

Abstract
Tumor heterogeneity has hampered the development of novel effective therapeutic options for aggressive cancers, including the deadly primary adult brain tumor glioblastoma (GBM). Intratumoral heterogeneity is partially attributed to the tumor initiating cell (TIC) subset that contains highly tumorigenic, stem-like cells. TICs display metabolic plasticity but can have a reliance on aerobic glycolysis. Elevated expression of GLUT1 and GLUT3 is present in many cancer types, with GLUT3 being preferentially expressed in brain TICs (BTICs) to increase survival in low nutrient tumor microenvironments, leading to tumor maintenance. Through structure-based virtual screening (SBVS), we identified potential novel GLUT inhibitors. The screening of 13 compounds identified two that preferentially inhibit the growth of GBM cells with minimal toxicity to non-neoplastic astrocytes and neurons. These compounds, SRI-37683 and SRI-37684, also inhibit glucose uptake and decrease the glycolytic capacity and glycolytic reserve capacity of GBM patient-derived xenograft (PDX) cells in glycolytic stress test assays. Our results suggest a potential new therapeutic avenue to target metabolic reprogramming for the treatment of GBM, as well as other tumor types, and the identified novel inhibitors provide an excellent starting point for further lead development.
Keywords: glioblastoma, tumor initiating cell, cancer stem cell, GLUT3, metabolism, Warburg Effect, drug discovery
Graphical Abstract
Due to their rapid growth, cancer cells present with an increased requirement for nutrients. These metabolic shifts are known as the Warburg effect and have been labeled as an emerging hallmark of cancer (1–5). Warburg recognized that cancer cells display enhanced utilization of glycolysis to generate ATP, even in environments with adequate oxygen (2, 5–7). Glycolysis also provides additional metabolic intermediates that can be used for multiple cellular processes necessary for tumor growth even under nutrient deficient conditions (1, 5–11). To sustain increased glycolysis, cancer cells express higher levels of glucose transporters, such as GLUT1 and GLUT3 (3, 10, 12). While glucose uptake is currently utilized for imaging many solid cancers, including glioblastoma (GBM), it has yet to be effectively targeted for cancer therapy.
GBM is the most common and deadly primary malignant adult brain tumor (13–15). Current therapeutic options for GBM include surgical resection, radiation therapy, and chemotherapy (14, 15). Despite research efforts, median patient survival has remained at approximately 14 months, with no profound advancements in the past decade (14, 15). Therapeutic development has been hampered by the heterogeneity within the tumors (14, 16–19). Tumor heterogeneity can be partially explained by the tumor initiating cell (TIC) hypothesis; TICs have some characteristics of stem cells and are thought to be at the apex of the tumor hierarchy (19–24). Brain TICs (BTICs) express stem cell markers such as CD133 and Sox2, are able to self-renew, and initiate a tumor with parental tumor characteristics in xenograft models (19–24). BTICs are also highly resistant to conventional therapies and therefore thought to contribute to recurrent GBM (19, 21, 22, 25–29).
Within the brain, GLUT1 and GLUT3 are believed to be the predominant isoforms modulating facilitative glucose uptake, with low expression of other glucose transporters in specific cell types (1, 3, 10, 30, 31). Previously, elevated expression of GLUT1 and GLUT3 was shown in GBM xenografts in comparison to non-tumorigenic brain, with GLUT3 expression being significantly higher in BTICs (3). Knockdown of GLUT3 in BTICs resulted in a decreased ability to form tumors in vivo, which suggests the possibility of targeting glucose uptake as a therapy (3). Silencing of GLUT1 or pharmacological inhibition with WZB117 has also been shown to decrease the tumor formation capabilities of TICs (32). Currently, there are few GLUT inhibitors and no GLUT3 specific inhibitors. The GLUT inhibitors that have been identified have not been extensively assessed for efficacy in GBM or for potential toxicity to normal cell types (5). As such, there is still a need to identify potent inhibitors of glucose transporters with strong efficacy but limited toxicity for potential novel therapeutic applications.
Both GLUT1 and GLUT3 are transmembrane proteins that belong to the major facilitator superfamily (MFS) (33). Each transporter molecule consists of a 12 transmembrane helices (TH) segment and an intracellular helices (ICH) bundle. The transmembrane segment is further divided into an N-terminal domain (TH 1–6) and a C-terminal domain (TH 7–12) (Figure 1a, b). Substrates are thought to be transported through an alternating access mechanism (34) that involves multiple conformational changes of the transporter (Figure 1c) (35). A recently published crystal structure of human GLUT1 bound to a sugar analog compound adopted an in-ward open conformation and provided detailed structural information regarding sugar-transporter interactions and a template for structure based drug discovery (Figure 1)(36). In the present study, we applied homology modeling and structure-based virtual screening (SBVS) to select putative small molecule GLUT3 inhibitors. Our investigation led to the identification of several compounds that blocked the uptake of glucose and decreased the growth of GBM patient-derived xenograft (PDX) cells in vitro.
Figure 1. Structural presentation of glucose transporters based on the crystal structure GLUT1 (PDB ID:4PYP).
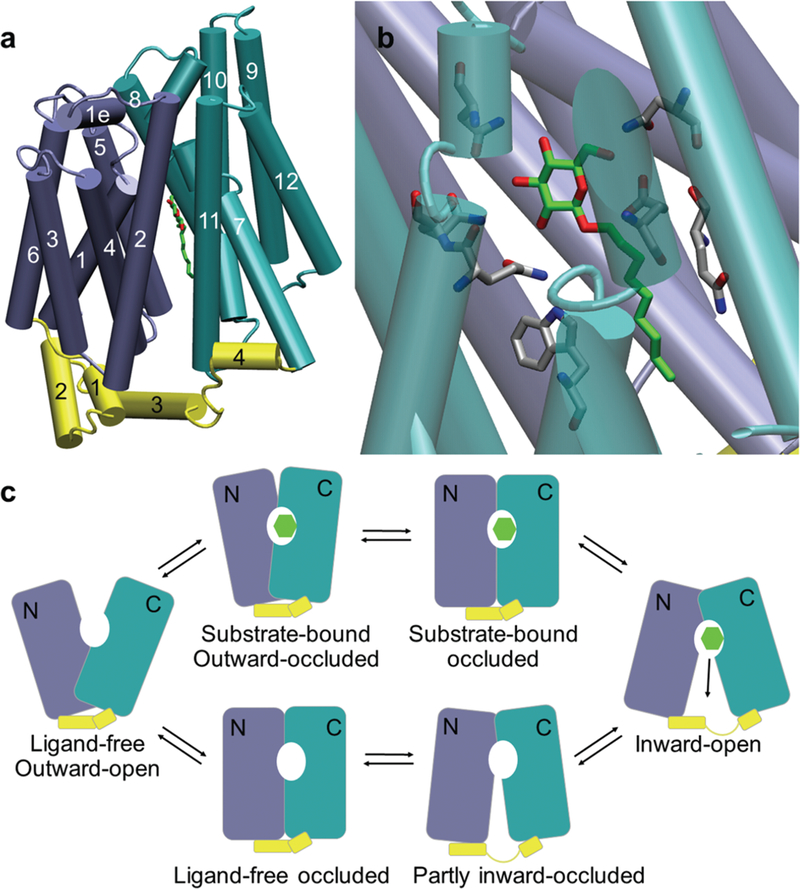
(a) Tube presentation of the GLUT1 crystal structure with the N-domain (transmembrane helices TH 1–6) colored in ice blue, C-domain (TH 7–12) colored in turquoise, and the intracellular helix bundle (ICH1–4) colored in yellow. The substrate (β-NG) is shown in solid green sticks. (b) The close-up view of the substrate binding site. The substrate is colored in green and the binding site residues are colored in grey. Nitrogen and oxygen atoms are colored blue and red respectively. (c) “Alternating access” mechanism of glucose transportation represented in a simplified six-conformation model. The N and the C domains are depicted in ice blue and turquoise respectively. The intracellular helix bundle is depicted in yellow. The green hexagon represents the substrate.
RESULTS AND DISCUSSION
Identification of Potential GLUT3 Inhibitors through Structure Based Virtual Screening.
To identify compounds that can block the transportation of glucose by GLUT3, we constructed a GLUT3 homology model based on the GLUT 1 crystal structure. We then conducted SBVS to target the glucose binding site of this GLUT3 model (Figure 2). A diverse library consisting of 500,000 structurally representative compounds was screened through a three-step docking/scoring process (Figure 2). The docked results of top-scored compounds at the final stage were then visually inspected to select potential GLUT3 inhibitors that showed both shape and electrostatic complementarities to the glucose binding site. Thirteen compounds were identified from the SBVS results and purchased for biological testing for further evaluation (Figure 3). These compounds can be separated into four groups: (1) indolinones/imidazolinones (Figure 3a); (2) dihydroquinolinones (Figure 3b); (3) isoflavones (Figure 3c); and (4) compounds with other distinct core structures (Figure 3d). Interestingly, all of these hits contain a bicyclic (5-,6- or 6-,6-) ring system with a carbonyl functionality.
Figure 2. Protocol for identifying potential small molecule inhibitors of GLUT3 using SBVS.

a) Compounds identified using computer modeling are tested in GBM PDX and non-malignant cells in vitro; hit compounds will then be used to re-evaluate modeling. b) Library construction and assessment using structure-based virtual screening.
Figure 3. Potential GLUT3 inhibitors identified by SBVS.
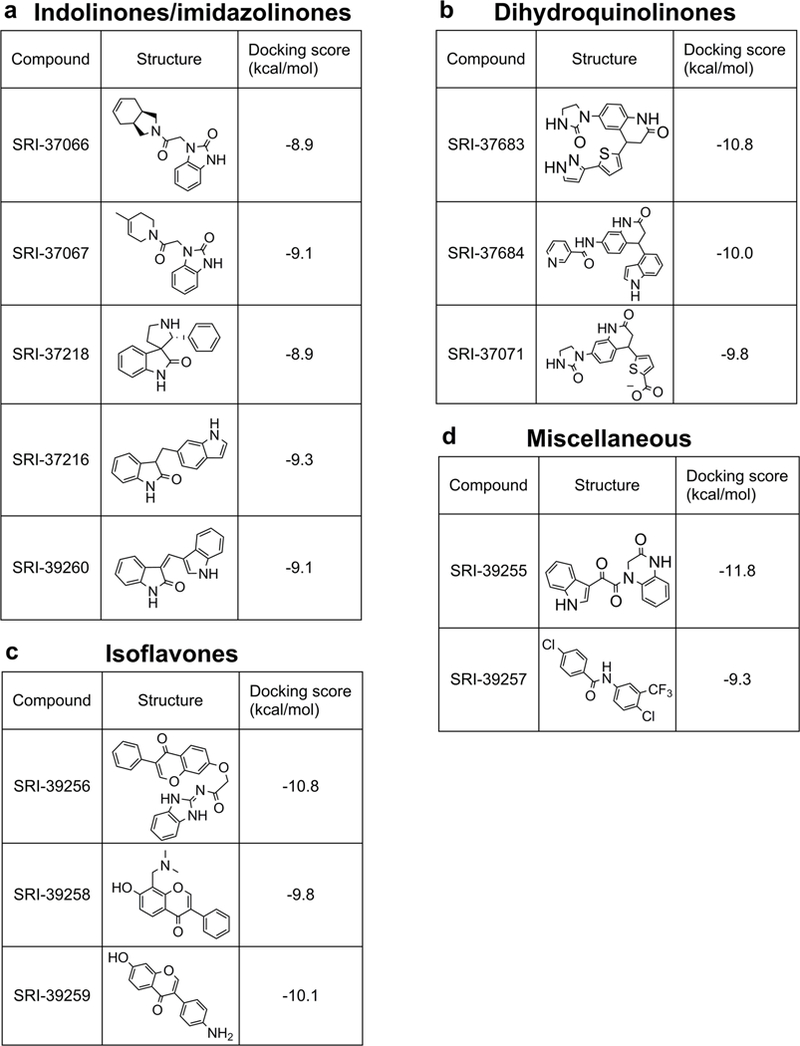
a) indolinones/imidazolinones b) dihydroquinolinones. c) Isoflavones d) miscellaneous core structures.
Small Molecules Inhibit GBM PDX Spheroid Growth in Vitro.
Since we aim to identify potential GLUT inhibitors that are toxic to GBM cells, we next sought to evaluate the ability of the 13 SBVS-identified compounds to inhibit the growth of GBM cells. Cells isolated from a pediatric primary (D456), adult primary (GBM157) and adult recurrent (1016) GBM PDX, were treated with compounds at a concentration of 50 μM as an initial screen (Supporting Information Figure 1 and data not shown). Treatment with 6 compounds (~50%) led to a significant decrease (>50%) in the growth of D456 GBM PDX cells (Supporting Information Figure 1). The 6 compounds were then tested at 5 μM, 20 μM, and 50 μM concentrations to estimate their IC50’s in D456 GBM PDX cells (Figure 4 a-f). As SRI-37683 almost completely inhibited D456 GBM PDX cell growth at 5 μM, analyses at lower concentrations were conducted to establish the IC50. The calculated IC50’s of the 6 compounds ranged from 1.69 μM to 41.11 μM (Figure 4 a-f). Candidate compounds were also tested in adult primary GBM PDX lines JX12 and/or JX14 with similar results to those for D456 GBM PDX (Supporting Information Figure 2). These hit compounds were also screened in GLUT1 or GLUT3 over-expressing D456 GBM PDX cells to assess potential selectivity. There was no significant difference in the response of these cells, indicating that these compounds are likely non-specific (data not shown).
Figure 4. Potential GLUT antagonists display differential effects on GBM PDX cells and NHAs.
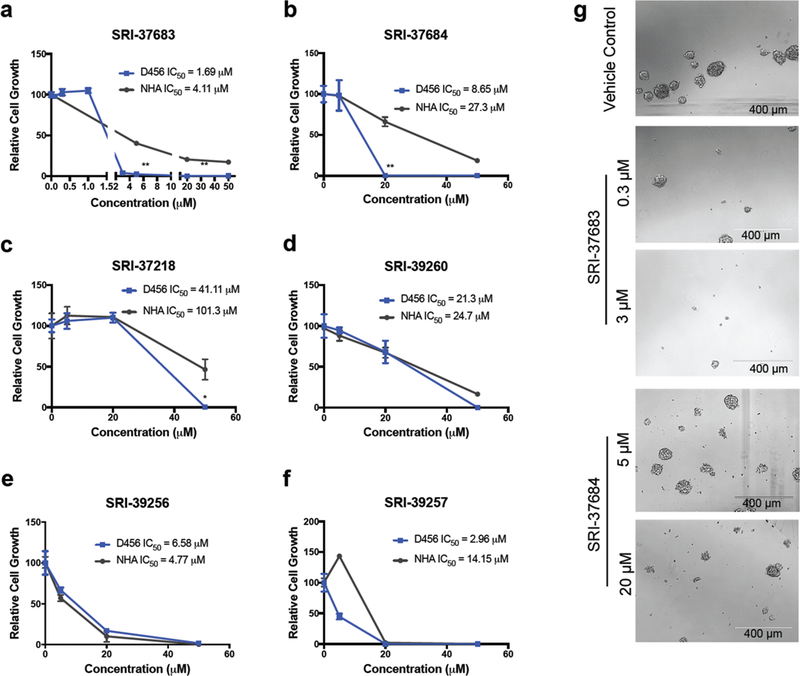
Cell growth of D456 GBM PDX and NHA cells following treatment with (a) SRI-37683 (**p < 0.001 one-ay ANOVA and Dunnett’s multiple comparisons between D456 and NHA treated with 5 μM and 20 μM) (b) SRI-37684 (**p < 0.001 one-ay ANOVA and Dunnett’s multiple comparisons between D456 and NHA treated with 5 μM) (c) SRI- 37218 (**p < 0.005 one-ay ANOVA and Dunnett’s multiple comparisons between D456 and NHA treated with 50 μM) (d) SRI-39260 (e) SRI-39256 (f) SRI-39257 at indicated concentrations for 7 days (g) Representative images of D456 GBM PDX cells on day 7 after treatment with indicated concentrations of antagonists. (Representative data from 3 experiments with n=3 per group)
Small Molecule GLUT Inhibitors Display Limited Toxicity to Normal Human Astrocytes and Neurons.
As GLUT1 and GLUT3 are extremely important to normal brain function, we assessed the toxicity of these compounds on normal human astrocytes (NHAs) which express high levels of GLUT1, and on neurons which express high levels of GLUT3. NHAs were treated in the same manner as the GBM PDX cells mentioned above to identify compounds with a potentially favorable therapeutic index. Both dihydroquinolinone compounds (SRI- 37683 and SRI-37684) and one indolinone compound (SRI-37218) displayed significantly stronger growth inhibition of GBM PDX cells compared to NHAs and thus were selected for further biological assessment (Figure 4 a-c). One other compound (SRI-39257) displayed a slight difference in growth between D456 GBM PDX cells and NHAs but was not selected for further analysis due to an insignificant decrease in growth at the 5 μM concentration in JX12 cells (Figure 4f and Supporting Information Figure 2d). Compounds without substantial IC50 differences between NHAs and GBM PDX cells were not evaluated further. Representative images of D456 GBM PDX cells treated with SRI-37683 and SRI-37684, the most efficacious compounds with minimal toxicity to NHAs, are shown in Figure 4g.
To assess the potential toxicity of the identified compounds against neurons, we tested SRI-37683, SRI- 37684, and SRI-37218 at 5 μM, 20 μM, and 50 μM concentrations in both mouse neurons and human induced pluripotent stem cell-derived neurons (data not shown and Figure 5). Following one week of treatment with the compounds, neurons had minimal decreases in cell number compared to GBM PDX cells and higher IC50 values, indicating the potential for GLUT targeting as a future therapeutic avenue (Figure 5). The IC50 values for GBM PDX cells and neurons for SRI-37218 were not substantially different, and therefore, this compound was not considered for future experiments or compound design (Figure 5). We realize that neurons cultured in vitro will behave differently than neurons in vivo. However, reports indicate that neurons can utilize lactate and ketone bodies as alternative fuel sources when glucose is unavailable (37–39). Neurons are also not mitotically active and therefore have a lower energy demand compared to that of the tumor, indicating that there is likely a window of opportunity for therapeutic intervention (3). As these compounds are not observed to have specificity for GLUT 1 or GLUT3, either through the analysis of non-malignant cells with endogenous expression of GLUT 1 or GLUT3 or with induced cDNA overexpression, modifications to these compounds can be made in the future to alter their specificity.
Figure 5. Dose-response analyses of neuronal growth in response to potential GLUT antagonists.
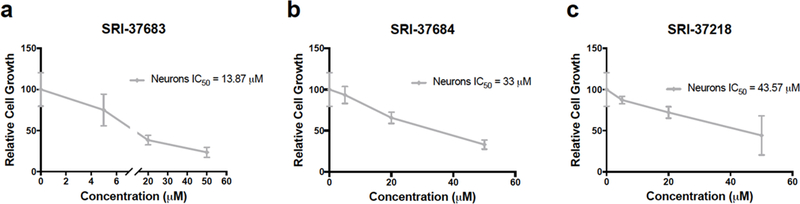
Dose-response of human iPSC-derived neurons treated with experimental compounds at indicated concentrations for 7 days. (Representative data from 3 experiments with n=3 per group; *, p<0.005, one-way ANOVA and Dunnett test for multiple comparisons.)
SBVS Identified Small Molecules Inhibit Glucose Uptake in D456 GBM PDX Cells and Decrease Glycolytic Capacity.
Glucose uptake inhibition by SRI-37683 and SRI 37684 was determined in three GBM PDX cell lines, D456, GBM157, and 1016 by 2-[N-(7-nitrobenz-2- oxa-1,3-diaxol-4-yl)amino]-2- deoxyglucose (2-NBDG), uptake (Figure 6a and Supporting Information Figures 3a,b). At a concentration of 50 μM for all compounds, we observed a consistent 40–50% inhibition of glucose uptake in all cells tested. Inhibition of glucose uptake was also determined using a 2-deoxyglucose (2-DG) based colorimetric glucose uptake kit that showed an approximately 40 and 50% decrease (60 and 50% glucose uptake compared to control) in D456 GBM PDX cells treated with SRI-37683 and SRI-37684, respectively (Figure 6b). Following confirmation of decreased glucose uptake, we sought to determine if this effect was dose-dependent for SRI-37683 and SRI-37684. The ability of SRI-37683 to inhibit glucose uptake plateaus near 50% at a threshold concentration of approximately 3 μM (Supporting Information Figure 3c). Since these data did not conform to the fit for an inhibition curve, we could not accurately calculate an IC50 for SRI-37683. SRI-37684 did conform to a predictable response curve, and the calculated IC50 for glucose uptake was 33 μM (Supporting Information Figure 3c) which, as expected for a metabolically active cell with a high dependence on glycolysis, is higher than the 8.6 μM IC50 for growth inhibition (Figure 4b).
Figure 6. Glucose uptake is inhibited by novel potential GLUT inhibitors.
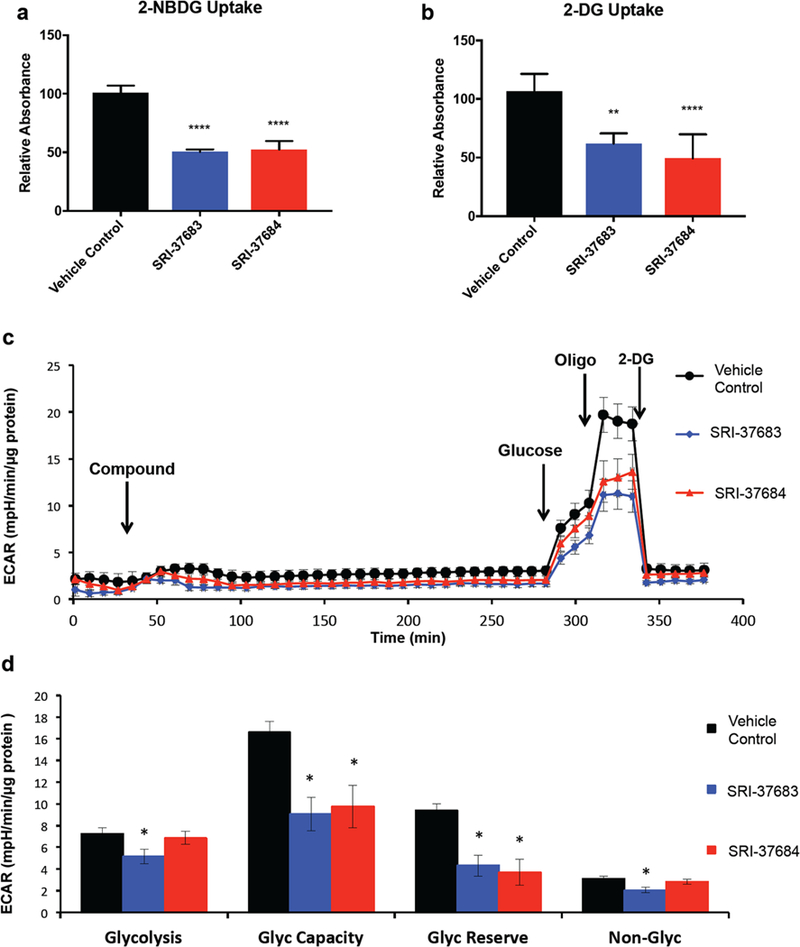
a) Compounds inhibited 2-NBDG uptake as measured in D456 GBM PDX cells. (Averaged data from 2 experiments with n=3 per group * p<0.0001 One-way ANOVA with Tukey’s multiple comparisons) b) 2-DG uptake is inhibited by identified compounds in D456 GBM PDX cells. (Averaged data from 2 experiments with n=3 per group * p<0.0001 One-way ANOVA with Tukey’s multiple comparisons). c) Representative traces of extracellular acidification rate (ECAR) over time, (Representative data (mean ± sem) from 2 experiments with n=6 per group) d) Quantification of glycolytic flux parameters after glycolysis stress test; glycolysis (ECAR after Glucose - basal ECAR), Glycolytic capacity (ECAR before 2-DG minus ECAR after 2-DG), Glycolytic Reserve (ECAR after Oligo minus ECAR before Oligo), and Non-Glycolytic after injection of 2-DG. (Representative data (mean ± sem) from 2 experiments with n=6 per group, * p<0.05 student’s t-test compared to control).
As a further test of the dependence of growth inhibition on glycolysis, we assessed the correlation between the ability of the compounds to reduce GBM PDX growth and inhibit glucose uptake. SRI-37218, which preferentially reduced GBM PDX cell growth in comparison to NHAs (Figure 4c), significantly inhibited glucose uptake (Supporting Information Figure 4). However, further investigation of this compound was discontinued due to toxicity to neurons. SRI-39257, which was broadly toxic (Figure 4f), also inhibited glucose uptake (Supporting Information Figure 4). SRI-37071, SRI-37066, SRI-37067, SRI-39259, and SRI-39255 were compounds that did not significantly inhibit GBM cell growth (Supporting Information Figure 1) and also did not inhibit glucose uptake (Supporting Information Figure 4). However, SRI-39260 (Figure 4d) and SRI-39256 (Figure 4e), which were toxic to all cells tested, did not inhibit glucose uptake (Supporting Information Figure 4), suggesting that an alternative mechanism was responsible for the observed toxicity. Taken together, these data, support our contention that SBVS coupled with growth inhibitory assays is a successful strategy to identify compounds that will inhibit glucose transport. Of course, specificity must be assessed independently as we have indicated.
Additionally, metabolic analysis to assess the extracellular acidification rate (ECAR) was performed using the extracellular flux analyzer with SRI-37683 and SRI-37684 (Figure 6c and 6d). Cells were glucose starved in the presence of the compounds at 50 μM for 4 hours prior to the addition of glucose. Injection of glucose resulted in the expected stimulation of extracellular acidification as indicated by the ECAR ascribed to glycolysis, which was significantly inhibited by SRI-37683 but not by SRI-37684. SRI-37683 and SRI-37684 both inhibited an increase in ECAR following the addition of oligomycin, an inhibitor of mitochondrial ATP production which stimulates glycolysis to compensate and is consistent with the proposed mechanism of action of the compounds. The final addition of 2-deoxy-D-glucose inhibits hexokinase and suppresses glycolytic flux. A more detailed analysis of these results is shown in Figure 6d and indicates inhibition of glycolytic capacity (ECAR before 2-DG minus ECAR after 2-DG) and glycolytic reserve capacity (ECAR after Oligo minus ECAR before Oligo) for all compounds tested, consistent with their proposed mechanism of action.
As glucose uptake is associated with a series of changes in the conformations of the GLUT proteins, we conjecture that the compounds could potentially bind to different transporter conformations leading to inhibition of glucose uptake (34). In the present study, we aimed to specifically target an inward-open conformation of GLUT3 and identified active compounds that inhibited glucose transport as well as cell growth. We also recognize that while our current study evaluated the efficacy of the glucose transporter inhibitors in vitro, it is critical to perform future studies evaluating tumor growth with drug treatment in vivo. Additionally, we recognize the need for further characterization studies to assess the binding affinity, specificity, and relationship between GLUT activity and growth inhibition following lead identification. Overall, we have shown that the compounds we identified can inhibit glucose uptake into GBM cells and selectively decrease GBM growth with limited toxicity to non-malignant cell types (Figure 7). These results suggest that preventing glucose uptake in GBM cells provides a therapeutic approach to be explored for the treatment of GBM and other brain tumors.
Figure 7. Proposed mechanism of action and IC50 concentrations in all cell types and glucose uptake inhibition in D456 GBM PDX cells.
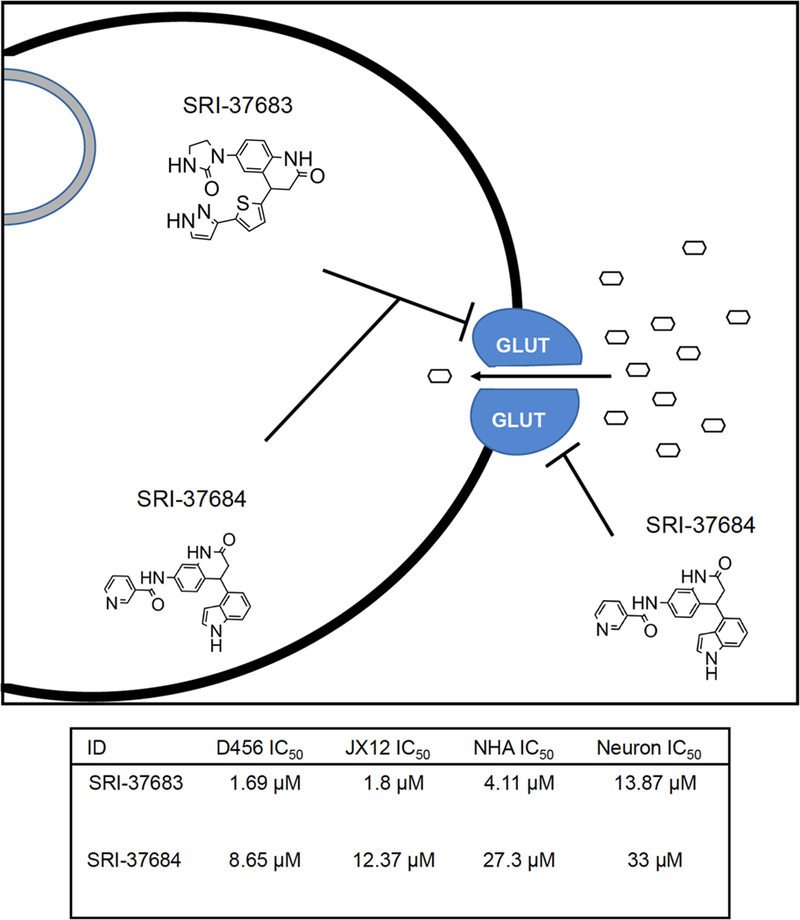
a) Summary of the proposed mechanism of action for hit compounds b) All IC50 values calculated by non-linear regression utilizing Prism by GraphPad from data represented in Figure 4, Figure 5, and Supplementary Figure2).
Predicted binding mode of the active compounds.
To explore the structural insights of GLUT3-inhibitor binding, we further conducted docking studies following a more sophisticated induced-fit docking (IFD) protocol which allows binding site residues to be flexible (40). The two identified GLUT inhibitors, SRI-37683 and SRI- 37684, were docked separately into the glucose binding site of the GLUT3 homology model (Figure 8a and 8b). The docked models were analyzed and compared with the crystal structure template which adopts an inward-open conformation and has a n-nonyl-β-D-glucopyranoside (β-NG) molecule bound at the glucose binding site (Figure 8c)(36). The main receptor/β-NG interactions in the crystal template are the hydrogen-bonds (H-bonds) between the sugar moiety and surrounding polar residues which are also observed in other sugar-transporter complex structures. Similarly, such H-bonds are also the major interaction contributors in our docked models. Specifically, despite their structural differences, all three docked compounds formed multiple H-bonds with the binding site residues Asn286, Gln280/GLN281 and Asn409. SRI-37684 and SRI-37683 share the same 3,4-dihydroquinolin-2-one backbone and possess a more extended linear chemical structure with one end forming ancillary H-bonds with Gln378/Asn315 and the other end filling an additional cavity surrounded by Trp386, Thr135, Gly406, and Met402. Interestingly, Met402 is a non-conserved residue which corresponds to Ile404 in GLUT1. GLUT3 and GLUT1 share high sequence similarity and the residues at glucose binding site are 100% conserved making it a challenge to develop GLUT3 selective inhibitors. Since the compounds SRI-37683 and SRI-37684 did not interact with variant residues between GLUT3 and GLUT 1, docking them to the GLUT 1 crystal structure indeed generated similar virtual binding modes and IFD scores to those of GLUT3. Fortunately, our modeling results showed that both variant residues Met402 (Ile404 in GLUT1) and Asn158 (His160 in GLUT1) are in close proximity (< 4Å) of the docked SRI-37683 and SRI-37684. It is, therefore, possible to design compounds which can maintain the critical H-bonds, and, at the same time, interact with those non-conserved residues to achieve GLUT3 selectivity. Additionally, beyond direct GLUT binding, other factors, including the physiochemical properties (solubility, stability, metabolism, etc.) and the inhibition mechanisms (reversible, competitive, etc.) of the compounds, can also contribute to the observed results in the cell growth and the glucose uptake assays. Also, our compounds were selected based on the docking studies using a relatively less comprehensive conformational sampling algorithm to facilitate high-throughput screening. Therefore, the resulted docking scores may not correlate well with the assay values. In our future optimization efforts, more sophisticated modeling methods such as induced fit docking, free energy perturbation or 3D pharmacophore models will be applied to develop a more robust quantitative structure-activity relationship (SAR) model to guide our compound design and synthesis of newly designed compounds.
Figure 8. Structural representation of predicted binding modes of the identified GLUT inhibitors.
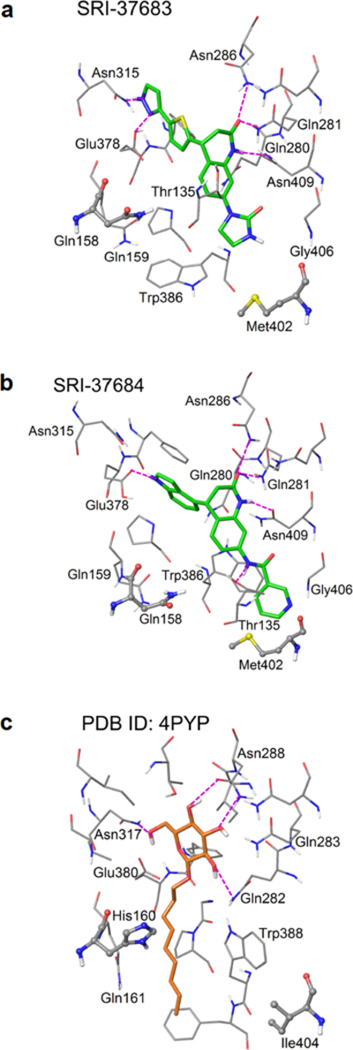
The predicted binding modes of (a) SRI-37683 and (b) SRI-37684 in the inward-open conformation of the modeled GLUT3 crystal structure. (c) The ligand (n-nonyl-β-D-glucopyranoside) binding mode in the crystal structure of GLUT1 in the inward-open conformation (PDB ID: 4PYP). Ligand molecules are represented in slide sticks. Binding site residues are shown in thin tubes. The two residues that are not conserved between GLUT1 and GLUT3 are illustrated in stick & ball mode. Hydrogen bonds are shown in dashed purple lines.
SRI-37683 and SRI-37684 possess a dihydroquinolinone framework which is not found in other known GLUT inhibitors, including the recently reported highly selective GLUT1 inhibitor BAY-876 that was not included in our virtual screening library (41–44). BAY-876 and BAY-588 (a non-specific control for BAY-876) both display similar docking scores to our identified compounds. However, BAY-876 is a relatively large molecule (MWT = 496) with a predicted brain/blood partition coefficient of −2.0 (44, 45). In comparison, dihydroquinolinones, SRI- 37683 (MW = 379) and SRI-37684 (MW = 382), are smaller compounds with better predicted brain/blood partition coefficients (−1.6 and −1.3, respectively) and are amenable to various structural modifications. Furthermore, genistein, an isoflavone reported to inhibit tyrosine kinases and glucose uptake that was present in our virtual library, was identified by our screen and has a docking score of −7.3 kcal/mol and predicted blood/brain partition coefficient of −1.27 (Supplementary Figure 5a) (46–49). As reported in the literature, genistein did reduce GBM PDX cell growth in our growth assays (Supplementary Figure 5b) (50–52). Additionally, genistein inhibited glucose uptake in the 2-NBDG uptake assay, further strengthening our compound identification strategy (Supplementary figure 5c). The low molecular weight, relatively high potency for inhibition of both GBM growth and glycolytic metabolism, and a core structure amenable to modifications to improve potency and selectivity as well as drug-like properties make these hit compounds a good starting point for SAR development.
Recently, a crystal structure of human GLUT3 was published in which the transporter adopted an outward-open conformation (53). We therefore also conducted docking studies of SRI-37683 and SRI-37684 using this newly available GLUT3 model (Supplementary Figure 6a and 6b). Based on virtual binding modes and best IFD scores, SRI-37683 might prefer inward-open conformation (IFD score is −12.1 kcal/mol for inward-open conformation vs. −9.6 for outward-open conformation). While SRI-37683 virtually formed more hydrogen bond interactions with binding site residues in inward-open conformation than outward-open conformation, this does not preclude its action from the outward-open conformation. In contrast, SRI-37684 may not have a conformational preference. IFD scores for SRI-37684 were −11.6 kcal/mol for the inward-open conformation vs. −10.1 kcal/mol for outward-open conformation. Nonetheless, the new GLUT3 structure offers the opportunity to identify additional GLUT3 inhibitors with novel chemical scaffolds. Specifically, from a drug discovery point of view, since the substrate binding site of an outward-open conformation is extracellularly accessible, compounds targeting outward-open conformation could have an advantage over inward-open targeting ones, because they do not need to cross the cell membrane. It would be interesting to see whether inhibitors targeting this outward-open GLUT3 conformation could be identified through a similar computer-aided drug discovery strategy as presented in this study.
CONCLUSION
GLUT3 is critical for the management of glucose influx in cancer cells, particularly due to their reliance on aerobic glycolysis. GLUT3 also represents a novel target for therapeutic intervention in the treatment of GBM. Through the use of SBVS, we have identified two hit compounds for the development of GLUT3 inhibitors. This study indicates that not only is GLUT inhibition a potential avenue for the treatment of GBM, but it also provides the ability to selectively target GBM. Many previous studies have been performed using cells overexpressing the human or rat GLUT family members but have failed to show selectivity for cancer cells (44, 54–57). Of interest, other studies have indicated that inhibition of GLUT1 could decrease the tumor initiating capacity of tumor initiating cells and that GLUT inhibitors enhance the efficacy of the chemotherapy temozolomide against GBM (32, 58). We acknowledge that at this stage of drug development, specific GLUT3 inhibitors have not been identified because the compounds screened thus far do not interact with variant residues. However, our results do suggest that development of GLUT3 specific inhibitors is possible. The identification of hit compounds is an important step in the development of compounds that can be ultimately optimized for human trials. Future research will focus on the design and synthesis of derivatives of selected hits and to develop a SAR profile that will guide the optimization of compounds toward the identification of lead candidates as inhibitors of GLUT3 for the treatment of GBM and other solid tumors.
METHODS
Additional details are available in the supplementary material.
Computer Aided Drug Discovery.
Molecular modeling:
Structural model generation and molecular docking studies were conducted using Schrödinger Suite 2015. Homology modeling of GLUT3 was conducted using the Prime program based on the crystal structure of human GLUT 1 (PDB ID: 4PYP). Molecular docking studies were performed using Glide. The 3D structures of small molecule compounds were prepared using LigPrep. The brain/blood partition coefficients of compounds were predicted using Qikprop.
Virtual library assembly:
A library of 500,000 virtual compounds was assembled from twelve commercial vendors (Asinex, Chembridge, ChemDiv, Enamine, FCH group, InterBioScreen, Life chemicals, Maybridge, Princeton, TIMTEC, SPECS, and Vitas-M). Using Pipeline Pilot (59), we identified a structurally diverse set of 100,000 representative compounds from a total of approximately eight million commercial compounds. We then selected four of the most structurally similar analogs, based on the Tanimoto coefficients calculated from the 2D structural fingerprints, for each of the 100,000 compounds.
Structure-based virtual screening:
A three-step docking/scoring protocol implemented in Glide was applied to screen the assembled library of 500,000 compounds, which consisted of a total of 923,408 conformers generated by Ligprep. Ultimately, the top scoring conformers were outputted and their docked models were visually examined to select compounds to be purchased for biological tests.
Materials, Cells, and Cell Growth.
GBM patient-derived xenografts (PDX) D456, 1016, JX12 and JX14 were obtained from Duke University and the UAB Brain Tumor Core Facility. Normal human astrocytes (NHAs) were purchased from Lonza. Primary wild-type cortical mouse neurons were derived from p0 pups as described previously (60). Dr. Marek Napierala provided human neurons differentiated from iPS cells by lentiviral overexpression of neurogenin 2 as previously reported (61). All cells were grown as reported previously (61, 62). The small molecule inhibitors were purchased from Chembridge or Vitas M Lab and resuspended in dimethyl sulfoxide (DMSO). Cell titer was performed as described previously (62) using compounds identified by SBVS.
Glucose Uptake.
Glucose uptake was determined using 2-NBDG (Fisher) and/or 2-DG (Sigma) according to the manufacturers’ protocols.
Extracellular Flux Analysis Glycolytic Stress Test.
A Seahorse Bioscience XF96 Extracellular Flux Analyzer was used to measure glycolytic flux in GBM PDX cells.
Statistical Analysis.
All statistics were performed with GraphPad Prism Version 7 (GraphPad Software Inc). One-way ANOVA and multiple comparison t-tests were performed and p values indicate a confidence level of 95% and significance of 0.05. IC50values were calculated using a curve-fit model in GraphPad Prism.
Supplementary Material
Acknowledgments
We thank Dr. Y. Gillespie and C. Langford for the guidance of the brain tumor core facility.
Financial Support: This work was supported by National Institutes of Health grant R21NS096531 to A.B.H., T32NS048039 to C.J.L, and in part by P30 G050886–01 to J.Z. and R01NS081366 to M.N., the UAB mitochondrial medicine laboratory and startup funds from the University of Alabama at Birmingham. These startup funds include contributions from the Department of Cell, Developmental and Integrative Biology, the Comprehensive Cancer Center, the Civitan International Research Center for Glial Biology in Medicine, the Center for Free Radical Biology, and the Neuro-Oncology Brain SPORE
Footnotes
Potential Conflicts of Interest: The authors declare no conflicts of interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary Methods. Supplementary Figure 1. Identification of potential GLUT inhibitors that significantly reduce growth. Supplementary Figure 2. Potential GLUT3 antagonists inhibit cell growth. Supplementary Figure 3 Small Molecule Antagonists inhibit glucose uptake in GBM157 and 1016 GBM PDX lines. Supplementary Figure 4. Small Molecule Antagonists inhibit glucose uptake in GBM PDX line D456. Supplementary Figure 5. Examples of docking poses at outward-open GLUT3. Supplementary Figure 6. Genistein, a known compound with glucose uptake inhibitory effects was identified through in silico screening and inhibits GBM growth and glucose uptake.
References
- 1.Barron CC, Bilan PJ, Tsakiridis T, and Tsiani E (2016) Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment, Metabolism 65, 124–139. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, and Negelein E (1927) The Metabolism of Tumors in the Body, J Gen Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, Day BW, Li M, Lathia JD, Rich JN, and Hjelmeland AB (2013) Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake, Nat Neurosci 16, 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, and Weinberg RA (2011) Hallmarks of cancer: the next generation, Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 5.Libby CJ, Tran AN, Scott SE, Griguer C, and Hjelmeland AB (2018) The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells, Biochim Biophys Acta 1869, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marie SK, and Shinjo SM (2011) Metabolism and brain cancer, Clinics (Sao Paulo) 66 Suppl 1, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuen CA, Asuthkar S, Guda MR, Tsung AJ, and Velpula KK (2016) Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: a new target gleaned from an old concept, CNS Oncol 5, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agnihotri S, and Zadeh G (2016) Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions, Neuro Oncol 18, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kathagen A, Schulte A, Balcke G, Phillips HS, Martens T, Matschke J, Gunther HS, Soriano R, Modrusan Z, Sandmann T, Kuhl C, Tissier A, Holz M, Krawinkel LA, Glatzel M, Westphal M, and Lamszus K (2013) Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells, Acta Neuropathol 126, 763–780. [DOI] [PubMed] [Google Scholar]
- 10.Labak CM, Wang PY, Arora R, Guda MR, Asuthkar S, Tsung AJ, and Velpula KK (2016) Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment, Am J Cancer Res 6, 1599–1608. [PMC free article] [PubMed] [Google Scholar]
- 11.Lunt SY, and Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation, Annu Rev Cell Dev Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- 12.Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, and Pajonk F (2011) Metabolic state of glioma stem cells and nontumorigenic cells, Proc Natl Acad Sci U S A 108, 16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Affronti ML, Heery CR, Herndon JE 2nd, Rich JN, Reardon DA, Desjardins A, Vredenburgh JJ, Friedman AH, Bigner DD, and Friedman HS (2009) Overall survival of newly diagnosed glioblastoma patients receiving carmustine wafers followed by radiation and concurrent temozolomide plus rotational multiagent chemotherapy, Cancer 115, 3501–3511. [DOI] [PubMed] [Google Scholar]
- 14.Hottinger AF, Stupp R, and Homicsko K (2014) Standards of care and novel approaches in the management of glioblastoma multiforme, Chin J Cancer 33, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO, European Organisation for, R., Treatment of Cancer Brain, T., Radiation Oncology, G., and National Cancer Institute of Canada Clinical Trials, G. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial, Lancet Oncol 10, 459–466. [DOI] [PubMed] [Google Scholar]
- 16.Singh SK, Clarke ID, Hide T, and Dirks PB (2004) Cancer stem cells in nervous system tumors, Oncogene 23, 7267–7273. [DOI] [PubMed] [Google Scholar]
- 17.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, and Dirks PB (2003) Identification of a cancer stem cell in human brain tumors, Cancer Res 63, 5821–5828. [PubMed] [Google Scholar]
- 18.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, and Dirks PB (2004) Identification of human brain tumour initiating cells, Nature 432, 396–401. [DOI] [PubMed] [Google Scholar]
- 19.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, and Rich JN (2015) Cancer stem cells in glioblastoma, Genes Dev 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altaner C (2008) Glioblastoma and stem cells, Neoplasma 55, 369–374. [PubMed] [Google Scholar]
- 21.Auffinger B, Spencer D, Pytel P, Ahmed AU, and Lesniak MS (2015) The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence, Expert Rev Neurother, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, and Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response, Nature 444, 756–760. [DOI] [PubMed] [Google Scholar]
- 23.Chalmers AJ (2007) Radioresistant glioma stem cells--therapeutic obstacle or promising target?, DNA Repair (Amst) 6, 1391–1394. [DOI] [PubMed] [Google Scholar]
- 24.Kang MK, and Kang SK (2007) Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma, Stem Cells Dev 16, 837–847. [DOI] [PubMed] [Google Scholar]
- 25.Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, Lesniak MS, and Ahmed AU(2014) Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy, Cell Death Differ 21, 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beier D, Schulz JB, and Beier CP (2011) Chemoresistance of glioblastoma cancer stem cells--much more complex than expected, Mol Cancer 10, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu J, Liu ZG, Liu XM, Chen FR, Shi HL, Pangjesse CS, Ng HK, and Chen ZP (2009) Glioblastoma stem cells resistant to temozolomide-induced autophagy, Chin Med J (Engl) 122, 1255–1259. [PubMed] [Google Scholar]
- 28.Rich JN (2007) Cancer stem cells in radiation resistance, Cancer Res 67, 8980–8984. [DOI] [PubMed] [Google Scholar]
- 29.Sakariassen PO, Immervoll H, and Chekenya M (2007) Cancer stem cells as mediators of treatment resistance in brain tumors: status and controversies, Neoplasia 9, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boado RJ, Black KL, and Pardridge WM (1994) Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors, Brain Res Mol Brain Res 27, 51–57. [DOI] [PubMed] [Google Scholar]
- 31.Macheda ML, Rogers S, and Best JD (2005) Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer, J Cell Physiol 202, 654–662. [DOI] [PubMed] [Google Scholar]
- 32.Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, and Kitanaka C (2015) Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells, Oncotarget 6, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pao SS, Paulsen IT, and Saier MH Jr. (1998) Major facilitator superfamily, Microbiol Mol Biol Rev 62, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jardetzky O (1966) Simple allosteric model for membrane pumps, Nature 211, 969–970. [DOI] [PubMed] [Google Scholar]
- 35.Quistgaard EM, Low C, Guettou F, and Nordlund P (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way, Nat Rev Mol Cell Biol 17, 123–132. [DOI] [PubMed] [Google Scholar]
- 36.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, and Yan N (2014) Crystal structure of the human glucose transporter GLUT1, Nature 510, 121–125. [DOI] [PubMed] [Google Scholar]
- 37.Wyss MT, Jolivet R, Buck A, Magistretti PJ, and Weber B (2011) In vivo evidence for lactate as a neuronal energy source, J Neurosci 31, 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamada A, Yamamoto K, Imamoto N, Momosaki S, Hosoi R, Yamaguchi M, and Inoue O (2009) Lactate is an alternative energy fuel to glucose in neurons under anesthesia, Neuroreport 20, 1538–1542. [DOI] [PubMed] [Google Scholar]
- 39.Katsu-Jimenez Y, Alves RM, and Gimenez-Cassina A (2017) Food for thought: Impact of metabolism on neuronal excitability, Exp Cell Res. [DOI] [PubMed] [Google Scholar]
- 40.Sherman W, Day T, Jacobson MP, Friesner RA, and Farid R (2006) Novel procedure for modeling ligand/receptor induced fit effects, J Med Chem 49, 534–553. [DOI] [PubMed] [Google Scholar]
- 41.Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, and Schimmer AD (2008) A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death, Mol Cancer Ther 7, 3546–3555. [DOI] [PubMed] [Google Scholar]
- 42.Zhang W, Liu Y, Chen X, and Bergmeier SC (2010) Novel inhibitors of basal glucose transport as potential anticancer agents, Bioorg Med Chem Lett 20, 2191–2194. [DOI] [PubMed] [Google Scholar]
- 43.Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, and Giaccia AJ (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality, Sci Transl Med 3, 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Muller T, Bauser M, and Buchmann B (2016) Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY- 876, ChemMedChem 11, 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlotte Kopitz LT, Carolyn Algire, Melanie Heroult, Anna-Lena Frisk, Kirstin Meyer, Arndt Schmitz, Eleni Lagkadinou, Heike Petrul, Iring Heisler, Roland Neuhaus, Bernd Buchmann, Herbert Himmel, Marcus Bauser, Andrea Haegebarth, Karl Ziegelbauer. (2016) Pharmacological characterization of BAY-876, a novel highly selective inhibitor of glucose transporter (GLUT)-1 in vitro and in vivo. [abstract]. AACR; Cancer Res 76, Abstract nr 4746. [Google Scholar]
- 46.Bazuine M, van den Broek PJ, and Maassen JA (2005) Genistein directly inhibits GLUT4- mediated glucose uptake in 3T3-L1 adipocytes, Biochem Biophys Res Commun 326, 511–514. [DOI] [PubMed] [Google Scholar]
- 47.Chou HF, Chuang KH, Tsai YS, and Chen YJ (2010) Genistein inhibits glucose and sulphate transport in isolated rat liver lysosomes, Br J Nutr 103, 197–205. [DOI] [PubMed] [Google Scholar]
- 48.Ganai AA, and Farooqi H (2015) Bioactivity of genistein: A review of in vitro and in vivo studies, Biomed Pharmacother 76, 30–38. [DOI] [PubMed] [Google Scholar]
- 49.Vera JC, Reyes AM, Carcamo JG, Velasquez FV, Rivas CI, Zhang RH, Strobel P, Iribarren R, Scher HI, Slebe JC, and Golde DW (1996) Genistein is a natural inhibitor of hexose and dehydroascorbic acid transport through the glucose transporter, GLUT1, J Biol Chem 271, 8719–8724. [DOI] [PubMed] [Google Scholar]
- 50.Atefeh Z, Vahid C, Hasan N, Saeed A, and Mahnaz H (2016) Combination Treatment of Glioblastoma by Low-Dose Radiation and Genistein, Curr Radiopharm 9, 258–263. [DOI] [PubMed] [Google Scholar]
- 51.Khoshyomn S, Nathan D, Manske GC, Osler TM, and Penar PL (2002) Synergistic effect of genistein and BCNU on growth inhibition and cytotoxicity of glioblastoma cells, J Neurooncol 57, 193–200. [DOI] [PubMed] [Google Scholar]
- 52.Liu X, Liu K, Qin J, Hao L, Li X, Liu Y, Zhang X, Liu X, Li P, Han S, Mao Z, and Shen L(2015) C/EBPbeta promotes angiogenesis through secretion of IL-6, which is inhibited by genistein, in EGFRvIII-positive glioblastoma, Int J Cancer 136, 2524–2534. [DOI] [PubMed] [Google Scholar]
- 53.Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W, Hirata K, Yamamoto M, Fan S, and Yan N (2015) Molecular basis of ligand recognition and transport by glucose transporters, Nature 526, 391–396. [DOI] [PubMed] [Google Scholar]
- 54.Siebeneicher H, Bauser M, Buchmann B, Heisler I, Muller T, Neuhaus R, Rehwinkel H, Telser J, and Zorn L (2016) Identification of novel GLUT inhibitors, Bioorg Med Chem Lett 26, 1732–1737. [DOI] [PubMed] [Google Scholar]
- 55.Ung PM, Song W, Cheng L, Zhao X, Hu H, Chen L, and Schlessinger A (2016) Inhibitor Discovery for the Human GLUT 1 from Homology Modeling and Virtual Screening, ACS Chem Biol 11, 1908–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra RK, Wei C, Hresko RC, Bajpai R, Heitmeier M, Matulis SM, Nooka AK, Rosen ST, Hruz PW, Schiltz GE, and Shanmugam M (2015) In Silico Modeling-based Identification of Glucose Transporter 4 (GLUT4)-selective Inhibitors for Cancer Therapy, J Biol Chem 290, 14441–14453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, and Carruthers A (2016) WZB117 (2-Fluoro- 6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) Inhibits GLUT1-mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site, J Biol Chem 291, 26762–26772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Azzalin A, Nato G, Parmigiani E, Garello F, Buffo A, and Magrassi L (2017) Inhibitors of GLUT/SLC2A Enhance the Action of BCNU and Temozolomide against High-Grade Gliomas, Neoplasia 19, 364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF, and Park DM (2009) Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha, Oncogene 28, 3949–3959. [DOI] [PubMed] [Google Scholar]
- 60.Redmann M, Wani WY, Volpicelli-Daley L, Darley-Usmar V, and Zhang J (2017) Trehalose does not improve neuronal survival on exposure to alpha-synuclein pre-formed fibrils, Redox Biol 11, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, and Sudhof TC (2013) Rapid single-step induction of functional neurons from human pluripotent stem cells, Neuron 78, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyd NH, Walker K, Fried J, Hackney JR, McDonald PC, Benavides GA, Spina R, Audia A, Scott SE, Libby CJ, Tran AN, Bevensee MO, Griguer C, Nozell S, Gillespie GY, Nabors B, Bhat KP, Bar EE, Darley-Usmar V, Xu B, Gordon E, Cooper SJ, Dedhar S, and Hjelmeland AB (2017) Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo, JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.