | 58±29 | 71% |
| 7 hours | 272±37 | 91% | 72±32 | 87% |
| 8 hours | 298±39 | 100% | 82±34 | 100% |
Although there are no prospective clinical trials designed to explore treatment strategies in patients with REM OSA, a few studies have reported changes in both REM and non-REM AHI. In a placebo-controlled randomized clinical trial of patients with mostly mild to moderate OSA, one week of nasal EPAP significantly decreased REM AHI from a median of 26.5 events/hour to 8.7 events/hour (p < 0.05) in 97 patients. At three months there was data on 66 participants and the REM AHI with nasal EPAP device off and on was 25.3 events/hour and 11.7 events/hour (p <0.01) [83]. In an observational long-term follow-up study of nasal EPAP, after one year of therapy the REM AHI decreased from 16.8 events/hour to 3.7 events/hour (p <0.001) in 30 participants [84].
Two randomized controlled trials for oral appliances have explored OSA improvement in both REM and non-REM sleep [85, 86]. The first one used a cross-over randomized trial design of 37 patients and found that therapeutic oral appliance decreased the non-REM AHI by an average of 58% whereas REM AHI decreased by 43% (from 34.2±19.4 to 19.3±15.5 events/hour; p=0.01) [85]. The second study randomized patients to either CPAP (n=18), therapeutic oral appliance (n=20), or a sham oral appliance (n=19). CPAP reduced both supine and nonsupine REM AHI by approximately 86% whereas therapeutic oral appliance decreased supine REM AHI by 51% (reduction in supine REM AHI of 12.5±34.8 events/hour from a baseline of 24.6±31.5 events/hour). The nonsupine REM AHI was reduced by 49% (reduction in non-supine REM AHI of 7.5±13.0 events/ hour from a baseline of 24.6±31.5 events/hour) [86]. The limited data from these two studies suggests that oral appliance therapy may be less effective in treating REM OSA than OSA during non-REM sleep.
The hypoglossal nerve stimulator was assessed in 126 carefully selected participants in the STAR randomized clinical trial [87]. After one year of therapy, both REM and non-REM AHI decreased to the same degree. The REM AHI decreased from 28.9±17.4 to 14.7±16.1 events/hour (p<0.0001). In contrast, the non-REM AHI decreased from 32.2±12.6 to 15.3±16.8 events/hour (p <0.0001).
Pharmacotherapy for OSA has consequently been a long sought-after goal, but most attempts at drug treatment of OSA have been ineffective [88]. It has been recently argued that greater success with pharmacotherapy might stem from appropriate OSA phenotyping where distinct physiological drivers of OSA and its repetitive, self-maintaining quality might be identified [89, 90]. In broad strokes, the two main approaches endorsed involve identifying targets that improve the pharyngeal dilator muscle activation and/or upper airway anatomy in general, and identifying targets that reduce the sensitivity of ventilatory control (or raise the arousal threshold). REM OSA is arguably a distinct phenotype of OSA [38], and given the atonia in REM, might be particularly suitable to the former approach. While the natural history of REM OSA is not well studied, approaches that target reducing the number of events in REM sleep would be useful even if an individual eventually converts from having REM OSA to nonspecific OSA.
Early studies suggested histamine increased tonic genioglossus muscle activity across vigilance states [91], but given its role in wake promotion, drugs targeting the histamine system might suffer from off-target wake-promoting effects. A major step forward in the understanding of the molecular mechanisms driving REM atonia of pharyngeal motoneurons was the identification of the requirement for muscarinic receptor-driven activation of G-protein coupled inward rectifying potassium channels (GIRK’s) [1]. Although there are numerous families of neuronal potassium channels [92], GIRK’s have particular biophysical properties, making drug targets that block these channels a feasible possibility. Furthermore, one member of the GIRK family, Kir2.4, is expressed almost exclusively in cranial motor nuclei [93, 94], and thus drug targets of Kir2.4 would offer some level of anatomical specificity. Although specific inhibitors of Kir2.4 remain under development, proof of concept of selectively pharmacologically manipulating cholinergic hypoglossal motoneurons was achieved with a chemogenetic approach using a virally transduced designer receptor exclusively activated by designer drugs (DREADD) [95, 96]. Systemic administration of clozapine-N-oxide, an otherwise biologically inert ligand for DREADD’s, resulted in sustained increases in tongue muscle activity and marked dilation of the pharynx without effect on sleep architecture or diaphragm and postural muscle activity in rodents.
Based on the expression of inhibitory cannabinoid receptors in the vagal nodose ganglion [97] and the theory that dampening vagal input to the medulla might stabilize respiratory pattern generation and raise activation of upper airway dilating muscles during sleep [98], interest in dronabinol, a nonselective agonist of cannabinoid type 1 and 2 receptors, as a potential treatment for OSA has risen. After some initial encouraging results in both rodents [99, 100] and human subjects [101], a fully blinded, parallel groups, placebo-controlled, randomized trial of dronabinol in people with moderate or severe OSA was completed [102]. Dronabinol was found to dose-dependently reduce the AHI3%a by 11–13 events/hour and reduce subjective scores on the Epworth sleepiness scale following 6 weeks of treatment. Of particular note to REM OSA, the effect of dronabinol at the higher dose (10 mg/day) on the REM apnea index was the largest treatment effect size for any event type in any sleep stage after controlling for age, gender, race, ethnicity, and baseline AHI3%a. Furthermore, analysis of treatment responders, arbitrarily defined as those subjects with a final on-treatment AHI3%a of ≤ 15 events/hour plus a reduction from baseline AHI3%a of ≥ 50%, demonstrated that responders had a significantly higher REM AHI3%a and ratio of REM AHI%3a to total AHI3%a versus non-responders. The mean decrease in REM AHI%3a in responders was 33 events/hour while the mean decrease in non-REM AHI3%a in responders was 10 events/hour. These tantalizing observations should prompt a further specific assessment of dronabinol in individuals with REM OSA.
The role of REM-sleep suppressing medications such as tricyclic antidepressants, monoamine oxidase inhibitors, or serotonin/norepinephrine reuptake inhibitors in the management of REM OSA has not been explored in human subjects. That said, combined use of the tricyclic antidepressant trazodone combined with L-tryptophan resulted in a dose-dependent decrease in OSA in an English bulldog model. Obstructive events during REM sleep in particular were reduced by 63% at the highest dose compared to placebo [103].
Conclusions, Knowledge Gaps, and Future Directions
REM OSA is quite prevalent and is associated with adverse cardiovascular, metabolic and neurocognitive outcomes. The literature suggests that obstructive apneas and hypopneas during REM sleep are more toxic than those in non-REM sleep. One ongoing gap in knowledge is whether this is related to the intermittently more severe obstructive events during REM, a fundamental property of REM physiology, or both. Evidence suggesting that, for example, supine OSA severity is associated with adverse health outcomes would at least lend credence to concept that intermittently severe OSA can be harmful. CPAP therapy of 3–4 hours per night may leave the majority of REM OSA untreated. While the concept of “effective AHI” has been explored [104], which estimates an individual’s residual AHI based on their baseline severity and hours of CPAP use, systematic study of timing of CPAP use in relation to sleep onset and offset is lacking. We suggest that this type of research should not only require time series analysis of nightly CPAP use, but also require concomitant actigraphy data in order to ascertain time of sleep onset. Further research is needed to explore novel therapeutic approaches, or combination of currently available non-CPAP therapies, in patients with REM OSA. Moreover, outcomes studies are necessary to demonstrate that effective treatment of REM OSA leads to better patient outcomes. While we await new research, clinicians should recognize the importance of REM OSA severity, even when overall OSA severity is significantly lower, and also emphasize the need for more prolonged CPAP usage in order to cover the second half of the sleep period when REM sleep predominates.
Acknowledgements and Funding:
We thank Ward D. Pettibone for assistance in creation of the figures. A.W.V is supported by the American Sleep Medicine Foundation Junior Faculty Award, an American Thoracic Society Foundation Unrestricted Grant, the Friedman Brain Institute Saint-Amand Award, and NIA awards R01AG056682 and R21AG059179. B.M. is supported by National Institutes of Health grant R01HL119161 and by the Merck Investigator Studies Program. These sponsors had no role in the design or conduct of this research.
Abbreviations
- AHI
Apnea-hypopnea index
- AHI4%
Apnea-hypopnea index using 4% oxygen desaturation criteria
- AHI3%a
Apnea-hypopnea index using 3% oxygen desaturation criteria and/or arousal
- BMI
Body mass index
- CPAP
Continuous positive airway pressure
- DREADD
Designer receptor exclusively activated by designer drugs
- EDS
Excessive daytime sleepiness
- EPAP
Expiratory positive airway pressure
- GIRK
G-protein coupled inward rectifying potassium channels
- MSLT
Multiple sleep latency test
- Non-REM
Non-rapid eye movement sleep
- PSG
Polysomnography
- REM
Rapid eye movement
Footnotes
Conflicts of Interest:
All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Ethical Approval:
For this type of study formal consent is not required.
REFERENCES
- 1.Grace KP, Hughes SW, and Horner RL, Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am J Respir Crit Care Med, 2013;187:311–9. [DOI] [PubMed] [Google Scholar]
- 2.Douglas NJ, White DP, Weil JV, Pickett CK, and Zwillich CW, Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis, 1982;126:758–62. [DOI] [PubMed] [Google Scholar]
- 3.Douglas NJ, White DP, Weil JV, Pickett CK, Martin RJ, Hudgel DW, et al. , Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis, 1982;125:286–9. [DOI] [PubMed] [Google Scholar]
- 4.Findley LJ, Wilhoit SC, and Suratt PM, Apnea duration and hypoxemia during REM sleep in patients with obstructive sleep apnea. Chest, 1985;87:432–6. [DOI] [PubMed] [Google Scholar]
- 5.Krieger J, Sforza E, Boudewijns A, Zamagni M, and Petiau C, Respiratory effort during obstructive sleep apnea: role of age and sleep state. Chest, 1997;112:875–84. [DOI] [PubMed] [Google Scholar]
- 6.Peppard PE, Ward NR, and Morrell MJ, The impact of obesity on oxygen desaturation during sleep-disordered breathing. Am J Respir Crit Care Med, 2009;180:788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brooks D, Horner RL, Kozar LF, Render-Teixeira CL, and Phillipson EA, Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest, 1997;99:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duce B, Kulkas A, Langton C, Toyras J, and Hukins C, The prevalence of REM-related obstructive sleep apnoea is reduced by the AASM 2012 hypopnoea criteria. Sleep Breath, 2017. [DOI] [PubMed]
- 9.Mokhlesi B and Punjabi NM, “REM-related” obstructive sleep apnea: an epiphenomenon or a clinically important entity? Sleep, 2012;35:5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Connor C, Thornley KS, and Hanly PJ, Gender differences in the polysomnographic features of obstructive sleep apnea. Am J Respir Crit Care Med, 2000;161:1465–72. [DOI] [PubMed] [Google Scholar]
- 11.Koo BB, Patel SR, Strohl K, and Hoffstein V, Rapid eye movement-related sleep-disordered breathing: influence of age and gender. Chest, 2008;134:1156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resta O, Carpanano GE, Lacedonia D, Di Gioia G, Giliberti T, Stefano A, et al. , Gender difference in sleep profile of severely obese patients with obstructive sleep apnea (OSA). Respir Med, 2005;99:91–6. [DOI] [PubMed] [Google Scholar]
- 13.Haba-Rubio J, Janssens JP, Rochat T, and Sforza E, Rapid eye movement-related disordered breathing: clinical and polysomnographic features. Chest, 2005;128:3350–7. [DOI] [PubMed] [Google Scholar]
- 14.Koo BB, Dostal J, Ioachimescu O, and Budur K, The effects of gender and age on REM-related sleep-disordered breathing. Sleep Breath, 2008;12:259–64. [DOI] [PubMed] [Google Scholar]
- 15.Conwell W, Patel B, Doeing D, Pamidi S, Knutson KL, Ghods F, et al. , Prevalence, clinical features, and CPAP adherence in REM-related sleep-disordered breathing: a cross-sectional analysis of a large clinical population. Sleep Breath, 2012;16:519–26. [DOI] [PubMed] [Google Scholar]
- 16.Mokhlesi B, Finn LA, Hagen EW, Young T, Hla KM, Van Cauter E, et al. , Obstructive sleep apnea during REM sleep and hypertension. results of the Wisconsin Sleep Cohort. Am J Respir Crit Care Med, 2014;190:1158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aurora RN, Crainiceanu C, Gottlieb DJ, Kim JS, and Punjabi NM, Obstructive Sleep Apnea During Rapid Eye Movement Sleep and Cardiovascular Disease. Am J Respir Crit Care Med, 2017. [DOI] [PMC free article] [PubMed]
- 18.Schutz SJ-L, G.; Rapoport DM; Ayappa I; Varga AW, Rem-related sleep apnea and cardiovascular risk. Sleep, 2016;39:A164–A165. [Google Scholar]
- 19.Khan A, Harrison SL, Kezirian EJ, Ancoli-Israel S, O’Hearn D, Orwoll E, et al. , Obstructive sleep apnea during rapid eye movement sleep, daytime sleepiness, and quality of life in older men in Osteoporotic Fractures in Men (MrOS) Sleep Study. J Clin Sleep Med, 2013;9:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acosta-Castro P, Hirotsu C, Marti-Soler H, Marques-Vidal P, Tobback N, Andries D, et al. , REM-associated sleep apnoea: prevalence and clinical significance in the HypnoLaus cohort. Eur Respir J, 2018. [DOI] [PubMed]
- 21.Dean DA, Wang R, Jacobs DR, Duprez D, Punjabi NM, Zee PC, et al. , A systematic assessment of the association of polysomnographic indices with blood pressure: the Multi-Ethnic Study of Atherosclerosis (MESA). Sleep, 2015;38:587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung J, Whitford EG, Parsons RW, and Hillman DR, Association of sleep apnoea with myocardial infarction in men. Lancet, 1990;336:261–4. [DOI] [PubMed] [Google Scholar]
- 23.Marin JM, Carrizo SJ, Vicente E, and Agusti AG, Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet, 2005;365:1046–53. [DOI] [PubMed] [Google Scholar]
- 24.Arzt M, Young T, Finn L, Skatrud JB, and Bradley TD, Association of sleep-disordered breathing and the occurrence of stroke. Am J Respir Crit Care Med, 2005;172:1447–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redline S, Yenokyan G, Gottlieb DJ, Shahar E, O’Connor GT, Resnick HE, et al. , Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med, 2010;182:269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peppard PE, Young T, Palta M, and Skatrud J, Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med, 2000;342:1378–84. [DOI] [PubMed] [Google Scholar]
- 27.Cano-Pumarega I, Duran-Cantolla J, Aizpuru F, Miranda-Serrano E, Rubio R, Martinez-Null C, et al. , Obstructive sleep apnea and systemic hypertension: longitudinal study in the general population: the Vitoria Sleep Cohort. Am J Respir Crit Care Med, 2011;184:1299–304. [DOI] [PubMed] [Google Scholar]
- 28.Cano-Pumarega I, Barbe F, Esteban A, Martinez-Alonso M, Egea C, Duran-Cantolla J, et al. , Sleep Apnea and Hypertension: Are There Sex Differences? The Vitoria Sleep Cohort. Chest, 2017;152:742–750. [DOI] [PubMed] [Google Scholar]
- 29.O’Connor GT, Caffo B, Newman AB, Quan SF, Rapoport DM, Redline S, et al. , Prospective study of sleep-disordered breathing and hypertension: the Sleep Heart Health Study. Am J Respir Crit Care Med, 2009;179:1159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Somers VK, Dyken ME, Clary MP, and Abboud FM, Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest, 1995;96:1897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somers VK, Dyken ME, Mark AL, and Abboud FM, Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med, 1993;328:303–7. [DOI] [PubMed] [Google Scholar]
- 32.Appleton SL, Vakulin A, Martin SA, Lang CJ, Wittert GA, Taylor AW, et al. , Hypertension Is Associated With Undiagnosed OSA During Rapid Eye Movement Sleep. Chest, 2016;150:495–505. [DOI] [PubMed] [Google Scholar]
- 33.Mokhlesi B, Hagen EW, Finn LA, Hla KM, Carter JR, and Peppard PE, Obstructive sleep apnoea during REM sleep and incident non-dipping of nocturnal blood pressure: a longitudinal analysis of the Wisconsin Sleep Cohort. Thorax, 2015;70:1062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarigianni M, Dimitrakopoulos K, and Tsapas A, Non-dipping status in arterial hypertension: an overview. Curr Vasc Pharmacol, 2014;12:527–36. [DOI] [PubMed] [Google Scholar]
- 35.Cuspidi C, Sala C, Tadic M, Rescaldani M, Grassi G, and Mancia G, Untreated Masked Hypertension and Subclinical Cardiac Damage: A Systematic Review and Meta-analysis. Am J Hypertens, 2015;28:806–13. [DOI] [PubMed] [Google Scholar]
- 36.Jouvet M, Michel F, and Mounier D, [Comparative electroencephalographic analysis of physiological sleep in the cat and in man]. Rev Neurol (Paris), 1960;103:189–205. [PubMed] [Google Scholar]
- 37.Cajochen C, Pischke J, Aeschbach D, and Borbely AA, Heart rate dynamics during human sleep. Physiol Behav, 1994;55:769–74. [DOI] [PubMed] [Google Scholar]
- 38.Zinchuk AV, Jeon S, Koo BB, Yan X, Bravata DM, Qin L, et al. , Polysomnographic phenotypes and their cardiovascular implications in obstructive sleep apnoea. Thorax, 2017. [DOI] [PMC free article] [PubMed]
- 39.Barbe F, Duran-Cantolla J, Sanchez-de-la-Torre M, Martinez-Alonso M, Carmona C, Barcelo A, et al. , Effect of continuous positive airway pressure on the incidence of hypertension and cardiovascular events in nonsleepy patients with obstructive sleep apnea: a randomized controlled trial. JAMA, 2012;307:2161–8. [DOI] [PubMed] [Google Scholar]
- 40.McEvoy RD, Antic NA, Heeley E, Luo Y, Ou Q, Zhang X, et al. , CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea. N Engl J Med, 2016;375:919–31. [DOI] [PubMed] [Google Scholar]
- 41.Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, et al. , A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature, 1996;380:243–7. [DOI] [PubMed] [Google Scholar]
- 42.Guan JL, Uehara K, Lu S, Wang QP, Funahashi H, Sakurai T, et al. , Reciprocal synaptic relationships between orexin- and melanin-concentrating hormone-containing neurons in the rat lateral hypothalamus: a novel circuit implicated in feeding regulation. Int J Obes Relat Metab Disord, 2002;26:1523–32. [DOI] [PubMed] [Google Scholar]
- 43.Tsuneki H, Wada T, and Sasaoka T, Role of orexin in the regulation of glucose homeostasis. Acta Physiol (Oxf), 2010;198:335–48. [DOI] [PubMed] [Google Scholar]
- 44.Vetrivelan R, Kong D, Ferrari LL, Arrigoni E, Madara JC, Bandaru SS, et al. , Melanin-concentrating hormone neurons specifically promote rapid eye movement sleep in mice. Neuroscience, 2016;336:102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alnaji A, Law GR, and Scott EM, The role of sleep duration in diabetes and glucose control. Proc Nutr Soc, 2016;75:512–520. [DOI] [PubMed] [Google Scholar]
- 46.Briancon-Marjollet A, Weiszenstein M, Henri M, Thomas A, Godin-Ribuot D, and Polak J, The impact of sleep disorders on glucose metabolism: endocrine and molecular mechanisms. Diabetol Metab Syndr, 2015;7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stoohs RA, Facchini F, and Guilleminault C, Insulin resistance and sleep-disordered breathing in healthy humans. Am J Respir Crit Care Med, 1996;154:170–4. [DOI] [PubMed] [Google Scholar]
- 48.Lindberg E, Theorell-Haglow J, Svensson M, Gislason T, Berne C, and Janson C, Sleep apnea and glucose metabolism: a long-term follow-up in a community-based sample. Chest, 2012;142:935–942. [DOI] [PubMed] [Google Scholar]
- 49.Reutrakul S and Mokhlesi B, Obstructive Sleep Apnea and Diabetes: A State of the Art Review. Chest, 2017;152:1070–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pamidi S, Wroblewski K, Broussard J, Day A, Hanlon EC, Abraham V, et al. , Obstructive sleep apnea in young lean men: impact on insulin sensitivity and secretion. Diabetes Care, 2012;35:2384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin QC, Zhang XB, Chen GP, Huang DY, Din HB, and Tang AZ, Obstructive sleep apnea syndrome is associated with some components of metabolic syndrome in nonobese adults. Sleep Breath, 2012;16:571–8. [DOI] [PubMed] [Google Scholar]
- 52.Bialasiewicz P, Pawlowski M, Nowak D, Loba J, and Czupryniak L, Decreasing concentration of interstitial glucose in REM sleep in subjects with normal glucose tolerance. Diabet Med, 2009;26:339–44. [DOI] [PubMed] [Google Scholar]
- 53.Thakkar M and Mallick BN, Rapid eye movement sleep-deprivation-induced changes in glucose metabolic enzymes in rat brain. Sleep, 1993;16:691–4. [PubMed] [Google Scholar]
- 54.Xu J, Long YS, Gozal D, and Epstein PN, Beta-cell death and proliferation after intermittent hypoxia: role of oxidative stress. Free Radic Biol Med, 2009;46:783–90. [DOI] [PubMed] [Google Scholar]
- 55.Wang N, Khan SA, Prabhakar NR, and Nanduri J, Impairment of pancreatic beta-cell function by chronic intermittent hypoxia. Exp Physiol, 2013;98:1376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shpirer I, Rapoport MJ, Stav D, and Elizur A, Normal and elevated HbA1C levels correlate with severity of hypoxemia in patients with obstructive sleep apnea and decrease following CPAP treatment. Sleep Breath, 2012;16:461–6. [DOI] [PubMed] [Google Scholar]
- 57.Bialasiewicz P, Czupryniak L, Pawlowski M, and Nowak D, Sleep disordered breathing in REM sleep reverses the downward trend in glucose concentration. Sleep Med, 2011;12:76–82. [DOI] [PubMed] [Google Scholar]
- 58.Chami HA, Gottlieb DJ, Redline S, and Punjabi NM, Association between Glucose Metabolism and Sleep-disordered Breathing during REM Sleep. Am J Respir Crit Care Med, 2015;192:1118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grimaldi D, Beccuti G, Touma C, Van Cauter E, and Mokhlesi B, Association of obstructive sleep apnea in rapid eye movement sleep with reduced glycemic control in type 2 diabetes: therapeutic implications. Diabetes Care, 2014;37:355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young LR, Taxin ZH, Norman RG, Walsleben JA, Rapoport DM, and Ayappa I, Response to CPAP withdrawal in patients with mild versus severe obstructive sleep apnea/hypopnea syndrome. Sleep, 2013;36:405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chervin RD and Aldrich MS, The relation between multiple sleep latency test findings and the frequency of apneic events in REM and non-REM sleep. Chest, 1998;113:980–4. [DOI] [PubMed] [Google Scholar]
- 62.Punjabi NM, Bandeen-Roche K, Marx JJ, Neubauer DN, Smith PL, and Schwartz AR, The association between daytime sleepiness and sleep-disordered breathing in NREM and REM sleep. Sleep, 2002;25:307–14. [PubMed] [Google Scholar]
- 63.Chami HA, Baldwin CM, Silverman A, Zhang Y, Rapoport D, Punjabi NM, et al. , Sleepiness, quality of life, and sleep maintenance in REM versus non-REM sleep-disordered breathing. Am J Respir Crit Care Med, 2010;181:997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Varga AW, Kishi A, Mantua J, Lim J, Koushyk V, Leibert DP, et al. , Apnea-induced rapid eye movement sleep disruption impairs human spatial navigational memory. J Neurosci, 2014;34:14571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SA, Paek JH, and Han SH, REM-related sleep-disordered breathing is associated with depressive symptoms in men but not in women. Sleep Breath, 2016;20:995–1002. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y, Su C, Liu R, Lei G, Zhang W, Yang T, et al. , NREM-AHI greater than REM-AHI versus REM-AHI greater than NREM-AHI in patients with obstructive sleep apnea: clinical and polysomnographic features. Sleep Breath, 2011;15:463–70. [DOI] [PubMed] [Google Scholar]
- 67.Pamidi S, Knutson KL, Ghods F, and Mokhlesi B, Depressive symptoms and obesity as predictors of sleepiness and quality of life in patients with REM-related obstructive sleep apnea: cross-sectional analysis of a large clinical population. Sleep Med, 2011;12:827–31. [DOI] [PubMed] [Google Scholar]
- 68.Stickgold R and Walker MP, Sleep-dependent memory triage: evolving generalization through selective processing. Nat Neurosci, 2013;16:139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai DJ, Mednick SA, Harrison EM, Kanady JC, and Mednick SC, REM, not incubation, improves creativity by priming associative networks. Proc Natl Acad Sci U S A, 2009;106:10130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karni A, Tanne D, Rubenstein BS, Askenasy JJ, and Sagi D, Dependence on REM sleep of overnight improvement of a perceptual skill. Science, 1994;265:679–82. [DOI] [PubMed] [Google Scholar]
- 71.Plihal W and Born J, Effects of early and late nocturnal sleep on declarative and procedural memory. J Cogn Neurosci, 1997;9:534–47. [DOI] [PubMed] [Google Scholar]
- 72.Rasch B, Gais S, and Born J, Impaired off-line consolidation of motor memories after combined blockade of cholinergic receptors during REM sleep-rich sleep. Neuropsychopharmacology, 2009;34:1843–53. [DOI] [PubMed] [Google Scholar]
- 73.Barsky MM, Tucker MA, and Stickgold R, REM sleep enhancement of probabilistic classification learning is sensitive to subsequent interference. Neurobiol Learn Mem, 2015;122:63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nishida M, Pearsall J, Buckner RL, and Walker MP, REM sleep, prefrontal theta, and the consolidation of human emotional memory. Cereb Cortex, 2009;19:1158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van der Helm E, Yao J, Dutt S, Rao V, Saletin JM, and Walker MP, REM sleep depotentiates amygdala activity to previous emotional experiences. Curr Biol, 2011;21:2029–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van der Helm E and Walker MP, Sleep and Emotional Memory Processing. Sleep Med Clin, 2011;6:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sloan MA, The effects of deprivation of rapid eye movement (REM) sleep on maze learning and aggression in the albino rat. J Psychiatr Res, 1972;9:101–11. [DOI] [PubMed] [Google Scholar]
- 78.Boyce R, Glasgow SD, Williams S, and Adamantidis A, Causal evidence for the role of REM sleep theta rhythm in contextual memory consolidation. Science, 2016;352:812–6. [DOI] [PubMed] [Google Scholar]
- 79.Cedernaes J, Osorio RS, Varga AW, Kam K, Schioth HB, and Benedict C, Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer’s disease. Sleep Med Rev, 2017;31:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma RA, Varga AW, Bubu OM, Pirraglia E, Kam K, Parekh A, et al. , Obstructive Sleep Apnea Severity Affects Amyloid Burden in Cognitively Normal Elderly: A Longitudinal Study. Am J Respir Crit Care Med, 2017. [DOI] [PMC free article] [PubMed]
- 81.Pase MP, Himali JJ, Grima NA, Beiser AS, Satizabal CL, Aparicio HJ, et al. , Sleep architecture and the risk of incident dementia in the community. Neurology, 2017;89:1244–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lettieri CJ, Williams SG, Collen JF, and Wickwire EM, Treatment of Obstructive Sleep Apnea: Achieving Adherence to Positive Airway Pressure Treatment and Dealing with Complications. Sleep Med Clin, 2017;12:551–564. [DOI] [PubMed] [Google Scholar]
- 83.Berry RB, Kryger MH, and Massie CA, A novel nasal expiratory positive airway pressure (EPAP) device for the treatment of obstructive sleep apnea: a randomized controlled trial. Sleep, 2011;34:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kryger MH, Berry RB, and Massie CA, Long-term use of a nasal expiratory positive airway pressure (EPAP) device as a treatment for obstructive sleep apnea (OSA). J Clin Sleep Med, 2011;7:449–53B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Naismith SL, Winter VR, Hickie IB, and Cistulli PA, Effect of oral appliance therapy on neurobehavioral functioning in obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med, 2005;1:374–80. [PubMed] [Google Scholar]
- 86.Aarab G, Lobbezoo F, Hamburger HL, and Naeije M, Oral appliance therapy versus nasal continuous positive airway pressure in obstructive sleep apnea: a randomized, placebo-controlled trial. Respiration, 2011;81:411–9. [DOI] [PubMed] [Google Scholar]
- 87.Strollo PJ Jr., Soose RJ, Maurer JT, de Vries N, Cornelius J, Froymovich O, et al. , Upper airway stimulation for obstructive sleep apnea. N Engl J Med, 2014;370:139–49. [DOI] [PubMed] [Google Scholar]
- 88.Mason M, Welsh EJ, and Smith I, Drug therapy for obstructive sleep apnoea in adults. Cochrane Database Syst Rev, 2013:CD003002. [DOI] [PMC free article] [PubMed]
- 89.Horner RL, Grace KP, and Wellman A, A resource of potential drug targets and strategic decision-making for obstructive sleep apnoea pharmacotherapy. Respirology, 2017;22:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sands SA, Edwards BA, Terrill PI, Taranto-Montemurro L, Azarbarzin A, Marques M, et al. , Phenotyping Pharyngeal Pathophysiology Using Polysomnography in Patients with Obstructive Sleep Apnea. Am J Respir Crit Care Med, 2018. [DOI] [PMC free article] [PubMed]
- 91.Bastedo T, Chan E, Park E, Liu H, and Horner RL, Modulation of genioglossus muscle activity across sleep-wake states by histamine at the hypoglossal motor pool. Sleep, 2009;32:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Trimmer JS, Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron, 2015;85:238–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grace KP, Hughes SW, Shahabi S, and Horner RL, K+ channel modulation causes genioglossus inhibition in REM sleep and is a strategy for reactivation. Respir Physiol Neurobiol, 2013;188:277–88. [DOI] [PubMed] [Google Scholar]
- 94.Topert C, Doring F, Wischmeyer E, Karschin C, Brockhaus J, Ballanyi K, et al. , Kir2.4: a novel K+ inward rectifier channel associated with motoneurons of cranial nerve nuclei. J Neurosci, 1998;18:4096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fleury Curado T., Fishbein K, Pho H, Brennick M, Dergacheva O, Sennes LU, et al. , Chemogenetic stimulation of the hypoglossal neurons improves upper airway patency. Sci Rep, 2017;7:44392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Horton GA, Fraigne JJ, Torontali ZA, Snow MB, Lapierre JL, Liu H, et al. , Activation of the Hypoglossal to Tongue Musculature Motor Pathway by Remote Control. Sci Rep, 2017;7:45860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burdyga G, Lal S, Varro A, Dimaline R, Thompson DG, and Dockray GJ, Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J Neurosci, 2004;24:2708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carley DW and Radulovacki M, Pharmacology of vagal afferent influences on disordered breathing during sleep. Respir Physiol Neurobiol, 2008;164:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carley DW, Paviovic S, Janelidze M, and Radulovacki M, Functional role for cannabinoids in respiratory stability during sleep. Sleep, 2002;25:391–8. [PubMed] [Google Scholar]
- 100.Calik MW, Radulovacki M, and Carley DW, Intranodose ganglion injections of dronabinol attenuate serotonin-induced apnea in Sprague-Dawley rat. Respir Physiol Neurobiol, 2014;190:20–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Prasad B, Radulovacki MG, and Carley DW, Proof of concept trial of dronabinol in obstructive sleep apnea. Front Psychiatry, 2013;4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carley DW, Prasad B, Reid KJ, Malkani R, Attarian H, Abbott SM, et al. , Pharmacotherapy of Apnea by Cannabimimetic Enhancement, the PACE Clinical Trial: Effects of Dronabinol in Obstructive Sleep Apnea. Sleep, 2018;41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Veasey SC, Fenik P, Panckeri K, Pack AI, and Hendricks JC, The effects of trazodone with L-tryptophan on sleep-disordered breathing in the English bulldog. Am J Respir Crit Care Med, 1999;160:1659–67. [DOI] [PubMed] [Google Scholar]
- 104.Boyd SB, Upender R, Walters AS, Goodpaster RL, Stanley JJ, Wang L, et al. , Effective Apnea-Hypopnea Index (“Effective AHI”): A New Measure of Effectiveness for Positive Airway Pressure Therapy. Sleep, 2016;39:1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]