

Ring sideroblasts are a hallmark of sideroblastic anemia, although little is known about their characteristics. Here, we first generated mutant mice by disrupting the GATA-1 binding motif at the intron 1 enhancer of the ALAS2 gene, a gene responsible for X-linked sideroblastic anemia (XLSA).
KEYWORDS: 5-aminolevulinic acid synthase 2, X-linked sideroblastic anemia, erythroid cells, heme, iron
ABSTRACT
Ring sideroblasts are a hallmark of sideroblastic anemia, although little is known about their characteristics. Here, we first generated mutant mice by disrupting the GATA-1 binding motif at the intron 1 enhancer of the ALAS2 gene, a gene responsible for X-linked sideroblastic anemia (XLSA). Although heterozygous female mice showed an anemic phenotype, ring sideroblasts were not observed in their bone marrow. We next established human induced pluripotent stem cell-derived proerythroblast clones harboring the same ALAS2 gene mutation. Through coculture with sodium ferrous citrate, mutant clones differentiated into mature erythroblasts and became ring sideroblasts with upregulation of metal transporters (MFRN1, ZIP8, and DMT1), suggesting a key role for ferrous iron in erythroid differentiation. Interestingly, holo-transferrin (holo-Tf) did not induce erythroid differentiation as well as ring sideroblast formation, and mutant cells underwent apoptosis. Despite massive iron granule content, ring sideroblasts were less apoptotic than holo-Tf-treated undifferentiated cells. Microarray analysis revealed upregulation of antiapoptotic genes in ring sideroblasts, a profile partly shared with erythroblasts from a patient with XLSA. These results suggest that ring sideroblasts exert a reaction to avoid cell death by activating antiapoptotic programs. Our model may become an important tool to clarify the pathophysiology of sideroblastic anemia.
INTRODUCTION
Red blood cells are the predominant consumer of iron during their differentiation. To acquire this large an amount of iron, erythroblasts mainly depend on the glycoprotein transferrin (Tf), which has two affinity sites for ferric iron (Fe3+). Thus, mice that lack transferrin receptor 1 (TfR1; also denoted TFRC and CD71) die due to severe effects on erythropoiesis (1). However, non-Tf iron can also be transported into erythroblasts through alternative metal transporters, such as divalent metal transporter 1 (DMT1 product) (2, 3) and Zrt- and Irt-like protein 8 (ZIP8 product; a transmembrane zinc transporter, until recently thought to be a ferrous iron transporter) (4–6). Although mice lacking these transporters have been shown to exhibit severe anemia (7, 8), the relative contribution of non-Tf iron to erythropoiesis remains unclear.
In erythroblasts, the majority of iron is incorporated into heme and is ultimately utilized for hemoglobin synthesis. However, some iron is also utilized for iron-sulfur (Fe-S) cluster synthesis within mitochondria; these clusters play a role in fundamental biological processes such as energy production, gene expression regulation, and protein translation (9). A deficiency in heme biosynthesis leads to sideroblastic anemia (SA), a term that encompasses a group of disorders which share common features, including mitochondrial iron accumulation in bone marrow erythroid precursors (i.e., ring sideroblasts) (10). In the final step of heme biosynthesis, ferrous iron is inserted into protoporphyrin IX by ferrochelatase (FECH) in mitochondria (11). Thus, mutations in genes responsible for porphyrin synthesis, such as ALAS2 (which encodes 5-aminolevulinic acid [ALA] synthase 2), lead to sideroblastic anemia. Mutations in genes involved in Fe-S clusters and mitochondrial tRNA metabolism are also associated with sideroblastic anemia (10, 12), although the detailed molecular mechanisms of how defects in each gene result in abnormal mitochondrial iron accumulation remain unclear. In adults, this syndrome is commonly observed in association with myelodysplastic syndrome (MDS; MDS with ring sideroblasts is termed MDS-RS) (10, 13). A recent study involving a large cohort revealed that SF3B1, a gene for a splicing factor, is frequently mutated in MDS-RS (13). Mutations in SF3B1 resulted in splicing errors of the ATP-binding cassette B7 (ABCB7) gene, which is involved in Fe-S cluster transport and is one of the causative genes of congenital sideroblastic anemia (CSA) (10, 12, 13).
The most common type of CSA is X-linked sideroblastic anemia (XLSA), which is caused by defects in the X-linked gene ALAS2 (10,12,14). ALAS2 encodes an enzyme that catalyzes the rate-limiting step in heme biosynthesis in erythroid cells (10, 12, 14). This pathway converts glycine and acetyl coenzyme A (acetyl-CoA) to ALA and requires pyridoxal 5′-phosphate (PLP; vitamin B6) as a cofactor (10, 12, 14). Most XLSA-associated mutations are missense substitutions that result in a loss of protein functionality (10, 12). In addition to mutations in the coding region of ALAS2, mutations in its regulatory region also have been identified. The ALAS2 gene is regulated by the transcription factor GATA-1, which is a master regulator of erythropoiesis (15, 16). Intriguingly, mutations involving the GATA-1 binding motif at the intron 1 enhancer of the gene can lead to the onset of XLSA (17, 18).
Although pyridoxal 5′-phosphate is commonly used to treat XLSA, nearly half of all XLSA cases are unresponsive to this treatment (19), necessitating the search for novel therapeutic strategies. However, evidence regarding the molecular characteristics of ring sideroblasts is scarce due to a lack of biological models. To date, several attempts have tried but failed to establish an XLSA model for further molecular analyses, such as Alas2 knockout mice (20), Alas2 knockout embryonic stem (ES) cells (21), CRISPR/Cas9-based target disruption of the GATA-1 binding motif at intron 1 of murine Alas2 both in vitro (22) and in vivo (23), and transgenic rescue of ALAS2 in Alas2 knockout mice (24). Although we recently succeeded in establishing ring sideroblasts from induced pluripotent stem cells with X-linked sideroblastic anemia, there were too few ring sideroblasts for further biochemical analyses (25). To overcome these limitations, we aimed to create a model of ring sideroblasts based on CRISPR/Cas9 genome editing.
RESULTS
Establishment of mutant mouse lines with disrupted GATA-1 binding site at the Alas2 intron 1 enhancer.
We generated a founder mouse showing a 5-bp deletion at the intron 1 enhancer region of Alas2 involving the GATA binding domain (Alas2del/X) (Fig. 1A). Quantitative reverse transcription-PCR (RT-PCR) analysis confirmed decreased Alas2 expression in Ter119-positive erythroblasts of the heterozygous Alas2del/X mice (Fig. 1B). Whereas the heterozygous Alas2del/X mice were viable and fertile, we could not obtain hemizygous male (Alas2del/Y) mice. To test if the hemizygous male genotype could lead to embryonic lethality, we also generated a founder mouse showing a 10-bp insertion at the intron 1 enhancer region of Alas2, resulting in the disruption of the GATA-1 binding domain (Alas2ins allele) (Fig. 2A). Similar to the Alas2del mice, we could not obtain hemizygous male (Alas2ins/Y) mice. While embryos of all genotypes (Alas2X/Y, Alas2X/X, Alas2ins/X, and Alas2ins/Y) were observed at embryonic day 10.5, the hemizygous male (Alas2ins/Y) embryos were severely anemic (Fig. 2B), implying that the hemizygous male (Alas2ins/Y) would lead to embryonic lethality. The finding was similar to findings in a previous report based on a 13-bp deletion in the same region (23). On the other hand, a detailed analysis of both the heterozygous Alas2del/X and Alas2ins/X female mice revealed that these mice exhibited anemia and increased red cell distribution width (RDW) (Fig. 1C and 2C), indicating bimodal red blood cell size distribution. Peripheral blood smears of the heterozygous Alas2ins/X female mice indicated the presence of hypochromic microcytic red blood cells as well as peripheral siderocytes (Fig. 2D and E). However, the heterozygous mice did not show ring sideroblasts by Prussian blue staining (Fig. 1D; data not shown for the heterozygous Alas2ins/X female mice). These findings, taken together, suggest that, unlike the case of patients with XLSA, it is impossible to obtain a mouse model for XLSA by Alas2 intron 1 mutation, indicating that there are differences among species, such as the contribution of the Alas2 intron 1 enhancer to its transcriptional activity or possibly iron metabolism. Thus, we aimed to establish a human model of ring sideroblasts based on CRISPR/Cas9-based genome editing.
FIG 1.

Generation of Alas2 knockdown mice by CRISPR/Cas9-mediated deletion of the Alas2 cis element. (A) Sequence analysis. (Upper) Core sequence of the Alas2 intron enhancer. (Lower) Sequence of the mutant allele (Alas2del allele). The GATA-1 binding motif is indicated by the red box. (B) Quantitative RT-PCR analysis for Alas2 expression in Ter119-positive erythroblasts in wild-type and heterozygous Alas2del/X female mice. Values presented are relative to those of GAPDH mRNA. Data are expressed as means ± standard deviations (SD) (n = 4). *, P < 0.05. (C) Hematological analysis of wild-type and heterozygous Alas2del/X female mice. Data are expressed as means ± SD (n = 7 and n = 4 for Alas2WT/WT and Alas2del/WT, respectively). *, P < 0.05. Hb, hemoglobin. (D) Prussian blue staining of Ter119-positive erythroblasts in wild-type and heterozygous Alas2del/X female mice.
FIG 2.

Generation of Alas2 knockdown mice by CRISPR/Cas9-mediated disruption of the Alas2 cis element. (A) Sequence analysis. (Upper) Core sequence of the Alas2 intron enhancer. (Lower) Sequence of the mutant allele (Alas2ins allele). The GATA-1 binding motif is indicated by the red box. (B) The panels show embryos without yolk sacs of wild-type and Alas2ins/y mice. (C) Hematological analysis of wild-type and heterozygous Alas2mt/X female mice. Data are expressed as means ± SD (n = 7 and n = 3 for Alas2WT/WT and Alas2ins/WT, respectively). *, P < 0.05. (D and E) Peripheral blood smear of May-Grünwald Giemsa (D) and Prussian blue (E) staining. Representative hypochromic microcytic red blood cells (D) and siderocytes (E) in the heterozygous Alas2ins/X female mice are indicated by arrows.
Erythroid differentiation was induced by coculture with OP9 cells in the presence of SFC but not holo-Tf.
Ring sideroblasts were observed predominantly in the orthochromatic and polychromatic erythroblast stages (26), whereas human iPS cell-derived erythroid progenitor (HiDEP) cells corresponded to the proerythroblast stage (27). Therefore, we attempted to differentiate HiDEP cells into more mature erythroblast stages by coculturing with OP9 cells. Interestingly, the addition of sodium ferrous citrate (SFC), but not holo-transferrin (holo-Tf), to the coculture medium induced erythroid differentiation, as reflected by chromatin condensation, decreased nuclear/cytosol ratio, and decreased basophilic features (Fig. 3A). Quantitative RT-PCR analysis demonstrated that ALAS2 and HBG expression was strongly induced after coculture with the addition of SFC, whereas moderate increases were seen under nontreated and holo-transferrin-treated conditions (Fig. 3B).
FIG 3.

SFC promotes erythroid differentiation. (A) May-Giemsa staining of wild-type (WT) HiDEP cells cocultured with OP9 cells for 6 days. NT, nontreated; Tf, iron-saturated transferrin; SFC, sodium ferrous citrate. (B) Quantitative RT-PCR analysis for ALAS2 and HBG expression in wild-type HiDEP cells cocultured with OP9 cells for 2, 4, and 6 days. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (C) FACS analysis of wild-type HiDEP cells cocultured with OP9 cells for 6 days. The percentages of TfR1-negative, GPA-positive fractions are summarized. Data represent the averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (D) BrdU incorporation assay of cocultured wild-type HiDEP cells by FACS. BrdU-positive and -negative fractions denote S and non-S phases, respectively. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (E and F) FACS analysis to evaluate TfR1 and GPA expression levels of primary erythroblasts. Erythroblasts were differentiated from human CD34+ cells derived from cord blood mononuclear cells based on serum-free medium (E) and subsequently cocultured with OP9 cells (F). T−G−, TfR1 negative and GPA negative; T+G−, TfR1 positive and GPA-negative; T+G+, TfR1 positive and GPA positive; T−G+, TfR1 negative and GPA positive. Data represent averages from five independent experiments and are expressed as means ± SD. *, P < 0.05. NT→SFC indicates that SFC was treated from the OP9 coculture stage.
We next performed fluorescence-activated cell sorting (FACS) analysis to test whether coculture with OP9 cells altered the erythroid differentiation stage. During erythroid differentiation, TfR1 expression precedes GPA (also denoted CD235a) expression and subsequently decreases in the terminal stages (28). The TfR1− GPA+ mature erythroid population significantly increased following the addition of SFC after coculturing in wild-type HiDEP cells (Fig. 3C). These findings were consistent with morphologic changes induced by SFC (Fig. 3A). Because the proliferation of erythroblasts decreases in the terminal stage of erythroid differentiation, we evaluated cell proliferation using a bromodeoxyuridine (BrdU) incorporation assay. The number of cells in S phase was significantly decreased by the addition of SFC (Fig. 3D). These results suggested that SFC promoted erythroid differentiation in HiDEP cells more efficiently than holo-transferrin.
We also evaluated whether coculture with OP9 and the addition of SFC promoted differentiation of cord blood CD34+ cell-derived primary erythroblasts. We confirmed that SFC increased the frequency of GPA+ cells and TfR1− GPA+ mature erythroid populations based on serum-free medium (Fig. 3E) and subsequent coculture with OP9 (Fig. 3F), respectively. However, we did not detect obvious morphologic changes (data not shown). These results indicated that ferrous iron exerts an additive effect on erythroid differentiation.
Generation of ALAS2-mutated erythroid progenitor cell lines.
To establish XLSA models, we generated HiDEP cells lacking a 19-bp fragment, including the GATA-1 binding motif, within the ALAS2 intron 1 enhancer region (Fig. 4A). The color of the cell pellet in ALAS2-mutated cells (XLSA clone Mu) changed to pale pink, unlike the color of wild-type cells, although their morphologies were similar (Fig. 4B). G-banding analysis of HiDEP cells demonstrated a female (XX) karyotype (data not shown), indicating that the mutation was homologously introduced. Quantitative chromatin immunoprecipitation (ChIP) demonstrated that the chromatin occupancies of GATA-1 and its cofactor TAL1 (16, 29, 30) were significantly abrogated by deletion at the intron 1 enhancer of the ALAS2 gene (Fig. 4C) despite no obvious change in GATA-1 protein expression in the XLSA clone (Fig. 4D). We confirmed significant decreases in the expression of ALAS2 and HBG (Fig. 4E) and intracellular heme concentration (Fig. 4F) in the XLSA clone cells. In addition, the XLSA clone cells exhibited decreased proliferation of viable cells under trypan blue staining (Fig. 4G). Decreased cell surface expression of TfR1 suggests an increase in intracellular iron content in mutant cells (Fig. 4H); however, ring sideroblast formation was not observed, even after the addition of SFC (Fig. 4I).
FIG 4.

Establishment of XLSA clones by CRISPR/Cas9-mediated ablation of ALAS2 cis elements in HiDEP cells. (A) Sequence analysis. WT, wild-type HiDEP cells; Mu, XLSA clones. (B) May-Giemsa staining (upper) and cell pellets (lower). (C) Quantitative ChIP analysis to detect endogenous GATA-1 (upper) and TAL1 (lower) chromatin occupancy in wild-type HiDEP cells and in the XLSA clone cells. HS2, which is the DNase I hypersensitivity site at the β-globin locus control region (6), and the NECDIN promoter were used as positive and negative controls, respectively. Data are expressed as means ± SD (n = 3). (D) Western blot to detect GATA-1 in wild-type HiDEP cells and XLSA clone cells. α-Tubulin was used as a loading control. Representative data from at least two independent experiments are shown. (E) Quantitative RT-PCR analysis for ALAS2 and HBG expression in wild-type HiDEP cells and XLSA clone cells. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (F) Intracellular heme concentration in wild-type HiDEP cells and XLSA clone cells. Data are presented as means ± SD (n = 3). *, P < 0.05. (G) Changes in total cell number by CRISPR/Cas9-mediated ablation of ALAS2 cis elements at intron 1 in HiDEP cells. (H) FACS analysis to evaluate TfR1 expression in wild-type HiDEP cells and XLSA clone cells. Representative data from at least two independent experiments are shown. Red, wild type; blue, XLSA clone cells. (I) Prussian blue staining (upper, without SFC; lower, with 100 μM SFC for 4 days) of wild-type HiDEP cells and XLSA clone cells.
Similar to the wild-type HiDEP cells (Fig. 3), coculture with OP9 cells and addition of SFC promoted erythroid differentiation in the XLSA clone cells in terms of their morphology (Fig. 5A), upregulation of ALAS2 and HBG (Fig. 5B), increased percentage of TfR1− GPA+ mature erythroid population (Fig. 5C), and decreased frequency of cells in S phase (Fig. 5D). Concomitantly, the cell pellets of the differentiated XLSA clone cells turned reddish (Fig. 5E).
FIG 5.

SFC promotes erythroid differentiation of XLSA clone cells. (A) May-Giemsa staining of XLSA clone cells cocultured with OP9 cells for 6 days. NT, nontreated; Tf, iron-saturated transferrin; SFC, sodium ferrous citrate. (B) Quantitative RT-PCR analysis for ALAS2 and HBG expression in XLSA clone cells that were cocultured with OP9 cells for 2, 4, and 6 days. Values presented are relative to those for GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± standard deviations. *, P < 0.05. (C) FACS analysis of XLSA clone cells cocultured with OP9 cells for 6 days. The percentages of TfR1-negative, GPA-positive fractions are summarized. Data represent averages from three independent experiments and are expressed as means ± standard deviations. *, P < 0.05. (D) BrdU incorporation assay by FACS. BrdU-positive and -negative fractions denote S phase and non-S phase, respectively. Data represent averages from three independent experiments and are expressed as means ± standard deviations. *, P < 0.05. (E) Cell pellets of cocultured HiDEP cells. Cell pellets of wild-type HiDEP cells and XLSA clone cells cocultured with OP9 cells for 2, 4, and 6 days. Representative data from at least two independent experiments are shown.
We next sought the molecular mechanism by which coculture with SFC induced the expression of the ALAS2 gene in the XLSA clone cells to a level that was comparable with that in wild-type cells (Fig. 3B, 5B, and 6A). In the XLSA clone cells cocultured with OP9 cells, GATA-1 occupancy remained abrogated at intron 1 but was significantly increased at intron 8 in the presence of SFC (Fig. 6B). Therefore, an increase in GATA-1 chromatin occupancy at intron 8 may compensate for the compromised ALAS2 expression in the XLSA clone cells.
FIG 6.
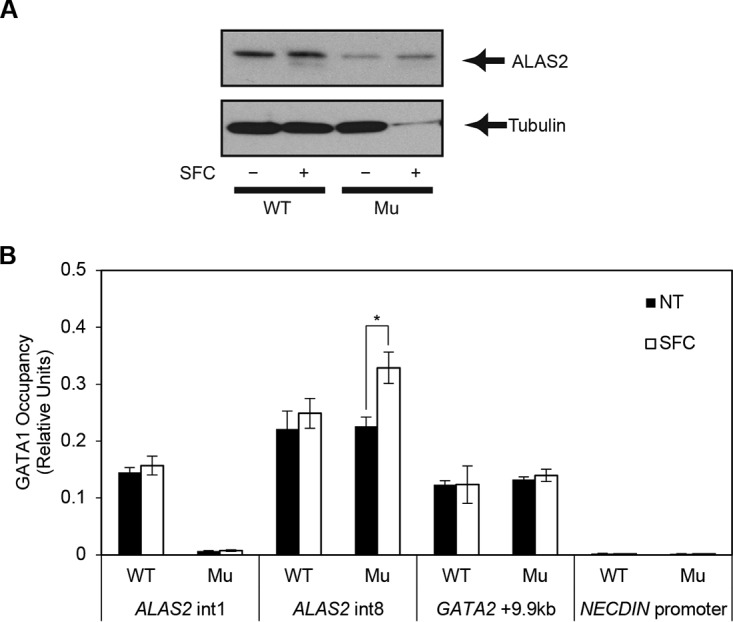
Increased ALAS2 expression in the ring sideroblasts. (A) Western blot analysis to detect endogenous ALAS2 protein from wild-type HiDEP cells and XLSA clone cells by coculturing with OP9 cells in the presence and absence of SFC. Tubulin was used as a loading control. (B) Quantitative ChIP analysis to detect endogenous GATA-1 chromatin occupancy in wild-type HiDEP cells and in XLSA clone cells cocultured with OP9 cells for 6 days in the presence or absence of SFC. GATA-2 + 9.9 kb, which is the GATA switch site during erythroid differentiation, and the NECDIN promoter were used as positive and negative controls, respectively (6). Data are expressed as means ± standard deviations (n = 3). *, P < 0.05. Average negative-control (IgG) signal did not exceed 0.003.
Ring sideroblasts were induced from the XLSA clone cells by coculture with OP9 cells in the presence of SFC but not holo-Tf.
Since erythroid differentiation was achieved by coculture with OP9 cells and SFC, we examined whether ring sideroblast formation was observed under these culture conditions. From day 4 of erythroid differentiation after coculture with OP9 cells in the presence of SFC, typical ring sideroblasts increased by up to 20% in the XLSA clone cells, but there were few in wild-type cells (Fig. 7A to C). To discriminate lysosomal iron from mitochondrial iron deposits, we quantified the distribution of iron deposits based on electron microscopy. Under SFC-treated conditions, the majority (80%) of XLSA clones had both lysosomal and mitochondrial iron deposition, while the iron deposition in the wild-type cells was exclusively observed in lysosome. Evaluation of the various metal transporters in erythroid cells then showed that the addition of SFC significantly increased the expression of mitoferrin 1 (MFRN1 product; a ferrous iron transporter in mitochondria) (31), DMT1 (2, 3), and ZIP8 (4–6) in the XLSA clone cells compared with the wild-type cells, which might have contributed to the formation of ring sideroblasts (Fig. 7D). Likewise, TfR1 expression in the XLSA clone cells was induced by the addition of SFC, but the expression level was lower than that seen in wild-type cells (Fig. 7D). In conjunction with the absence of an obvious effect of holo-Tf on erythroid differentiation and ring sideroblast formation (Fig. 3A and 7A), increased expression of MFRN1, DMT1, and ZIP8 might have a stronger effect on iron uptake than that of TfR1 in the XLSA clone cells.
FIG 7.

Induction of ring sideroblasts by coculture of XLSA clone cells with OP9 cells and addition of SFC. (A) Prussian blue staining of wild-type HiDEP cells and XLSA clones cocultured with OP9 cells for 6 days. (B) The percentage of ring sideroblasts is shown based on wild-type HiDEP cells and XLSA clone cells cocultured with OP9 cells for 2, 4, and 6 days in the presence of SFC. Data represent the averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (C) Electron microscopy of wild-type HiDEP cells and XLSA clones cocultured with OP9 cells for 4 days in the presence or absence of SFC. (D) Quantitative RT-PCR analysis for TfR1, MFRN1, DMT1, and ZIP8 in wild-type HiDEP cells and in XLSA clone cells that were cocultured with OP9 cells for 0, 2, 4, and 6 days. NT, nontreated; Tf, iron-saturated transferrin; SFC, sodium ferrous citrate. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05.
We also established and evaluated another XLSA clone (Mu2). Each allele showed 13- and 15-bp deletions involving GATA binding motifs (Fig. 8A). Similar to the XLSA clone (Mu), the color of the Mu2 cell pellet changed to pale pink (Fig. 8B). Again, we confirmed decreased expression for ALAS2 and HBG (see Fig. S8C in the supplemental material), emergence of ring sideroblasts by coculture with OP9 cells in the presence of SFC (Fig. 8D), and significantly increased expression for MFRN1, DMT1, and ZIP8 on day 6 (Fig. 8E).
FIG 8.

Another clone of ALAS2-mutated HiDEP cells (Mu2). (A) Sequence analysis of another XLSA clone (Mu2). Each allele showed 13- and 15-bp deletions involving GATA binding motifs. (B) Cell pellets of mutant clones. Mu is presented in Fig. 3. (C) Quantitative RT-PCR analysis for ALAS2 and HBG expression in wild-type HiDEP cells and in XLSA clone cells. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± standard deviations. *, P < 0.05. (D) Prussian blue staining of Mu2 cocultured with OP9 cells supplemented with SFC. (E) Quantitative RT-PCR analysis for TfR1, MFRN1, DMT1, and ZIP8 in wild-type HiDEP cells and in another XLSA clone (Mu2) that were cocultured with OP9 cells for 0 and 6 days. NT, nontreated; Tf, iron-saturated transferrin; SFC, sodium ferrous citrate. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05.
Increased antiapoptotic activity in ring sideroblasts.
To reveal the biochemical characteristics of ring sideroblasts, we performed expression analyses on wild-type cells and the XLSA clone cells (Fig. 9A and B). Before coculture, a 19-bp deletion at the intron 1 enhancer region of the ALAS2 gene caused a more than 2-fold upregulation of 36 genes and downregulation of 61 genes (Fig. 9A and Table S2). The downregulated genes included ALAS2, globin genes (HBZ, HBG, HBE, HBD, HBM, and HBQ), and genes involved in iron/heme metabolism (TfR1, CPOX, and MFRN1). Gene ontology (GO) analysis revealed significant enrichment of cellular iron ion homeostasis (P = 0.018) and regulation of transcription (P = 0.0021) (Fig. 9C and Table S2). On the other hand, we noticed significant upregulation of genes involved in antiapoptosis activity (P = 0.0015), as represented by HSPA1A (heat shock protein family A member 1A) (Fig. 9A and C). Additionally, on day 6 after coculture in the presence of SFC, a 19-bp deletion at the intron 1 enhancer region of the ALAS2 gene caused a more than 2-fold upregulation of 287 genes and downregulation of 143 genes (Fig. 9B and Table S3). Interestingly, compared to the status before coculture (Fig. 9A), we noticed more prominent upregulation of antiapoptosis genes (P = 0.000772), including HSPA1A, SOD1 (superoxide dismutase 1), and SOD2 (Fig. 9A, B, and D) (32, 33).
FIG 9.

Expression profiling analysis of ring sideroblasts. (A and B) The heat map depicts the degree of change resulting from ALAS2 mutation in HiDEP cells before coculture (A) and after coculture in the presence of SFC for 6 days (B). The top 30 differentially upregulated or downregulated genes are shown. (C and D) Gene ontology analysis showed genes were upregulated or downregulated by more than 2-fold following ALAS2 mutation, before coculture (C), and after coculture in the presence of SFC (D).
To test if the genes involved in antiapoptotic processes were also induced in patient-derived erythroblasts, we conducted a microarray analysis based on GPA-positive erythroblasts from a patient with XLSA and a healthy individual. This analysis demonstrated a more than 2-fold upregulation of 622 genes and downregulation of 339 genes (Fig. 10A and Table S4). We found significant upregulation of genes involved in the apoptosis process, as represented by AEN (apoptosis-enhancing nuclease) (34), DDX47 (DEAD box helicase 47) (35), and GAAD45A (growth arrest and DNA damage-inducible 45 alpha) (36), as well as antiapoptotic genes, such as HSPA1A and SOD2 (Fig. 10B), which were commonly upregulated in in vitro-derived ring sideroblasts (Fig. 10C and D). These results suggested that our in vitro model of ring sideroblasts reflected the phenotypic characteristics of XLSA patient-derived erythroblasts and supported our observation that ring sideroblasts might avoid cell death by inducing antiapoptotic properties.
FIG 10.

Expression profiling analysis of a patient with XLSA. (A) The heat map depicts the degree of change of an ALAS2 intron 1 mutated in a patient with XLSA. The top 20 differentially upregulated or downregulated genes are shown. (B) Gene ontology analysis shows the genes upregulated or downregulated by more than 2-fold in the patient with XLSA. (C and D) Venn diagram showing the relationship between upregulated genes in in vitro-derived ring sideroblasts (Fig. 5B and Table S3) and in XLSA patient-derived erythroblasts (Table S4). We focused on upregulated genes (>2-fold; global normalization of >100) involved in apoptosis processes (C) and antiapoptosis processes (D).
To test this hypothesis, we first evaluated the frequency of annexin V-positive cells using FACS, which reflected the expression of the apoptosis marker phosphatidylserine (37). The number of viable cells was significantly increased, and the rate of early and late apoptosis processes, which were quantified as propidium iodide-negative (PI−) annexin V+ and PI+ annexin V+, respectively, was decreased in SFC-treated XLSA clones (ring sideroblasts) compared to the level with nontreated and holo-Tf-treated XLSA clones (Fig. 11A and B). On the other hand, compared with the wild-type cells, the percentage of apoptotic cells in SFC-treated cells was only marginally higher. We further confirmed the decreased frequency of late apoptosis by evaluating DNA fragmentation (38). DNA smear was induced in nontreated and holo-Tf-treated XLSA clone cells and was obviously diminished in SFC-treated XLSA clones, whereas a DNA ladder was not evident in the wild type (Fig. 11C). Apoptosis is known to be caused by increased reactive oxygen species (ROS) levels (39, 40). As with a previous study that indicated erythroblasts derived from Alas2 knockout embryonic stem cells exhibited increased ROS levels (21), ROS levels significantly decreased after the addition of SFC in wild-type and ring sideroblasts, and the levels between them were comparable (Fig. 11D). Erythroblasts in the late differentiation stage accumulate many antioxidant enzymes to protect the cells against oxygen radicals before enucleation (41). In our study, the promotion of erythroid differentiation by SFC treatment could lead to the accumulation of antioxidant enzymes and decreased ROS levels. Additionally, upregulation of antiapoptotic genes in ring sideroblasts could also contribute to the equal ROS levels between wild-type and ALAS2 mutant cells.
FIG 11.

Antiapoptotic activity is induced in ring sideroblasts. (A) Apoptosis assay by FACS (representative data). (B) Percentage of viable cells, early apoptosis (PI negative and annexin V positive), and late apoptosis (PI positive and annexin V positive) are summarized in wild-type HiDEP cells and XLSA clone cells that were cocultured with OP9 cells for 6 days. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (C) DNA fragmentation assay in wild-type HiDEP cells and in XLSA clone cells that were cocultured with OP9 cells for 6 days. HiDEP cells treated with actinomycin D according to the manufacturer’s recommendations were used as a positive control. (D) Reactive oxygen species activity based on CellROX deep red reagent (Thermo Fisher Scientific). Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. NT, nontreated; Tf, iron-saturated transferrin; SFC, sodium ferrous citrate.
From the above results, taken together, it appears that ring sideroblast formation was accompanied by increased antiapoptosis characteristics and protection from cellular stress molecules, such as ROS.
ALA restores defective heme synthesis caused by ALAS2 mutation.
Because ALAS2 catalyzes the biosynthesis of 5-aminolevulinic acid (ALA) (10), ALA supplementation could be an alternative therapeutic strategy to restore heme synthesis in CSA caused by ALAS2 defects (42). Therefore, we evaluated the effect of ALA on established ring sideroblasts. When the culture medium was supplemented with ALA on day 0 of coculture, we observed an obvious increase in intracellular heme concentration in the XLSA clone cells but not in wild-type cells (Fig. 12A and B). Quantitative RT-PCR analysis confirmed that the addition of ALA restored the expression of HBG and HO1 (Fig. 12C), supporting our observation that ALA restored heme biosynthesis. However, when ALA was supplemented in the presence of SFC, we observed no significant change in heme concentration compared to that under the SFC-only condition (Fig. 12A and B).
FIG 12.
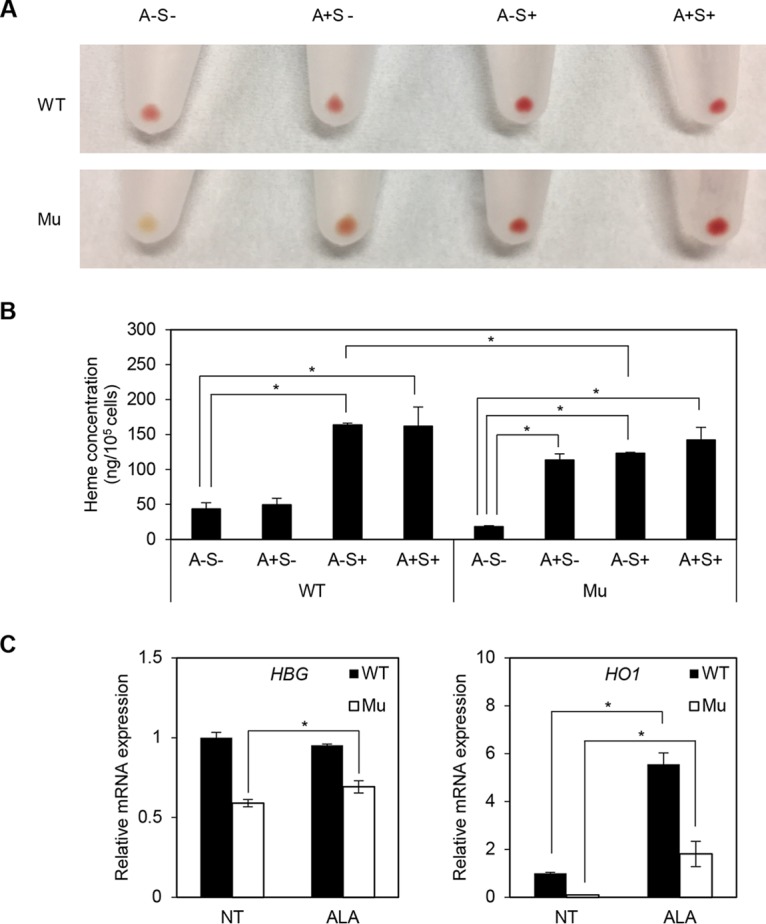
ALA restores heme synthesis in XLSA clones. (A and B) Representative cell pellets (A) and heme concentration (B) of wild-type HiDEP cells and XLSA clone cells that were cocultured with OP9 cells for 6 days. A, 1 mM ALA; S, 100 μM SFC. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05. (C) Quantitative RT-PCR analysis for HBG and HO1 expression in wild-type HiDEP cells and XLSA clone cells that were cocultured with OP9 cells for 6 days. NT, nontreated; ALA, ALA treated. Values presented are relative to those of GAPDH mRNA. Data represent averages from three independent experiments and are expressed as means ± SD. *, P < 0.05.
DISCUSSION
First, we first attempted to establish an in vivo model of XLSA. While we succeeded in disrupting the GATA-1 binding motif at the Alas2 intron 1 enhancer, we could not obtain hemizygous male (Alas2del/Y, Alas2ins/Y) mice (Fig. 1 and 2). Similarly, Zhang et al. recently demonstrated that a 13-bp deletion involving the GATA-1 binding motif led to embryonic lethality (23). Thus, whereas disruption of the ALAS2 intron 1 enhancer led to the onset of XLSA in humans (17, 18), this sequence may be indispensable for maintaining the minimal Alas2 expression required for erythroid maturation in mice. Interestingly, we found that heterozygous Alas2del/X and Alas2ins/X female mice exhibited anemia (Fig. 1C and 2B) and decreased Alas2 expression in Ter119-positive erythroblasts (Fig. 1B). It is known that patients with XLSA are predominantly hemizygous males while most heterozygous female carriers are asymptomatic, but female patients with the heterozygous ALAS2 mutation can exhibit sideroblastic anemia (43, 44). In these cases, the sideroblastic anemia is attributed to a severe loss-of-function mutation in ALAS2, resulting in the intramedullary apoptosis of ALAS2-mutated erythroid precursors and enabling only wild-type ALAS2-expressing erythroid cells to enter circulation (45, 46). Taking these findings together, we speculate that mutant Alas2-expressing hematopoietic stem cells are not able to differentiate into mature erythrocytes in mice. Nevertheless, the reason why the heterozygous Alas2del/X and Alas2ins/X female mice did not exhibit typical ring sideroblasts remains unknown.
Thus, we next sought to establish an in vitro model of XLSA by inducing a deletion at the ALAS2 intron 1 enhancer region based on HiDEP cells. However, we could not observe ring sideroblasts in the ALAS2 mutant clones (Fig. 4I), presumably because the differentiation stage of parental HiDEP cells is not mature enough to induce abnormal mitochondrial iron accumulation. Our study revealed that non-Tf iron (i.e., SFC) exerts an additive effect on erythroid differentiation. It could not be ruled out that additional non-Tf iron was incorporated into transferrin and functioned as holo-Tf. However, this may be less likely because the concentration of SFC added to the medium was sufficient to saturate transferrin, and the addition of holo-Tf did not work on erythroid differentiation as SFC did. These results were surprising, because holo-Tf has been considered important for human CD34-positive cell-derived erythroid differentiation (47). Interestingly, the importance of the non-Tf form of iron has been suggested in the context of erythroblastic islands (48, 49) in which macrophages provide iron to promote erythroid differentiation via the iron exporter ferroportin (to provide inorganic iron) (50), the heme exporter (heme-iron) (51), or TfR1 (ferritin-bound iron) (52). In addition, the macrophage-specific disruption of ferroportin in mice exhibited severe anemia by an iron-deficient diet, implying the importance of non-Tf iron in the pathogenesis of anemia (53), which warrants further investigation.
We demonstrated that the addition of SFC, but not holo-Tf, induced ring sideroblast formation (Fig. 7A and C and 8D). We noticed that holo-Tf appeared to promote erythroid differentiation, which was not seen under the nontreated condition (Fig. 3A). Thus, we speculated that holo-Tf failed to induce erythroid differentiation of HiDEP cells to the stages corresponding to ring sideroblast formation (Fig. 3A). Also, we speculated that absorption of SFC was accelerated in the XLSA clones, presumably mediated by increased expression of proteins encoded by DMT1 and ZIP8, responsible for the incorporation of ferrous iron (Fig. 7D and 8E) (2–5). Subsequently, the increased amount of intracellular iron might be transported into the mitochondria by increased expression of the mitochondrial iron transporter, MFRN1 (33), leading to mitochondrial iron accumulation combined with impaired protoporphyrin synthesis by decreased expression of ALAS2 (Fig. 13). However, the molecular mechanisms by which SFC induces expression of iron transporters in the context of the XLSA clones remain to be elucidated.
FIG 13.

Model of iron acquisition, trafficking, and utilization in XLSA clone cells. Shown is a schematic representation of the iron pathway and iron use in wild-type (left) and ALAS2-mutated (right) erythroblasts. In ring sideroblasts, iron uptake was increased by the increased expression of ZIP8 and DMT1 as a compensatory mechanism against impaired heme biosynthesis. Increased amounts of intracellular iron might be transported into mitochondria by increased expression of the mitochondrial iron transporter, MFRN1. Mitochondrial iron, which could not be utilized as a heme due to low ALAS2 expression, was aberrantly deposited in the mitochondria.
Based on analysis of clinical samples from patients with myelodysplastic syndrome with ring sideroblasts (MDS-RS), defective heme biosynthesis has been assumed to induce apoptotic death of erythroblasts, which could cause ineffective erythropoiesis and lead to the onset of sideroblastic anemia (54–56). We found that nontreated and even holo-Tf-treated XLSA clone cells underwent apoptosis before forming ring sideroblasts. We speculated that although OP9 cells may release some signaling factors that promote erythroid differentiation, the XLSA clone cells could not generate enough heme in the absence of SFC, resulting in disturbed differentiation and cell death. Thus, it is possible that ineffective erythropoiesis due to heme deficiency occurs during differentiation in XLSA via the same mechanism as MDS-RS. On the other hand, ring sideroblast formation following the addition of SFC significantly decreased the number of apoptotic cells (Fig. 11B and C). Consistent with these findings, the expression of antiapoptotic genes became more prominent after coculture and addition of SFC (Fig. 9B). In the microarray analysis of XLSA patient-derived erythroblasts, we also noticed upregulation of apoptosis-inducing genes, which was in line with our in vitro results (Fig. 10D). These results suggested that once XLSA clone cells differentiated into ring sideroblasts following SFC supplementation, they could avoid cell death by activating the antiapoptotic system.
On the other hand, the fate of ring sideroblasts beyond terminal differentiation remains unclear. It is possible that the enucleation process is impaired in ring sideroblasts or that the survival time of ring sideroblasts is shorter than that in the wild-type cells after enucleation. However, our erythroid differentiation system did not yield erythroblasts in the terminal stage, including enucleated erythroid cells, even after prolonged coculture (data not shown). Therefore, this issue should be clarified by further refining the in vitro culture conditions that can yield enucleated erythroid cells or by establishing an in vivo model of XLSA.
Given that ALAS2 uses glycine and succinyl-CoA to form ALA, ALA supplementation may be an effective therapeutic strategy in restoring heme synthesis in patients with XLSA. Previous studies have demonstrated that the addition of ALA partially restored the expression of globins as well as HO1, which were downregulated by short interfering RNA-mediated transient knockdown of ALAS2 in HiDEP cells (42). However, the effect of ALA on restoring heme concentration has not been confirmed in HiDEP cells. Based on the coculture system with OP9 cells, we demonstrated that the addition of ALA increased heme concentration (Fig. 12A and B). Considering that ALA is an endogenous amino acid that has been shown to be safe in clinical settings (57), ALA may be a promising drug for XLSA, although a pharmacokinetics study showed that serum ALA concentration did not reach the sufficient levels seen in this study when administered at the currently used dose (58). Therefore, a dose escalation clinical trial is necessary to confirm the efficacy of ALA for XLSA. Nevertheless, the potential usefulness of our established ring sideroblast is not limited to a tool to evaluate the efficacy of ALA. A large number of ring sideroblasts can be induced based on coculturing with OP9 cells, and these cells would be applicable to a wide variety of screening systems to identify and explore novel therapeutic strategies for XLSA.
To our knowledge, this is the first report of a detailed molecular characterization of human ring sideroblasts in vitro. Our findings should help to clarify the molecular etiology of XLSA as well as helping to establish novel therapeutic strategies.
MATERIALS AND METHODS
Mice.
Cas9 mRNA and guide RNA targeting Alas2 intron 1 were injected into fertilized eggs from BDF1 mice. The target sequence used to disrupt the GATA-1 binding regions of intron 1 in a human ALAS2 gene was 5′-AGCTCTAGGGGCTTTATCTA-3′. All mice were maintained in a specific-pathogen-free facility. PCR genotyping using amplified mouse tail genomic DNA was performed to assess the Alas2 intron 1 deletion. Prior to the analysis, all mice were sacrificed under isoflurane-induced anesthesia (Wako, Japan). Mouse embryo images were investigated by Leica MZ F16 (Leica Biosystems, Newcastle, UK). All animal studies were approved by the Tohoku University Animal Welfare Committee.
Cell culture.
Human iPS cell-derived erythroid progenitor (HiDEP) cells were established by viral transduction of transcription factor TAL1 and human papillomavirus E6/E7 (27) and were cultured in StemSpan serum-free expansion medium (SFEM) (STEMCELL Technologies, Vancouver, BC, Canada) containing 3 U/ml erythropoietin (EPO; Kyowa Hakko Kirin, Tokyo, Japan), 1 μg/ml doxycycline (DOX; Sigma-Aldrich Corp., St. Louis, MO), and 1 μM dexamethasone (DEX; Sigma-Aldrich Corp.) (27). OP9 cells were cultured in α-minimum essential medium (α-MEM; Gibco, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) and 1% penicillin-streptomycin (Life Technologies). To induce erythroid differentiation, the HiDEP cells were seeded onto mitomycin C-treated OP9 cells in Iscove’s modified Dulbecco’s medium (IMDM; Sigma-Aldrich Corp.) supplemented with 20% FBS, insulin-transferrin-selenium (ITS; Gibco), 50 μg/ml l-ascorbic acid 2-phosphate (Sigma-Aldrich Corp.), 0.45 mM 1-thioglycerol (MTG; Sigma-Aldrich Corp.), 1% penicillin-streptomycin, 3 IU/ml EPO, 1 μg/ml DOX, and 1 μM DEX. 5-Aminolevulinic acid (ALA) hydrochloride and sodium ferrous citrate (SFC) were treated at concentrations of 1 mM and 100 μM, respectively (42). Holo-transferrin (holo-Tf; Sigma-Aldrich Corp.) was treated at a concentration of 100 μM (as an iron). ALA and SFC were kindly gifted by SBI Pharmaceuticals Co., Ltd. (Tokyo, Japan). OP9 cells were obtained from the American Type Culture Collection (Manassas, VA). Mitomycin C was obtained from Wako (Tokyo, Japan).
Human CD34+ cord blood cells were obtained from the Kanto-Koshinetsu Block Blood Center (Tokyo, Japan). Use of cord blood samples for the study was approved by the Ethics Committee of Tohoku University. Technical details of this method have been described previously (59). Briefly, CD34+ cord blood cells were cultured in expansion medium for 6 days, and then cells were cultured in differentiation medium for 8 days. Subsequently, the erythroblasts were seeded onto OP9 cells. The expansion medium was StemMACS HSC expansion medium XF human (magnetic-activated cell sorting, or MACS) with StemMACS HSC expansion cocktail human (MACS). The differentiation medium was StemSpan SFEM (STEMCELL Technologies) with human SCF (hSCF; 100 ng/ml; Peprotech, Rocky Hill, NJ), hFLT3-L (33.3 ng/ml; Peprotech), human interleukin-3 (13.3 ng/ml; Peprotech), hBMP4 (13.3 ng/ml, Peprotech), EPO (2.67 IU/ml; Kyowa Hakko Kirin, Tokyo, Japan), and hydrocortisone (1 μM; Sigma-Aldrich Corp.).
Generation of ALAS2-mutated cell lines using the CRISPR/Cas9 system.
The target sequence used to disrupt the GATA-1 binding regions of intron 1 in a human ALAS2 gene was 5′-AACTCTGGCAACTTTATCTG-3′. To introduce the mutation into HiDEP cells, the PrecisionX LentiCas9 SmartNuclease system (System Bioscience, Palo Alto, CA) was used according to the manufacturer’s protocol. Clonal cell lines were isolated by serial dilution, and mutations were detected by direct sequencing of the PCR products. The study protocol was approved by the Committee for Safe Handling of Living Modified Organisms of Tohoku University Graduate School of Medicine.
Isolation of bone marrow erythroblasts.
Bone marrow aspirates were overlaid on Ficoll-Paque Plus (GE Healthcare UK, Ltd., Buckinghamshire, England) to obtain mononuclear cells, and glycophorin A (GPA)-positive erythroblasts were then separated using the MACS system (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). In this study, bone marrow samples were obtained from a patient with XLSA harboring a mutation at the intron 1 enhancer region of the ALAS2 gene (16) and a healthy individual, after obtaining the written informed consent of both individuals. The genetic analysis was approved by the Ethical Committee of Tohoku University Graduate School of Medicine.
To separate murine erythroblasts from bone marrow, Ter119-positive erythroblasts were separated using a magnetic-activated cell sorting system (Miltenyi Biotec, Auburn, CA).
Quantitative RT-PCR analysis.
Total RNA was purified using TRIzol reagent (Invitrogen, Carlsbad, CA), followed by cDNA synthesis using the ReverTra Ace quantitative PCR RT master mix with guide RNA remover (Toyobo, Osaka, Japan). Quantitative RT-PCR was performed using QuantiTect SYBR green PCR master mix (Qiagen, Hilden, Germany). The primer sequences used are listed in Table S1 in the supplemental material. Data were normalized to the GAPDH mRNA expression levels.
Flow cytometry.
Cells were analyzed using a FACSAria II flow cytometer (Becton, Dickinson, Franklin Lakes, NJ), and data were analyzed using FACSDiva (Becton, Dickinson) or FlowJo software (TreeStar, Ashland, OR).
BrdU analysis.
Cell proliferation was analyzed using an allophycocyanin (APC) BrdU flow kit (BD Pharmingen, San Jose, CA) according to the manufacturer’s protocol. Briefly, cells were incubated in culture medium containing 10 μM BrdU for 1 h, stained with anti-BrdU antibody for 20 min at room temperature, and subsequently analyzed using a FACSAria II.
Measurement of intracellular ROS.
Cells were incubated in a medium containing 500 nM CellROX deep red reagent (Thermo Fisher Scientific, Waltham, MA) for 30 min at 37°C, followed by SYTOX blue dead cell staining (Thermo Fisher Scientific), and subsequently analyzed by flow cytometry. Fluorescence intensities were calculated by subtraction of nonstained from stained intensities.
Cytospin.
Cultured cells were centrifuged at 500 rpm for 3 min onto glass slides using a Shandon Cytospin 4 cytocentrifuge (Thermo, Pittsburgh, PA) and analyzed by microscopy after staining with May-Grünwald-Giemsa (Merck KGaA, Darmstadt, Germany) or Prussian blue (Sigma-Aldrich Corp.).
Microarray analysis.
Human oligonucleotide chip 25k (Toray, Tokyo, Japan) was used for expression profiling (59). GO analysis was performed using GeneCodis (http://genecodis.cnb.csic.es/).
Apoptosis analysis.
For FACS-based apoptosis analysis, cells were washed with cold phosphate-buffered saline and resuspended in annexin V binding buffer (eBioscience, San Diego, CA), followed by incubation with APC-annexin V (eBioscience) and propidium iodide (PI; eBioscience). To evaluate DNA fragmentation, DNA was extracted and stained using an apoptosis ladder detection kit (Wako, Tokyo, Japan).
Quantitative ChIP analysis.
Quantitative ChIP analysis was performed as previously described (16). The primer sequences used are listed in Table S1.
Western blotting.
Western blotting was conducted as previously described (59).
Antibodies.
Anti-GATA-1 (3535) antibody was obtained from Cell Signaling Technology (Danvers, MA). Anti-α-tubulin was obtained from Carbiochem (Darmstadt, Germany). Anti-TAL1 antibody (C-20) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-ALAS2 and control IgG were purchased from Abcam (Cambridge, MA). APC-labeled CD71 (551374) and peridinin chlorophyll protein/Cy 5.5-labeled CD235a (306614) were obtained from BD Biosciences (San Jose, CA) and BioLegend (San Diego, CA), respectively.
Intracellular heme content.
Intracellular heme content was determined by fluorescence (42).
Electron microscopy.
HiDEP cells were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer. The cells were then postfixed in 1% osmium tetroxide for 30 min at 4°C, rinsed in 0.1 M cacodylate buffer containing 8% sucrose, dehydrated in a graded series of alcohol and propylene oxide, and finally embedded in epoxy resin. Ultrathin (75 nm) sections were prepared using an ultramicrotome (UC-7; Leica, Heerbrugg, Switzerland) and stained with uranyl acetate and lead citrate before viewing with an electron microscope (H-7600; Hitachi, Tokyo, Japan).
Statistical analysis.
Statistical significance was assessed using a 2-sided Student t test. In all analyses, a P value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Department of Hematology and Rheumatology, Tohoku University Graduate School of Medicine, for helpful discussions and the staff of the Biomedical Research Core and Pathology Platform of Tohoku University and the Center for Laboratory Animal Research of Tohoku University for technical support. We appreciate SBI Pharmaceuticals Co., Ltd. (Tokyo, Japan), for providing ALA and SFC.
The study was supported by JSPS KAKENHI grant no. 18K08314 to T.F., 16K19564 to S.H., and 26293225/17H04668 to H.H.
K.S., T.F., and H.H. conceived and designed the experiments. K.S., T.F., S.H., M.M., K.O., and C.S. performed the experiments and analyzed the data. T.F., N.F., Y.O., Y.N., S.K., R.S., M.Y., and H.H. contributed reagents, materials, and/or analytical tools. K.S., T.F., and H.H. wrote the manuscript.
We have no conflicts of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00387-18.
REFERENCES
- 1.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. 1999. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet 21:396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- 2.Andrews NC. 1999. The iron transporter DMT1. Int J Biochem Cell Biol 31:991–994. [DOI] [PubMed] [Google Scholar]
- 3.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. 1997. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 4.Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD. 2012. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals 25:643–655. doi: 10.1007/s10534-012-9526-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeong J, Eide DJ. 2013. The SLC39 family of zinc transporters. Mol Aspects Med 34:612–619. doi: 10.1016/j.mam.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD. 2012. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem 287:34032–34043. doi: 10.1074/jbc.M112.367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC. 2005. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Investig 115:1258–1266. doi: 10.1172/JCI24356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gálvez-Peralta M, He L, Jorge-Nebert LF, Wang B, Miller ML, Eppert BL, Afton S, Nebert DW. 2012. ZIP8 zinc transporter: indispensable role for both multiple-organ organogenesis and hematopoiesis in utero. PLoS One 7:e36055. doi: 10.1371/journal.pone.0036055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rouault TA, Maio N. 2017. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J Biol Chem 292:12744–12753. doi: 10.1074/jbc.R117.789537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harigae H, Furuyama K. 2010. Hereditary sideroblastic anemia: pathophysiology and gene mutations. Int J Hematol 92:425–431. doi: 10.1007/s12185-010-0688-4. [DOI] [PubMed] [Google Scholar]
- 11.Fujiwara T, Harigae H. 2015. Biology of heme in mammalian erythroid cells and related disorders. Biomed Res Int 2015:278536. doi: 10.1155/2015/278536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bottomley SS, Fleming MD. 2014. Sideroblastic anemia: diagnosis and management. Hematol Oncol Clin North Am 28:653–670. v. doi: 10.1016/j.hoc.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Malcovati L, Cazzola M. 2013. Refractory anemia with ring sideroblasts. Best Pract Res Clin Haematol 26:377–385. doi: 10.1016/j.beha.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Yew NS, Federspiel M, Dodgson JB, Hayashi N, Engel JD. 1985. Isolation of recombinant cDNAs encoding chicken erythroid delta-aminolevulinate synthase. Proc Natl Acad Sci U S A 82:3702–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pevny L, Simon MC, Robertson E, Klein WH, Tsai S-F, D'Agati V, Orkin SH, Costantini F. 1991. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349:257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 16.Fujiwara T, O'Geen H, Keles S, Blahnik K, Linnemann AK, Kang Y-A, Choi K, Farnham PJ, Bresnick EH. 2009. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell 36:667–681. doi: 10.1016/j.molcel.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Campagna DR, de Bie CI, Schmitz-Abe K, Sweeney M, Sendamarai AK, Schmidt PJ, Heeney MM, Yntema HG, Kannengiesser C, Grandchamp B, Niemeyer CM, Knoers NV, Swart S, Marron G, van Wijk R, Raymakers RA, May A, Markianos K, Bottomley SS, Swinkels DW, Fleming MD. 2014. X-linked sideroblastic anemia due to ALAS2 intron 1 enhancer element GATA-binding site mutations. Am J Hematol 89:315–319. doi: 10.1002/ajh.23616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaneko K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Harigae H, Shibahara S. 2014. Identification of a novel erythroid-specific enhancer for the ALAS2 gene and its loss-of-function mutation which is associated with congenital sideroblastic anemia. Haematologica 99:252–261. doi: 10.3324/haematol.2013.085449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohba R, Furuyama K, Yoshida K, Fujiwara T, Fukuhara N, Onishi Y, Manabe A, Ito E, Ozawa K, Kojima S, Ogawa S, Harigae H. 2013. Clinical and genetic characteristics of congenital sideroblastic anemia: comparison with myelodysplastic syndrome with ring sideroblast (MDS-RS). Ann Hematol 92:1–9. doi: 10.1007/s00277-012-1564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakajima O, Takahashi S, Harigae H, Furuyama K, Hayashi N, Sassa S, Yamamoto M. 1999. Heme deficiency in erythroid lineage causes differentiation arrest and cytoplasmic iron overload. EMBO J 18:6282–6289. doi: 10.1093/emboj/18.22.6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harigae H, Nakajima O, Suwabe N, Yokoyama H, Furuyama K, Sasaki T, Kaku M, Yamamoto M, Sassa S. 2003. Aberrant iron accumulation and oxidized status of erythroid-specific delta-aminolevulinate synthase (ALAS2)-deficient definitive erythroblasts. Blood 101:1188–1193. doi: 10.1182/blood-2002-01-0309. [DOI] [PubMed] [Google Scholar]
- 22.Tanimura N, Miller E, Igarashi K, Yang D, Burstyn JN, Dewey CN, Bresnick EH. 2016. Mechanism governing heme synthesis reveals a GATA factor/heme circuit that controls differentiation. EMBO Rep 17:249–265. doi: 10.15252/embr.201541465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Zhang J, An W, Wan Y, Ma S, Yin J, Li X, Gao J, Yuan W, Guo Y, Engel JD, Shi L, Cheng T, Zhu X. 2017. Intron 1 GATA site enhances ALAS2 expression indispensably during erythroid differentiation. Nucleic Acids Res 45:657–671. doi: 10.1093/nar/gkw901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakajima O, Okano S, Harada H, Kusaka T, Gao X, Hosoya T, Suzuki N, Takahashi S, Yamamoto M. 2006. Transgenic rescue of erythroid 5 aminolevulinate synthase-deficient mice results in the formation of ring sideroblasts and siderocytes. Genes Cells 11:685–700. doi: 10.1111/j.1365-2443.2006.00973.x. [DOI] [PubMed] [Google Scholar]
- 25.Hatta S, Fujiwara T, Yamamoto T, Saito K, Kamata M, Tamai Y, Kawamata S, Harigae H. 2018. A defined culture method enabling the establishment of ring sideroblasts from induced pluripotent cells of X-linked sideroblastic anemia. Haematologica 103:e188–e191. doi: 10.3324/haematol.2017.179770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amos RJ, Miller AL, Amess JA. 1988. Autosomal inheritance of sideroblastic anemia. Clin Lab Haematol 10:347–353. [DOI] [PubMed] [Google Scholar]
- 27.Kurita R, Suda N, Sudo K, Miharada K, Hiroyama T, Miyoshi H, Tani K, Nakamura Y. 2013. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondo A, Fujiwara T, Okitsu Y, Fukuhara N, Onishi Y, Nakamura Y, Sawada K, Harigae H. 2016. Identification of a novel putative mitochondrial protein FAM210B associated with erythroid differentiation. Int J Hematol 103:387–395. doi: 10.1007/s12185-016-1968-4. [DOI] [PubMed] [Google Scholar]
- 29.Wadman IA, Osada H, Grütz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J 16:3145–3157. doi: 10.1093/emboj/16.11.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikkola HK, Klintman J, Yang H, Hock H, Schlaeger TM, Fujiwara Y, Orkin SH. 2003. Haematopoietic stem cells retain long-term repopulating activity and multipotency in the absence of stem-cell leukaemia SCL/tal-1 gene. Nature 421:547–551. doi: 10.1038/nature01345. [DOI] [PubMed] [Google Scholar]
- 31.Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, Gwynn B, Lambert AJ, Wingert RA, Traver D, Trede NS, Barut BA, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss MJ, Peters LL, Kaplan J, Zon LI, Paw BH. 2006. Mitoferrin is essential for erythroid iron assimilation. Nature 440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 32.Mosser DD, Caron AW, Bourget L, Meriin AB, Sherman MY, Morimoto RI, Massie B. 2000. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol 20:7146–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zelko IN, Mariani TJ, Folz RJ. 2002. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med 33:337–349. [DOI] [PubMed] [Google Scholar]
- 34.Kawase T, Ichikawa H, Ohta T, Nozaki N, Tashiro F, Ohki R, Taya Y. 2008. p53 target gene AEN is a nuclear exonuclease required for p53–dependent apoptosis. Oncogene 27:3797–3810. doi: 10.1038/onc.2008.32. [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, Rho SB, Chun T. 2005. GABAA receptor-associated protein (GABARAP) induces apoptosis by interacting with DEAD (Asp-Glu-Ala-Asp/His) box polypeptide 47 (DDX47). Biotechnol Lett 27:623–628. doi: 10.1007/s10529-005-3628-2. [DOI] [PubMed] [Google Scholar]
- 36.Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. 2012. GADD45 proteins: central players in tumorigenesis. Curr Mol Med 12:634–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segawa K, Nagata S. 2015. An apoptotic “eat me” signal: phosphatidylserine exposure. Trends Cell Biol 25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Nagata S. 2000. Apoptotic DNA fragmentation. Exp Cell Res 256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 39.Simon HU, Haj-Yehia A, Levi-Schaffer F. 2000. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 5:415–418. [DOI] [PubMed] [Google Scholar]
- 40.Circu ML, Aw TY. 2010. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghaffari S. 2008. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal 10:1923–1940. doi: 10.1089/ars.2008.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujiwara T, Okamoto K, Niikuni R, Takahashi K, Okitsu Y, Fukuhara N, Onishi Y, Ishizawa K, Ichinohasama R, Nakamura Y, Nakajima M, Tanaka T, Harigae H. 2014. Effect of 5-aminolevulinic acid on erythropoiesis: a preclinical in vitro characterization for the treatment of congenital sideroblastic anemia. Biochem Biophys Res Commun 454:102–108. doi: 10.1016/j.bbrc.2014.10.050. [DOI] [PubMed] [Google Scholar]
- 43.Furuyama K, Kaneko K. 2018. Iron metabolism in erythroid cells and patients with congenital sideroblastic anemia. Int J Hematol 107:44–54. doi: 10.1007/s12185-017-2368-0. [DOI] [PubMed] [Google Scholar]
- 44.Fujiwara T, Harigae H. 2018. Molecular pathophysiology and genetic mutations in congenital sideroblastic anemia. Free Radic Biol Med 2018:S0891-5849(18)31355-8. doi: 10.1016/j.freeradbiomed.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Sankaran VG, Ulirsch JC, Tchaikovskii V, Ludwig LS, Wakabayashi A, Kadirvel S, Lindsley RC, Bejar R, Shi J, Lovitch SB, Bishop DF, Steensma DP. 2015. X-linked macrocytic dyserythropoietic anemia in females with an ALAS2 mutation. J Clin Investig 125:1665–1669. doi: 10.1172/JCI78619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujiwara T, Fukuhara N, Ichikawa S, Kobayashi M, Okitsu Y, Onishi Y, Furuyama K, Harigae H. 2017. A novel heterozygous ALAS2 mutation in a female with macrocytic sideroblastic anemia resembling myelodysplastic syndrome with ring sideroblasts: a case report and literature review. Ann Hematol 96:1955–1957. doi: 10.1007/s00277-017-3106-7. [DOI] [PubMed] [Google Scholar]
- 47.Byrnes C, Lee YT, Meier ER, Rabel A, Sacks DB, Miller JL. 2016. Iron dose-dependent differentiation and enucleation of human erythroblasts in serum-free medium. J Tissue Eng Regen Med 10:E84–E89. doi: 10.1002/term.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chasis JA, Mohandas N. 2008. Erythroblastic islands: niches for erythropoiesis. Blood 112:470–478. doi: 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klei TR, Meinderts SM, van den Berg TK, van Bruggen R. 2017. From the cradle to the grave: the role of macrophages in erythropoiesis and erythrophagocytosis. Front Immunol 8:73. doi: 10.3389/fimmu.2017.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nairz M, Theurl I, Swirski FK, Weiss G. 2017. “Pumping iron”—how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch 469:397–418. doi: 10.1007/s00424-017-1944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korolnek T, Hamza I. 2015. Macrophages and iron trafficking at the birth and death of red cells. Blood 125:2893–2897. doi: 10.1182/blood-2014-12-567776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakamoto S, Kawabata H, Masuda T, Uchiyama T, Mizumoto C, Ohmori K, Koeffler HP, Kadowaki N, Takaori-Kondo A. 2015. H-ferritin is preferentially incorporated by human erythroid cells through transferrin receptor 1 in a threshold-dependent manner. PLoS One 10:e0139915. doi: 10.1371/journal.pone.0139915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, Wu Q, Zhang Y, Yu Y, Ning B, Nie G, Knutson MD, Anderson GJ, Wang F. 2011. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 118:1912–1922. doi: 10.1182/blood-2011-01-330324. [DOI] [PubMed] [Google Scholar]
- 54.Matthes TW, Meyer G, Samii K, Beris P. 2000. Increased apoptosis in acquired sideroblastic anaemia. Br J Haematol 111:843–852. [PubMed] [Google Scholar]
- 55.Hellström-Lindberg E, Schmidt-Mende J, Forsblom AM, Christensson B, Fadeel B, Zhivotovsky B. 2001. Apoptosis in refractory anaemia with ringed sideroblasts is initiated at the stem cell level and associated with increased activation of caspases. Br J Haematol 112:714–726. [DOI] [PubMed] [Google Scholar]
- 56.Tehranchi R, Invernizzi R, Grandien A, Zhivotovsky B, Fadeel B, Forsblom AM, Travaglino E, Samuelsson J, Hast R, Nilsson L, Cazzola M, Wibom R, Hellström-Lindberg E. 2005. Aberrant mitochondrial iron distribution and maturation arrest characterize early erythroid precursors in low-risk myelodysplastic syndromes. Blood 106:247–253. doi: 10.1182/blood-2004-12-4649. [DOI] [PubMed] [Google Scholar]
- 57.Ishizuka M, Abe F, Sano Y, Takahashi K, Inoue K, Nakajima M, Kohda T, Komatsu N, Ogura S, Tanaka T. 2011. Novel development of 5-aminolevurinic acid (ALA) in cancer diagnoses and therapy. Int Immunopharmacol 11:358–365. doi: 10.1016/j.intimp.2010.11.029. [DOI] [PubMed] [Google Scholar]
- 58.Ishida H, Imamura T, Morimoto A, Fujiwara T, Harigae H. 2018. Five-aminolevulinic acid: new approach for congenital sideroblastic anemia. Pediatr Int 60:496–497. doi: 10.1111/ped.13558. [DOI] [PubMed] [Google Scholar]
- 59.Fujiwara T, Saitoh H, Inoue A, Kobayashi M, Okitsu Y, Katsuoka Y, Fukuhara N, Onishi Y, Ishizawa K, Ichinohasama R, Harigae H. 2014. 3-Deazaneplanocin A (DZNep), an inhibitor of S-adenosylmethionine-dependent methyltransferase, promotes erythroid differentiation. J Biol Chem 289:8121–8134. doi: 10.1074/jbc.M114.548651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.