

Abstract
Tumor mutational burden correlates with improved survival and immunotherapy response in some malignancies, and with tumor aggressiveness in others. To study the link between mutational burden and survival, we analyzed survival effects of tumor exonic missense mutation burden (TEMMB) across 6947 specimens spanning 31 cancers which have undergone whole exome sequencing as part of TCGA. We adjusted TEMMB for age, sex, stage, and recruitment center, and computed Cox-proportional models of TEMMB survival effects. We assigned a recurrence score (RS) to each cohort, defining RS as the burden of recurrent mutations exceeding 1% population prevalence. High TEMMB was associated with improved survival in cutaneous melanoma: hazard ratio (HR) = 0.71 [0.60–0.85], p = 0.0002, urothelial bladder carcinoma: HR = 0.74 [0.59–0.93], p = 0.01, and ovarian carcinoma: HR = 0.80 [0.70–0.93], p = 0.003. High TEMMB was associated with decreased survival in colorectal adenocarcinoma: HR = 1.32 [1.00–1.74], p < 0.05. We identified that TEMMB survival effects were governed by the balance of recurrent and non-recurrent mutations. In cancers with a low RS, high TEMMB was correlated with better survival outcomes (r = 0.49, p = 0.02). In conclusion, TEMMB effects on survival depend on recurrent mutation enrichment; tumor types that are highly enriched in passenger mutations show a survival benefit in the setting of high tumor mutational burden.
Introduction
Tumor mutational burden has been described as a predictor of tumor behavior and immunological response1–3. At its core, mutation formation promotes carcinogenesis via activation or inactivation of genes and associated pathways, thus generating novel peptide sequences which can stimulate immune response. High mutational burden may in some cases represent a high underlying number of drivers, and indicate a higher-risk tumor: for example patients with high mutational burden lung adenocarcinoma tumors showed a 14-month survival decrease4, supporting that high mutation burden may be a harbinger of poor clinical outcomes. Alternatively, highly mutated tumors may develop many novel peptides and thus display more neoantigens, rendering them more susceptible T-cell targets5. For example, patients with melanomas with a high mutational load showed improved survival with ipilimumab6 and improved overall survival7; patients with highly mutated ovarian cancer had improved postoperative chemotherapy response and higher overall survival2.
Here, we systematically analyzed mutational burden survival effects across multiple cancer types. We hypothesized that tumor exonic missense mutational burden (TEMMB) is predictive of underlying total exonic mutational burden (TEMB), and that TEMMB is independent of critical demographic and tumor-specific factors. Furthermore, we hypothesized that TEMMB is a predictive marker of tumor immune surveillance and clinical outcomes. We sought to test these hypotheses, and to describe the potential genetic underpinnings for the impact of TEMMB on survival. We focused on somatic missense mutation burden in subsequent analyses. Missense mutations represent the most common observed oncogenic variants8, and are known to alter sequences of expressed transcripts and thus lead to downstream translation of mutated proteins9. Furthermore, missense variants specifically have been suggested to be the most frequent class of alterations to carry the potential for neoepitope generation in chronic lymphocytic leukemia malignancy (as compared to frameshift or splice-site variants)10. In multiple myeloma, missense mutational load was found to be highly correlated with predicted neoantigen loads11. Missense mutations produce specific amino acid changes in a known pattern, allowing for a systematic way to characterize mutational profiles by defining recurrent and non-recurrent mutations.
Results
Tumor missense mutational burden (TEMMB) variability among cancers
Total missense mutational burden across all cohorts ranged from a low of 8 (median) missense mutations among acute myeloid leukemia (LAML) and thymoma (THYM), to 256 median mutations among the skin cutaneous melanoma (SKCM) cohort (Fig. S1). 10 individuals were removed as TEMMB outliers (Fig. S2). Total (TEMB) and missense (TEMMB) tumor exonic mutational burden were found to be closely correlated among all cohorts: Pearson’s r ranged from 0.95–1.00 for all cohorts other than uveal melanoma (UVM) which also revealed a strong positive correlation with r = 0.88 likely due to a small (N = 79) sample size (p < 2.2 × 10−16 for all cohorts) (Fig. S3).
TEMMB relations to age, sex and tumor stage
Increasing patient age was significantly correlated with high TEMMB among 17 of 31 (55%) cohorts (Table 1). Male sex was significantly associated with high TEMMB in renal papillary cell carcinoma (KIRP), sarcoma (SARC), and cutaneous melanoma (SKCM). Female sex was significantly associated with high TEMMB in colorectal adenocarcinoma (COAD) and glioblastoma multiforme (GBM). High tumor stage (Stage III and above) was observed to be significantly associated with both high TEMMB in 3 cohorts and low TEMMB in 7 cohorts.
Table 1.
Contributions of age, sex, and tumor stage to tumor exonic missense mutational burden (TEMMB). Each model was additionally adjusted by recruitment center (IRR and p-values not shown).
| Age | Sex | Tumor Stage | ||||||
|---|---|---|---|---|---|---|---|---|
| Age, yrs (IRRa) | p b | Fem (ref) | Male (IRRa) | p b | I-II (ref) | III + (IRRa) | p b | |
| ACC | 1.02 [1.01–1.03] | <0.05/31** | 1 | 0.78 [0.57–1.09] | 0.14 | 1 | 1.35 [0.99–1.85] | 0.06 |
| BLCA | 1.00 [0.99–1.02] | 0.56 | 1 | 1.30 [0.95–1.76] | 0.08 | 1 | 1.37 [1.03–1.80] | <0.05* |
| BRCA | 1.01 [1.01–1.02] | <0.05/31** | 1 | 0.64 [0.35–1.33] | 0.19 | 1 | 0.78 [0.67–0.91] | <0.05/24** |
| CESC | 1.02 [1.01–1.03] | <0.05/31** | 1 | — | — | 1 | 1.09 [0.76–1.61] | 0.63 |
| CHOL | 1.01 [1.00–1.01] | 0.07 | 1 | 0.93 [0.80–1.08] | 0.32 | 1 | 0.81 [0.68–0.97] | <0.05* |
| COAD | 1.00 [0.98–1.01] | 0.76 | 1 | 0.57 [0.39–0.83] | <0.05* | 1 | 0.50 [0.34–0.74] | <0.05/24** |
| DLBC | 1.00 [0.99–1.02] | 0.70 | 1 | 1.26 [0.80–1.97] | 0.31 | 1 | 0.57 [0.33–1.01] | <0.05* |
| ESCA | 1.01 [1.00–1.02] | <0.05* | 1 | 0.89 [0.71–1.11] | 0.32 | 1 | 0.99 [0.83–1.19] | 0.95 |
| GBM | 1.01 [1.00–1.01] | <0.05/31** | 1 | 0.87 [0.80–0.94] | <0.05/26** | 1 | — | — |
| HNSC | 1.01 [1.01–1.02] | <0.05/31** | 1 | 1.04 [0.85–1.27] | 0.69 | 1 | 1.00 [0.80–1.23] | 0.98 |
| KICH | 1.00 [1.00–1.01] | 0.09 | 1 | 0.93 [0.79–1.09] | 0.37 | 1 | 1.25 [1.06–1.47] | <0.05* |
| KIRC | 1.01 [1.01–1.02] | <0.05/31** | 1 | 1.04 [0.91–1.18] | 0.57 | 1 | 1.01 [0.89–1.16] | 0.87 |
| KIRP | 1.01 [1.01–1.02] | <0.05/31** | 1 | 1.20 [1.07–1.34] | <0.05/26** | 1 | 1.11 [1.00–1.25] | 0.06 |
| LAML | 1.01 [1.00–1.02] | <0.05/31** | 1 | 1.11 [0.91–1.36] | 0.30 | 1 | — | — |
| LGG | 1.02 [1.02–1.03] | <0.05/31** | 1 | 0.94 [0.85–1.04] | 0.21 | 1 | — | — |
| LIHC | 1.00 [1.00–1.01] | 0.24 | 1 | 1.06 [0.86–1.29] | 0.58 | 1 | 0.94 [0.76–1.17] | 0.59 |
| LUAD | 1.00 [0.99–1.01] | 0.99 | 1 | 1.18 [0.92–1.50] | 0.19 | 1 | 0.81 [0.62–1.07] | 0.13 |
| LUSC | 0.99 [0.98–1.01] | 0.35 | 1 | 1.04 [0.83–1.28] | 0.75 | 1 | 1.01 [0.80–1.29] | 0.91 |
| OV | 1.01 [1.00–1.01] | <0.05/31** | 1 | — | — | 1 | 0.67 [0.50–0.89] | <0.05* |
| PAAD | 1.00 [0.99–1.01] | 0.92 | 1 | 1.11 [0.97–1.27] | 0.14 | 1 | 1.11 [0.82–1.53] | 0.51 |
| PCPG | 1.01 [1.01–1.02] | <0.05/31** | 1 | 1.06 [0.93–1.22] | 0.38 | 1 | — | — |
| PRAD | 1.00 [0.99–1.01] | 0.84 | 1 | — | — | 1 | — | — |
| READ | 0.98 [0.96–1.00] | 0.05 | 1 | 0.92 [0.61–1.38] | 0.68 | 1 | 0.56 [0.37–0.85] | <0.05* |
| SARC | 1.02 [1.01–1.03] | <0.05/31** | 1 | 1.43 [1.17–1.75] | <0.05/26** | 1 | — | — |
| SKCM | 1.01 [1.00–1.02] | <0.05* | 1 | 1.39 [1.11–1.72] | <0.05* | 1 | 0.93 [0.74–1.17] | 0.51 |
| STAD | 1.02 [1.00–1.03] | <0.05* | 1 | 0.95 [0.68–1.32] | 0.72 | 1 | 0.95 [0.69–1.30] | 0.72 |
| THCA | 1.01 [1.01–1.02] | <0.05/31** | 1 | 0.96 [0.85–1.08] | 0.49 | 1 | 1.14 [1.00–1.30] | <0.05* |
| THYM | 1.03 [1.02–1.05] | <0.05/31** | 1 | 1.19 [0.85–1.66] | 0.31 | 1 | — | — |
| UCEC | 0.98 [0.96–0.99] | 0.14 | 1 | — | — | 1 | 0.79 [0.47–1.37] | 0.31 |
| UCS | 1.00 [0.98–1.02] | 0.89 | 1 | — | — | 1 | 1.20 [0.88–1.63] | 0.21 |
| UVM | 1.00 [1.00–1.01] | 0.43 | 1 | 1.13 [0.96–1.34] | 0.14 | 1 | 0.81 [0.67–0.98] | <0.05** |
Older age correlated with high TEMMB in 17 of 31 cancers studied. Effects of male sex and high tumor stage (defined as Stage III or greater) were variable.
aIRR: Incidence rate ratio calculated with multivariate binomial regression, reported with 95% confidence intervals.
bp-values displayed with conventional 0.05 significance cutoff and with cutoff using Bonferroni correction for multiple comparisons (n = 31, 26, 24 for age, sex, stage respectively).
Melanoma, ovarian carcinoma, and bladder carcinoma benefit from high mutational load
Following multivariate adjustment for age, sex, stage, and patient recruitment center and exclusion of seven cohorts with a low number of non-censored events, TEMMB was found to be significantly correlated with survival in 4 of 24 TCGA cohorts (Fig. 1). High TEMMB correlated with improved survival in skin cutaneous melanoma (SKCM): HR = 0.71 [0.60–0.85], p = 0.0002, bladder urothelial carcinoma (BLCA): HR = 0.74 [0.59–0.93], p = 0.01, and ovarian carcinoma (OV): HR = 0.80 [0.70–0.93], p = 0.003. High TEMMB was associated with decreased survival in colorectal adenocarcinoma (COAD): HR = 1.32 [1.00–1.74], p < 0.05 (p = 0.0497). Following Bonferroni adjustment of α = 0.05 for 24 comparisons, yielding a p-value cutoff of α/24 = 0.0021, only cutaneous melanoma retained a significant correlation between TEMMB and survival.
Figure 1.

Survival effects of tumor exonic missense mutational burden (TEMMB). Survival is expressed as hazard ratio (HR) per effective multivariate-adjusted TEMMB. Raw two-sided Wald-test p-values are reported with *indicating p < 0.05 and **indicating Bonferroni-adjusted significance for 24 multiple comparisons. Ovarian carcinoma, cutaneous melanoma, bladder carcinoma, and colorectal adenocarcinoma showed significant survival benefit with high TEMMB.
Relative burden of recurrent and non-recurrent mutations expressed with recurrence score (RS)
To characterize the somatic mutational profile of each cancer, we determined the relative burden of recurrent mutations to total mutations within each cohort, expressed as a recurrence score (RS). Recurrent mutations were defined as specific amino acid changes observed among greater than 1% of each cohort’s population. Mutational profiles, and thus RS, varied significantly between distinct cancers (Fig. S4). Several cohorts, notably adrenocortical carcinoma (ACC), acute myeloid leukemia (LAML), brain lower grade glioma (LGG), pheochromocytoma and paraganglioma (PCPG), thyroid carcinoma (THCA), thymoma (THYM), and uveal melanoma (UVM), revealed mutations occurring at high prevalence among the sequenced population. The recurrent mutations can be readily visualized as sharp peaks in the cancers’ mutational profiles. Such cohorts were found to have high recurrence scores (RS). Other cohorts, such as skin cutaneous melanoma (SKCM) and ovarian carcinoma (OV), displayed mutational profiles with fewer pronounced recurring mutations (sharp peaks). These cohorts carried a higher enrichment of non-recurrent mutations, and thus were found to have lower RS (Fig. 2).
Figure 2.

Recurrence scores (RS) of all cancer cohorts, calculated as the fraction of recurrent missense mutations to total missense mutations in the pool. Recurrent mutations were defined as those which exceeded 1% prevalence in the cohort.
We catalogued the specific tumor mutations observed among these cohorts displaying a highly-recurrent mutational landscape. In the adrenocortical carcinoma (ACC) cohort, 0.29% of all pooled missense mutations were in the ZNF517 gene (p.V349A), and 0.29% of missense mutations were recurrent GARS (p.P42A) mutations. In uveal melanoma (UVM) cohort, 2.54% were recurrent GNA11 (p.Q209P) mutations, 2.01% were recurrent GNAQ p.Q209P, and 0.75% GNAQ p.Q1209L. In thyroid carcinoma (THCA), 5.23% were BRAF p.V600E, 0.65% were NRAS p.Q61R, and 0.25% HRAS p.Q61R. In the acute myeloid leukemia (LAML) cohort, 1.38% of missense mutations were in DNMT3A gene (p.R882H), 1.05% were IDH2 p.R140Q, and 0.79% were IDH1 p.R132C. In pheochromocytoma and paraganglioma (PCPG), 0.64% of mutations were recurrent HRAS p.Q61R, and 0.36% were CHEK2 p.K152E.
Cancers with low recurrence scores (RS) show survival benefit from high TEMMB
We identified a significant positive correlation (r = 0.49, p = 0.016) among all cancer cohorts between the survival effect, or Hazard Ratio (HR), of adjusted-TEMMB and cancer recurrence score (RS) (Fig. 3). Cancers with low RS tended to exhibit survival benefit (HR < 1) with increased adjusted-TEMMB. Conversely, cancers with high RS were observed to have a decrease in survival (HR > 1) with increased adjusted-TEMMB. Testing an alternate recurrence cutoff of 5% (traditional cutoff for minor allele frequency) confirmed a significant positive correlation: r = 0.66, p = 0.002 (Table S1).
Figure 3.
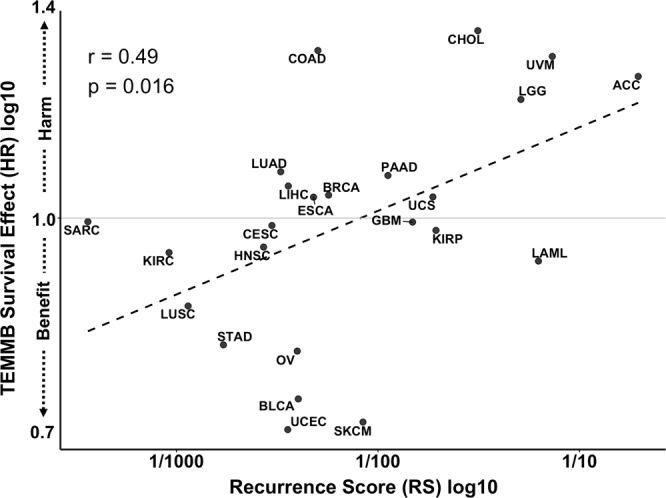
Correlation of log-adjusted mutational burden survival Hazard Ratios (HR) with cohorts’ log-adjusted recurrence scores (RS). Cancers with high recurrent mutation enrichment showed survival harm with increasing TEMMB, while tumors with low recurrent missense mutation burden tended to show survival benefit (r = 0.49, p = 0.016).
Discussion
Exonic missense mutation distribution displays considerable variability among cancers studied in TCGA. We identified cutaneous melanoma and lung squamous cell carcinoma as the tumors with the highest TEMMB, and acute myeloid leukemia and thyroid carcinoma as among the lowest. These results were consistent with previously-reported mutational burden distribution12. Somatic missense mutations strongly contribute to the generation of novel tumor epitopes. Understanding whether a more highly-immunogenic tumor carries a direct link to mutational burden could provide a mechanistic explanation for observed clinical survival patterns. In our results, TEMMB was closely correlated with TEMB among all TCGA cohorts, supporting TEMMB’s role as a robust proxy for TEMB.
Exonic missense mutational burden showed strong consistent positive association with age, supporting current understanding of human mutagenesis. While age-related mutagenesis rates do vary between individuals and tissue types, a consistent positive correlation between mutational load and age has been supported by animal and human research13–18. Several “clock-like” mutational signatures may be contributory to this chronological mutagenesis phenomenon19.
Interestingly, low tumor stage was correlated (after Bonferroni adjustment) with high TEMMB in breast carcinoma, colon and rectal adenocarcinoma, and uveal melanoma. Chromosomal and microsatellite instability (MSI) are observed in early stages of adenomas, and significant chromosomal instability has been proposed as an underlying feature present prior to malignant transformation20–22. Low-stage adenocarcinoma tumors may thus carry higher mutational loads due to the pronounced underlying genomic instability. Although the role of immune therapy is not yet strongly established in colorectal cancer (CRC), the immune tumor microenvironment in CRC is an important factor in disease progression23,24. Likewise, breast carcinogenesis has been proposed to be regulated by innate and adaptive inflammatory responses25. It is possible that during progression towards high-stage adenocarcinoma tumors in breast and colorectal cancers, highly-immunogenic or high-TEMMB cells are cleared through immune targeting and elimination, thus selecting for a population of low-TEMMB cells with low neoantigen loads. Uveal melanoma has a low mutational burden which has been suggested as a possible reason for low success of immunotherapy in its treatment26. Given the high propensity for rapid metastasis in uveal melanoma, it is possible that intercepting such tumors at an early stage may partially be explained by a higher mutational load and thus more favorable immune response.
Driver mutations impart tumor growth advantage and are positively selected in cancer evolution, while biologically inert passengers accumulate without directional selection over the tumor growth timespan27. Many established bioinformatics methods to study drivers rely on techniques that identify recurrent mutations28, and thus we quantified recurrent and non-recurrent mutations to serve as proxy for relative amounts of drivers and passengers within a cancer type. Our results suggest high TEMMB tends to confer survival benefit in cancers with more non-recurrent (likely passenger) mutations, and decreased survival in cancers with high recurrent (likely driver) fractions. We propose that in malignancies with large enrichments of non-recurrent mutations, high TEMMB marks a high passenger count, and increasing passenger mutation load increases neoantigen presentation29 without imparting additional growth advantage or aggressiveness. Our observed benefit with high TEMMB supports literature findings for melanoma6 and ovarian carcinoma2. In cases of malignancies with higher relative amounts of recurrent or driver mutations, for instance in adrenocortical carcinoma (ACC), uveal melanoma (UVM), and brain lower grade glioma (LGG), high mutational burden correlates with increased drivers of aggressiveness and invasion. In our study, increasing TEMMB showed a trend towards survival harm in these highly somatically-recurrent tumors.
Recent work has suggested a “double-edged” effect of increased DNA variants, noting that on the one hand, high DNA variation increases accumulation of drivers which are beneficial to tumor adaptation; conversely, high concurrent passenger loads may outweigh the driver effects30. Our results suggest a model for improved understanding of the variable manifestations of this molecular tug-of-war among a variety of cancer types. We found the underlying mutational landscape of DNA changes to be quite variable among malignancies documented in TCGA. A group of cancers such as adrenocortical carcinoma, uveal melanoma, and brain glioma emerged as a “driver-enriched” class, while a second group – including cutaneous melanoma and ovarian carcinoma – emerged as a “passenger-enriched” class. Increasing DNA variation in these two classes, quantified as TEMMB, yielded opposing survival effects. Our findings highlight TEMMB as an independent survival biomarker with potential utility for risk-stratification and identification of those patients who may benefit from immunotherapy. Classification of malignancies into driver- or passenger-rich classes may also aid in identifying suitable candidate cancers for immune therapy trials.
The study was limited by the following factors: first, TCGA describes exome sequences, and thus mutations in noncoding regions could not be analyzed. Thus, TEMMB reflects specifically the exonic mutational burden rather the full genome scale. It is possible that non-coding DNA contributes significantly to survival, and further study with comprehensive full genome sequencing may help elucidate such effects. Second, details of therapy and treatment course were available not for all patients, and thus we were unable to systematically study effect modification and confounding by treatment differences. Third, in-silico findings are important for discovery of novel relationships and insights in tumor biology, but in-vivo studies are required to further analyze mechanisms by which TEMMB affects tumor immune surveillance, metabolic, and growth properties. Future work will focus on analysis of immunological mechanisms responsible for clearing high-TEMMB tumors with a low enrichment of recurrent mutations. Lastly, the study is also significantly limited by a lack of controlled population-based recruitment among the TCGA cohorts. We adjusted TEMMB to account for recruitment center to partially address this limitation. However, future work would benefit from a study with more clearly and regularly ascertained cohorts.
Our overall analyses suggest that positive and negative TEMMB effects on survival may depend on the enrichment of underlying recurrent mutations. Cancers with higher proportions of non-recurrent and thus likely passenger mutations showed survival benefit with high TEMMB, while cancers with higher recurrent mutation fractions (likely drivers) revealed a decrease in survival. Mutational signatures for some cancers might contribute significantly to overall TEMMB (e.g. UV-signature in the cutaneous melanoma cohort), thus, in part environmental effects contribute to the TEMMB survival effect. These findings highlight the relationship of tumor mutational burden to driver and passenger effects. Understanding how tumor mutational burden correlates with clinical outcomes for certain classes of malignancies will help guide clinical decisions regarding TEMMB as a useful biomarker for predicting survival and response to immunotherapy.
Methods
R statistical language (Version 3.4.4)31 with ‘RTCGAToolbox’32, ‘MASS’33, ‘survminer’34, ‘forestplot’35 were used for analysis and plotting. We obtained somatic mutation and clinical data for 31 cancer cohorts in The Cancer Genome Atlas (TCGA). 6947 individuals had available mutation data; 6717 of the set had complete clinical data on age, sex, and stage; 2113 patients were deceased and had available time-to-death survival data.
We examined individuals with maximum TEMMB value in each cohort, excluding those with TEMMB greater than triple of the next largest TEMMB value. As an initial quality control (QC) step, 10 (0.1% of total) samples were excluded as outliers potentially representing technical batch effects in tumor DNA analysis. Pearson’s correlation was used to examine the relationship between TEMMB and TEMB across all cohorts. We then analyzed the relationship between TEMMB and patients’ clinical factors. Negative binomial regression was used to model TEMMB as a function of age (continuous variable: “years”), sex (categorical variable: “male” and “female”), tumor stage (categorical variable: “low” defined as Stage 0, I, II, “high” defined as Stage III, IV), and recruitment center (categorical variable). Sex was omitted from the model for those cancers affecting exclusively one gender – cervical squamous cell carcinoma (CESC), ovarian carcinoma (OV), prostate adenocarcinoma (PRAD), uterine corpus endometrial carcinoma (UCEC), and uterine carcinosarcoma (UCS). Staging data was not available for glioblastoma multiforme (GMB), acute myeloid leukemia (LAML), brain lower grade glioma (LGG), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), sarcoma (SARC), and thymoma (THYM).
Next, we examined the effect of TEMMB on survival. We considered the residuals obtained from the negative binomial regression models as the effective TEMMB adjusted for age, sex, stage, and recruitment center. We used these residuals as inputs to Cox-proportional hazards models to predict survival (in days) as a function of effective TEMMB. Survival effects were expressed as hazard ratios (HR), which can be defined as the effective hazard per day conferred by effective TEMMB. Because TEMMB is an overdispersed count variable, it was adjusted well through negative binomial regression. The significance of Cox-proportional hazards models was calculated with two-sided Wald tests. Survival analysis for all 31 cohorts is reported in Figure S5A. We observed that in certain cohorts, such as pheochromocytoma and paraganglioma (PCPG), fewer than 10 patients were tracked until death, with the majority lost to follow-up. In such cases, we suspected that the survival analysis was dominated by censored data points (Fig. S5B). Thus, we performed an additional QC step by excluding cohorts in the bottom 5th, 10th, and 20th percentiles of number of non-censored events. Results upon stringent exclusion of the bottom 20th percentile of cohorts are reported in the main text.
We aggregated all nonsynonymous missense mutations among all individuals in each cancer. Missense variants resulting in identical amino acid changes were aggregated as one specific variant type. Recurrent mutations were defined as those variants exceeded 1% prevalence in the cohort, which is the traditional allele frequency cutoff for eliminating rare DNA variation36,37. A somatic recurrence score (RS) was calculated as the fraction of total mutations in the cohort’s pool comprised by recurrent mutations as defined above:
A RS was assigned to each cancer type, and the correlation between log10-adjusted survival HR and log10-adjusted RS was computed with Pearson’s correlation. To demonstrate robustness to parameter choice, an additional recurrent mutation prevalence definition of 5% (traditional Minor Allele Frequency cutoff for common DNA variation38) was tested.
Supplementary information
Acknowledgements
NIH K24CA149202 (to HT). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
Nikolai Klebanov: conceptualization, methodology, data curation, formal analysis, investigation, software, writing – original draft, review, and editing. Mykyta Artomov: conceptualization, methodology, investigation, writing – review and editing. William Goggins: methodology, supervision. Emma Daly: methodology, data curation. Mark Daly: conceptualization, supervision. Hensin Tsao: funding acquisition, conceptualization, supervision, writing – review and editing.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-41015-5.
References
- 1.Goodman AM, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017;16:2598–2608. doi: 10.1158/1535-7163.MCT-17-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birkbak NJ, et al. Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS One. 2013;8:e80023. doi: 10.1371/journal.pone.0080023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao D, et al. Analysis of ultra-deep targeted sequencing reveals mutation burden is associated with gender and clinical outcome in lung adenocarcinoma. Oncotarget. 2016;7:22857–22864. doi: 10.18632/oncotarget.8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalmers ZR, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta, S., Artomov, M., Goggins, W., Daly, M. & Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. J Natl Cancer Inst107, 10.1093/jnci/djv221 (2015). [DOI] [PMC free article] [PubMed]
- 8.Brown SD, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24:743–750. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srivastava PK. Neoepitopes of Cancers: Looking Back, Looking Ahead. Cancer Immunol Res. 2015;3:969–977. doi: 10.1158/2326-6066.CIR-15-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajasagi M, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;124:453–462. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller A, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017;7:e612. doi: 10.1038/bcj.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolle ME, et al. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 14.Dolle ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci USA. 2000;97:8403–8408. doi: 10.1073/pnas.97.15.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA. 2013;110:1999–2004. doi: 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson AK, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330–337. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blokzijl F, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milholland B, Auton A, Suh Y, Vijg J. Age-related somatic mutations in the cancer genome. Oncotarget. 2015;6:24627–24635. doi: 10.18632/oncotarget.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexandrov LB, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shih IM, et al. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–822. [PubMed] [Google Scholar]
- 21.Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43–49. doi: 10.1016/j.semcancer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Armaghany T, Wilson JD, Chu Q, Mills G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012;5:19–27. [PMC free article] [PubMed] [Google Scholar]
- 23.Pernot S, et al. Colorectal cancer and immunity: what we know and perspectives. World J Gastroenterol. 2014;20:3738–3750. doi: 10.3748/wjg.v20.i14.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointest Oncol. 2015;6:208–223. doi: 10.3978/j.issn.2078-6891.2014.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeNardo DG, Coussens LM. Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9:212. doi: 10.1186/bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helgadottir H, Hoiom V. The genetics of uveal melanoma: current insights. Appl Clin Genet. 2016;9:147–155. doi: 10.2147/TACG.S69210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trevino V, Martinez-Ledesma E, Tamez-Pena J. Identification of outcome-related driver mutations in cancer using conditional co-occurrence distributions. Sci Rep. 2017;7:43350. doi: 10.1038/srep43350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016;28:22–27. doi: 10.1016/j.smim.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McFarland CD, et al. The Damaging Effect of Passenger Mutations on Cancer Progression. Cancer Res. 2017;77:4763–4772. doi: 10.1158/0008-5472.CAN-15-3283-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria., 2018).
- 32.Samur MK. RTCGAToolbox: a new tool for exporting TCGA Firehose data. PLoS One. 2014;9:e106397. doi: 10.1371/journal.pone.0106397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.W N. Venables B. D. R. Modern Applied Statistics with S. Fourth Edition. Springer, New York., (2002).
- 34.survminer: Drawing Survival Curves using ‘ggplot2’ v. 0.4.2 (2018).
- 35.forestplot: Advanced Forest Plot Using ‘grid’ Graphics v. 1.7.2 (2017).
- 36.Primrose, S. B. & Twyman, R. M. Principles of genome analysis and genomics. 3rd edn, (Blackwell Pub., 2003).
- 37.Clancy S. Genetic mutation. Nature Education. 2008;1:187. [Google Scholar]
- 38.Hinds DA, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.