

Abstract
Studies using omics-based approaches have advanced our knowledge of metabolic remodeling in cardiac hypertrophy and failure. Metabolomic analysis of the failing heart has revealed global changes in mitochondrial substrate metabolism. Peroxisome proliferator-activated receptor-α (PPARα) plays a critical role in synergistic regulation of cardiac metabolism through transcriptional control. Metabolic reprogramming via PPARα signaling in heart failure ultimately propagates into myocardial energetics. However, emerging evidence suggests that the expression level of PPARα per se does not always explain the energetic state in the heart. The transcriptional activities of PPARα are dynamic, yet highly coordinated. An additional level of complexity in the PPARα regulatory mechanism arises from its ability to interact with various partners, which ultimately determines the metabolic phenotype of the diseased heart. This review summarizes our current knowledge of the PPARα regulatory mechanisms in cardiac metabolism and the possible role of PPARα in epigenetic modifications in the diseased heart. In addition, we discuss how metabolomics can contribute to a better understanding of the role of PPARα in the progression of cardiac hypertrophy and failure.
Keywords: fatty acids, deoxyribonucleotide, metabolism, peroxisome proliferator-activated receptor-α
heart failure (HF) is characterized by substantial defects in cardiac oxidative capacity (18, 35, 59, 80, 102). Increasing lines of evidence suggest that metabolic dysregulation and energy metabolic derangements contribute to the pathogenesis of HF (55, 85, 108). Some clinical trials and animal studies suggest that metabolism is a promising therapeutic target for HF (14, 69, 74, 76, 81, 114). Understanding the metabolic phenotype of HF and the role of metabolic changes in the progression of the pathology is critical for developing the concept of metabolic intervention in HF.
Recent advances in omics-based analyses have begun to provide a glimpse into the molecular framework underpinning the altered fuel metabolism and mitochondrial energetics in the hypertrophied and failing heart. Metabolomics is the most recently added omics discipline and is a powerful method to define disturbed biochemical pathways in complex physiological and disease states. Metabolic profiling of HF through unbiased metabolomic screening has revealed that metabolic remodeling occurs globally in various pathways, including fatty acid oxidation (FAO), glucose metabolism, branched-chain amino acid degradation (113), ketone body metabolism (5, 10), and the pentose phosphate pathway (57). These complex changes in metabolic networks, particularly in the advanced stages of HF, are the cumulative consequences of primary and compensatory responses to hemodynamic stress, as well as adaptive and maladaptive myocardial remodeling associated with cardiac dysfunction. Emerging evidence suggests that pressure overload (PO) primarily induces impaired FAO, which precedes the onset of HF (26). Unbiased transcriptomic analysis at the compensated stage of hypertrophy showed that prominent expression changes occurred only in FAO genes (59), suggesting that reprogramming of FAO is the initial step in metabolic remodeling during HF.
Peroxisome proliferator-activated receptor-α (PPARα) is a master regulator of FAO and plays an important role in ensuring coordinated expression in the pathway through transcriptional control. It has been believed that downregulation of PPARα leads to impaired FAO in HF. However, there is reason to believe that the regulatory mechanisms of PPARα in the diseased heart are extremely complex. First, impaired FAO in HF was observed in the presence of various levels of PPARα expression (8, 18, 52, 77, 80). Second, the role of PPARα in cardiac function is controversial. Both constitutive knockout and overexpression of PPARα in the mouse heart led to the development of cardiac dysfunction (34, 130). Third, the effects of PPARα agonists on cardiac function depend on the pathological stage of HF. The activation of PPARα using WY-14643 in hypertrophied rat hearts led to severe impairment of cardiac function at the early stage (136). In contrast, the induction of PPARα at the advanced stage of HF preserved cardiac function and the expression of FAO genes in mice (51). Last, our recent studies suggest that PPARα can act as both an activator and a repressor of transcription (77, 78).
This review summarizes the recent developments concerning the regulatory mechanisms of PPARα and downstream signaling to metabolic networks. In particular, we discuss 1) how expression of PPARα is altered and 2) how PPARα differentially regulates genes known to be involved in FAO and previously defined as PPARα targets during the development of cardiac hypertrophy and failure. This review also examines how we can apply metabolomics to better understand the complex regulatory mechanisms of PPARα in the heart. An extensive discussion of the biological effects of PPARα is beyond the scope of this article, and we refer the reader to several excellent reviews on this subject (33, 43, 68).
PPARα EXPRESSION IN HEART FAILURE
Upstream physiological cues for maintaining energetic status in the heart are integrated with the actions of cardiac nuclear receptors (126). PPARα, a member of the PPAR nuclear receptor subfamily, is a ligand-regulated transcription factor that primarily regulates lipid metabolism (68). Together with its dimerization partner, retinoid X receptor (RXR) and its coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), PPARα binds to the PPAR response element (PPRE), also called direct repeat (DR) 1 (120, 138), in genes involved in fatty acid (FA) degradation (43, 65, 70, 100, 107) (Fig. 1A).
Fig. 1.

Schematic representation of epigenetic regulation of peroxisome proliferator-activated receptor-α (PPARα) in fatty acid oxidation (FAO) genes (A–C) and PPARα signaling to the FAO metabolome (D). A: PPARα heterodimerizes with retinoid X receptor-α (RXRα) and binds to the specific region of DNA sequence known as the peroxisome proliferator receptor response element (PPRE)/direct repeat 1 (DR1) located in the promoters of PPARα target genes, which leads to initiation of transcription by recruiting RNA polymerase II (RNA POL II) and other transcription factors. Thus, PPARα positively regulates expression of FAO genes throughout the pathway. B: during pressure overload (PO), PPARα also interacts with sirtuin 1 (Sirt1), which is a class III histone deacetylase (HDAC). PPARα recruits Sirt1 to the promoter regions of a subset of PPARα target genes that contain an “imperfect” PPRE/DR1, leading to transcriptional repression, presumably through deacetylation of histones. It is hypothesized that PPARα also interacts with other epigenetic modulators, which possibly affect the methylation (Me) and acetylation (Ac) of histones surrounding the PPARα-binding region. (“Me” in pink, associated with gene activation, e.g., H3K4; Me in gray, associated with gene repression, e.g., H3K9). C: promoters of genes that are upregulated by PPARα principally harbor the “perfect” PPRE/DR1, which contains the consensus sequence of two typical hexads with no more than one nucleotide mismatch (yellow). In contrast, the imperfect PPRE/DR1 contains one typical hexad and one nonconserved (atypical) hexad [red, bottom left, modified from Oka et al. (78)] and is found in genes involved in β-oxidation, the tricarboxylic acid (TCA) cycle, and the electron transport chain (ETC) in the FAO downstream pathway. Thus, the PPARα-Sirt1 complex preferentially targets the FAO downstream pathway in the diseased heart, and the unique DNA sequences of PPREs allow PPARα to differentially regulate its target genes during PO. D: epigenetic modifications regulated by PPARα ultimately determine the metabolic phenotype of the diseased heart, which includes the FAO metabolome (i.e., free fatty acids, fatty acyl-CoA, carnitine, acylcarnitines, acetyl-CoA), fatty acid (FA) storage (triglycerides), and the redox status (cellular levels of NAD+, NADH, and FADH2). Free fatty acids are byproducts of lipolysis from triglycerides and are the ligands of PPARα. The altered myocardial contents of fatty acids and carnitine/acylcarnitine species (bold) are the signature of the metabolomic phenotype of the failing heart. OCTN2, organic cation/carnitine transporter 2; FABP, fatty acid-binding protein; FACS, fatty acyl-CoA synthetase; MCAD, medium-chain acyl-CoA dehydrogenase; PPi, inorganic pyrophosphate.
Pathological stress conditions influence the expression of PPARα (6, 43, 56, 94). How, then, is the expression of this master regulator of cardiac energetics controlled in the hypertrophied and failing heart? Drosatos et al. showed that cardiac-specific deletion of Krüppel-like family of transcription factor 5, a zinc finger DNA-binding protein, in mice led to remarkable downregulation of PPARα at the mRNA level and impaired FAO (27), suggesting its potential role as an activator of PPARα transcription. Expression of PPARα is also regulated by a PPRE located in its own enhancer. In murine skeletal muscle, Tateishi et al. showed that the histone demethylase JHDM2a directly stimulates gene expression of PPARα by binding to its PPRE and demethylating histone H3 Lys9 (H3K9me, a marker of gene repression) in response to β-adrenergic stimulation (118). In addition, our recent study showed that cardiac-specific ablation of the histone methyltransferase Smyd1 in mice led to downregulation of PPARα at the mRNA level (103), although the turnover rates of mRNA need to be taken into account. To date, it is largely unknown what determines PPARα expression during the development of hypertrophy and HF. Clarifying the role of epigenetic modification in regulating PPARα expression is of great interest.
More importantly, cardiac hypertrophy and failure lead to variable changes in PPARα expression. Several studies have reported that PPARα is downregulated in HF, in association with suppression of FAO (8, 52, 97). However, other studies showed that the expression level of PPARα did not change in PO-induced HF in mice or in a canine model of pacing-induced HF, despite downregulation of FAO proteins (18, 80). Upregulation of PPARα at the protein level was also reported in PO-induced HF in mice (77). The expression level of PPARα presumably varies depending on the severity and the timing of PO. This varying pattern of PPARα expression in the hypertrophied and failing heart, where FAO is consistently suppressed, indicates that the expression level of PPARα may not be an absolute determinant of FAO function and/or that more complex regulatory mechanisms exist.
DOWNSTREAM TARGETS OF PPARα
Studies using gain- and loss-of-function strategies in mice have shown that the activation of PPARα increases the expression of genes involved in virtually every step of cardiac FA utilization, including 1) FA transport and esterification (fatty acid transporter, CD36, fatty acid-binding protein, and acetyl-CoA synthase, 2) FA mitochondrial import [carnitine palmitoyltransferase (CPT) 1β], and 3) mitochondrial FA β-oxidation [medium-chain acyl-CoA dehydrogenase (MCAD), long-chain acyl-CoA dehydrogenase, and very-long-chain acyl-CoA dehydrogenase] (43, 65, 70, 100, 107). PPARα knockout (PPARα−/−) mice exhibited coordinated downregulation of FAO genes in the heart (2, 63), suggesting that PPARα is a master regulator of cardiac FAO. Carnitine is required for mitochondrial FA uptake and is the substrate of CPT1, which catalyzes formation of acylcarnitine species from carnitine and fatty acyl-CoA. The organic carnitine transporter 2 (OCTN2) is responsible for the cellular uptake of carnitine and was found to be downregulated in patients with idiopathic hypertrophic cardiomyopathy (73). Recent studies showed that PPARα directly regulates gene expression of OCTN2 in cardiomyocytes (66, 132). Thus, PPARα coordinately modulates a transcriptional program that governs FAO from upstream to downstream, and the altered PPARα expression in HF globally affects the FA degradation pathway, including the expression of PPARα target genes and the myocardial contents of FAO substrates/intermediates (Fig. 1D).
PPARα Regulatory Mechanisms to Target Specific FAO Genes
The initial complexity of PPARα regulatory mechanisms comes from the fact that FAO is suppressed in HF in the presence of varying levels of PPARα expression. An additional layer of complexity arises from the downregulation of only a subset of PPARα target genes in HF, rather than global downregulation of FAO genes. To date, the molecular mechanisms through which the presence of cardiac hypertrophy and HF affects specific steps of FAO are not clearly understood. Our recent studies uncovered some potential mechanisms for conferring specificity to PPARα-mediated gene expression, as summarized below.
Functional significance of DR1 sequences.
The known downstream targets of PPARα signaling can be differentially regulated by PPARα due to subtle differences in the nucleotide sequence at which PPARα binds to their promoters. Our previous study showed that upregulation of PPARα can either stimulate or inhibit FAO genes previously recognized as targets of PPARα (77). The function of PPARα may be altered depending on its binding partner. It is widely accepted that PPARα forms a heterodimer with RXRα, which activates transcription of genes involved in FAO through binding to the PPRE (107) (Fig. 1A). However, PPARα can also interact with silent information regulator 1 (Sirt1), a NAD-dependent protein deacetylase, which in turn leads to repression of some of the genes involved in FAO (77) (Fig. 1B). What, then, induces the interaction of PPARα with Sirt1 in HF instead of the heterodimerization with RXRα? It was discovered that a subset of genes, which are suppressed by PPARα, contain characteristic DNA sequences in their promoters to which PPARα binds. Although the consensus sequence of the PPARα-binding site has been reported as RGSWVANAGGTCA (23) (R = A or G, S = G or C, W = A or T, and V = C, G, or A), the actual nucleotide sequence exhibits considerable variation from gene to gene. Some PPARα target genes possess PPARα-binding sites that are a nearly perfect match with the consensus PPRE/DR1 sequence (<1 nucleotide mismatch from the conserved sequence, dubbed “perfect DR1”), whereas others contain less-conserved sequences (>1 nucleotide mismatch from the conserved sequence, dubbed “imperfect DR1”; see Fig. 1C). We found that the former group is generally upregulated, whereas the latter group is downregulated in the heart in the presence of PO, where the expression of PPARα is increased. Because FAO is mediated through the coordinated actions of many molecules, downregulation of some genes would be sufficient to lead to impaired FAO, as indicated by a reduced mitochondrial respiration rate (18, 80) and altered levels of acylcarnitines (59, 99). These findings suggest that the upregulated PPARα in the failing heart specifically targets the genes involved in the downstream pathway of FAO, which are distinguished by the unique DNA sequences of their PPARα-binding sites (imperfect DR1).
Determinants of selective target FAO genes in PO.
Why are PPARα target genes containing the imperfect1 DR1 downregulated when PPARα is upregulated? We found that the DNA sequence of the PPARα-binding site significantly affects heterodimerization of PPARα with its partner (78). Although PPARα forms a stable dimer with RXRα on the perfect DR1 (Fig. 1, A–C), it fails to do so on the imperfect DR1. Namely, while PPARα can stably bind to a typical hexad sequence, AGGTCA, on one part of the imperfect DR1 (Fig. 1C), the mismatches in other areas of the imperfect DR1 make the binding of RXRα relatively unstable. Accordingly, PPARα can easily switch its heterodimerization partner from RXRα to another molecule, such as Sirt1 (Fig. 1B). Importantly, structurally homologous regions exist in both RXRα and Sirt1 that compete for binding to PPARα. In addition, RXRα is downregulated, whereas Sirt1 is upregulated in mice that underwent transverse aortic constriction (TAC) (77). Taken together, it appears that PPARα localized on the imperfect DR1 preferentially switches its partner from RXRα to Sirt1 in HF, whereas the PPARα-RXRα heterodimer is relatively stable on the perfect DR1. The recruitment of Sirt1 induces histone deacetylation of the promoter, thereby causing suppression of gene expression. Thus, the recruitment of Sirt1 to the imperfect DR1s, which is presumably mediated by upregulation of PPARα, leads to transcriptional suppression of specific genes (e.g., MCAD, VLDL) during PO. In contrast, RXRα has a high affinity for the perfect DR1 found in the genes involved in myocardial FA uptake (e.g., CD36) and mitochondrial FA uptake (CPT1), so that these genes are upregulated/unchanged by upregulation of PPARα during PO. Thus, impairment of FAO in PO-induced HF (mainly in downstream FAO) can be attributed to transcriptional repression of these specific PPARα targets through the recruitment of Sirt1 to their promoter regions. In support of this notion, inhibition of the PPARα-Sirt1 interaction restored FAO and rescued cardiac function during PO (78). In summary, the identity of the heterodimerization partner of PPARα specifies the downstream signaling of PPARα under pathological conditions. Additionally, the expression levels of PPARα partners and the nucleotide sequence of the PPARα-binding site are probably the critical determinants of how PPARα selects its binding partners. These mechanisms are similar to the dynamic regulation of PPARγ in stress resistance of adipocytes where a subset of PPARγ target genes, but not its classical targets (e.g., adiponectin), was repressed by Sirt1 in vitro (129). In addition, transcriptional suppression through imperfect DR1-like sequences has been reported for both PPARβ/δ (1) and PPARα (119).
Other Possible Regulatory Mechanisms of PPARα
Our study implies that the changes in expression of PPARα, Sirt1, and RXRα are involved in differential regulation of FAO genes in HF (77, 78). Other mechanisms may also contribute to dynamic regulation of PPARα under pathological conditions. Below, we discuss posttranslational modifications (PTMs) of PPARs and the role of PPARα in chromatin remodeling, and further assess the role of PTM and epigenetics in PPARα regulation.
Posttranslational modifications of PPARs.
The activity of the PPAR family proteins is regulated by PTMs, including phosphorylation, SUMOylation, ubiquitylation, GlcNAcylation, and acetylation (20, 128) (Fig. 2A). The roles of PTMs in fine tuning PPAR activity have been described in several studies. Below, we describe examples in which PTM of PPARs provides selectivity and examine the possibility that PTM of PPARα is involved in regulating its activity during HF.
Fig. 2.

Hypothetical regulatory mechanisms of PPARα activity through posttranslational modifications (PTMs, A) and epigenetic regulation (B). A: during PO, PPARα undergoes various PTMs, including phosphorylation (P), Ac, SUMOylation [small ubiquitin-like modifier (SUMO)ylation], and ubiquitination (Ubi). Consequently, PPARα changes in its sensitivity to ligands and/or its affinity for its binding proteins, leading to the modifications in PPARα downstream signaling. Ubiquitination of PPARs can result in protein degradation. B: PO induces the recruitment of epigenetic modulators to the promoter regions of PPARα target genes. Depending on which modulators are recruited, PPARα can trigger histone PTMs, DNA methylation, or posttranscriptional modifications. In general, histone acetylation leads to gene activation, whereas histone deacetylation leads to gene repression. The functional consequence of histone methylation is complex, since transcriptional control depends on the position of the lysine residue that undergoes methylation. Direct DNA methylation by methyltransferase (DNMTs) generally leads to gene silencing. A micro-RNA (miR) is a small noncoding RNA molecule that may alter expression of PPARα targets. HAT, histone acetylase; HDAC, histone deacetylase; HMT, histone methyltransferase; HDM, histone demethylase.
Phosphorylation of PPAR is associated with enhanced ligand-dependent transcriptional activity. PPARα is a substrate of various kinases that mainly target serine residues. The stress-activated p38 mitogen-activated protein kinase (MAPK) phosphorylates the serine residues located within the NH2-terminal A/B domain of PPARα, which enhances functional cooperation with PGC-1 and ligand-dependent transactivation in rat neonatal cardiomyocytes (9). A study by Yoon et al. suggests that adiponectin signaling causes sequential activation of AMP-activated protein kinase, p38 MAPK, and PPARα in C2C12 cells (134). PKCα and -β phosphorylate PPARα in human liver cells at Ser179 and Ser230, which increases ligand-induced PPARα transcriptional activity (12), whereas activators of protein kinase A, such as cholera toxin, enhance PPARα activity regardless of the presence of exogenous ligands (60). In contrast, phosphorylation of PPARα by glycogen synthase kinase 3 (GSK3) at Ser73 decreases the transcriptional activity and increases ubiquitination and protein turnover of PPARα (42). Inhibition of GSK3β activity in hepatocytes increases expression of PPARα (93). It is important to note that inhibition of the ERK-MAPK pathway, but not of the p38-MAPK pathway, prevents the repression of CPT1 through the PPRE observed in α1-adrenergic agonist-induced hypertrophy in cultured rat neonatal cardiac myocytes (8). Whether and how phosphorylation/dephosphorylation regulates PPARα in PO-induced hypertrophy and HF would be an important subject for future investigation (50).
The deacetylation activity of Sirt1 is involved in various biological and pathological processes, such as stimulation of autophagy (62), improving insulin sensitivity (112), and glucose homeostasis (96). Sirt1 is responsible for deacetylation of a variety of proteins, including PGC-1α (75, 96), p53 (67, 125), and forkhead box O3 (15, 72). In white adipocytes, Sirt1-dependent deacetylation of PPARγ at Lys268 and Lys293 is required to recruit coactivator Prdm16 for reprogramming to brown adipocyte tissue, leading to selective induction of genes involved in energy expenditure and repression of genes associated with insulin resistance (90). Although increased interaction between PPARα and Sirt1 in PO hearts (77) suggests a possible role of deacetylation in the transcriptional regulation of PPARα, no significant increase in the acetylation status of PPARα was detected in mice in which Sirt1 was knocked out (77). Therefore, it is unlikely that deacetylation of PPARα itself is a functional consequence of the heterodimerization with Sirt1 during PO.
The conjugation of ubiquitin (a 76-amino acid polypeptide) and small ubiquitin-like modifier (SUMO, a 101-amino acid polypeptide) to PPAR family members negatively regulates their transcriptional activity. The role of the ubiquitin-proteasome system in regulating PPARs in cancer pathogenesis has been described in several studies (36). In the mouse heart, the muscle-specific E3 ubiquitin ligase muscle ring finger 2 (MuRF2) monoubiquitylates PPARs and thereby inhibits transcription of downstream targets, possibly by inducing the nuclear exit of PPARs (41). Downregulation of MuRF2 exacerbates the development of diabetic cardiomyopathy in vivo, but the targets of PPARs are not necessarily all altered uniformly, raising the possibility that monoubiquitylation of PPARs differentially regulates known targets of PPARs. SUMOylation of PPARγ leads to modification of genes that do not possess classical PPREs by converting the PPAR/RXR heterodimer to a PPAR monomer, potentiating a novel interaction with corepressor complexes in macrophages (82). These findings suggest that SUMOylation may convert PPARα from a stimulator to an inhibitor of transcription by changing its interacting partners.
These studies highlight the importance of PTMs as part of the dynamic regulatory mechanisms of PPAR family proteins. It is of great interest to us to determine whether these PTMs occur in response to pathological stress and whether differential regulation of FAO genes during PO depends on PTMs of PPARα. Identification of specific PTMs of PPARα in HF might provide new insight into the mechanisms by which PPARα selectively targets FAO genes in the diseased heart.
Epigenetic regulation of PPARα target genes.
Transcription of PPARα targets is subject to various types of epigenetic regulation. PPARα is a transcription factor and directly binds to DNA. DNA-binding proteins often interact with epigenetic modulators for reprogramming in various developmental and pathological stages and in response to environmental changes (54). Epigenetics in modern terms refers to the study of molecular processes that regulate genomic activities independent of the DNA sequence. Epigenetic regulation plays an important role in ensuring that functional gene modules are synchronized to produce coordinated expression. There are three major types of epigenetic modifications: histone modification, DNA methylation, and noncoding RNA (Fig. 2B).
Chromatin can undergo posttranslational covalent modification of the NH2-terminal tails of histones, including histone methylation, acetylation, phosphorylation, ubiquitination, and SUMOylation (141). Well-known histone modulators are histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases, and histone demethylases (141). Our recent finding that PPARα recruits Sirt1 to a specific region in the promoter of its target genes during PO-induced HF (78) suggests a potential role of PPARα in epigenetic remodeling in response to stress. Sirt1, a class III NAD+-dependent HDAC, can modulate chromatin function through direct deacetylation of histones [histone H4 Lys9 (123), H3 Lys14 (47), and H1 Lys26 (123)], leading to chromatin silencing and transcriptional repression. Thus, it is conceivable that the interaction of PPARα with Sirt1 in PO-induced HF is involved in chromatin remodeling of its target genes. Indeed, histone acetylation/deacetylation has been shown to play a role in the development of cardiac hypertrophy and failure (131, 139). The HAT activity of CREB-binding protein and p300 is required for the induction of hypertrophic changes in cardiomyocytes by phenylephrine (40), whereas the inhibition of HDAC activity results in an increase in the size of myoblasts (44). It is of great interest to further clarify how interaction of PPARα with Sirt1 alters the acetylation status of PPARα target genes and how the histone acetylation profile is linked to impaired FAO.
In HF patients and a rat model of high-salt diet-induced HF, the histone methylation profile, rather than the histone acetylation profile, was significantly modified (53). The inhibition of histone H3K9 methyltransferase maintained left ventricular systolic function and mitochondrial function in association with restoration of the genes involved in energetics, such as PGC-1α, Acadm, and Ndufs4 (79). In our recent study, cardiac-specific deletion of the histone H3K4 methyltransferase Smyd1 led to downregulation of PPARα and FAO genes (103), which may indicate a role of histone methylation in PPARα’s transcriptional activity in the heart. Notably, histone acetylation and histone methylation are often coordinately regulated (29). Strahl and Allis have proposed that epigenetic changes regulate gene transcription not through a single modification but through a combination of various markers, known as the “histone code” (111). Thus, it is plausible that PPARα recruits various epigenetic modulators to the promoters of its target genes under pathological conditions, leading to dynamic chromatin remodeling secondary to coordinated reprogramming of genes involved in energetics.
Direct methylation of DNA in promoter regions often causes gene silencing by inhibiting transcription factor binding or recruiting chromatin-inactivation complexes. Interestingly, DNA methylation in the body of a gene can activate transcription through an unknown mechanism (46, 117). This type of epigenetic modification is relatively stable and can be maintained throughout the lifetime and be heritable (48) because DNA methyltransferases recognize hemimethylated 5′-C-phosphate-G-3′ generated during DNA replication (38) and dependably copy cytosine methylation from the parental to the daughter strand. For instance, in rats that were fed protein-restricted diets during pregnancy, there was a decrease in PPARα promoter methylation in both maternal rats and their offspring, suggesting that prenatal nutrition alters the future transcriptional regulation of cardiac energy metabolism in the next generation through changes in epigenetic regulation of specific genes (106). Ligand-activated PPARα-dependent DNA demethylation of FAO genes occurs in the postnatal mouse liver (28). Hepatic FAO is activated progressively during the neonatal period to produce energy from breast milk lipids (83, 84), which appears to be correlated with the increased levels of DNA demethylation of a specific subset of genes involved in FAO in the liver of pups. This may represent an adaptive mechanism to the marked changes in nutritional environment after birth. It is speculated that ligand-activated PPARα recruits a protein involved in DNA demethylation, such as thymine DNA glycosylase, to the promoter regions of FAO genes via the PPRE, thus leading to the gene-specific DNA demethylation. If so, this would be a novel mechanism by which PPAR regulates only a specific subset of genes in a ligand-dependent manner. In patients with HF, free fatty acids (FFA), the ligands of PPARα, particularly saturated FA, are elevated in plasma, and the levels of circulating FFA are positively associated with incidence of HF (25). Several n-3 polyunsaturated FAs have been shown to have positive effects on survival of patients with HF (49). It would be interesting to determine whether DNA methylation occurs in a subset of PPARα target genes in response to the changes in circulating FFA.
Evidence is emerging that noncoding RNA plays a role in reprogramming lipid metabolism (31). Micro-RNAs (miRs) are small RNAs (~22 nucleotides) encoded in the genome that bind to complementary target sites in the 3′-untranslated region of their target transcripts, causing translational repression or mRNA degradation. Several miRs have been identified that act as posttranscriptional regulators of lipid metabolism genes involved in cholesterol homeostasis and FAO, including miR-122, miR-370, miR-378/378*, miR-335, miR-125a-5p, and miR-33 (31). Among them, miR-33 specifically targets CPT1α (116), and inhibition of miR-33 promoted FAO in the mouse liver (22). Increased expression of miR-199α and miR-214 was found in human and mouse HF, leading to the reduction of cardiac PPARδ expression and mitochondrial FAO capacity in mice (29). miR-27 inhibits the expression of PPARγ and C/EBPα (64). On the other hand, unlike most miRs, which inhibit gene expression, miR-378/378*, an intronic miR located within the PGC-1α genomic sequence, increases the transcriptional activity of C/EBPα and C/EBPβ on adipocyte gene promoters (37). To date, a miR that directly targets PPARα has not been reported, and it is unknown whether the PPARα genomic sequence contains a noncoding RNA that regulates FAO genes.
PPARα SIGNALING TO METABOLOMICS
Metabolomics is the comprehensive analysis of the small molecules (<1,500 Da) of a cell, tissue, biofluid, or organism, measuring a wide range of metabolites in various biological pathways. The metabolome represents the downstream effects of modifications of the genome and proteins in metabolic networks. Plasma metabolomics is commonly used to discover a biomarker or to predict diseases in patients, whereas tissue metabolomic analysis enables metabolic phenotyping of the heart and provides insights into global metabolic remodeling that occurs in response to physiological and pathological stimuli.
Metabolomic analysis includes FFAs, acylcarnitines, carnitine, glycerol, cholesterol, glycolysis substrates/intermediates, tricarboxylic acid (TCA) cycle intermediates, purines, and amino acids, whereas lipidomics measures triacylglycerides, long-chain acylcarnitines, phospholipids, ceramides, and diacylglycerol. Recent studies using unbiased metabolomic analysis of the failing heart suggest that the changes in the myocardial contents of FFA and carnitine/acylcarnitines (FAO substrates) represent unique metabolic signatures of HF (Fig. 1D). Several studies reported that the myocardial contents of carnitine, acylcarnitines, and FFA were most reduced in the advanced stage of HF (99, 104, 124), suggesting a deficiency of FAO substrates. Because metabolomic analysis per se is a “snapshot” of metabolites in the heart and does not provide an assessment of substrate flux in pathways, recent studies have integrated metabolomic analysis with either transcriptomics or proteomics (59, 104, 113), known as a multisystems approach. Integrating transcriptomic and metabolomic analyses of mice that had undergone TAC revealed that the levels of myocardial long-chain acylcarnitines species were increased during the progression of HF, in association with downregulation of very-long-chain acyl-CoA dehydrogenase (ACADVL) but no change in the expression level of CPT1β (59). This study also supports the notion that not every target of PPARα is equally affected by pathological stress but rather that downstream FAO is specifically targeted during PO, resulting in the accumulation of long-chain acylcarnitines, presumably because of the mismatch in metabolic flux between the upstream and downstream FAO pathways. Furthermore, varying levels of myocardial long-chain acylcarnitines at different time points during PO [increased at 4 wk of PO (59), decreased at 8 wk of PO (99)] may reflect dynamic regulation by PPARα of its target genes through the aforementioned mechanisms. Interestingly, in contrast to the decreased levels of myocardial acylcarnitines, plasma levels of carnitine and acylcarnitines are elevated in HF patients (30) and in a canine model of pacing-induced HF (104). The plasma levels of some acylcarnitine species significantly correlate with the New York Heart Association functional class, supporting a link between disturbed carnitine metabolism and the severity of HF (121). Although PPARα expression was not reported in those studies, it might be interesting to determine the correlation between plasma levels of carnitine/acylcarnitines and the myocardial expression of PPARα and its target genes at different stages of HF.
It is possible that differential regulation of PPARα during PO reflects changes in metabolites in global networks beyond FAO substrates and intermediates. For instance, prominent changes in branched-chain amino acids, the mitochondrial fuel substrates, were observed after 1 wk of TAC, whereas longer durations of TAC and myocardial infarction led to a decrease in purines, acylcarnitines, FFAs, and several lysolipid and sphingolipid species (99). This metabolomic phenotype might be correlated with differential regulation of PPARα at various stages of HF. Unbiased metabolomic analysis of cardiac tissue from MuRF2 knockout mice identified taurine as being associated with the development of diabetic cardiomyopathy, in which PPARα was upregulated (41). Although it is unclear whether taurine affects the function of PPARα, interplay between taurine and PPARγ regulation has been reported. Taurine enhances the transcriptional activity of PPARγ in the HEK 293 cell line (110), and PPARγ positively regulates the taurine transporter in placentas (19). More importantly, taurine has a cardioprotective effect, attenuating oxidative stress and alleviating HF in diabetic rats (97). Thus, metabolomics might be a useful tool for identifying novel mechanisms of differential regulation of PPARα targets and downstream signaling.
From an energetic standpoint, the changes in PPARα regulation during PO alter metabolic flux and redox status in the heart. Through the catabolism of FAs, reducing equivalents are transferred to NAD+ in FAO and the TCA cycle, forming NADH, which enters the electron transport chain and ultimately generates ATP in complex IV (Fig. 1D). Palmitate (16:0), for instance, yields 129 ATP molecules that come from seven cycles of FAO [7 NADH, 7 flavin adenine dinucleotide 2 (FADH2)] and eight cycles of acetyl-CoA (24 NADH, 8 FADH2, 8 GTP), with 2 ATP molecules consumed to form fatty acyl-CoA from FFAs and CoA by fatty acyl-CoA synthetase. How do changes in PPARα affect the NAD+-to-NADH ratio in the TAC heart, where PPARα increases its interaction with Sirt1? It is important to note that there have been conflicting reports regarding the change in the cellular NAD+/NADH ratio in response to PO, with both increased (77) and decreased (61, 86) ratios having been reported. In the hypertrophied rat heart, there is initially a large imbalance between NADH production and consumption rates because of a rapid fall in NADH, and this imbalance is compensated for by the enhanced Ca2+-dependent recovery (13). Also, a decline in the total pool of NAD+ appears to occur in association with the progression of HF (86, 104). Complex I deficiency leads to NADH accumulation and a decrease in the NAD+/NADH ratio, which inhibits Sirt3 activity, resulting in hyperacetylation of mitochondrial proteins secondary to the reduced activities of the malate-aspartate shuttle and the ATP synthase complex (55). In contrast, in our study, the myocardial level of NAD+ was significantly increased after 4 wk of TAC in mice, whereas the level of NADH was slightly decreased, resulting in elevation of the NAD+/NADH ratio (77), which favors the activation of Sirt1. NAD+ is essential for the deacetylase and mono-ADP-ribosyltransferase activity of Sirt1 (47). Thus, the increased interaction of PPARα with Sirt1 under this condition likely enhances the repression of transcriptional activity on PPARα target genes, which may further inhibit NADH generation because of the decreased β-oxidative flux. Taken together, these observations suggest that the redox imbalance in PO leads to a negative feedback loop among PPARα, Sirt1, and mitochondrial energetics (Fig. 3). Normalization of the NAD+/NADH ratio via administration of nicotinamide mononucleotide (NMN, a source of NAD+) blunts hyperacetylation of mitochondrial proteins and prevents the development of HF (61). In sharp contrast, cardiac-specific overexpression of Nampt, an enzyme that produces NMN, induces spontaneous development of cardiac hypertrophy (88). Moreover, haploinsufficiency of Nampt protects against angiotensin II-induced cardiac hypertrophy (87). Thus, one possible therapeutic strategy targeting metabolism is to disrupt this negative feedback loop by maintaining the NAD+ pool and redox balance, although it remains unclear whether the changes in PPARα regulation in HF are the cause or consequence of metabolic perturbations and energy starvation.
Fig. 3.
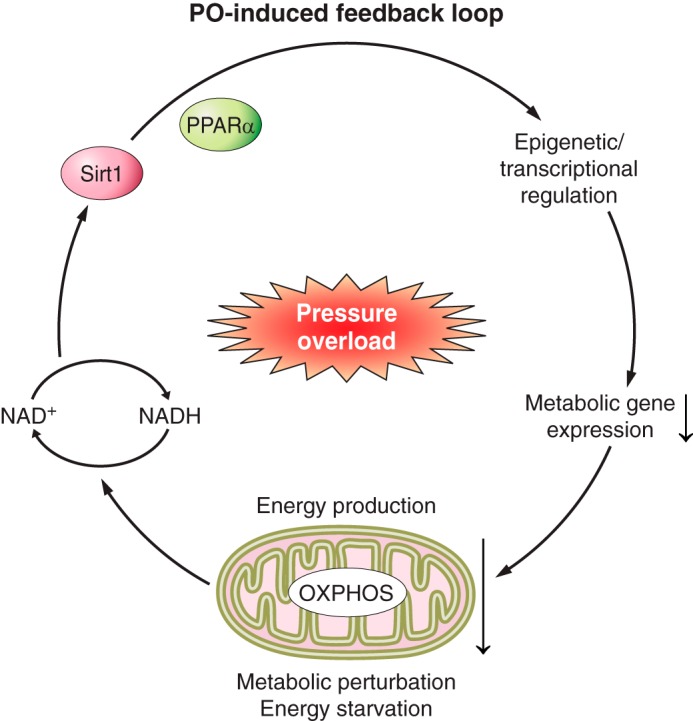
PO-induced feedback loop in the heart in association with PPARα regulation, redox balance, and energetics. The elevation of cellular NAD+ during PO-induced heart failure (HF) enhances the activity of Sirt1 (i.e., histone deacetylation), which targets a subset of PPARα target genes by interacting with PPARα during PO, presumably through epigenetic/transcriptional modifications. The downregulation of a subset of PPARα targets in the TCA cycle and ETC aggravates the impaired mitochondrial energetics in the PO heart, which further causes a redox imbalance. One possible therapeutic strategy targeting metabolism is to disrupt this negative feedback loop.
Furthermore, metabolic flux analysis of the TAC heart, in addition to multisystem analysis, would be informative in delineating the mechanisms by which changes in PPARα regulation lead to energetic perturbations. The pivotal advances in metabolic flux analysis are network-wide stable isotope balancing methods (mostly using 13C) and appropriate computational methods (137).
Another important feature of PPARα regulation in terms of metabolic flux and the metabolome is that PPARα is highly involved in the regulation of triacyglyceride (TAG) turnover (7), which can be estimated by prelabeling the TAG pool with [14C]palmitate and measuring the rate of endogenous TAG-derived FAO during a chase perfusion with [3H]palmitate (98). The TAG turnover rates of palmitoyl units are 3.75-fold faster than palmitate oxidation (7); thus, cardiomyocytes preferentially use FAs derived from TAG for oxidation. The TAG turnover rate of palmitate through long-chain FA (LCFA) oxidation is accelerated in mice with cardiac-specific overexpression of PPARα (myosin heavy chain-PPARα), in association with the upregulation of genes involved in both TAG synthesis and lipolysis (7), suggesting a fundamental role of PPARα in the regulation of TAG turnover. The accumulation of TAG through high LCFA import in myosin heavy chain-PPARα mice is rescued by crossing these mice with CD36 knockout mice (133). Interestingly, TAG turnover in PO-induced HF is differentially regulated by saturated and unsaturated FAs. Palmitate (16:0) decreases the TAG turnover rate in the PO heart, whereas oleate (18:1) maintains the basal rate of TAG turnover (58). More importantly, the basal transcript levels of PPARα target genes were maintained in the presence of supplementation with oleate and were significantly downregulated by palmitate perfusion although the protein levels of PPARα were decreased to a similar degree in TAC hearts that were perfused with palmitate and oleate (58). It is of interest to determine whether the modifications in PPARα signaling during PO include TAG turnover homeostasis, including the expression levels of TAG turnover genes (e.g., glycerol-3-phosphate acyltransferase 1, diglyceride acyltransferase 1, and Agpat3) and the rates of TAG synthesis and degradation.
CROSS TALK BETWEEN PPARα AND INSULIN AND THYROID RECEPTOR SIGNALING
Activation of the muscle PPARα regulatory pathway parallels the development of insulin resistance. PPARα-null mice are protected from high-fat diet-induced insulin resistance (39), whereas chronic activation of PPARα in muscle leads to a reduced glucose uptake capacity and insulin resistance (32). Glucose intolerance in muscle overexpressing PPARα is associated with repression of GLUT4 gene transcription, albeit in the absence of significant decrements in insulin-stimulated insulin receptor substrate-1, protein kinase B-1 phosphorylation, or PI3K activity (32), suggesting that PPARα-mediated insulin resistance is mainly associated with suppression of the downstream signaling of GLUT4. Another possible mechanism of PPARα-mediated insulin resistance is that PPARα increases expression of the insulin-signaling interfering protein suppressor of cytokine signaling-3 (4). Insulin itself also modulates PPARα’s transcriptional activity (101). Treatment with insulin induces a time-dependent increase in PPAR phosphorylation in primary rat adipocytes and human CV-1 cells, in parallel with enhanced transcriptional activity of PPARα and PPARγ (101). Whether insulin-mediated phosphorylation of PPARα occurs in the heart remains unknown. However, given that insulin activates p38 MAPK (45, 89, 109) and that phosphorylation of PPARα by p38 MAPK was observed in cardiomyocytes as well as in CV-1 cells (9), it may be that p38 MAPK-mediated phosphorylation of PPARα occurs in diabetic hearts that are exposed to chronic hyperinsulinemia.
Varying levels of PPARα expression in the diabetic heart have been reported in the literature. Downregulation of PPARα was reported in streptozotocin (STZ)-induced type 1 diabetes (27), 6-mo-old ob/ob mice (91), and 3-mo-old Akita mice (17). However, no significant change in PPARα expression was observed in 4-wk-old ob/ob and db/db mice (16) or in 10-wk-old db/db mice (34). PPARα transcript levels were shown to be decreased to an equal extent in nondiabetic and diabetic HF patients (92). A significantly increased level of PPARα was observed in ob/ob mice at the age of 15 wk (16) and STZ-induced diabetic mice (34). The conflicting results regarding PPARα expression in the diabetic heart might be attributed to the aforementioned complex PPARα regulation, and, thus, PPARα expression per se may not completely explain metabolic derangements in diabetes. Supporting this notion, abnormal PPARα regulatory signaling was reported in some animal models of diabetes. The responses of PPARα-regulated genes (e.g., MCAD, CPT1) to fasting were altered in obese Zucker rats compared with lean rats despite similar levels of PPARα expression in both groups (135). Conversely, the reduced mRNA levels of PPARα in STZ-induced diabetic rats were not reflected in the expression level of CPT1, which is a well-known PPARα target gene (24). Dissociation between the expression levels of PPARα and its target genes and FAO capacity in the diabetic heart might be attributable to altered transcriptional control by PPARα through changes in its dimerization partners or its sensitivity to ligands because of posttranslational modifications.
There are conflicting reports regarding myocardial insulin resistance in a mouse model of PO-induced hypertrophy and HF. The insulin-signaling pathway was upregulated in the heart after 7 and 14 days of TAC (105), whereas insulin resistance was induced by 2 wk of postabdominal aortic constriction (140). Despite the discrepancy, these studies suggest that modification of insulin signaling occurs as an initial response to PO, which presumably affects PPARα regulation.
The other important hormone-induced signaling pathway that is closely coupled to PPAR transactivation involves thyroid hormone receptor-α1 (TRα1). TRα1 is a member of another nuclear hormone receptor superfamily that is activated by binding thyroid hormone, and not only do TRs and PPARs share binding sites and heterodimeric partners such as RXRs but TRα1 and PPAR also compete for DNA binding at PPRE (3, 71). Interestingly, TR monomers or heterodimers with RXRα specifically activate transcription of genes containing direct repeats of AGGTCA separated by four base pairs (DR4) (122), whereas PPARα binds to core elements separated by one base pair (DR1) (120, 138). Given that upregulation and disruption of thyroid hormone signaling contributes to maladaptive cardiac hypertrophy (95, 115, 127), PPARα-mediated derangements of cardiac metabolism in the progression of hypertrophy may be linked to TR signaling.
CONCLUSIONS AND PERSPECTIVES
Despite the powerful influences of PPARα in metabolic remodeling, studies to define its regulatory mechanisms in cardiac hypertrophy and failure have just begun. Impaired FAO is a hallmark of HF. The functional assessments of FAO include several steps in the upstream (i.e., cellular uptake, mitochondrial import) and downstream (i.e., mitochondrial β-oxidation, TCA cycle, electron transport chain) pathways. Although PPARα coordinately modulates FAO genes in the degradation pathway, our studies suggest that only a subset of PPARα’s target genes (mainly in the FAO downstream pathway) are affected by pathological stress. These PPARα target genes contain imperfect DR1 sequences in their promoter regions, where the PPARα-Sirt1 complex preferentially binds under PO stress. The recruitment of Sirt1 to the specific DNA element in these PPARα targets, therefore, may direct the pathospecific PPARα signaling in metabolism, targeting the downstream pathway of FAO. This may explain the PPARα-mediated maladaptive metabolic remodeling in HF that leads to impaired FAO. Whether inhibition of the Sirt1-PPARα interaction ameliorates HF, irrespective of the etiology, warrants investigation.
The progression of HF is associated with transcriptional changes in PPARα target genes, suggesting that PPARα is a promising therapeutic target. However, given the complex roles of PPARα in cardiac energetics, manipulating the expression or activity of PPARα alone may not be sufficient to prevent metabolic derangements in the diseased heart. How does pathological stress alter PPARα expression and its downstream signaling? How does PPARα selectively control a subset of its target genes? Perhaps most importantly, the fundamental question still remains as to whether the changes in PPARα are a cause or consequence of HF. Better understanding of these mechanisms may lead to the development of a novel HF therapy that selectively and effectively modulates PPARα’s downstream signaling. Chromatin immunoprecipitation sequencing analysis of PPARα at various time points during cardiac hypertrophy and HF may allow for elucidation of the unique function of PPARα. Moreover, because PPARα is also closely linked to lipid metabolism in the body, it is important to identify a therapeutic method that specifically targets cardiac PPARα to prevent maladaptive remodeling in systemic metabolism, such as insulin resistance. Last, the modifications of PPARα expression in HF probably vary depending on the severity and timing of PO, etiology, and epigenetics. For instance, the specific expression pattern of PPARα target genes might be dictated by epigenetic modifications acquired during the long-term history of a patient. Thus, future interventions designed to improve the energetics of the failing heart by targeting the PPARα pathway may need to be tailored to the individual patient. Metabolomics has been used as part of personalized medicine to identify populations that will respond to a particular drug intervention (21). Thus pharmaco-metabolomic phenotyping of HF may allow the development of personalized drugs that block the specific PPARα signaling that dysregulates cardiac metabolism in diseases. Metabolomics might enable subclassification of HF patients and become a useful tool for applying precision medicine in HF patients (11).
GRANTS
This work was supported by the Nora Eccles Treadwell Foundation (J. S. Warren), American Heart Association Grant in Aid 17GRNT33440031 (S. Oka), a New Jersey Health Foundation Research Grant (S. Oka), National Institutes of Health Grants HL-67724, HL-91469, HL-102738, HL-112330, and AG-23039 (J. Sadoshima), and by the Leducq Foundation Transatlantic Networks of Excellence (J. Sadoshima).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.S.W., S.O., D.Z., and J.S. prepared figures; J.S.W., S.O., D.Z., and J.S. drafted manuscript; J.S.W., S.O., D.Z., and J.S. edited and revised manuscript; J.S.W., S.O., D.Z., and J.S. approved final version of manuscript.
REFERENCES
- 1.Adhikary T, Kaddatz K, Finkernagel F, Schönbauer A, Meissner W, Scharfe M, Jarek M, Blöcker H, Müller-Brüsselbach S, Müller R. Genomewide analyses define different modes of transcriptional regulation by peroxisome proliferator-activated receptor-β/δ (PPARβ/δ). PLoS One : e16344, 2011. doi: 10.1371/journal.pone.0016344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem : 5678–5684, 1998. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 3.Araki O, Ying H, Furuya F, Zhu X, Cheng SY. Thyroid hormone receptor beta mutants: Dominant negative regulators of peroxisome proliferator-activated receptor gamma action. Proc Natl Acad Sci USA : 16251–16256, 2005. doi: 10.1073/pnas.0508556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asrih M, Lerch R, Papageorgiou I, Pellieux C, Montessuit C. Differential regulation of stimulated glucose transport by free fatty acids and PPARα or -δ agonists in cardiac myocytes. Am J Physiol Endocrinol Metab : E872–E884, 2012. doi: 10.1152/ajpendo.00427.2011. [DOI] [PubMed] [Google Scholar]
- 5.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation : 698–705, 2016. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta : 840–847, 2013. doi: 10.1016/j.bbamcr.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res : 233–241, 2010. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest : 1723–1730, 2000. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem : 44495–44501, 2001. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 10.Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation : 706–716, 2016. doi: 10.1161/CIRCULATIONAHA.115.017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beger RD, Dunn W, Schmidt MA, Gross SS, Kirwan JA, Cascante M, Brennan L, Wishart DS, Oresic M, Hankemeier T, Broadhurst DI, Lane AN, Suhre K, Kastenmüller G, Sumner SJ, Thiele I, Fiehn O, Kaddurah-Daouk R; for “Precision Medicine and Pharmacometabolomics Task Group”-Metabolomics Society Initiative . Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics : 149, 2016. doi: 10.1007/s11306-016-1094-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase C signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol : 1906–1918, 2004. doi: 10.1210/me.2003-0327. [DOI] [PubMed] [Google Scholar]
- 13.Brandes R, Maier LS, Bers DM. Regulation of mitochondrial [NADH] by cytosolic [Ca2+] and work in trabeculae from hypertrophic and normal rat hearts. Circ Res : 1189–1198, 1998. doi: 10.1161/01.RES.82.11.1189. [DOI] [PubMed] [Google Scholar]
- 14.Bristow M. Etomoxir: a new approach to treatment of chronic heart failure. Lancet : 1621–1622, 2000. doi: 10.1016/S0140-6736(00)03149-4. [DOI] [PubMed] [Google Scholar]
- 15.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science : 2011–2015, 2004. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 16.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology : 5341–5349, 2005. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 17.Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes : 1986–1997, 2009. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bugger H, Schwarzer M, Chen D, Schrepper A, Amorim PA, Schoepe M, Nguyen TD, Mohr FW, Khalimonchuk O, Weimer BC, Doenst T. Proteomic remodelling of mitochondrial oxidative pathways in pressure overload-induced heart failure. Cardiovasc Res : 376–384, 2010. doi: 10.1093/cvr/cvp344. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, He P, Ding X, Huang Y, Gu H, Ni X. PPARγ stimulates expression of L-type amino acid and taurine transporters in human placentas: the evidence of PPARγ regulating fetal growth. Sci Rep : 12650, 2015. doi: 10.1038/srep12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi SS, Park J, Choi JH. Revisiting PPARγ as a target for the treatment of metabolic disorders. BMB Rep : 599–608, 2014. doi: 10.5483/BMBRep.2014.47.11.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost JP, Le Net JL, Baker D, Walley RJ, Everett JR, Nicholson JK. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature : 1073–1077, 2006. doi: 10.1038/nature04648. [DOI] [PubMed] [Google Scholar]
- 22.Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suárez Y, Lai EC, Fernández-Hernando C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA : 9232–9237, 2011. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degenhardt T, Matilainen M, Herzig KH, Dunlop TW, Carlberg C. The insulin-like growth factor-binding protein 1 gene is a primary target of peroxisome proliferator-activated receptors. J Biol Chem : 39607–39619, 2006. doi: 10.1074/jbc.M605623200. [DOI] [PubMed] [Google Scholar]
- 24.Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, Davies PJ, Taegtmeyer H. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J Mol Cell Cardiol : 985–996, 2000. doi: 10.1006/jmcc.2000.1139. [DOI] [PubMed] [Google Scholar]
- 25.Djoussé L, Benkeser D, Arnold A, Kizer JR, Zieman SJ, Lemaitre RN, Tracy RP, Gottdiener JS, Mozaffarian D, Siscovick DS, Mukamal KJ, Ix JH. Plasma free fatty acids and risk of heart failure: the Cardiovascular Health Study. Circ Heart Fail : 964–969, 2013. doi: 10.1161/CIRCHEARTFAILURE.113.000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doenst T, Pytel G, Schrepper A, Amorim P, Färber G, Shingu Y, Mohr FW, Schwarzer M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res : 461–470, 2010. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- 27.Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, Trent CM, Hu Y, Guo S, Aifantis I, Goldberg IJ. Cardiac myocyte KLF5 regulates ppara expression and cardiac function. Circ Res : 241–253, 2016. doi: 10.1161/CIRCRESAHA.115.306383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehara T, Kamei Y, Yuan X, Takahashi M, Kanai S, Tamura E, Tsujimoto K, Tamiya T, Nakagawa Y, Shimano H, Takai-Igarashi T, Hatada I, Suganami T, Hashimoto K, Ogawa Y. Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid β-oxidation genes in the postnatal liver. Diabetes : 775–784, 2015. doi: 10.2337/db14-0158. [DOI] [PubMed] [Google Scholar]
- 29.el Azzouzi H, Leptidis S, Dirkx E, Hoeks J, van Bree B, Brand K, McClellan EA, Poels E, Sluimer JC, van den Hoogenhof MM, Armand AS, Yin X, Langley S, Bourajjaj M, Olieslagers S, Krishnan J, Vooijs M, Kurihara H, Stubbs A, Pinto YM, Krek W, Mayr M, da Costa Martins PA, Schrauwen P, De Windt LJ. The hypoxia-inducible microRNA cluster miR-199a∼214 targets myocardial PPARδ and impairs mitochondrial fatty acid oxidation. Cell Metab : 341–354, 2013. doi: 10.1016/j.cmet.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 30.El-Aroussy W, Rizk A, Mayhoub G, Aleem SA, El-Tobgy S, Mokhtar MS. Plasma carnitine levels as a marker of impaired left ventricular functions. Mol Cell Biochem : 37–41, 2000. doi: 10.1023/A:1007142919941. [DOI] [PubMed] [Google Scholar]
- 31.Fernández-Hernando C, Suárez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol : 86–92, 2011. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, Kelly DP. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab : 133–144, 2005. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor alpha (PPARalpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol : 1249–1257, 2002. doi: 10.1006/jmcc.2002.2061. [DOI] [PubMed] [Google Scholar]
- 34.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest : 121–130, 2002. doi: 10.1172/JCI0214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garnier A, Fortin D, Deloménie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol : 491–501, 2003. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genini D, Carbone GM, Catapano CV. Multiple interactions between peroxisome proliferators-activated receptors and the ubiquitin-proteasome system and implications for cancer pathogenesis. PPAR Res : 195065, 2008. doi: 10.1155/2008/195065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerin I, Bommer GT, McCoin CS, Sousa KM, Krishnan V, MacDougald OA. Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am J Physiol Endocrinol Metab : E198–E206, 2010. doi: 10.1152/ajpendo.00179.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruenbaum Y, Stein R, Cedar H, Razin A. Methylation of CpG sequences in eukaryotic DNA. FEBS Lett : 67–71, 1981. doi: 10.1016/0014-5793(81)80055-5. [DOI] [PubMed] [Google Scholar]
- 39.Guerre-Millo M, Rouault C, Poulain P, André J, Poitout V, Peters JM, Gonzalez FJ, Fruchart JC, Reach G, Staels B. PPAR-alpha-null mice are protected from high-fat diet-induced insulin resistance. Diabetes : 2809–2814, 2001. doi: 10.2337/diabetes.50.12.2809. [DOI] [PubMed] [Google Scholar]
- 40.Gusterson RJ, Jazrawi E, Adcock IM, Latchman DS. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J Biol Chem : 6838–6847, 2003. doi: 10.1074/jbc.M211762200. [DOI] [PubMed] [Google Scholar]
- 41.He J, Quintana MT, Sullivan J, Parry TL, Grevengoad T, Schisler JC, Hill JA, Yates CC, Mapanga RF, Essop MF, Stansfield WE, Bain JR, Newgard CB, Muehlbauer MJ, Han Y, Clarke BA, Willis MS. MuRF2 regulates PPARgamma1 activity to protect against diabetic cardiomyopathy and enhance weight gain induced by a high fat diet. Cardiovasc Diabetol : 97, 2015. doi: 10.1186/s12933-015-0252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin reductase a attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem : 25179–25191, 2016. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res : 568–578, 2004. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- 44.Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Maklan E, Minetti G, Zhao P, Hoffman EP, Puri PL, Sartorelli V. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell : 673–684, 2004. doi: 10.1016/S1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- 45.Igarashi M, Yamaguchi H, Hirata A, Daimon M, Tominaga M, Kato T. Insulin activates p38 mitogen-activated protein (MAP) kinase via a MAP kinase kinase (MKK) 3/MKK 6 pathway in vascular smooth muscle cells. Eur J Clin Invest : 668–677, 2000. doi: 10.1046/j.1365-2362.2000.00671.x. [DOI] [PubMed] [Google Scholar]
- 46.Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev : 612–619, 1989. doi: 10.1101/gad.3.5.612. [DOI] [PubMed] [Google Scholar]
- 47.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature : 795–800, 2000. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 48.Jaeger K, Saben JL, Moley KH. Transmission of metabolic dysfunction across generations. Physiology (Bethesda) : 51–59, 2017. doi: 10.1152/physiol.00017.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang W, Oken H, Fiuzat M, Shaw LK, Martsberger C, Kuchibhatla M, Kaddurah-Daouk R, Steffens DC, Baillie R, Cuffe M, Krishnan R, O’Connor C; SADHART-CHF Investigators . Plasma omega-3 polyunsaturated fatty acids and survival in patients with chronic heart failure and major depressive disorder. J Cardiovasc Transl Res : 92–99, 2012. doi: 10.1007/s12265-011-9325-8. [DOI] [PubMed] [Google Scholar]
- 50.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A, Chin WW, Burger AG, Meier CA. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor alpha by phosphorylation of a ligand-independent trans-activating domain. J Biol Chem : 10505–10510, 1999. doi: 10.1074/jbc.274.15.10505. [DOI] [PubMed] [Google Scholar]
- 51.Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K, Uchihashi M, Fukai K, Iwai-Kanai E, Matoba S.. Activation of PPARα in the early stage of heart failure maintained myocardial function and energetics in pressure overload heart failure. Am J Physiol Heart Circ Physiol : H305–H313, 2016. doi: 10.1152/ajpheart.00553.2016. [DOI] [PubMed] [Google Scholar]
- 52.Kanda H, Nohara R, Hasegawa K, Kishimoto C, Sasayama S. A nuclear complex containing PPARalpha/RXRalpha is markedly downregulated in the hypertrophied rat left ventricular myocardium with normal systolic function. Heart Vessels : 191–196, 2000. doi: 10.1007/s003800070022. [DOI] [PubMed] [Google Scholar]
- 53.Kaneda R, Takada S, Yamashita Y, Choi YL, Nonaka-Sarukawa M, Soda M, Misawa Y, Isomura T, Shimada K, Mano H. Genome-wide histone methylation profile for heart failure. Genes Cells : 69–77, 2009. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 54.Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol : 49, 2014. doi: 10.3389/fcell.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab : 239–250, 2013. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karbowska J, Kochan Z, Smoleński RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett : 49–53, 2003. [PubMed] [Google Scholar]
- 57.Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail : 420–430, 2010. doi: 10.1161/CIRCHEARTFAILURE.109.888479. [DOI] [PubMed] [Google Scholar]
- 58.Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation : 1790–1799, 2014. doi: 10.1161/CIRCULATIONAHA.114.011687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail : 1022–1031, 2014. doi: 10.1161/CIRCHEARTFAILURE.114.001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lazennec G, Canaple L, Saugy D, Wahli W. Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol Endocrinol : 1962–1975, 2000. doi: 10.1210/mend.14.12.0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation : 883–894, 2016. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA : 3374–3379, 2008. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol : 3012–3022, 1995. doi: 10.1128/MCB.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. FEBS J : 2348–2358, 2009. doi: 10.1111/j.1742-4658.2009.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev : 207–258, 2010. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 66.Luo H, Zhang Y, Guo H, Zhang L, Li X, Ringseis R, Wen G, Hui D, Liang A, Eder K, He D. Transcriptional regulation of the human, porcine and bovine OCTN2 gene by PPARα via a conserved PPRE located in intron 1. BMC Genet : 90, 2014. doi: 10.1186/s12863-014-0090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell : 137–148, 2001. doi: 10.1016/S0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 68.Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol : 968–975, 2008. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 69.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation : 135–142, 1996. doi: 10.1161/01.CIR.93.1.135. [DOI] [PubMed] [Google Scholar]
- 70.Mistry NF, Cresci S. PPAR transcriptional activator complex polymorphisms and the promise of individualized therapy for heart failure. Heart Fail Rev : 197–207, 2010. doi: 10.1007/s10741-008-9114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miyamoto T, Kaneko A, Kakizawa T, Yajima H, Kamijo K, Sekine R, Hiramatsu K, Nishii Y, Hashimoto T, Hashizume K. Inhibition of peroxisome proliferator signaling pathways by thyroid hormone receptor. Competitive binding to the response element. J Biol Chem : 7752–7758, 1997. doi: 10.1074/jbc.272.12.7752. [DOI] [PubMed] [Google Scholar]
- 72.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell : 551–563, 2004. doi: 10.1016/S0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 73.Nakamura T, Sugihara H, Kinoshita N, Ito K, Adachi Y, Hirasaki S, Matsuo A, Azuma A, Kodo N, Nakagawa M. Serum carnitine concentrations in patients with idiopathic hypertrophic cardiomyopathy: relationship with impaired myocardial fatty acid metabolism. Clin Sci (Lond) : 493–501, 1999. doi: 10.1042/cs0970493. [DOI] [PubMed] [Google Scholar]
- 74.Nakamura Y, Yamada Y, Shimomura H, Nagayoshi Y, Tsujita K, Yamashita T, Fukuda M, Ohba K, Nako H, Ogura Y, Chitose T, Yamaguchi M, Nagata T, Soejima H, Kaikita K, Sugiyama S, Ogawa H. Effect of edaravone on plasma monocyte chemoattractant protein-1 levels in patients with acute myocardial infarction. J Cardiol : 416–424, 2009. doi: 10.1016/j.jjcc.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 75.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem : 16456–16460, 2005. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 76.Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation : 955–961, 2004. doi: 10.1161/01.CIR.0000139339.85840.DD. [DOI] [PubMed] [Google Scholar]
- 77.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, Sadoshima J. PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab : 598–611, 2011. doi: 10.1016/j.cmet.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oka S, Zhai P, Yamamoto T, Ikeda Y, Byun J, Hsu CP, Sadoshima J. Peroxisome proliferator activated receptor-α association with silent information regulator 1 suppresses cardiac fatty acid metabolism in the failing heart. Circ Heart Fail : 1123–1132, 2015. doi: 10.1161/CIRCHEARTFAILURE.115.002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ono T, Kamimura N, Matsuhashi T, Nagai T, Nishiyama T, Endo J, Hishiki T, Nakanishi T, Shimizu N, Tanaka H, Ohta S, Suematsu M, Ieda M, Sano M, Fukuda K, Kaneda R. The histone 3 lysine 9 methyltransferase inhibitor chaetocin improves prognosis in a rat model of high salt diet-induced heart failure. Sci Rep : 39752, 2017. doi: 10.1038/srep39752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-α in pacing-induced heart failure. Circulation : 606–612, 2002. doi: 10.1161/01.CIR.0000023531.22727.C1. [DOI] [PubMed] [Google Scholar]
- 81.Palaniswamy C, Mellana WM, Selvaraj DR, Mohan D. Metabolic modulation: a new therapeutic target in treatment of heart failure. Am J Ther : e197–e201, 2011. doi: 10.1097/MJT.0b013e3181d70453. [DOI] [PubMed] [Google Scholar]
- 82.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature : 759–763, 2005. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perez-Castillo A, Schwartz HL, Oppenheimer JH. Rat hepatic mRNA-S14 and lipogenic enzymes during weaning: role of S14 in lipogenesis. Am J Physiol Endocrinol Metab : E536–E542, 1987. [DOI] [PubMed] [Google Scholar]
- 84.Périchon R, Bourre JM. Aging-related decrease in liver peroxisomal fatty acid oxidation in control and clofibrate-treated mice. A biochemical study and mechanistic approach. Mech Ageing Dev : 115–126, 1996. doi: 10.1016/0047-6374(96)01705-8. [DOI] [PubMed] [Google Scholar]
- 85.Phillips D, Ten Hove M, Schneider JE, Wu CO, Sebag-Montefiore L, Aponte AM, Lygate CA, Wallis J, Clarke K, Watkins H, Balaban RS, Neubauer S. Mice over-expressing the myocardial creatine transporter develop progressive heart failure and show decreased glycolytic capacity. J Mol Cell Cardiol : 582–590, 2010. doi: 10.1016/j.yjmcc.2009.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pillai JB, Chen M, Rajamohan SB, Samant S, Pillai VB, Gupta M, Gupta MP. Activation of SIRT1, a class III histone deacetylase, contributes to fructose feeding-mediated induction of the α-myosin heavy chain expression. Am J Physiol Heart Circ Physiol : H1388–H1397, 2008. doi: 10.1152/ajpheart.01339.2007. [DOI] [PubMed] [Google Scholar]
- 87.Pillai JB, Gupta M, Rajamohan SB, Lang R, Raman J, Gupta MP. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol : H1545–H1553, 2006. doi: 10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- 88.Pillai VB, Sundaresan NR, Kim G, Samant S, Moreno-Vinasco L, Garcia JG, Gupta MP. Nampt secreted from cardiomyocytes promotes development of cardiac hypertrophy and adverse ventricular remodeling. Am J Physiol Heart Circ Physiol : H415–H426, 2013. doi: 10.1152/ajpheart.00468.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, Dostal DE, White MF, Baker KM, Guo S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes : 3887–3900, 2013. doi: 10.2337/db13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, Accili D. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell : 620–632, 2012. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rame JE, Barouch LA, Sack MN, Lynn EG, Abu-Asab M, Tsokos M, Kern SJ, Barb JJ, Munson PJ, Halushka MK, Miller KL, Fox-Talbot K, Zhang J, Hare JM, Solomon MA, Danner RL. Caloric restriction in leptin deficiency does not correct myocardial steatosis: failure to normalize PPARalpha/PGC1alpha and thermogenic glycerolipid/fatty acid cycling. Physiol Genomics : 726–738, 2011. doi: 10.1152/physiolgenomics.00088.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Razeghi P, Young ME, Cockrill TC, Frazier OH, Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation : 407–411, 2002. doi: 10.1161/01.CIR.0000026392.80723.DC. [DOI] [PubMed] [Google Scholar]
- 93.Ren F, Zhang L, Zhang X, Shi H, Wen T, Bai L, Zheng S, Chen Y, Chen D, Li L, Duan Z. Inhibition of glycogen synthase kinase 3β promotes autophagy to protect mice from acute liver failure mediated by peroxisome proliferator-activated receptor α. Cell Death Dis : e2151, 2016. doi: 10.1038/cddis.2016.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Riehle C, Abel ED. PGC-1 proteins and heart failure. Trends Cardiovasc Med : 98–105, 2012. doi: 10.1016/j.tcm.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ripoli A, Pingitore A, Favilli B, Bottoni A, Turchi S, Osman NF, De Marchi D, Lombardi M, L’Abbate A, Iervasi G. Does subclinical hypothyroidism affect cardiac pump performance? Evidence from a magnetic resonance imaging study. J Am Coll Cardiol : 439–445, 2005. doi: 10.1016/j.jacc.2004.10.044. [DOI] [PubMed] [Google Scholar]
- 96.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature : 113–118, 2005. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 97.Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA : 6438–6443, 1997. doi: 10.1073/pnas.94.12.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem : 8162–8170, 1991. [PubMed] [Google Scholar]
- 99.Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail : 634–642, 2014. doi: 10.1161/CIRCHEARTFAILURE.114.001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res : 907–925, 1996. [PubMed] [Google Scholar]
- 101.Shalev A, Siegrist-Kaiser CA, Yen PM, Wahli W, Burger AG, Chin WW, Meier CA. The peroxisome proliferator-activated receptor alpha is a phosphoprotein: regulation by insulin. Endocrinology : 4499–4502, 1996. doi: 10.1210/endo.137.10.8828512. [DOI] [PubMed] [Google Scholar]
- 102.Sharov VG, Goussev A, Lesch M, Goldstein S, Sabbah HN. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. J Mol Cell Cardiol : 1757–1762, 1998. doi: 10.1006/jmcc.1998.0739. [DOI] [PubMed] [Google Scholar]
- 103.Shibayama J, Barton DW, Wang L, Yuzyuk TN, Cox J, Zaitsev AV, Franklin S. Cardiac-Specific Knock-Out of the Histone Methyltransferase Smyd1 Leads to Coordinated Downregulation of Energy Metabolism (Abstract). Orlando, FL: Am Heart Assoc Scientific Sessions 2015, 2015, p. e3. [Google Scholar]
- 104.Shibayama J, Yuzyuk TN, Cox J, Makaju A, Miller M, Lichter J, Li H, Leavy JD, Franklin S, Zaitsev AV. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: integrated metabolomics and proteomics study. PLoS One : e0118974, 2015. doi: 10.1371/journal.pone.0118974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, Komuro I. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest : 1506–1514, 2010. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Slater-Jefferies JL, Lillycrop KA, Townsend PA, Torrens C, Hoile SP, Hanson MA, Burdge GC. Feeding a protein-restricted diet during pregnancy induces altered epigenetic regulation of peroxisomal proliferator-activated receptor-α in the heart of the offspring. J Dev Orig Health Dis : 250–255, 2011. doi: 10.1017/S2040174410000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smeets PJ, de Vogel-van den Bosch HM, Willemsen PH, Stassen AP, Ayoubi T, van der Vusse GJ, van Bilsen M. Transcriptomic analysis of PPARalpha-dependent alterations during cardiac hypertrophy. Physiol Genomics : 15–23, 2008. doi: 10.1152/physiolgenomics.90296.2008. [DOI] [PubMed] [Google Scholar]
- 108.Smeets PJ, Teunissen BE, Willemsen PH, van Nieuwenhoven FA, Brouns AE, Janssen BJ, Cleutjens JP, Staels B, van der Vusse GJ, van Bilsen M. Cardiac hypertrophy is enhanced in PPAR alpha-/- mice in response to chronic pressure overload. Cardiovasc Res : 79–89, 2008. doi: 10.1093/cvr/cvn001. [DOI] [PubMed] [Google Scholar]
- 109.Somwar R, Perreault M, Kapur S, Taha C, Sweeney G, Ramlal T, Kim DY, Keen J, Côte CH, Klip A, Marette A. Activation of p38 mitogen-activated protein kinase alpha and beta by insulin and contraction in rat skeletal muscle: potential role in the stimulation of glucose transport. Diabetes : 1794–1800, 2000. doi: 10.2337/diabetes.49.11.1794. [DOI] [PubMed] [Google Scholar]
- 110.Song MK, Salam NK, Roufogalis BD, Huang TH. Lycium barbarum (Goji Berry) extracts and its taurine component inhibit PPAR-γ-dependent gene transcription in human retinal pigment epithelial cells: Possible implications for diabetic retinopathy treatment. Biochem Pharmacol : 1209–1218, 2011. doi: 10.1016/j.bcp.2011.07.089. [DOI] [PubMed] [Google Scholar]
- 111.Strahl BD, Allis CD. The language of covalent histone modifications. Nature : 41–45, 2000. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 112.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab : 307–319, 2007. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 113.Sun H, Wang Y. Branched chain amino acid metabolic reprogramming in heart failure. Biochim Biophys Acta : 2270–2275, 2016. doi: 10.1016/j.bbadis.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 114.Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation : 894–896, 2004. doi: 10.1161/01.CIR.0000139340.88769.D5. [DOI] [PubMed] [Google Scholar]
- 115.Takano APC, Munhoz CD, Moriscot AS, Gupta S, Barreto-Chaves MLM. S100A8/MYD88/NF-κB: a novel pathway involved in cardiomyocyte hypertrophy driven by thyroid hormone. J Mol Med (Berl) : 671–682, 2017. doi: 10.1007/s00109-017-1511-y. [DOI] [PubMed] [Google Scholar]
- 116.Tarling EJ, Ahn H, de Aguiar Vallim TQ. The nuclear receptor FXR uncouples the actions of miR-33 from SREBP-2. Arterioscler Thromb Vasc Biol : 787–795, 2015. doi: 10.1161/ATVBAHA.114.304179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev : 226–231, 1993. doi: 10.1016/0959-437X(93)90027-M. [DOI] [PubMed] [Google Scholar]
- 118.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature : 757–761, 2009. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tu AY, Albers JJ. DNA sequences responsible for reduced promoter activity of human phospholipid transfer protein by fibrate. Biochem Biophys Res Commun : 802–807, 1999. doi: 10.1006/bbrc.1999.1597. [DOI] [PubMed] [Google Scholar]
- 120.Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J : 433–439, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ueland T, Svardal A, Øie E, Askevold ET, Nymoen SH, Bjørndal B, Dahl CP, Gullestad L, Berge RK, Aukrust P. Disturbed carnitine regulation in chronic heart failure--increased plasma levels of palmitoyl-carnitine are associated with poor prognosis. Int J Cardiol : 1892–1899, 2013. doi: 10.1016/j.ijcard.2012.04.150. [DOI] [PubMed] [Google Scholar]
- 122.Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell : 1255–1266, 1991. doi: 10.1016/0092-8674(91)90020-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vaquero A, Scher M, Lee D, Erdjument-Bromage H, Tempst P, Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol Cell : 93–105, 2004. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 124.Vary TC, Neely JR. Sodium dependence of carnitine transport in isolated perfused adult rat hearts. Am J Physiol : H247–H252, 1983. [DOI] [PubMed] [Google Scholar]
- 125.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell : 149–159, 2001. doi: 10.1016/S0092-8674(01)00527-X. [DOI] [PubMed] [Google Scholar]
- 126.Vega RB, Kelly DP. Cardiac nuclear receptors: architects of mitochondrial structure and function. J Clin Invest : 1155–1164, 2017. doi: 10.1172/JCI88888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wadosky KM, Berthiaume JM, Tang W, Zungu M, Portman MA, Gerdes AM, Willis MS. MuRF1 mono-ubiquitinates TRα to inhibit T3-induced cardiac hypertrophy in vivo. J Mol Endocrinol : 273–290, 2016. doi: 10.1530/JME-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wadosky KM, Willis MS. The story so far: post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am J Physiol Heart Circ Physiol : H515–H526, 2012. doi: 10.1152/ajpheart.00703.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang H, Qiang L, Farmer SR. Identification of a domain within peroxisome proliferator-activated receptor gamma regulating expression of a group of genes containing fibroblast growth factor 21 that are selectively repressed by SIRT1 in adipocytes. Mol Cell Biol : 188–200, 2008. doi: 10.1128/MCB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem : 22293–22299, 2000. doi: 10.1074/jbc.M000248200. [DOI] [PubMed] [Google Scholar]
- 131.Wei JQ, Shehadeh LA, Mitrani JM, Pessanha M, Slepak TI, Webster KA, Bishopric NH. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation : 934–946, 2008. doi: 10.1161/CIRCULATIONAHA.107.760488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wen G, Ringseis R, Eder K. Mouse OCTN2 is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha) via a PPRE located in the first intron. Biochem Pharmacol : 768–776, 2010. doi: 10.1016/j.bcp.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 133.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res : 1208–1217, 2007. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- 134.Yoon MJ, Lee GY, Chung JJ, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes : 2562–2570, 2006. doi: 10.2337/db05-1322. [DOI] [PubMed] [Google Scholar]
- 135.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes : 2587–2595, 2002. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 136.Young ME, Laws FA, Goodwin GW, Taegtmeyer H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem : 44390–44395, 2001. doi: 10.1074/jbc.M103826200. [DOI] [PubMed] [Google Scholar]
- 137.Zamboni N, Fendt SM, Rühl M, Sauer U. (13)C-based metabolic flux analysis. Nat Protoc : 878–892, 2009. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- 138.Zhang B, Marcus SL, Miyata KS, Subramani S, Capone JP, Rachubinski RA. Characterization of protein-DNA interactions within the peroxisome proliferator-responsive element of the rat hydratase-dehydrogenase gene. J Biol Chem : 12939–12945, 1993. [PubMed] [Google Scholar]
- 139.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell : 479–488, 2002. doi: 10.1016/S0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, Lopaschuk GD. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail : 1039–1048, 2013. doi: 10.1161/CIRCHEARTFAILURE.112.000228. [DOI] [PubMed] [Google Scholar]
- 141.Zhang QJ, Liu ZP. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics : 321–330, 2015. doi: 10.2217/epi.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]