

Abstract
Systematic assessment of tyrosine kinase-substrate relationships is fundamental to a better understanding of cellular signaling and its profound alterations in human diseases such as cancer. In human cells, complex signaling networks, feedback loops, conditional activity and intra-kinase redundancy combine to confound systematic attempts to address this topic. We leveraged the model organism S. cerevisiae to individually express human non-receptor tyrosine kinases (NRTKs) and exploited the full yeast proteome as an in vivo model substrate. For 16 NRTKs, we recorded 3,279 kinase-substrate relationships involving 1,351 yeast pY-sites. From this data we generated a set of new linear kinase motifs and assigned ~1,300 known human pY-sites to specific NRTKs. Furthermore, experimentally defined pY-sites for each individual kinase were shown to cluster within the yeast interactome network irrespective of linear motif information. We therefore applied a network inference approach to predict kinase-substrate relationships for more than 3,500 human proteins, marking a substantial step forward in our understanding of kinase biology.
Introduction
Cells of all organisms store and transmit information via post-translational modification (PTM) of proteins such as phosphorylation of serine, threonine or tyrosine side chains by protein kinases. Phosphorylation regulates a vast array of cellular processes and its deregulation is central to disease such as cancer. In order to understand how cellular signaling impacts upon normally functioning or disease processes, it is necessary to define kinase-substrate relationships. However, less than ten percent of over 200,000 human phospho-sites are linked to responsible protein kinases (Hornbeck et al., 2012). How the set of ~500 human protein kinases (Manning et al., 2002) specifically phosphorylates ~400 times the number of phospho-sites on proteins is a largely unanswered question.
Major obstacles in defining kinase-substrate relationships stem from the fact that at any point in time, kinases are differentially expressed dependent on cell type or cell cycle phase or subcellular localization, exhibit partly overlapping substrate specificity and have magnitude differences in enzymatic activity. Furthermore, kinases form complex signaling networks containing layers of redundancy and feedback loops, hindering identification of kinase targets using traditional perturbation approaches in the context of endogenous cellular signaling. Therefore kinase specificity determinants were mainly assayed with purified kinases and synthetic peptide substrates in vitro. As a result, while specificity determinants such as docking sites have been reported (Ubersax and Ferrell, 2007; Zeke et al., 2015), the primary amino acid sequence surrounding the phosphorylation site, referred to as kinase motif, is the predominant specificity determinant studied to date (Miller et al., 2008; Mok et al., 2010; Deng et al., 2014; Duarte et al., 2014). As alternatives to synthetic peptide-based approaches proteome-derived peptide libraries constructed from tryptic digests of human cell lysates were used to probe CK2 kinase specificity (Wang et al., 2013), and a micro array of 4191 full-length human proteins was used to assay kinase-substrate relationships for 289 kinases (Newman et al., 2013). To bypass endogenous kinase activity, motifs were also revealed by mass spectrometry for recombinant serine/threonine kinases PKA and CK2 expressed in bacteria (Chou et al., 2012). Utilizing experimentally derived motif data, a variety of computational approaches for scoring linear kinase motifs in the proteome to predict putative substrate sites have been developed (Miller et al., 2008; Xue et al., 2008; Hu et al., 2014). Linding et al. improved motif-based predictions by including contextual information, mainly protein-protein interaction (PPI) networks, in a machine learning approach (Linding et al., 2007). On top of phospho-sites that match known kinase motifs, they demonstrated that network context of kinases and phosphoproteins can contribute up to 60–80% to kinase-substrate specificity. However 80%, of human phosphorylated sites do not match any currently known kinase motifs, limiting the predictive power of such approaches. Here we use the in vivo yeast proteome as a model substrate for individual human tyrosine kinases to characterize pY-sites that elude kinase-substrate prediction with linear motif based approaches.
In contrast to serine/threonine signaling, tyrosine signaling can be regarded as a hallmark of multi-cellularity and has not evolved in yeast. Bona-fide protein tyrosine kinase (PTK) sequences - 58 cell membrane-spanning receptor tyrosine kinases and 32 non-receptor tyrosine kinases (NRTKs) in human - were not detected and tyrosine kinase orthologs are absent in fungi (Manning et al., 2002). Protein tyrosine kinase activity in yeast is low (Schieven et al., 1986) and only few phosphorylated tyrosine residues are known in yeast (Gnad et al., 2009) likely due to a few dual-specificity kinases. As such yeast can be leveraged as a background-free, eukaryotic expression system in which to study tyrosine kinase activity. Early indications of heterologous PTK activity in yeast were observed through growth inhibition upon overexpression of v-SRC, later explained by aberrant phosphorylation of yeast proteins (Brugge et al., 1987; Kornbluth et al., 1987; Cooper and MacAuley, 1988; Florio et al., 1994). The toxic effect of PTK activity upon overproduction in yeast was exploited for screening of kinase inhibitors or phosphatases which restore yeast growth (Montalibet and Kennedy, 2004; Koyama et al., 2006; Harris et al., 2013). Despite these overexpression toxicity issues, Nada and colleagues (Nada et al., 1991) effectively used a heterologous yeast system to discover that CSK negatively regulates SRC by C-terminal tyrosine phosphorylation. In addition SRC- FES- and HCK-kinase regulatory mechanisms were further investigated in Saccharomyces cerevisiae (e.g. (Murphy et al., 1993; Superti-Furga et al., 1993; Takashima et al., 2003; Lerner et al., 2005)). C-ABL auto-inhibition was analyzed in Schizosaccharomyces pombe (Pluk et al., 2002) exploiting the absence of inhibitory factors. Finally, the first systematic use of low level human NRTK expression in yeast enabled screening of phospho-tyrosine dependent interactions on a proteome scale (Grossmann et al., 2015).
Here, we describe an alternative approach of individually-expressing active human NRTKs in yeast to comprehensively record pY-sites on the yeast proteome using mass spectrometry. We exploit the complete proteome in living S. cerevisiae as a model substrate for individual human NRTKs. In our approach the yeast proteome serves as a fully folded substrate space that is phosphorylated by specific human kinases in vivo. pY-sites can be recorded from a crowded, competitive, cellular context and directly attributed to the kinase expressed. The yeast proteome is one of the best characterized, spanning more than 5 orders of magnitude in protein concentration (Wang et al., 2012) and 30% of the yeast proteins have homologous proteins in human. Furthermore, the yeast protein interaction network is very well mapped, with more than 60,000 high confidence interactions reported to date (Gavin et al., 2006; Krogan et al., 2006; Yu et al., 2008) and serves as a reliable basis for network analyses. Finally, yeast can be grown in large amount enabling robust phospho-proteomics data set recording.
We take three different routes to exploit the recorded pY-data and infer human kinase-substrate relationships for known human pY-sites (Figure 1a). Firstly, we directly transfer kinase-substrate relationships through sequence homology to human proteins. Secondly, we de novo define linear sequence motifs for 16 non-receptor human kinases to score known human pY sites. Thirdly, we use a network inference approach to assign human kinases to a large fraction of known human pY-sites independently of linear motif signatures. As such we infer thousands of kinase-substrate relationships in human, marking a large step forward in our understanding of phosphorylation specificity.
Figure 1. Assaying human tyrosine kinases in yeast.
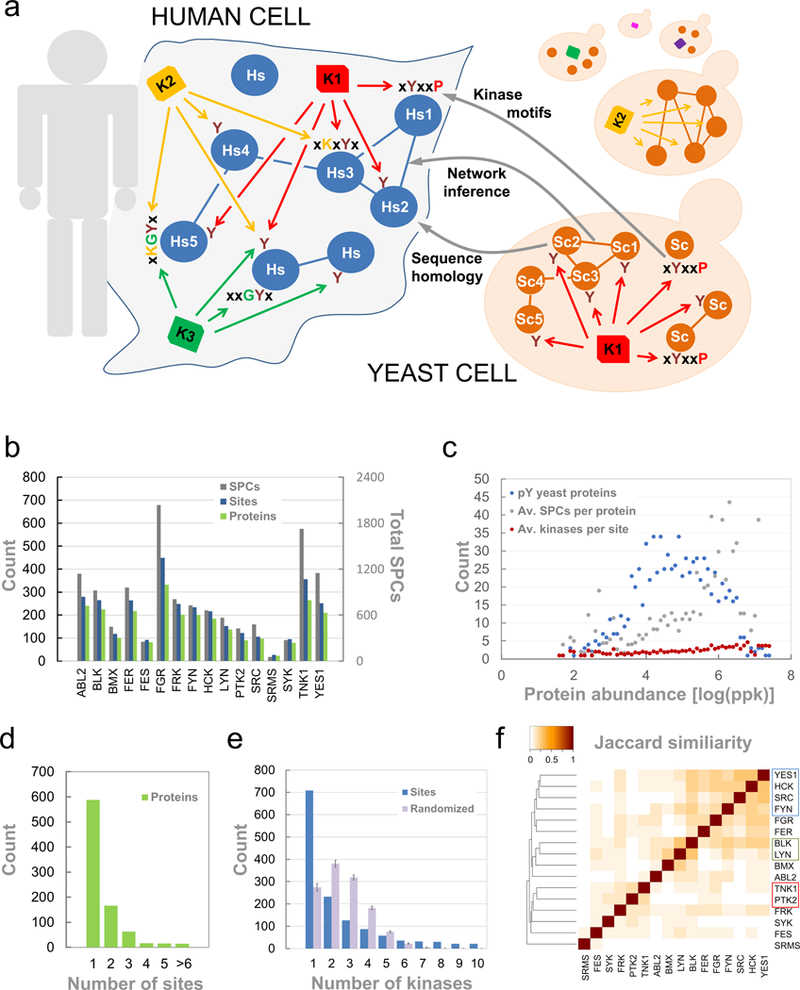
a. Workflow of the analyses. In human kinases have overlapping substrate specificity and conditional enzymatic activity. Therefore kinase-substrate relationships are difficult to assess. Human NRTKs were individually expressed in S.cerevisiae and tyrosine phosphorylation was detected via mass spectrometry. Data analysis provided clues to human kinase-substrate relationships via i) sequence homology, ii) kinase motifs and iii) network inference. b. Overview of the pY-data determined by mass spectrometry in yeast. 16 different kinases were assayed and the number of spectral counts (grey), pY-sites (blue) and phosphorylated proteins (green) per kinase are given. In total, the data involved 12,625 SPCs covering 1,351 pY-sites on 862 yeast proteins. c. Abundance distribution of all pY-modified proteins is shown according to “whole organism-SC (PeptideAtlas)” data set from PaxDB (Wang et al., 2012). Blue: number of proteins measured (843/862 mapped). Grey: Average number of spectral counts for proteins as a function of relative protein abundance. Red. Average number of kinases per sites as a function of relative protein abundance. d. Overview of the pY-data determined by mass spectrometry in yeast. Number of pY-sites per protein shows that 2/3 of the proteins were modified at a single site. e. Overview of the pY-data determined by mass spectrometry in yeast. Number of kinases modifying pY-sites (blue). More than 700 pY-sites (52%) were modified by a single kinase. Randomized: An equal number of phosphorylation sites as present in the experimental data were randomly sampled from the total dataset for every kinase. The average number of kinases per site was then calculated from 100 randomized dataset (grey). Error bars represent the standard deviation. f. Pairwise overlap of pY-sites between different kinases. Hierarchical clustering of the Jaccard indices for the pairwise pY-site overlap revealed similarities between subset of kinases. Src kinase family members clustered together (blue) as well as BLK and LYN (red) and TNK1 with PTK2 (green), respectively.
Results
Measuring tyrosine phosphorylation by human kinases in yeast
In a recent study, we generated yeast strains individually expressing human NRTKs for the systematic analysis of phospho-tyrosine (pY) dependent interactions (Grossmann et al., 2015). We expressed active full-length NRTKs at very low levels and did not observe toxicity of the L40c-Y2H S. cerevisiae strain under conditions of fast growth (Suppl. Figure 1a). When we probed yeast lysates via western blotting with a pan pY antibody, we observed that human NRTKs modify large sets of yeast proteins (Suppl. Figure 1b). Human kinases show distinct activities, as indicated by the pattern of pY-proteins that was observed on the blots. We then set out to comprehensively map pY-sites on the yeast proteome for a set of human NRTKs by phospho-peptide immunoaffinity enrichment followed by tandem mass spectrometry. As such, the yeast proteome served as a fully folded, dynamically-expressed substrate space reflecting a crowded, competitive, cellular context that was phosphorylated by specific kinases in vivo. Analyses of pY-sites recorded in yeast may thus provide clues to kinase-substrate specificity via at least three different routes (Figure 1a).
In order to comprehensively record pY-sites on the yeast proteome we applied a commercial phospho-tyrosine enrichment protocol (Rush et al., 2005). We tailored the protocol specifically: starting with liter cultures expressing a single human NRTK (i.e. up to 100 mg wet protein), tryptic pY-peptides were enriched subsequently by applying pTyr-100-AB and 4G10-AB conjugates. pY-peptides were measured on a liquid chromatography-coupled LTQ-Orbitrap tandem mass spectrometer and mapped to the yeast proteome using the SEQUEST algorithm (Ballif et al., 2008; Eng et al., 2008). Overall yeast strains expressing 16 of the 32 NRTKs known in human were successfully assayed (Suppl. Table 1). Except for the JAK and CSK family of kinases, at least one member representative of each of the 10 NRTK families showed activity in yeast (Suppl. Figure 1c).
Our final dataset included a total of 12,625 quality-filtered pY-peptides (SPCs, Suppl. Table 2a) mapping to 1,351 unique pY-sites on 862 yeast proteins and 3,279 kinase-substrate relationships (Suppl. Table 2b). For this final dataset we excluded known endogenous pY-sites in yeast (Gnad et al., 2009; Tan et al., 2009a; Hornbeck et al., 2012) and sites that were found with most kinases (>10) and with high number of spectral counts (a total of 7710 SPCs for 55 sites were removed; Suppl. Table 2c). The dataset is characterized by a median number of 694 spectral counts (SPCs) of pY-containing peptides per kinase, with the majority of kinases reporting well over 100 phosphorylation sites (Figure 1b). As the concentration of proteins in yeast is distributed over at least 5 orders of magnitude (Wang et al., 2012), we characterized the phosphorylated proteins recorded here to observe any abundance bias present in the data. As would be expected from any mass spectrometry based measurement our measured proteins showed a shift towards more abundant proteins (Suppl. Figure 1d). Furthermore, the SPCs per protein (number of pY-peptides measured per protein) increased with protein abundance (Figure 1c), which prevented us from using any quantitative information on the identified sites; rather we took each pY-site as a binary annotation in all further analyses. Importantly however, the number of kinases per site (median = 1, average = 2.4) shows minimal increase over at least 4 orders of magnitude of protein abundance, covering the vast majority of our data (Figure 1c, red dots). As such, using this in vivo model system we covered several orders of magnitude of cellular concentrations, with very little evidence to suggest global protein abundance drives the recorded Y-phosphorylation.
As expected, the number of tyrosine phosphorylation sites per protein showed a tailed distribution (Figure 1d) as well as the number of kinases found to phosphorylate any given site (Figure 1e) or protein (Suppl. Figure1e). Two thirds of all identified substrates were modified on one tyrosine and about half of the identified sites (52%; 708/1351) were modified by a single kinase only. In order to investigate whether this distribution is expected we performed a computational permutation analysis, maintaining the data structures present in the original dataset. For each kinase we randomly sampled the same number of pY sites as annotated in the experimental dataset from the total list of 1351 sites and plotted the randomized data distribution for both kinases per site (Figure 1e) and kinases per protein (Suppl. Figure 1e). Our data contain both a higher number of sites only phosphorylated by a single kinases and a higher number of sites modified by a larger number of kinases. Therefore, in addition to a small number of pY-hubs, kinases generally targeted more distinct protein sites in S. cerevisiae than would be expected by random chance, showing relatively low substrate overlap.
Visualizing the pairwise overlap of kinase targets highlighted some similarities between kinases. Related SRC-kinase family members YES1, SRC, FYN and HCK cluster together (Figure 1f). LYN, BMX and BLK share several target sites in agreement with their similarities (e.g. in domain content and organization). The relatively high target overlap between the FAK-family kinase PTK2 and the ACK-family kinase TNK1 is less expected as they have different domain structures (Suppl. Figure 1c).
Previous observation report that tyrosine phosphorylation, in comparison to serine/threonine phosphorylation, shows lower propensity to cluster in disordered regions of protein sequence in vivo (Woodsmith et al., 2013). We compared protein disorder of both modified and unmodified Y and S/T sites in yeast to general protein disorder, utilizing IUPred for disorder prediction (Dosztanyi et al., 2005). The percentages of unmodified tyrosines predicted to adopt a disordered conformation were smaller (8.5%) than for all residues (17.5%) and pY-sites (9.7%) showed a modest increase (Suppl. Figure 1f). These are both substantially lower than both modified (63.4%) and unmodified S/T residue disorder (31.3%). The pY disorder increase is comparatively small, but in generally these trends in the yeast proteome recapitulated the characteristic structural property of human tyrosine phosphorylation and are similar using alternative disorder prediction methods (Suppl. Figure 1f).
Transfer of pY kinase target sites from yeast to human via sequence homology
Approximately 30% of proteins are conserved between yeast and human and we would expect that some of the pY-sites in yeast may have homologous pY-sites in human (Figure 1a). Using the InParanoid database (Remm et al., 2001) we obtained sequence alignments from up to 20 species without tyrosine specific kinases (nonTK-group, including S. cerevisiae) and from up to 17 species with an evolved tyrosine signaling protein repertoire with tyrosine kinases (TK-group, including human). For 479 yeast ORFs with at least one measured pY-site we obtained alignments covering 942 phosphorylated and 9,644 non-phosphorylated tyrosine residues. To compare the conservation of any tyrosine through phylogeny between the TK-group and nonTK-group we calculated a Y-score that indicates the preference in tyrosine conservation between the non-TK and the TK-species (Figure 2a). Values at zero reflected no preference in tyrosine conservation, typically characteristic of very well-conserved pY-sites within an almost invariant sequence stretch. An example is Y654 in yeast protein cdc48 in line with the possibility that the kinases targeting this site could modify the corresponding Y644 site in human VCP (Figure 2a). A somewhat stronger inference can be made for pY-sites that are better conserved in the TK group than the non-TK group, pointing to a functional constraint on the tyrosine that may be linked to phosphorylation. For example, Y13 on RPL21 is phosphorylated by TNK1 and has a high Y-score of 1.65, indicating a strong conservation of the tyrosine in species capable of modifying it. We also detected pY-sites with negative scores and additionally many tyrosine-to-phenylalanine substitutions in the TK-group (e.g. ssa1/HSPA8 with a Y-score of −1.4; Figure 2a).
Figure 2. Homology analysis.
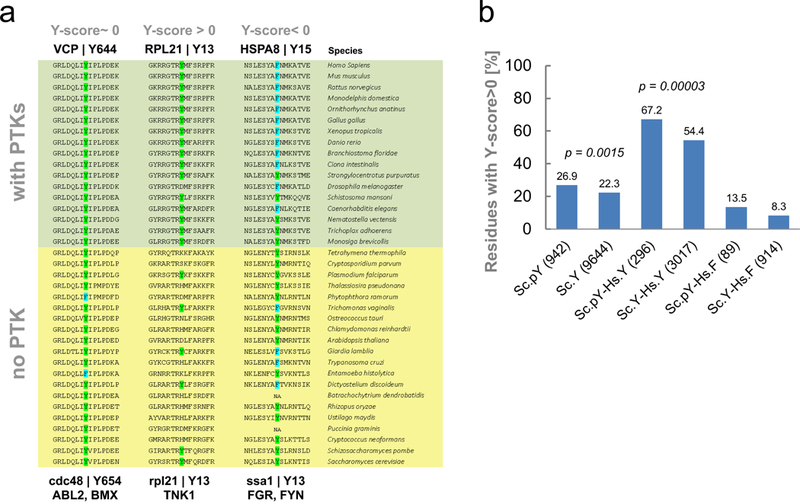
a. Three selected multiple sequence alignments. 479 sequence alignments for yeast proteins with pY-sites were retrieved including up to 17 species with annotated NRTKs (TK-group) and up 20 species without NRTK (nonTK-group). The nonTK-group of species (yellow) includes S. cerevisiae at the bottom and the TK-group of species (green) includes human at the top. A Y-score determined the fraction of tyrosines in the TK-group over the nonTK-group. The cdc48 (S.c.) Y654 is well conserved throughout to human (VCP, Y644) with a Y-score close to zero. Y13 in rpl21 (S.c.) is better conserved in the TK-group, Y-score > 0, and Y13 in ssa1 (S.c.) is largely replaced by a phenylalanine (cyan) in species of the TK-group. A Y-score < 0 may indicate counter selection of the tyrosine residue. b. Statistical analyses of the Y conservation between yeast and human. Percentage of Y-sites in the alignments with Y-scores larger than zero are shown. A score for a total of 942 pY-sites in yeast (Sc.pY) and 9644 non phosphorylated tyrosines on the same proteins (Sc.Y) were calculated. The fraction of positive Y-scores is higher for phosphorylated sites in yeast than for non-phosphorylated sites. This is also observed for pY-sites in yeast that also have a tyrosine in human (Sc.pY-Hs.Y) showing a significantly higher fraction of positive Y-scores than the non-modified tyrosine residues with a tyrosine in human (Sc.Y-Hs.Y) in the corresponding set of proteins (chi-square test). For yeast tyrosine residues that align to a phenylalanine in human (Sc.Y-Hs.F), phenylalanine residues are more prominent in the TK-group of species for phosphorylated residues (Sc.pY-Hs.F; non-significant).
Overall, the 942 aligned phosphorylated yeast tyrosine residues are significantly more conserved within the TK-species group than the non-phosphorylated tyrosines in the same proteins (Sc.pY vs Sc.Y, Figure 2b). This observation also holds for two thirds of the 296 cases where the pY-site in yeast locally aligns to a tyrosine in the human ortholog (Figure 2b, Sc.pY-Hs.Y = 67.2% and Sc.Y-Hs.Y=54.4%). With this analysis we provide evidence for previously unreported human kinase-substrate relationships for 63 of the 296 yeast sites that locally align to a tyrosine in human as they are reported to be phosphorylated in human (Suppl. Table 3).
The fraction of phenylalanine residue with higher conservation in the TK-group is almost identical to the fraction of conserved Ys (Sc.F-H.s.F= 54.2%). However, 89 of the phosphorylated tyrosines in yeast are phenylalanine residues in human. These sites also had a much lower fraction of Ys in the TK-group (Sc.pY-Hs.F = 13.5%) and a median Y-score below zero. Tan et al. (Tan et al., 2009b) observed negative correlation of total protein tyrosine content in the proteomes of organisms with increasing number of cell types or increasing number of predicted tyrosine kinases from yeast to human. It remains controversial if this apparent counter-selection of tyrosine residues in species with tyrosine signaling can be attributed to beneficial reduction of adventitious tyrosine phosphorylation (Tan et al., 2009b) or to other reasons (Pandya et al., 2015). Our data provide 89 testable cases for further investigation into this topic as candidate sites which may have been selected against in species with tyrosine kinases.
Linear sequence motif analysis
Kinase specificities are modelled through degenerate linear sequence motifs flanking the P-site from known kinase-substrate relationships (Miller et al., 2008). For about 28 NRTKs or NRTK subfamilies, between 4 and 400 pY-sites can be collected from the literature, however kinase motifs typically represent the averaged specificity of several kinases within a certain kinase family (Miller et al., 2008; Wagih et al., 2015). We measured between 27 (SRMS) and 449 (FGR) pY-sites per kinase and used iceLogo (Colaert et al., 2009) to determine over- and under-represented amino acids from the alignment of seven amino acids flanking the pY-sites for all 16 kinases (Figure 3a and Suppl. Figure 2). To evaluate the predictive performance of the motifs using receiver operating characteristic (ROC) analysis, we computed the area under the curve (AUC) in a 10-fold cross validation procedure recalling pY-sites for the corresponding kinase in a background of all other tyrosine residues contained in the set of 862 modified proteins (Figure 3b). 10-fold cross-validation produced AUC values in the range of 0.66–0.78 (Figure 3c and Suppl. Figure 3a). Importantly, available motifs in the literature agree with the motifs obtained in our approach. For example a strong preference for a proline at the +3 position for ABL2 is recapitulated (Colicelli, 2010; Wagih et al., 2015) (Figure 3a) as well as a preference for acidic amino acids (D and E) at the −3 position for SRC kinase (Songyang and Cantley, 1995) or the preference for aspartic acid at the −1 position for SYK (Deng et al., 2014; Shah et al., 2016) (Suppl. Figure 2). Additionally, we refined our motif by discarding sequences with low motif score and redrew the linear motif for every kinase using the 20% best matching peptides only (Figure 3a and Suppl. Figure 2). Similar approaches for motif refinement have been used in an iterative manner before (Schwartz and Gygi, 2005; Wagih et al., 2015) as this procedure enriches for residues that are more likely contributing to binding. Importantly, at very high accuracy cut off values (i.e. combined sensitivity and specificity) the false discovery rate dropped substantially when scoring the pY-sites of the yeast proteome with the refined motifs. This drop in FDR is not due the refinement procedure as such, because it is not observed in randomized controls (Suppl. Figure 3b). Scoring the yeast proteome at an accuracy value of 0.995, we retrieved 918 kinase-site pairs and predicted 663 (72%) correctly (FDR=0.278).
Figure 3. Kinase motifs.
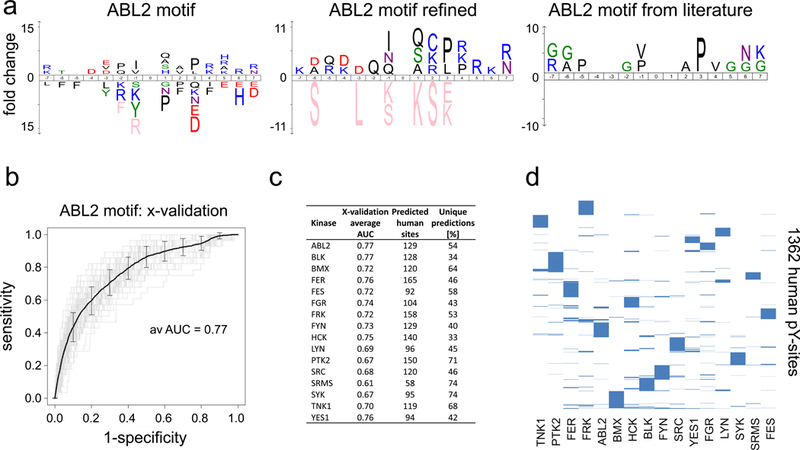
a. Motif generation. Using ico-Logo (Colaert et al., 2009) linear sequence motifs covering 7 amino acids N- and and C-terminal of the phosphorylated tyrosine residues (position 0, Y not shown). Novel motifs were generated for all 16 NTRKS and compared to the literature in ROC analyses (Suppl. Figure 2), ABL2 motifs are exemplarily shown. b. 10-fold cross-validation for kinase motifs derived from yeast pY-sites resulted AUC values in the range of 0.66–0.78. The ROC curve for ABL2 motif is exemplarily shown (see Suppl. Figure 3a). c. Summary of motif analyses and motif predictions for human phospho-tyrosine sites. AUC values from the cross-validation (x-validation average AUC) are listed. At an accuracy cut-off of 0.995 between 165 and 92 human pY-sites were scored (Predicted human sites). About 50% of the motif based kinase-substrate assignments are unique for a single kinase (Unique predictions). d. Human pY-sites with motif based kinase assignments. Graphical representation showing motif based kinase-substrate relationships for 1362 human phospho-tyrosine sites.
We quantitatively compared the published linear motifs for six kinases from Deng et al. (Deng et al., 2014) and three kinases from Wagih et al. (Wagih et al., 2015) using ROC analyses (Suppl. Figure 2). The analysis demonstrated that with the exception of SYK kinase in all comparisons the refined motifs generated from the yeast data perform comparable or better than the reported motifs when benchmarked with independent human pY-data (Suppl. Figure 2).
We next set out to use the new motifs to link kinases to ~13,240 human phospho-tyrosine-sites recorded in multiple studies from human cells (Hornbeck et al., 2012) predicting potential kinase-substrate relationships. At the accuracy cut off of 0.995 we assigned kinase-substrate relationships for 1,362 human pY-sites (Suppl. Table 4) with roughly half of the 1,105 predicted human target proteins were assigned to a single NRTK (Figure 3d). The number of predicted human targets varied from 58 for SRMS to 165 for FER (Figure 3c) with an average of 3% of the proteins containing two sites for one kinase. 20 kinase-substrate pairs were confirmed through reports in the literature (Suppl. Table 5). In summary, we defined motifs for 16 individual NRTKs from the analysis of our yeast data, benchmarked the motifs against known human kinase-substrate relationship and predicted about 1,900 kinase-substrate relationships for more than 1,100 human phospho-proteins.
Validation of human p-Y site prediction
In order to confirm predictions experimentally we expressed putative human target proteins in the yeast strains with the corresponding kinases. PGAM1 (phosphoglycerate mutase 1), EIF2S1 (eukaryotic translation initiation factor 2 subunit 1) and PGK1 (phosphoglycerate kinase 1) were successfully purified via immunoprecipitation as determined by coomassie stained SDS-page and subjected to mass spectrometry based phospho-peptide identification. Peptides for the phosphorylated and non-phosphorylated form were unambiguously identified and validated in comparison to reference spectra from peptide atlas (Suppl. Figure 3c). Four predicted sites were confirmed in this approach: One ABL2 pY-site in PGAM1 (Y92) and three FGR sites across EIF2S1 (Y147 and Y150) and PGK1 (Y76) respectively. On the other hand Y196 in PGK1 was predicted to be phosphorylated by FGR but was not found in the analyses. However, in this validation approach we cannot distinguish true negative from false negative results as some tryptic peptides may not be suitable for MS identification. Rather, in support of our approach these experiments demonstrate that known pY-sites in human proteins can be phosphorylated by the assigned kinases.
Network inference analyses of pY-sites of the yeast model substrate
Deriving new kinase motifs from our data, we have extended the motif based substrate scoring and attributed about 10% of the human pY-sites to specific kinases. However, on a proteome scale, the local properties of the pY-site, i.e. the amino acids surrounding the site, are alone insufficient to define the substrate specificities of protein kinases as the majority of phospho-sites recorded in living cells do not resemble any known kinase motif.
In our in vivo experimental system we did not determine tyrosine phosphorylation of substrates in isolation, but in the context of interaction networks that reflect the organization of protein assemblies and cellular processes. In yeast, where tyrosine phosphorylation does not play a role in bona fide cellular processes, phosphorylation will occur preferentially at sites that match recognition determinants in individual proteins but we would expect it to occur randomly, i.e. equally distributed, with respect to protein interaction networks. We therefore asked whether yeast proteins modified by a human tyrosine kinase are more closely connected in protein interaction networks than expected randomly. We calculated the average shortest path between all nodes targeted by each kinase in an established yeast protein interaction network (63,545 PPIs, 5,804 proteins, Data S1) in comparison to 100 networks where the nodes were randomized keeping their degree. For all kinases, except SRMS that only targeted less than 20 proteins in the network, the average shortest path was significantly smaller than in the randomized network versions (Figure 4a, Z-score > 2). Scrutinizing the null hypothesis again to corroborate this observation, we also tested whether two proteins that were modified by a kinase were more likely to interact than expected randomly. The average number of interacting kinase target pairs was much higher for most kinases than in the corresponding networks with randomized links that have the same size and degree distribution, keeping the number of interactions for each protein constant (Z-score > 2; Figure 4b). Both these analyses demonstrate clustering of kinase substrates in the yeast interaction network.
Figure 4. Network inference.
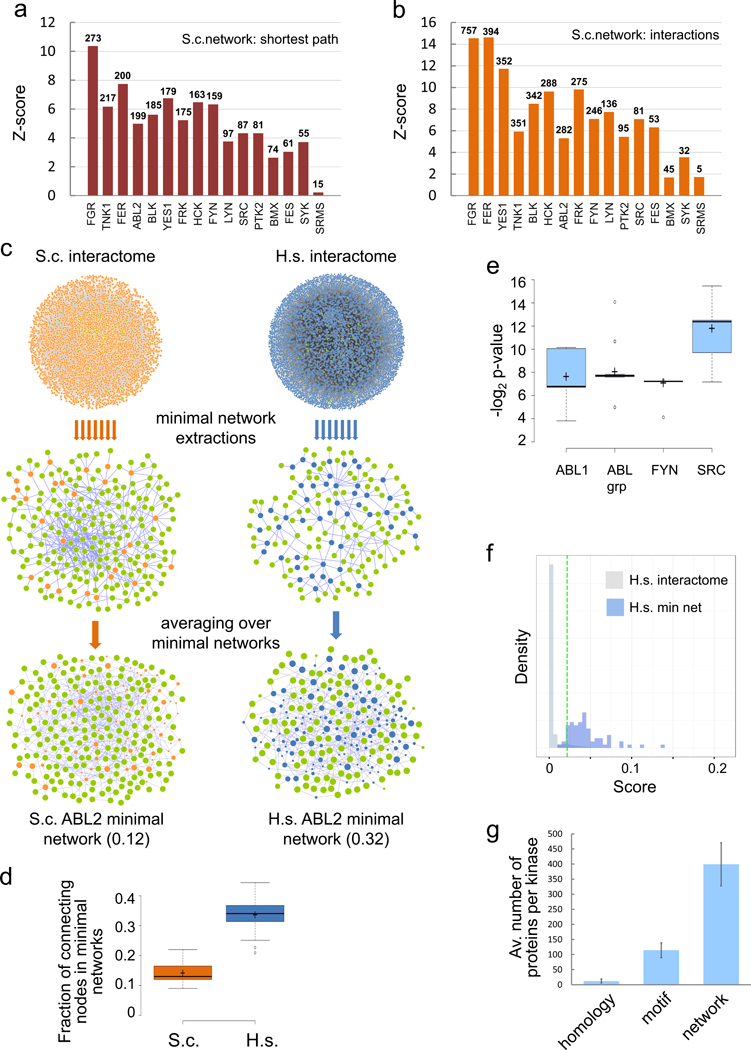
a. Phosphorylated proteins are close in the yeast interactome network. The average shortest path between proteins phosphorylated by a human NRTK in yeast is significantly shorter in the yeast PPI network than in 100 randomized networks (node randomization). Number of pY-proteins in the network is indicated. b. Phosphorylated proteins preferentially interact in the yeast interactome network. Number of interacting protein pairs which were phosphorylated by a human NRTK in yeast is significantly higher in the yeast PPI network than in 100 randomized versions (link randomization). Number of interacting pY-protein pairs is indicated. c. Extraction of minimal networks from global interactome maps. Left, interactome of S. cerevisiae with proteins phosphorylated by Human ABL2 kinase colored in green. Minimal networks containing all phosphorylated proteins and on average 12% non-phosphorylated proteins (orange) were extracted. Right, the network extraction approach is applied to the human interactome using seed proteins for human ABL2 NRTK. Minimal networks contain about 32% non-seed proteins, and statistical analyses of 20 minimal networks provides a measure (size of blue nodes) to assign putative substrates among non-seed proteins which are reported to be phosphorylated in human. d. Average fraction of non-seed nodes contained in minimal networks for all 16 NRTKs in S.c. networks and 19 NRTKs in H.s. networks, respectively. e. Benchmark of network extraction. Known substrates for ABL1, ABLgrp, FYN and SRC were omitted from the group of seed proteins in the minimal network extraction approach. ABLgrp refers to seeds not specifically defined to either ABL1 or ABL2. From 20 extractions each, p-Values for the recovery of known substrates were calculated (Fishers Exact test). The large majority of network extractions recovered a statistical significant number of known kinase-substrate target proteins among connecting proteins. f. Network propagation, distributions of the network propagation score. Exemplarily shown for ABL2, the distribution of the score in the complete network is shown in comparison to the non-seed nodes contained in minimal networks. The latter served as positive data to determine a cut-off systematically for each of the NRTKs. The green dashed line shows the optimal cut off for ABL2 (sensitivity = 0.956 and specificity = 0.946). g. Summary of average kinase-substrate assignments. Average numbers of tyrosine phosphorylated protein for human NRTKs are given. Average number of kinase-substrate relationships per kinase predicted from homology transfer, motif scoring and network inference are given and demonstrate a substantial increase in putative relationships.
For multiple different PTMs, groups of highly modified functionally coherent protein complexes were previously characterized in human (Woodsmith et al., 2013). Phospho-tyrosine enriched complexes (Suppl. Figure 4a) were strongly associated with GO terms relating to extra-cellular stimulation, cell migration, adhesion and immune cell functions (-logP range from 3 to 25; Suppl. Figure 4b). When we analyzed the pY-sites obtained in yeast, controlling for both protein size and frequency in the protein complex dataset, we also found these groups of highly modified complexes separated from the majority distribution suggesting that complexes were in general targeted by tyrosine phosphorylation in yeast (Suppl. Figure 4a). In contrast to the human dataset, we observed weak signals only when performing a GO-term enrichment analysis on highly modified complexes sampled across a variety of different functions (-logP range 2 to 6; Suppl. Figure 4b). This is in agreement with tyrosine phosphorylation not playing a role in yeast bona fide cellular processes. We also analyzed kinase targets in the framework of likely physical protein complexes using COMPLEAT (Vinayagam et al., 2013), a tool to identify preferentially targeted protein complexes with diverse proteomics inputs. COMPLEAT analysis using the pY-protein data as input revealed that a total of 282 yeast protein complexes (169 non redundant; -logP >1.3) were significantly modified by one or more human tyrosine kinases. Between 10 and 200 complexes were found per kinase with a median of 60 complexes. Each kinase showed a very unique set of complexes in this analysis and no obvious functional cluster appeared (Suppl. Figure 4c). These analyses suggest that tyrosine kinases preferentially phosphorylate multiple substrates in physical assemblies such as protein complexes.
Our global network analyses showed that the phosphorylated yeast proteins cluster in protein complex and binary interactome networks. To directly reveal the connectivity of kinase substrates, we next sought to extract the actual subnetworks modified by the individual kinases from global yeast interactome maps. Several related algorithms that search networks to retrieve active subnetworks have been developed in the context of expression analysis (Alcaraz et al., 2014), disease associations (Vanunu et al., 2010) and cancer mutational analysis (Hofree et al., 2013; Creixell et al., 2015b). We overlaid the phosphorylation values on the corresponding proteins in the yeast interactome network as seeding points to search for subnetworks with a maximum number of phospho-proteins. Specifically, using a greedy search algorithm (Alcaraz et al., 2014) we extracted subnetworks including all mapped pY-proteins for a given kinase, and a minimal number of proteins not phosphorylated that were required to connect the subnetworks. The number of non-phosphorylated proteins can then act as an indicator of how clustered target proteins are within these networks (Figure 4c). For 15 kinases with more than 55 proteins mapped to the yeast interactome, extraction resulted in sets of minimal networks which involved on average 14% and not more than 22% non-phosphorylated nodes (for ABL2: 12% Figure 4c; Suppl. Table 6). In agreement with our analysis demonstrating shorter average paths between pY-proteins and preferential phosphorylation of interacting proteins, minimal network generation from a comparable number of randomly selected seed nodes required a much higher percentage of additional proteins (~32%).
Network inference to identify putative human pY kinase substrates
As the extracted yeast subnetworks contained a very high fraction of phosphorylated proteins, similar subnetworks that could be built around known pY-substrates in human may be informative to revealing potential kinase-substrate relationships. Specifically, we proposed that minimal subnetworks that contain many substrates of one specific kinase would be useful in assigning other pY-sites with unknown kinase-substrate relationships to this kinase. Therefore, we initially applied the same minimal network extraction technique used in yeast to a high quality human interactome on the basis of known human kinase substrates (seed nodes) (Figure 4c). We used a global binary human protein interaction network (9,412 proteins, 33,646 PPIs, Data S2 (Woodsmith and Stelzl, 2014)), and collected known kinase-substrate relationships from literature databases (Dinkel et al., 2011; Hornbeck et al., 2012). The number of known kinase-substrate pairs varied from 0 (TNK1) to about 200 substrates reported for Src kinase in the databases. However, for half of the kinases less than 15 human substrates were known (Suppl. Table 7). To establish a set of human seed nodes for each tyrosine kinase, we combined the known kinase substrates with direct protein interaction partners of the kinases and homology or linear motif inferred targets derived from our experimental yeast approach. This yielded sets with 60 to 320 human seed nodes in the human interactome for 18 kinases plus ABLgrp (ABL1 or ABL2), respectively (Suppl. Table 7). We extracted 20 minimal networks for every kinase using the greedy search approach for minimal network extraction around the defined human target seeds (Figure 4c). In contrast to yeast minimal networks that contained around 14% non-seed nodes, extracted minimal networks for human contained on average 32% non-seed nodes. This difference is consistent with less well defined human seeds and observed for all kinases (Figure 4d). Non-seed nodes point towards potential kinase-substrate relationships and those that occurred more often in the 20 extracted networks received higher scores (Suppl. Table 7).
The number of seed nodes was large enough to benchmark the search procedure for ABL1, ABLgrp (ABL1 or ABL2), FYN and SRC kinases. When omitting known targets from the seed nodes in the search, a statistically significant number of known kinase substrates was recovered in the minimal networks with all four kinases (Figure 4e), highlighting the potential of this approach to identify kinase-substrate relationships.
In general, the number of known kinase-target relationships of NRTKs are limiting (median of 8 database known and 146 seed proteins, Suppl. Table 7) to the network extraction approach. We thus applied a network propagation algorithm to infer potential kinases targets and extend the minimal network approach. With this iterative network propagation method, flow originating from the seed proteins is simulated throughout the network generating a smooth scoring function over larger network areas (Vanunu et al., 2010; Hofree et al., 2013). For every kinase, the propagation score distribution over all nodes in the human interactome was systematically compared with the score distribution over non-seed nodes in the minimal networks to determine a threshold for kinase-substrate prioritization (Figure 4f and Suppl. Figure 4d). Applying this signal propagation approach, we generated a scoring matrix of kinase-substrate relationships for 3,323 human phospho-proteins (Suppl. Table 8).
In summary, we have leveraged growing yeast as in vivo model substrate for characterizing human protein tyrosine kinase activity. From the collected data set involving 3,279 kinase-substrate relationships we took three approaches, homology transfer, motif scoring and network inference (Figure 1a), to assign kinase-substrate relations for 3,653 known human pY-proteins and 18 kinases. Approximately half the tyrosine modified proteins were specifically assigned to one NRTK, with an average of 12 predictions per kinase based on homology transfer, 114 predictions from motif assignment and about 399 relationships inferred through network extraction and propagation (Figure 4g).
Discussion
We used an experimental setup assaying the in vivo proteome of yeast as a model substrate for human NRTKs and recorded a large set of pY-sites on yeast proteins, each attributed unambiguously to a specific human kinase via mass spectrometry (3,279 kinase-substrate pairs). This one to one assignment is prohibitively difficult in any human cell system, due to hugely variable kinase activities, kinases cascades and overlapping specificities.
The data enabled the assignment of human kinase-substrate relationships via homology transfer (Fig. 2). We also derived linear sequence motifs for 16 kinases from the data and provide performance benchmarks with sets of known kinase-substrate pairs (Fig. 3). ROC analyses demonstrated that motifs generated from the yeast proteome identify known sites from independent human data with similar specificity and sensitivity as known motifs from the literature (Deng et al., 2014; Wagih et al., 2015) (Fig. S2). This shows that reliable data reflecting kinase specificity have been recorded, validating our in vivo data generation in a heterologous system and the approach used. We used the 16 new motifs to score about 10% of known human pY-sites, substantially expanding the current literature.
Protein interaction networks can refine motif based approaches to improve specificity in kinase-substrate assignment (Linding et al., 2007). However, the vast majority of measured pY-sites in human do not show motif signatures to begin with, and how networks in general influence kinase-substrate relations has not been scrutinized. Therefore it is important to develop tools that can address kinase-substrate specificity features independently of linear peptide motifs. As our phosphorylation platform is assaying the functional yeast proteome in the context of in vivo interaction networks, it can go significantly beyond motif based approaches and additional specificity determinants that can be attributed to substrates in isolation (Bhattacharyya et al., 2006; Ubersax and Ferrell, 2007; Creixell et al., 2015a).
We hypothesized that in yeast, which does not utilize tyrosine phosphorylation for bona fide signaling processes, pY-sites ought to be distributed equally across the yeast interactome map unless network structures strongly influence kinase-substrate targeting. When investigating the global yeast interactome we observed clustering of kinase targets in binary and protein complex networks. Phospho-proteins have been shown to cluster in binary networks and on protein complexes in human, as protein networks reflect cellular processes (Beltrao et al., 2012; Woodsmith et al., 2013; Duan and Walther, 2015). Whether clustering is dependent upon individual kinases could not be tested. In four different analyses (Figs. 4a,4b, S4a, S4c), we showed that the targets of each human tyrosine kinase cluster in a well-defined yeast protein-protein interactome network.
Network clustering, a key feature of biological networks, has been very successfully exploited in confining expression profiles, establishing new disease gene associations and in prioritization of cancer mutations (Creixell et al., 2015b). Here we defined minimal networks that best represent the clustered phospho-signal using subnetwork extraction procedures (Alcaraz et al., 2014). For each of the 16 kinases, we extracted minimal subnetworks that contained all phospho-proteins and on average 12% connecting, non-phosphorylated proteins from a global yeast interactome of 5,804 proteins and 63,545 PPIs (Fig. 4d). In analogy to the work flow in yeast (Fig. 4c), the next step for network based inference of human kinase-substrate pairs was to build minimal subnetworks around assigned substrates from human phospho- and interactome data. To this end, we exploited assignments resulting from motif and homology analysis in this study as starting nodes in subnetwork construction, together with known substrates and kinase interaction partners. Importantly, as a validation of this approach the known literature substrates were identified when omitted from the set of starting seeds (Fig. 4f). Finally, we applied the network inference approach further using network propagation (Vanunu et al., 2010; Hofree et al., 2013) (Fig. 4g) to score tyrosine phosphorylated proteins in the human interactome map as potential substrates for each of the 18 tyrosine kinases. In total we provide candidate kinases for 3,653 phospho-tyrosine modified human proteins for future in depth investigation. The predictive power of our approach is limited by relatively small sets of known substrate-kinases relationships, spurious phospho-sites in big data collections and incomplete human protein networks. As these data become better defined, the reliability of network inferences will increase.
Unlike in vitro systems to screen for kinase-substrate relationships, our yeast model substrate has the potential to account for kinase specificity determinants that are not necessarily encoded in the phospho-proteins as such. Cantley and coworkers recently reported crystal structures of the EGFR kinase domain with a bound peptide substrate where the peptide residues did not have well-defined electron density despite the use of an optimized linear sequence and concluded from their structural observations that, other than the +1 residue, the primary sequence surrounding the phosphorylation site may have little influence on EGFR specificity (Begley et al., 2015). Other work has shown involvement of docking sites, targeting subunits and scaffolds, which better explain kinase-substrate specificity through additional protein interactions (Bhattacharyya et al., 2006; Ubersax and Ferrell, 2007; Zeke et al., 2015). However, since those additional protein interactions may not all exist in the yeast model substrate, the physical and topological constraints on substrates in the cell could also contribute. Investigation into this question through cellular biophysics studies may shed further light on how the relatively small number of tyrosine kinases can address a major part of the proteome and how local interaction networks mediate broad dynamic cellular phospho responses.
STAR * METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Ulrich Stelzl (ulrich.stelzl@uni-graz.at).
EXPERIMENTAL MODEL DETAILS
Yeast cell culture
Yeast strain L40c (MATa his3Δ200 trp1–901 leu2–3,112 ade2 lys2–801am can1 URA3:: (lexAop)8-GAL1TATA-lacZ LYS2::(lexAop)4-HIS3TATA-HIS3) (Worseck et al., 2012) expressing human NRTKs under a copper-inducible yeast promoter using pASZ-DM (Grossmann et al., 2015) was grown in two liter liquid selective media (-Ade). After six hours of growth, human NRTK expression was induced by addition of CuSO4 to a final concentration of 20 to 100 μM (dependent on the observed activity of NRTKs in yeast via Western blotting using 4G10 antibody) growth was continued over-night. After centrifugation at 4°C at 4,300 × g for 15 min, aliquots of 1 ml dry yeast pellets were frozen and stored until lysis at −80°C.
METHOD DETAILS
Yeast lysis
An equal volume of zirconia beads (Carl Roth GmbH & Co. KG) and 500 μl lysis buffer (20 mM HEPES pH 8.0, 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate) (Cell Signaling Technology Inc., Danvers, MA, USA) containing 9 M urea (Biomol GmbH, Hamburg, Germany) was added to each frozen dry yeast pellet and cells were lysed using a FastPrep24 (MP Biomedicals, Santa Ana, CA, USA) homogenizer for 20 seconds at its highest speed (6.5 Ms-1).
Phospho-peptide enrichment from yeast lysate
Dependent on observed NRTK activity in yeast, two to six 1 ml dry yeast pellets were lyzed. Yeast lysates were cleared on a cooled (4°C) table-top centrifuge for 15 min at 20,000 g and the supernatants were transferred to a 50 ml centrifugation tube. The lysis was repeated twice by adding each time 500 μl lysis buffer to the cell debris/pellets. 1/10th volume of 45 mM DTT (Cell Signaling Technology Inc., Danvers, MA, USA) was added to the combined cleared lysates and incubated for 20 min in a 60°C water bath. After cooling the solution to room temperature (RT) for 10 min, 1/10th volume of 110 mM iodoacetamide (I-6125, Sigma, St.Louis, MO, USA) was added and the solution was incubated for 10 min at RT in the dark. For Trypsin digestion, the solution was increased in volume 4 times and diluted with HEPES buffer such that the final concentration was 1 M urea and 10 mM HEPES, pH 8.0. Finally, 1/100th volume of 1 mg/ml trypsin-TPCK solution (Worthington Biochemical Corporation, Lakewood, NJ, USA / Roche Diagnostics GmbH, Mannheim, Germany) was added and the proteins in solution were digested overnight at room temperature. The tryptic digest was acidified by the addition of 1/20th volume of 20% tri-fluoroacetic acid (TFA) solution (AppliChem GmbH, Darmstadt, Germany) for 10 min at RT. The acidified peptide solution was centrifuged for 5 min at 1,800 × g and the supernatant decanted into a fresh tube. Peptides were desalted using a reversed-phase Sep-Pak solid phase extraction column (Waters, Milford, MA, USA). After the column was pre-wetted with 5 ml 100% acetonitrile (MeCN) (51101, Thermo Fisher Scientific Inc., Waltham, MA, USA) and washed twice with 3.5 ml 0.1% TFA, the entire acidified peptide solution was passed through the column by gravity flow or by the use of a plunger. Subsequently, the column was washed applying 1 ml, then 5 ml and finally 6 ml of 0.1% TFA before eluting the peptides into a polypropylene tube in 2 ml 0.1% TFA, 40% MeCN thrice. The eluate was frozen in liquid nitrogen and subsequently lyophilized.
For anti-phospho-tyrosine immunoaffinity enrichment we built on the protocol first established by Rush et al. (Rush et al., 2005) and our previous work (Ballif et al., 2008; Doubleday and Ballif, 2014). The final protocol and reagents used were from the P-Tyr-100 PhosphoScan Kit (Cell Signaling Technology, Danvers, MA, USA). Lyophilized peptides resuspended in 1.4 ml “IAP buffer plus detergent” (50 mM MOPS pH 7.2, 10 mM sodium phosphate, 50 mM sodium chloride, detergent (proprietary formulation; Cell Signaling Technology Inc., Danvers, MA), kept at RT for 5 min and briefly sonicated in an ultrasound bath. The pH was controlled adjustments using 1 M Tris Base to be neutral. All of the following steps were conducted at 4°C. The peptide solution was clarified via centrifugation at for 15 min and transferred directly onto P-Tyr-100-conjugated beads and incubated on a rotator for 2 hours. After subsequent centrifugation at 2,700 × g for 1 minute the beads were washed twice with 1 ml IAP buffer. In order to capture peptides unbound in the immuno-precipitation (IP) using the anti-phospho-tyrosine P-Tyr-100 antibody conjugated beads the supernatant was again applied to anti-phospho-tyrosine 4G10 antibody conjugated beads, incubated on a rotator for 2 hours, and washed twice with 1 ml IAP buffer. Consecutive processing steps were identical for both IPs. The beads were again washed 5 times by applying 1ml purified water. Peptides were eluted from the beads by the addition of 55 μl of 0.15% TFA for 10 minutes at RT twice. The eluate was divided into two aliquots of 50 μl and purified on ZipTips using solvent A (0.1% TFA) and solvent B (0.1% TFA, 40% MeCN) and dried in a vacuum concentrator for 60 min.
Mass spectrometry analyses
LC-MS/MS analyses were set up and conducted as described previously (Doubleday and Ballif, 2014) using a MicroAs autosampler, a Surveyor PumpPlus HPLC and a linear ion trap-orbitrap (LTQ-Orbitrap) platform (Thermo Electron, Waltham, MA, USA). To identify tyrosine phosphorylated peptides, we performed a SEQUEST search of the MS/MS data using yeast proteome downloaded from SGD database (Jan. 2011). The search parameters required a precursor mass tolerance of 10 PPM, required peptides to be tryptic, and allowed dynamic modification of methionine (+15.99491 Da for oxidation), cysteine (+57.02146 Da for carbamidomethylation) and serines, threonines and tyrosines (+79.9663 Da for phosphorylation). By using the Ascore algorithm, we could determine the precise position of the phosphorylated residue with a confidence above 95% for 1433 pY sites.
Immuno-precipitation of predicted human NRTK targets expressed in yeast
The method is an adoption of a chromatin immuno-precipitation protocol of Grably and Engelberg (Grably and Engelberg, 2010). In brief, selected human targets and NRTKs were picked from an open reading frame (ORF) collection of gateway entry clones and were shuttled into the yeast expression vectors pRS425_GDP_TAP (Addgene). Co-transformed yeast was grown and lyzed as stated above however, with an additional step. Zirconia beads were removed manually by poking a 0.4 mm hole using a Bunsen burner heated needle in each 2 ml tube spinning at 3,220×g for 1 min and directly into a 15 ml tube. The bead-free lysate was sonicated (5 cycles for 30 sec) and cleared by centrifugation. 110 μl slurry of washed IgG beads was added to the cleared 10 ml lysate and incubated over-night at 4°C. Beads were washed four to six times with 1 ml wash buffer (50 mM ammonium carbonate; pH 8), and the proteins were eluted with 110 μl of 2.5 × SDS gel loading buffer (200 mM Tris-Cl (pH 6.8), 1% SDS, 10% glycerol, 0.1% bromphenol blue, 50 mM DTT) for 5 min at 95 °C. After separation of the proteins on 10–12% SDS polyacrylamide gels bands with the expected molecular weight were excised. The gel slices were grinded using a micro-pistil within protein low-binding reaction tubes (LoBind, Eppendorf AG, Hamburg, Germany) and proteins in-gel digested with MS-grade trypsin (Roche Diagnostics GmbH, Mannheim, Germany). The resulting peptides were alkylated, reduced and thereafter purified using a C18 column and finally desiccated in a vacuum concentrator. Tyrosine phosphorylation was measured on a Q-Exactive mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) and peptides identified using the MaxQuant environment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Homology transfer
Using the Inparanoid database (Remm et al., 2001) 479 sequence alignments to NRTK targeted yeast proteins were retrieved including up to 20 species having NRTK signaling evolved (TK-group) and up 20 species having not NRTK signaling evolved (nonTK-group) - with the prerequisite that a human orthologous sequence exists. Using a custom-made Python script, orthologous positions to tyrosine residues in the yeast proteins sequences were analyzed. A “Y-score” was calculated indicating the fixed-position conservation for each tyrosine residue by comparing the occurrence of tyrosine residues among the two groups of species in each position:
A Y-score above above zero hence indicated higher conservation of tyrosine residues among species having NRTK signaling evolved whereas a Y-score close to zero indicated full conservation of the residue between all species of both groups.
Motif analysis
Tryptic pY-peptides from the mass spectrometry (MS) output were mapped to the yeast proteome and processed to 15 mer sequences where seven amino acids each are flanking a central tyrosine residue. For each kinase a list of aligned 15 mers was analysed with the iceLogo stand-alone application (Colaert et al., 2009). The background set used was a list of 15 mers capturing all non-phosphorylated tyrosine residues of the proteins identified in the MS measurements (“expressed yeast proteome”). Fold change was set as the enrichment/significance parameter. The default color scheme was used and the enrichment axis adjusted manually to show all enriched residues at appropriate scale.
Using a custom made python script all phosphorylation sites were scored additively from the enrichment value (EV) matrix obtained via iceLogo. The R package ROCR (Sing et al., 2005) was used for performance analysis. The program inputs a list of scores with assigned binaries and outputs a graphical display of the performance as ROC (Receiver Operating Characteristic) curve. The “expressed yeast proteome” was scored and targeted sites labeled for each NRTK separately. Due to the limited number of reported kinase-substrate relationships in public databases for the majority of NRTKs, it was not possible to retrieve sufficiently large independent positive sets for systematic motif performance testing involving all kinases. Therefore, a hundred-fold cross-validation was performed. Ten percent of the kinase target sets were randomly removed and a new sequence motif generated using the remaining 90 percent of hits for each kinase. The reference set was subsequently scored applying the new motif and binaries assigned labeling the omitted, independent ten percent of targeted pY-sites. ROCR also outputs average accuracy values for each scored site over all randomized performance tests which were used to both normalize the score between NRTKs and for annotation of NRTKs to human substrates. Using the ROCR package, motif comparison with literature was performed with positive data set (>10 sites annotated for a given kinase) from phosphosite plus (Jan 2017) (Hornbeck et al., 2012) and the negative data were all other tyrosine residues of the respective proteins.
Network analysis
Union of the ConsensusPathDB “binary network”, the SGD “physical network” and STRING “high confidence” network removing all proteins with a degree larger than 150 (5798 proteins, 63542 PPIs; Data S1). Human binary interactome map combined 16 high quality yeast two hybrid studies (Woodsmith and Stelzl, 2014) excluding proteins with a degree higher than 150 (9412 proteins, 33646 PPIs; Data S2).
Randomized networks were generated with custom made Perl scripts. Node randomization: Nodes in the network were sorted by their degrees into bins of 3%. 100 randomized networks were created with the same number of nodes from the same bins and the average shortest paths between modified nodes were calculated. Link randomization: Networks were rewired by shuffling the interactions (Fisher Yates shuffle) but keeping the number of interactions for each protein as in the experimental network.
Optimal subnetworks were generated using cytoscape (Version 3.2.1 / Java environment 1.8.0_51, (Shannon et al., 2003)) app “KeyPathMiner” (KPM 4, (Alcaraz et al., 2014)) and network propagation preformed using a liner integer program (Vanunu et al., 2010) implemented in the cytoscape app “Propagate”.
KPM was set up to retrieve optimal subnetworks including the maximum number of phosphorylated proteins (seeds) (variable L=0) and step-wise increasing number of exceptions k. The minimal k was chosen at the point where no further seeds were included in the subnetworks. Using a custom-made python script, optimal subnetworks were processed and analyzed. Non phospho-proteins mapped to the periphery of the subnetworks were excluded. An “exception score” was generated which delineates how often a non-phospho-protein was included in 20 optimal subnetworks to account for the heuristic approach. A comparison between repeated runs showed that proteins with a score below 0.2 were not reproducible and not considered as predictions.
Using the app “Propagate” the entire PPI network was scored using the same seeds (“priors”) as previously. Visualization of the propagate scores over the entire network in comparison to high scoring KPM nodes was performed using R package ggplot2 (Wickham, 2009). A “Propagate” score cut-off was selected via AUC analysis using the R package “OptimalCutpoints” (López-Ratón et al., 2014).
Annotation, visualization
GO enrichment was performed at consensuspathdb.org/ (Kamburov et al., 2013) and COMPLEAT analysis was performed at www.flyrnai.org/compleat (Vinayagam et al., 2013). Network visualization was performed with cytoscape (Shannon et al., 2003).
Supplementary Material
A. List of 12,625 quality-filtered pY-peptides.
B. List of 3279 pY-sites measured in yeast.
C. List of 55 pY-peptides that were filtered from the dataset. Sites reported by Gnad et al. (Gnad et al., 2009), Tan et al. (Tan et al., 2009a) or reported in BIOGRID (https://thebiogrid.org/) are indicated. 46 peptides were removed because they were detected with more than 10 kinases and high spectral counts.
(related to Figure 2): List of conserved yeast phospho-tyrosine sites which locally align to a tyrosine in human. 63 of the 296 yeast sites that locally align to a tyrosine in human and are reported to be phosphorylated.
(related to Figure 3): Motif-based prediction of kinase substrate relationships for known human pY sites.
(related to Figure 4): List of Network based predictions for 4,158 human proteins of which 3,323 arse known phosphoproteins.
(separate file, related to Figure 4): Yeast protein interaction network (63,545 PPIs).
(separate file, related to Figure 4): Human protein interaction network (33,646 PPIs).
Acknowledgements
The work was supported by the Max-Planck Society, by the University of Graz, by the U.S. National Science Foundation IOS grant 1021795 and the Vermont Genetics Network through U. S. NIH grant 8P20GM103449 from the INBRE program of the NIGMS.
Footnotes
References
- Alcaraz N, Pauling J, Batra R, Barbosa E, Junge A, Christensen, Anne GL, Azevedo V, Ditzel HJ, and Baumbach J. (2014). KeyPathwayMiner 4.0: condition-specific pathway analysis by combining multiple omics studies and networks with Cytoscape. BMC systems biology 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Carey GR, Sunyaev SR, and Gygi SP (2008). Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. Journal of proteome research 7, 311–318. [DOI] [PubMed] [Google Scholar]
- Begley MJ, Yun C. h., Gewinner CA, Asara JM, Johnson JL, Coyle AJ, Eck MJ, Apostolou I, and Cantley LC (2015). EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nature structural & molecular biology 22, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrao P, Albanese V, Kenner LR, Swaney DL, Burlingame A, Villen J, Lim WA, Fraser JS, Frydman J, and Krogan NJ (2012). Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya RP, Reményi A, Yeh BJ, and Lim WA (2006). Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annual review of biochemistry 75, 655–680. [DOI] [PubMed] [Google Scholar]
- Brugge JS, Jarosik G, Andersen J, Queral-Lustig A, Fedor-Chaiken M, and Broach JR (1987). Expression of Rous sarcoma virus transforming protein pp60v-src in Saccharomyces cerevisiae cells. Molecular and cellular biology 7, 2180–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MF, Prisic S, Lubner JM, Church GM, Husson RN, and Schwartz D (2012). Using bacteria to determine protein kinase specificity and predict target substrates. PloS one 7, e52747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N, Helsens K, Martens L, Vandekerckhove J, and Gevaert K (2009). Improved visualization of protein consensus sequences by iceLogo. Nature methods 6, 786–787. [DOI] [PubMed] [Google Scholar]
- Colicelli J (2010). ABL tyrosine kinases: evolution of function, regulation, and specificity. Science signaling 3, re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, and MacAuley A (1988). Potential positive and negative autoregulation of p60c-src by intermolecular autophosphorylation. Proceedings of the National Academy of Sciences of the United States of America 85, 4232–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creixell P, Palmeri A, Miller CJ, Lou HJ, Santini CC, Nielsen M, Turk BE, and Linding R (2015a). Unmasking determinants of specificity in the human kinome. Cell 163, 187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creixell P, Reimand J, Haider S, Wu G, Shibata T, Vazquez M, Mustonen V, Gonzalez-Perez A, Pearson J, and Sander C, et al. (2015b). Pathway and network analysis of cancer genomes. Nature methods 12, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Alicea-Velázquez NL, Bannwarth L, Lehtonen SI, Boggon TJ, Cheng H-C, Hytönen VP, and Turk BE (2014). Global analysis of human nonreceptor tyrosine kinase specificity using high-density peptide microarrays. Journal of proteome research 13, 4339–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, and Diella F (2011). Phospho.ELM: a database of phosphorylation sites--update 2011. Nucleic acids research 39, D261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, and Simon I (2005). IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics (Oxford, England) 21, 3433–3434. [DOI] [PubMed] [Google Scholar]
- Doubleday PF, and Ballif BA (2014). Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics. Proteomes 2, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan G, and Walther D (2015). The roles of post-translational modifications in the context of protein interaction networks. PLoS computational biology 11, e1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte ML, Pena DA, Ferraz Nunes, Augusto Felipe, Berti DA, Sobreira Paschoal, José Tiago, Costa-Junior HM, Baqui Abdel, Muhammad Munira, Disatnik M-H, Xavier-Neto J, and de Oliveira Lopes, Sérgio Paulo, et al. (2014). Protein folding creates structure-based, noncontiguous consensus phosphorylation motifs recognized by kinases. Science signaling 7, ra105. [DOI] [PubMed] [Google Scholar]
- Eng JK, Fischer B, Grossmann J, and Maccoss MJ (2008). A fast SEQUEST cross correlation algorithm. Journal of proteome research 7, 4598–4602. [DOI] [PubMed] [Google Scholar]
- Florio M, Wilson LK, Trager JB, Thorner J, and Martin GS (1994). Aberrant protein phosphorylation at tyrosine is responsible for the growth-inhibitory action of pp60v-src expressed in the yeast Saccharomyces cerevisiae. Molecular biology of the cell 5, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A-C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, and Dumpelfeld B, et al. (2006). Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636. [DOI] [PubMed] [Google Scholar]
- Gnad F, de Godoy, Lyris MF, Cox J, Neuhauser N, Ren S, Olsen JV, and Mann M. (2009). High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 9, 4642–4652. [DOI] [PubMed] [Google Scholar]
- Grably M, and Engelberg D (2010). A detailed protocol for chromatin immunoprecipitation in the yeast Saccharomyces cerevisiae. Methods in molecular biology (Clifton, N.J.) 638, 211–224. [DOI] [PubMed] [Google Scholar]
- Grossmann A, Benlasfer N, Birth P, Hegele A, Wachsmuth F, Apelt L, and Stelzl U (2015). Phospho-tyrosine dependent protein-protein interaction network. Molecular systems biology 11, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LK, Frumm SM, and Bishop AC (2013). A general assay for monitoring the activities of protein tyrosine phosphatases in living eukaryotic cells. Analytical biochemistry 435, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofree M, Shen JP, Carter H, Gross A, and Ideker T (2013). Network-based stratification of tumor mutations. Nature methods 10, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, and Sullivan M (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic acids research 40, D261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Rho H-S, Newman RH, Zhang J, Zhu H, and Qian J (2014). PhosphoNetworks: a database for human phosphorylation networks. Bioinformatics (Oxford, England) 30, 141–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Stelzl U, Lehrach H, and Herwig R (2013). The ConsensusPathDB interaction database: 2013 update. Nucleic acids research 41, D793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornbluth S, Jove R, and Hanafusa H (1987). Characterization of avian and viral p60src proteins expressed in yeast. Proceedings of the National Academy of Sciences of the United States of America 84, 4455–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Saito S, Nakagawa R, Katsuyama I, Hatanaka M, Yamamoto T, Arakawa T, and Tokunag M (2006). Expression of human tyrosine kinase, Lck, in yeast Saccharomyces cerevisiae: growth suppression and strategy for inhibitor screening. Protein and peptide letters 13, 915–920. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, and Tikuisis AP, et al. (2006). Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643. [DOI] [PubMed] [Google Scholar]
- Lerner EC, Trible RP, Schiavone AP, Hochrein JM, Engen JR, and Smithgall TE (2005). Activation of the Src family kinase Hck without SH3-linker release. The Journal of biological chemistry 280, 40832–40837. [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Ostheimer GJ, van Vugt, Marcel ATM, Jørgensen C, Miron IM, Diella F, Colwill K, Taylor L, and Elder K, et al. (2007). Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Ratón M, Rodríguez-Álvarez MX, Suárez CC, and Sampedro FG (2014). OptimalCutpoints. An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. J. Stat. Soft. 61. [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S (2002). The protein kinase complement of the human genome. Science (New York, N.Y.) 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jørgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, and Sicheritz-Ponten T, et al. (2008). Linear motif atlas for phosphorylation-dependent signaling. Science signaling 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam, Hugo YK, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, and Jwa M, et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Science signaling 3, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalibet J, and Kennedy BP (2004). Using yeast to screen for inhibitors of protein tyrosine phosphatase 1B. Biochemical pharmacology 68, 1807–1814. [DOI] [PubMed] [Google Scholar]
- Murphy SM, Bergman M, and Morgan DO (1993). Suppression of c-Src activity by C-terminal Src kinase involves the c-Src SH2 and SH3 domains: analysis with Saccharomyces cerevisiae. Molecular and cellular biology 13, 5290–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nada S, Okada M, MacAuley A, Cooper JA, and Nakagawa H (1991). Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 351, 69–72. [DOI] [PubMed] [Google Scholar]
- Newman RH, Hu J, Rho H-S, Xie Z, Woodard C, Neiswinger J, Cooper C, Shirley M, Clark HM, and Hu S, et al. (2013). Construction of human activity-based phosphorylation networks. Molecular systems biology 9, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya S, Struck TJ, Mannakee BK, Paniscus M, and Gutenkunst RN (2015). Testing whether metazoan tyrosine loss was driven by selection against promiscuous phosphorylation. Molecular biology and evolution 32, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluk H, Dorey K, and Superti-Furga G (2002). Autoinhibition of c-Abl. Cell 108, 247–259. [DOI] [PubMed] [Google Scholar]
- Remm M, Storm CE, and Sonnhammer EL (2001). Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. Journal of molecular biology 314, 1041–1052. [DOI] [PubMed] [Google Scholar]
- Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha X-M, Polakiewicz RD, and Comb MJ (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nature biotechnology 23, 94–101. [DOI] [PubMed] [Google Scholar]
- Schieven G, Thorner J, and Martin GS (1986). Protein-tyrosine kinase activity in Saccharomyces cerevisiae. Science (New York, N.Y.) 231, 390–393. [DOI] [PubMed] [Google Scholar]
- Schwartz D, and Gygi SP (2005). An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nature biotechnology 23, 1391–1398. [DOI] [PubMed] [Google Scholar]
- Shah NH, Wang Q, Yan Q, Karandur D, Kadlecek TA, Fallahee IR, Russ WP, Ranganathan R, Weiss A, and Kuriyan J (2016). An electrostatic selection mechanism controls sequential kinase signaling downstream of the T cell receptor. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, and Lengauer T (2005). ROCR: visualizing classifier performance in R. Bioinformatics (Oxford, England) 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Songyang Z, and Cantley LC (1995). Recognition and specificity in protein tyrosine kinase-mediated signalling. Trends in biochemical sciences 20, 470–475. [DOI] [PubMed] [Google Scholar]
- Superti-Furga G, Fumagalli S, Koegl M, Courtneidge SA, and Draetta G (1993). Csk inhibition of c-Src activity requires both the SH2 and SH3 domains of Src. The EMBO journal 12, 2625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Delfino FJ, Engen JR, Superti-Furga G, and Smithgall TE (2003). Regulation of c-Fes tyrosine kinase activity by coiled-coil and SH2 domains: analysis with Saccharomyces cerevisiae. Biochemistry 42, 3567–3574. [DOI] [PubMed] [Google Scholar]
- Tan C, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, Jørgensen C, Bader GD, Aebersold R, Pawson T, and Linding R (2009a). Comparative analysis reveals conserved protein phosphorylation networks implicated in multiple diseases. Science signaling 2, ra39. [DOI] [PubMed] [Google Scholar]
- Tan C, Pasculescu A, Lim WA, Pawson T, Bader GD, and Linding R (2009b). Positive selection of tyrosine loss in metazoan evolution. Science (New York, N.Y.) 325, 1686–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, and Ferrell JE (2007). Mechanisms of specificity in protein phosphorylation. Nature reviews. Molecular cell biology 8, 530–541. [DOI] [PubMed] [Google Scholar]
- Vanunu O, Magger O, Ruppin E, Shlomi T, and Sharan R (2010). Associating genes and protein complexes with disease via network propagation. PLoS computational biology 6, e1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayagam A, Hu Y, Kulkarni M, Roesel C, Sopko R, Mohr SE, and Perrimon N (2013). Protein complex-based analysis framework for high-throughput data sets. Science signaling 6, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O, Reimand J, and Bader GD (2015). MIMP: predicting the impact of mutations on kinase-substrate phosphorylation. Nature methods 12, 531–533. [DOI] [PubMed] [Google Scholar]
- Wang C, Ye M, Bian Y, Liu F, Cheng K, Dong M, Dong J, and Zou H (2013). Determination of CK2 specificity and substrates by proteome-derived peptide libraries. Journal of proteome research 12, 3813–3821. [DOI] [PubMed] [Google Scholar]
- Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, and Mering C von (2012). PaxDb, a database of protein abundance averages across all three domains of life. Molecular & cellular proteomics : MCP 11, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). Ggplot2. Elegant graphics for data analysis (Dordrecht, New York: Springer; ). [Google Scholar]
- Woodsmith J, Kamburov A, and Stelzl U (2013). Dual coordination of post translational modifications in human protein networks. PLoS computational biology 9, e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsmith J, and Stelzl U (2014). Studying post-translational modifications with protein interaction networks. Current opinion in structural biology 24, 34–44. [DOI] [PubMed] [Google Scholar]
- Worseck JM, Grossmann A, Weimann M, Hegele A, and Stelzl U (2012). A stringent yeast two-hybrid matrix screening approach for protein-protein interaction discovery. Methods in molecular biology (Clifton, N.J.) 812, 63–87. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, and Yao X (2008). GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics : MCP 7, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, and Simonis N, et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science (New York, N.Y.) 322, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeke A, Bastys T, Alexa A, Garai A, Meszaros B, Kirsch K, Dosztanyi Z, Kalinina OV, and Remenyi A (2015). Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Molecular systems biology 11, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. List of 12,625 quality-filtered pY-peptides.
B. List of 3279 pY-sites measured in yeast.
C. List of 55 pY-peptides that were filtered from the dataset. Sites reported by Gnad et al. (Gnad et al., 2009), Tan et al. (Tan et al., 2009a) or reported in BIOGRID (https://thebiogrid.org/) are indicated. 46 peptides were removed because they were detected with more than 10 kinases and high spectral counts.
(related to Figure 2): List of conserved yeast phospho-tyrosine sites which locally align to a tyrosine in human. 63 of the 296 yeast sites that locally align to a tyrosine in human and are reported to be phosphorylated.
(related to Figure 3): Motif-based prediction of kinase substrate relationships for known human pY sites.
(related to Figure 4): List of Network based predictions for 4,158 human proteins of which 3,323 arse known phosphoproteins.
(separate file, related to Figure 4): Yeast protein interaction network (63,545 PPIs).
(separate file, related to Figure 4): Human protein interaction network (33,646 PPIs).