

Abstract
Idiopathic pulmonary fibrosis (IPF) is a lethal lung disease of unknown etiology. The development of pulmonary hypertension (PH) is considered the single most significant predictor of mortality in patients with chronic lung diseases. The processes that govern the progression and development of fibroproliferative and vascular lesions in IPF are not fully understood. Using human lung explant samples from patients with IPF with or without a diagnosis of PH as well as normal control tissue, we report reduced BMPR2 expression in patients with IPF or IPF+PH. These changes were consistent with dampened P-SMAD 1/5/8 and elevated P-SMAD 2/3, demonstrating reduced BMPR2 signaling and elevated TGF-β activity in IPF. In the bleomycin (BLM) model of lung fibrosis and PH, we also report decreased BMPR2 expression compared with control animals that correlated with vascular remodeling and PH. We show that genetic abrogation or pharmacological inhibition of interleukin-6 leads to diminished markers of fibrosis and PH consistent with elevated levels of BMPR2 and reduced levels of a collection of microRNAs (miRs) that are able to degrade BMPR2. We also demonstrate that isolated bone marrow-derived macrophages from BLM-exposed mice show reduced BMPR2 levels upon exposure with IL6 or the IL6+IL6R complex that are consistent with immunohistochemistry showing reduced BMPR2 in CD206 expressing macrophages from lung sections from IPF and IPF+PH patients. In conclusion, our data suggest that depletion of BMPR2 mediated by a collection of miRs induced by IL6 and subsequent STAT3 phosphorylation as a novel mechanism participating to fibroproliferative and vascular injuries in IPF.
Keywords: interleukin-6, miRNA, STAT-3, TGF, vascular remodeling
idiopathic pulmonary fibrosis (IPF) is a lethal lung disease defined by chronic, progressive, and irreversible interstitial fibrosis limited to the lungs (6, 61). Median survival after diagnosis is 2–3 years (61) and the aggregate cost due to IPF is estimated to be over $1 billion per year (39). In IPF fibroproliferative injury is due to relentless and extensive extracellular matrix deposition that occurs when normal repair processes in the lung, following stimuli, are unable to deactivate (6, 61). The development of pulmonary hypertension (PH) is considered the single most significant predictor of mortality in patients with chronic lung diseases (38, 52, 54). PH is defined by a mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg that is observed in 30–80% of patients with IPF (4, 52). PH in the setting of IPF is classified as WHO Group III, which also includes PH associated with other chronic lung diseases such as chronic obstructive pulmonary disease or sleep apnea (57). PH is characterized by remodeling of the pulmonary vasculature leading to vessel occlusion, muscularization of previously nonmuscular vessels, and the formation of complex vascular lesions (70) that ultimately lead to right ventricular dysfunction and right-sided heart failure (17, 51, 52, 54, 56, 67).
Despite the strong association of PH with lethality in IPF, the mechanisms leading to increased disease severity and vascular remodeling in IPF remain elusive. Our limited understanding in the pathogenesis of IPF and its cardiovascular complications has resulted in limited therapies to treat IPF outside of lung transplantation. Thus there is a critical need to identify the mechanisms that lead to overactive repair processes contributing to the development of fibrosis and vascular complications in IPF to develop new therapies to interrupt the progression of disease and reduce mortality.
Bone morphogenic protein receptor 2 (BMPR2) is a ubiquitously expressed member of the transforming growth factor (TGF)-β superfamily (53) that has a major role in suppressing TGF-β signaling (21). Under normal signaling conditions, BMPR2 associates with BMPR1A. The resulting complex is then able to bind to BMP ligands such as BMP2, BMP4, and BMP7, leading to phosphorylation of SMADs 1/5/8, which act to suppress TGF-β signaling (21). TGF-β signals through phosphorylation of SMAD 2/3 resulting from TGFβR1 and TGFβR2 complexes (44). Thus reduced levels of P-SMAD 1/5/8 and elevated levels of P-SMAD 2/3 are associated with reduced BMPR2 expression and enhanced TGF-β signaling. Loss of function of BMPR2 leads to dampened phosphorylation of the downstream SMAD 1/5/8 proteins and an enhanced phosphorylation of the SMAD 2/3 that are activated by TGF-β (40), a master regulator of fibrosis (18, 26, 36, 66). Gene knockout and silencing studies have demonstrated that BMPR2 is a master regulator of the lung arteriole structure, affecting both cell apoptosis and proliferation (46). As such, intact BMPR2 signaling is considered an essential component of the arteriole wound healing response that provides a stop signal preventing aberrant tissue repair processes (73). Loss-of-function mutations of BMPR2 were first described in patients with a heritable form of PH, a disease characterized by aberrant proliferation of pulmonary artery endothelial and smooth muscle cell layers where increased inflammation and fibrotic deposition are observed (14, 34). These observations implicate a central role for BMPR2 in modulating repair processes; however, the role of this receptor in modulating aberrant repair processes leading to lung fibrosis has not been identified. Our hypothesis is that BMPR2 loss-of-function in the lungs promotes aberrant tissue repair in IPF.
In this paper we show depleted levels of BMPR2 in IPF consistent with reduced levels of P-SMAD1/5/8 yet elevated levels of P-SMAD 2/3 demonstrating heightened TGF-β signaling. Reduced levels of BMPR2 correlate with increased mPAP levels and the presence of Group III PH. These findings are also echoed in our experimental model of lung fibrosis and PH. We also demonstrate that elevated levels of interleukin (IL)6 and P-STAT3 correlate with depleted BMPR2 levels through the generation of microRNAs (miRs) that are predicted to target BMPR2. Remarkably, genetic or pharmacological blockade of interleukin-6 (IL6) resulted in reduced levels of miRs targeting BMPR2 that translated to expression of BMPR2 and attenuated fibrogenic and features of PH in our experimental model of lung fibrosis. Finally, we also show elevated levels of miRs in patients with IPF and Group III PH that can target BMPR2 expression. In conclusion, our results show that BMPR2 depletion in the lungs is an important mechanism promoting lung and vascular remodeling that can be regulated by heightened IL6 levels. Using isolated bone marrow-derived macrophages (BMDM) from bleomycin (BLM)-exposed mice we demonstrate that both IL6 and the IL6+IL6R complex lead to phosphorylation of STAT3 and subsequent depletion of BMPR2. These findings are consistent with immunohistochemistry (IHC) showing reduced BMPR2 signals in CD206 positive macrophages in BLM-exposed mice or in lung sections from patients with IPF or IPF+PH.
METHODS
Subjects.
The use of human material for this study was approved by the University of Texas Health Science Center at Houston Committee for the Protection of Human Subjects HSC-MS-08-0354. Deidentified explanted lung samples IPF patients were obtained from the Methodist Hospital Debakey Heart & Vascular Center. Collection and use of these tissues for research were in accordance with the guidelines approved by the Methodist Hospital Institutional Review Board. Normal lung tissue was obtained from the International Institution for the Advancement of Medicine (IIAM; Edison, NJ). The diagnosis of IPF was performed by a board-certified pulmonologists upon admittance to the Methodist Hospital Transplant Center and tissue was collected from the same anatomical region for each patient including controls. To account for the heterogeneity of IPF, sections were stained for Masson's trichrome to identify fibrotic regions. Our methods to identify patients with IPF and methods to isolate human tissue are described in detail in Garcia-Morales et al. (19).
Animals.
Male C57/BL6 mice 4–5 wk old were purchased from Envigo (formerly Harlan Industries) (Indianapolis, IN). IL6−/− mice were bred in house and were congenic on a C57/BL background. All mice were housed in ventilated cages equipped with microisolator lids and maintained under strict containment protocols. Mice were kept at an ambient temperature of 22°C and in a 12-h dark/light cycle. Animal care was in accordance with institutional and NIH guidelines. All studies were reviewed and approved by the University of Texas Health Science Center at Houston Animal Welfare Committee.
Experimental design.
Mice were treated with 0.035 U/g of BLM (TEVA Pharmaceuticals, Sellersville, PA) or vehicle [phosphate-buffered-saline (PBS); Life Technologies, Grand Island, NY] twice a week for 4 wk intraperitoneally (IP). On day 15 mice were provided with recombinant mouse sGP130 chimera (R&D Systems, Minneapolis, MN) or vehicle (saline) for the remainder of the experiment. On day 33, physiological readouts were performed and the animals were then euthanized for the collection of tissues and fluids for analysis.
Arterial oxygen saturation.
Physiological assessment measuring arterial oxygen saturation was conducted on conscious mice with the pulse MouseOx software analysis (STARR Life Sciences, Oakmont, PA). The hair around the neck was removed from mice to use the collar clip light sensor. The MouseOx provides real-time percent oxygen saturation of functional arterial hemoglobin by utilizing pulse oximetry measurements of light absorption from the red and infrared LEDs (light-emitting diodes).
Hemodynamic measurements.
The right ventricle (RV) systolic pressure (RVSP), heart rate, and RV hypertrophy procedure was performed as previously described (29). Briefly, mice were given 0.75 mg/g of 2.5% Avertin (a mixture of tert-amyl alcohol and 2-2-2 tribromoethanol, Sigma-Aldrich, St. Louis, MO) to induce a surgical plane of anesthesia. Mice were placed on a heated surgical workstation (Molecular Imaging Products, Bend, OR) and secured with surgical tape. Mice were then tracheotomized with a 19G blunt needle (BRICO, Dayton, NJ), attached to a small animal ventilator (MiniVent, Hugo-Sachs Elektronik, March-Hugstetten, Germany), and ventilated at a stroke volume of 250 μl at 200 strokes per minute. The surgical site is viewed by use of a surgical microscope (SMZ-2B, Nikon, Tokyo, Japan). An incision of ∼1 cm in length was made just below the xiphoid process. An Alm retractor (ALM-112, Braintree Scientific, Braintree, MA) was used to expose the abdominal cavity to visualize the diaphragm and the liver. An incision was then made on the diaphragm to expose the heart and the pericardium was then removed. The right ventricle was then identified and a puncture was made with a 28G needle. A 1-French pressure catheter (SPR-1000, Millar Instruments, Houston, TX) was then inserted through the puncture. The heart rate results were continuously recorded by using a PowerLab 8-SP A/D (AD Instruments) converter, acquired at 1,000 Hz. All RVSP results were recorded to a PC utilizing Chart8 software. After completion of the measurements, blood was collected and the lungs were excised and flash frozen in liquid nitrogen for RNA extraction. The heart was excised and the atria were removed. The RV was then surgically removed and the dry weights of the RV were used to determine the extent of RV hypertrophy (RV/left ventricle+septum). Systemic blood pressure and pulse were measured via a tail cuff and pulse transducer run through a PowerLab noninvasive blood pressure controller (AD Instruments, Colorado Springs, CO).
Bone marrow macrophage isolation.
Bilateral femurs were isolated and removed from male C57BL/6 mice (5–6 wk old, inbred, Envigo Laboratories) treated with biweekly IP injections of PBS or BLM for 4 wk. The bone marrow cavity was flushed with complete macrophage medium, DMEM (Fisher Scientific) supplemented with 10% FBS and 20% L929 medium supplement. To make L929 medium supplement, L929 cells were plated a density of 5 × 105 cells per T75 flask in 55-ml medium of DMEM with 1% HEPES, 1% penicillin-streptomycin, 1% l-glutamine, and 10% FBS. L929 cells were then cultured for 7 days and medium was harvested then sterile filtered for addition to macrophage culture medium. Bone marrow cells were pelleted, resuspended in complete macrophage medium, counted, and plated in 100-mm bacterial dishes at a density of 5 × 106 cells in 10 ml medium per dish. Cells were incubated at 37°C, 5% CO2 for 4 days and then supplemented with 5 ml of medium. On day 7, adherent BMDM were detached, collected, pelleted, and resuspended in macrophage medium for seeding in six-well tissue culture plates at a density of 1.5 × 106 cells/ml and allowed to adhere for 2–3 h. Cells were then used for IL6 and interleukin-6 receptor alpha (IL6Ra) experiments.
IL6 and IL6R-mediated depletion of BMPR2 in macrophages.
Bone marrow-derived macrophages were incubated with medium alone, IL6 (50 ng/ml), or IL6 (50 ng/ml) and IL6Ra (100 ng/ml). Macrophages were lysed in RIPA lysis buffer containing protease inhibitors, and 10 μg protein per sample was subjected to Western blot analysis to determine expression of BMPR2.
RT-PCR and protein expression.
Total RNA was isolated from frozen lung tissue by using TRIzol reagent (Life Technologies). RNA samples were then DNase treated (ArticZymes, Tromso, Norway) and subjected to quantitative real-time RT-PCR, except for miRNA studies in which no DNase treatment was performed. The specific primer sequences used are shown in Table 1.
Table 1.
List of primers used
| Gene | Target Sequence | Provider |
|---|---|---|
| 18 srRNA | Fwd: GTAACCCGTTGAACCCCATT; Rev: CCATCCAATCGGTAGTAGCG. | Sigma Aldrich |
| mus IL6 | Fwd:TAGTCCTTCCTACCCCAATTTCC; Rev: TTGGTCCTTAGCCACTCCTTC. | Sigma Aldrich |
| miR-7 | UGGAAGACUAGUGAUUUUGUUGU | Exiqon (Vedbaek, Denmark) |
| miR-21 | UAGCUUAUCAGACUGAUGUUGA | Exiqon |
| miR-23a | AUCACAUUGCCAGGGAUUUCC | Exiqon |
| miR-23b | AUCACAUUGCCAGGGAUUACC | Exiqon |
| miR-26a | CCUAUUCUUGGUUACUUGCACG | Exiqon |
| miR-103a | AGCAGCAUUGUACAGGGCUAUGA | Exiqon |
| miR-103b | UCAUAGCCCUGUACAAUGCUGCU | Exiqon |
| miR-181 | ACCAUCGACCGUUGAUUGUACC | Exiqon |
| U6 snRNA | Product no. 203907 | Exiqon |
Fwd, forward; Rev, reverse.
Histology and IHC.
Our standard IHC protocol was used for all of our staining; briefly, lung sections were dewaxed by using Histo-Clear (National Diagnostics, Atlanta, GA) and rehydrated with a gradient of ethanol. Sections were then subject to high-temperature antigen retrieval using a citrate buffer, endogenous peroxidase and alkaline phosphatase were inactivated with BLOXALL (Vector Laboratories, Burlingame, CA), and 2.5% normal horse serum (Vector Laboratories) was used as a blocking solution prior to incubation with the primary antibody. Following overnight incubation with the primary antibody, sections were treated with the ImmPRESS polymer detection kits for alkaline phosphatase or peroxidase (Vector Laboratories) based on the host of the primary antibody and developed with Vector red or Vector Blue (Vector Laboratories). For immunofluorescence chicken anti-mouse Alexa Fluor 488 (Life Technologies Carlsbad, CA) was used and counterstained with DAPI (Abcam). Specific information on the antibodies used for IHC is described in Table 2. Lungs stained for Masson's trichrome were analyzed on a modified Ashcroft scale optimized for mouse lung sections (25).
Table 2.
List of primary antibodies used
| α-SMA | A2547 Sigma Aldrich 1:1,000 (IHC) |
| β-Actin | A5316 Sigma Aldrich 1:2,000 (IB) |
| BMPR2 | MA5-15826 Thermo Fisher 1:200 (IHC) |
| BMPR2 | 612292 BD Laboratories 1:1,000 (IB) |
| GAPDH | LifeSpan BioSciences (Seattle, WA) (IB 1:500) |
| ID1 | GTX5326 GeneTex 1:1,000 (IB) |
| P-STAT-3 | ab76315 Abcam 1:5,000 (IB) |
| P-SMAD 1/5/8 | 9511S Cell Signaling 1:1,000 (IB) |
| P-SMAD 2/3 | 8828S Cell Signaling 1:1,000 (IB) |
| SMAD 2/3 | 8685S Cell Signaling 1:1,000 (IB) |
| STAT3 | ab5073 Abcam 1:2,000 (IB) |
IHC, immunohistochemistry; IB, immunoblot.
Morphometry.
Muscularized arterioles of the lung parenchyma were observed under ×40 magnification and noted as being different from both airways and nonmuscularized arterioles. Muscularized arterioles were then photographed under ×40 magnifications. Micropictographs were then analyzed by use of Image Pro-Plus software (MediaCybernetics, Bethesda, MD). In short, the overall area of the muscularized portion was measured for each arteriole. To account for size the largest diameter for each arteriole was also measured. The area of the arteriole was then divided by the largest diameter to give a relative measurement of muscularization.
Immunoblots.
Protein samples in SDS-sample buffer (Boston Bioproducts BP-111R) were separated on Criterion TGX Stain-Free 4–20% gels (Bio-Rad) for 1 h at 200 V. The Stain-Free dye was activated and imaged with the ChemiDoc Touch Imaging System (Bio-Rad). The activated gel was transferred to low-fluorescence PVDF membrane by using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were imaged with the ChemiDoc Touch Imaging System (Bio-Rad) to obtain total protein measures. The membranes were then blocked for 1 h in 5% fat-free dried milk in TBST buffer (0.1% Tween 20 and 150 mM NaCl in 10 mM Tris·HCl, pH 7.4). Primary antibodies were incubated overnight with the concentrations listed in table 2 in 5% milk in TBST. After incubation with the primary antibody the membrane was washed with TBST. The rabbit or mouse secondary antibodies were diluted 1:2,000 in 5% fat-free dried milk in TBST (rabbit: Cell Signaling 7074S, mouse: Cell Signaling 7076S). The membranes were then washed and exposed to Clarity ECL (Bio-Rad) and imaged using the ChemiDoc Touch Imaging System. These images were used in the densitometry analysis of each protein band by normalizing to the total protein seen in the Stain-Free image. These measurements were performed by using the software ImageLab (Bio-Rad).
Statistical analysis.
A one-way analysis of variance (ANOVA) with a Newman-Keuls post hoc test was performed for all experiments. The Student's unpaired t-test was used to compare two groups. Statistical significance was defined as P < 0.05 by use of GraphPad Prism version 5 (GraphPad Software, La Jolla, CA). Densitometry analyses from immunoblots were performed using ImageJ (National Institutes of Health, Bethesda, MD) or ChemiDoc Touch Imaging System.
RESULTS
BMPR2 is depleted in fibrotic lesions and remodeled vessels of patients with IPF.
We performed dual IHC in lung sections from patients with IPF, IPF + PH, and normal controls for BMPR2 and α-smooth muscle actin (αSMA). Here we observed positive staining for BMPR2 in both the parenchyma and pulmonary arterioles from normal lung tissue (Fig. 1A). However, in fibrotic areas rich in myofibroblasts (positive for αSMA) of patients with IPF and in remodeled arterioles from patients with IPF+PH, we report reduced signals for BMPR2 (Fig. 1A). Interestingly, our staining showed reduced levels of BMPR2 signals in the tunica externa of remodeled vessels compared with normal (Fig. 1A). These observations were in line with immunoblots showing reduced signals for BMPR2 in IPF and IPF+PH lung samples, consistent with diminished BMP signaling, highlighted by reduced P-SMAD 1/5/8 in IPF and IPF+PH groups compared with normal human lungs (Fig. 1, B and C). In addition, our results show enhanced TGF-β signaling, demonstrated by elevated P-SMAD 2/3 levels in IPF and IPF+PH groups compared with normal lungs (Fig. 1, B and C).
Fig. 1.

Bone morphogenic protein receptor 2 (BMPR2) expression is depleted in idiopathic pulmonary fibrosis (IPF). A: lung sections double immunofluorescently (IF) stained for BMPR2 (violet/purple signals) and α-smooth muscle actin (αSMA; green signals) and counterstained with DAPI (white/gray signals) from normal lung tissue (left) or patients with IPF (top right) or IPF + PH (bottom left). Scale bar represents 100 μm. B: immunoblots for BMPR2, P-SMAD 1/5/8, P-SMAD 2/3, and GAPDH from isolated lung tissue from normal donor tissue, or lung explants from patients with IPF or IPF+PH. C: densitometries from immunoblots for BMPR2, P-SMAD 1/5/8, and P-SMAD 2/3 normalized to GAPDH. *P ≤ 0.05, comparisons between normal and IPF or IPF+PH.
BMPR2 depletion is also present in experimental models of lung fibrosis.
We next examined the expression of BMPR2 and the BMP signaling pathway in our model of experimental lung fibrosis. Masson's trichrome staining in mice revealed increased fibrotic lesions following BLM (0.035 U IP biweekly for 4 wk) exposure in mice compared with vehicle (PBS)-exposed mice (Fig. 2A). Staining for BMPR2 in these mice revealed increased signals in the parenchyma of PBS exposed and diminished signals in fibrotic lesions in BLM-exposed mice (Fig. 2B). These results were consistent with immunoblots for BMPR2 showing reduced expression in BLM-exposed mice compared with PBS exposure (Fig. 2C). In line with this, immunoblots for ID1 (a marker for BMPR2 signaling) and P-SMAD 2/3 demonstrated reduced BMPR2 signaling but enhanced TGF-β signaling (Fig. 2C). These observations are consistent with our human data and suggest a role for loss of BMPR2 signaling in the pathogenesis of lung fibrosis. Densitometries for BMPR2, ID1, and P-SMAD 2/3 revealed reduced BMPR2 and ID1 yet elevated P-SMAD 2/3 levels (Fig. 2D).
Fig. 2.

Bone morphogenic protein receptor 2 (BMPR2) expression is depleted in experimental lung fibrosis. Masson's trichrome (A) or immunofluorescently (IF) stained lung sections (B) from BMPR2 (violet/purple signals) and α-smooth muscle actin (αSMA/green signals) from phosphate-buffered saline (PBS)- or bleomycin (BLM)-exposed mice, showing increased fibrotic deposition in BLM-exposed mice. A, Airway structures; asterisks identify fibrotic areas in the lung. Scale bar = 100 μm. C: immunoblots for BMPR2, ID1, P-SMAD 2/3, and β-actin from PBS- and BLM-exposed mice. D: densitometries for BMPR2, ID1, and P-SMAD2/3. *P ≤ 0.05.
IL6 and STAT-3 activation is present in IPF and experimental lung fibrosis.
Elevated IL6 and subsequent activation of STAT-3 has been implicated as a profibrotic mediator in several fibroproliferative diseases that affect the lung (32, 58), the cardiovascular system (22), and other organs such as the liver and pancreas (37, 48). Interestingly, in these diseases, alterations of the BMPR2 pathway are also observed (9, 13, 68, 74). Here we report elevated IL6 transcripts in IPF compared with normal lung tissue that are in turn higher in patients with IPF+PH compared with IPF alone (Fig. 3A). A linear regression and correlation analysis between IL6 transcript levels and mPAP revealed a significant correlation between elevated IL6 transcripts and increased mPAP in patients with IPF with and without a secondary diagnosis of PH (Fig. 3B). Examination of STAT3 activation levels revealed increased signals for P-STAT3 from lung tissue from patients with IPF that were in turn greater in patients with IPF+PH (Fig. 3C). Densitometries for P-STAT3 and STAT3 demonstrated increased levels and activation of STAT3 in IPF or IPF+PH compared with normal lungs (Fig. 3D). Our observations in human data are replicated in our animal model of lung fibrosis where increased IL6 protein levels in bronchoalveolar lavage fluid were consistent with increased P-STAT3 signals in BLM-exposed mice compared with PBS exposure (Fig. 3E). These results show that elevated IL6 and subsequent P-STAT3 signaling are present both in patients with IPF and in an experimental model of fibrosis, and increased levels of IL6 and P-STAT3 signaling are increased in more severe forms of IPF that are accompanied by the presence of PH.
Fig. 3.
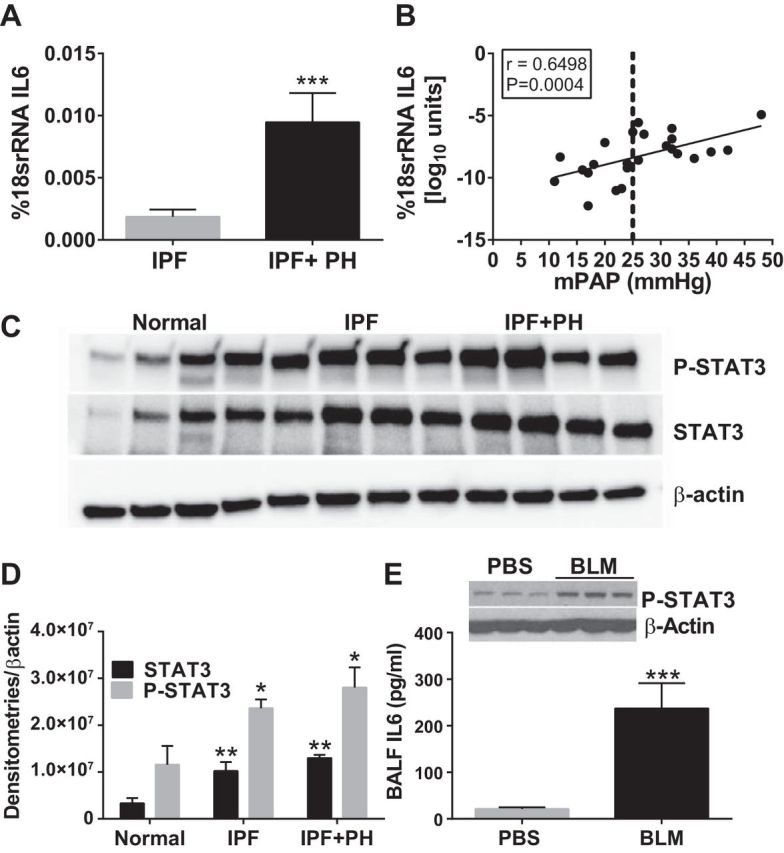
Increased interleukin (IL) 6 and STAT3 activation in IPF and experimental lung fibrosis. A: transcript levels for IL6 from normal lung tissue and from lung explants from patients with IPF and IPF+PH. B: linear regression and Spearman correlation analysis between IL6 transcript levels and mean pulmonary arterial pressures (mPAP) from patients with IPF. C: immunoblots for P-STAT3 and β-actin from normal lung tissue and from lung explants from IPF and IPF+PH. D: densitometries for P-STAT3 and STAT3. E: IL6 levels in bronchoalveolar lavage fluid and immunoblots for P-STAT3, STAT3, and β-actin from isolated lung tissue from PBS- or BLM-exposed mice (E). ***P ≤ 0.01, comparisons between normal and IPF or IPF+PH or between PBS and BLM groups.
Genetic or pharmacological ablation of IL6 signaling halts the development of pulmonary hypertension.
We next examined whether mice deficient in IL6 (IL6−/− mice) were protected from the development of PH or whether treatment with soluble GP130 (sGP130) could be used as new therapy to treat lung fibrosis and PH (Fig. 4A). Recombinant sGP130 has been shown to bind to circulating IL6/IL6R complexes thus inhibiting IL6 trans-signaling in models of lung fibrosis (35). Our results show that indeed IL6−/− mice exposed to BLM presented with reduced αSMA deposition, compared with BLM-exposed mice (Fig. 4B). Similarly, sGP130-treatment led to reduced αSMA deposition compared with BLM-exposed mice (Fig. 4B). These observations are consistent with morphometric analyses of vascular smooth muscle remodeling, showing a greater smooth muscle area per vessel diameter in BLM-exposed mice that is significantly reduced in BLM+ IL6−/− and BLM+sGP130 groups (Fig. 4C). In line with these observations, we report heightened RVSP levels and evidence of right ventricle hypertrophy (RVH) in BLM-exposed mice compared with controls that are attenuated in BLM-exposed IL6−/− mice and in BLM-exposed mice treated with sGP130 (Fig. 4, D and E). No significant differences were observed in systemic blood pressure and heart rate between experimental groups (Fig. 4, F and G). These experiments demonstrate that preventing IL6 signaling can attenuate the development of PH associated with lung fibrosis.
Fig. 4.

Blocking IL6 attenuates pulmonary hypertension. A: experimental model. Mice are treated biweekly with 0.035 U BLM or PBS vehicle intraperitoneally twice weekly for 4 wk in C57/BL6 or IL6−/− mice. Soluble GP130 (sGP130) or vehicle was administered starting on day 15 until the end of the experiment. B: immunohistochemistry for smooth muscle actin (αSMA/magenta signals) and counterstained with methyl green. V, vessel. Morphometric quantification of vascular αSMA deposition (C), right ventricle systolic pressure (RVSP) (D), right ventricle hypertrophy (RVH) determined from measuring dry RV and left ventricle (LV) + septum weights (E), heart rate (F), and systemic blood pressure (BP; G) from PBS, BLM, BLM+IL6−/−, and BLM+SGP130 groups. *P ≤ 0.05, **0.001 < P < 0.01, ***P ≤ 0.01, ANOVA comparisons between PBS and experimental treatment groups. ###P < 0.001, ##0.001 < P < 0.01, #P < 0.05, ANOVA comparisons between BLM and BLM+IL6−/− or BLM+sGP130 treatment groups.
Pulmonary fibrosis is attenuated in IL6−/− mice and following treatment with sGP130.
In line with the role of IL6 as a profibrotic cytokine (32, 35, 58), our data showed that IL6−/− mice are protected from the development of BLM-induced lung fibrosis, a phenomenon replicated following treatment with sGP130 (Fig. 5, A and B). These results are supported by IHC for αSMA showing increased presence of myofibroblasts in BLM-exposed mice compared with control mice, which were attenuated in IL6−/− mice or following sGP130 treatment (Fig. 5C). Consistent with these results and our data in Fig. 4, we show reduced arterial oxygenation levels in BLM-exposed mice compared with normal controls that are rescued in IL6−/− or following sGP130 treatment (Fig. 5D).
Fig. 5.

Blocking IL6 attenuates lung fibrosis. Masson's trichrome stain (A), Ashcroft scores (B), IHC (C) for smooth muscle actin (αSMA/magenta signals) and counterstained with methyl green. V, vessel, arrowheads denote myofibroblasts; from PBS, BLM, BLM+IL6−/− and BLM+sGP130 treatment groups. D: arterial oxygenation (SpO2) determined by pulse oximetry from PBS, BLM, BLM+IL6−/−, and BLM+sGP130 groups. ***P ≤ 0.01, ANOVA comparisons between PBS and experimental treatment groups. ###P < 0.001, ##0.001 < P < 0.01, #0.05 < P, ANOVA comparisons between BLM and BLM+IL6−/− or BLM+sGP130 treatment groups.
Blockade of IL6 signaling rescues BMPR2 expression.
In line with our hypothesis, BMPR2 protein levels were attenuated in whole lung tissue from mice exposed to BLM, consistent with enhanced P-STAT3 signals, demonstrating increased IL6 signaling compared with PBS-exposed mice (Fig. 6A). These observations were accompanied by reduced P-STAT3 levels consistent with the genetic deletion of IL6 (Fig. 6A). Immunoblots for ID1 and fibronectin (FN) also revealed reduced signals for ID1 yet elevated levels of FN in BLM-exposed mice compared with PBS that were consistent with reduced BMPR2 expression following BLM exposure (Fig. 6B). Interestingly, IL6−/− mice exposed to BLM presented with increased BMPR2 levels compared with BLM exposure alone (Fig. 6, A and B). Double immunofluorescence for BMPR2 and αSMA revealed reduced BMPR2 signals in fibrotic areas and remodeled vessels in BLM-exposed mice compared with control (PBS-exposed) mice (Fig. 6C). Interestingly, IL6-deficient mice presented with increased signals for BMPR2 in the lung parenchyma and in pulmonary arterioles that also appeared less remodeled than BLM-exposed mice (Fig. 6C). These observations suggest that signaling through IL6 leads to depletion of BMPR2 levels that are in turn rescued following strategies aimed at preventing IL6 signaling.
Fig. 6.

BMPR2 signals are maintained in IL6-deficient mice. Immunoblot for BMPR2, P-STAT3, and β-actin (A); immunoblot for BMPR2, inhibitor of differentiation (ID) 1, fibronectin (FN), and β-actin (B) from PBS, BLM, and BLM+IL6−/− treatment groups. C: immunofluorescence for BMPR2 (magenta signals) or α-smooth muscle actin (αSMA/green signals) and counterstained with DAPI (white/gray signals) from PBS, BLM, and BLM+IL6−/− treatment groups. Arrowheads denote signals for BMPR2. V, vessel; F, fibroproliferative lesions. Scale bar = 50 μm.
Depletion of BMPR2 during the development of lung injury.
We next assessed levels of BMPR2, ID1, P-SMAD 2/3, and FN at days 14, 21, 25, and 33 of BLM exposure (Fig. 7A). Our data show a progressive decline in BMPR2 levels reaching the lowest levels on days 21 and 33 of BLM exposure (Fig. 7, A and B). Consistent with reduced BMPR2 signaling, we report a decline in ID1 signals on day 14 that appears to reduce further on day 21 and remain attenuated compared with PBS levels throughout the remainder of the experiment (Fig. 7, A and C). Increased evidence of TGF-β signaling is observed through elevated P-SMAD 2/3 on day 21 that appears to remain elevated throughout the remainder of the study (Fig. 7, A and D). Consistent with the development of lung fibrosis, we report a steady increase in FN levels, reaching a maximum on day 33 (Fig. 7, A and E), that is in line with our data showing fibrotic deposition on day 33 (Fig. 5). These results show that loss of BMPR2 signaling correlates with enhanced TGF-β signaling and the development of fibrotic lesions in the lung.
Fig. 7.
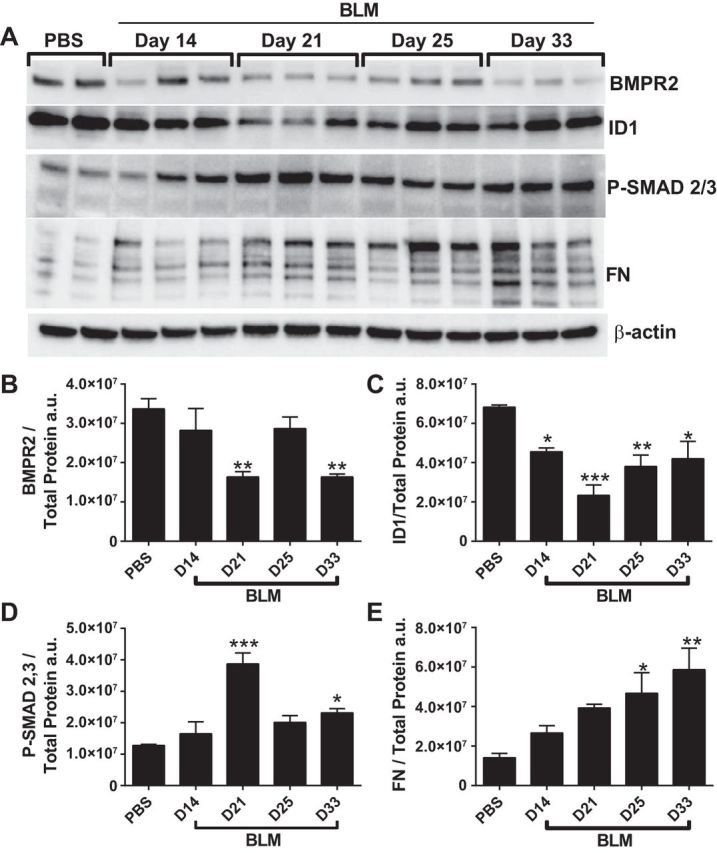
Changes in BMPR2 expression during the development of fibrosis. Immunoblot for BMPR2, ID1, P-SMAD 2/3, fibronectin (FN), and β-actin (A) and densitometries for BMPR2 (B), ID1 (C), P-SMAD 2/3 (D), and FN (E) from PBS- and BLM-exposed mice on days 14, 21, 25, and 33.
Differential miR expression is associated with BMPR2 downregulation.
IL6 and subsequent STAT3 activation has been shown to lead to the differential expression of several miRs (8, 11, 78), some of which can target the 3′-untranslated region (3′UTR) sequence of BMPR2 (8, 10, 78). Thus, to study whether BMPR2 depletion correlated with differential miR expression, we assessed BMPR2 protein- and P-STAT3-induced miR levels at 14, 21, and 25 days of BLM exposure. Here we show that BMPR2 levels begin to decline starting at day 21 and remain reduced at day 25 (Fig. 8A). These levels correlate with differential miR expression, showing time-dependent increases in miR-7 and miR-21 levels reaching a maximum on day 25 (Fig. 8, B and C). We also report significantly increased expression levels of miR-23a, -23b, -130, -130b, and -181 on day 25 of BLM exposure (Fig. 8, D–H). No significant changes were seen for miR-26a expression albeit a tendency for elevated levels on day 25 (Fig. 8 I). These results suggest a role for differential miR expression as a mechanism leading to depletion of BMPR2 expression.
Fig. 8.

Changes in microRNA (miR) expression during the development of fibrosis. A: immunoblot for BMPR2 and β-actin for PBS, BLM day 14 (D14), BLM day 21 (D21), and BLM day 25 (D25) treatment groups. Expression levels of miR-7 (B), miR-21 (C), miR-23a (D), miR-23b (E), miR-130 (F), miR-130b (G), miR-181 (H), and miR-26 (I), using U6 small nuclear RNA as a control from PBS, BLM day 14 BLM day 21 and BLM day 25 treatment groups. **0.001 < P < 0.01, ***P ≤ 0.01, ANOVA comparisons between PBS and BLM treatment groups.
Deletion of IL6 attenuates expression of miRs that are predicted to target BMPR2.
We next examined the expression of miRs that are known to bind to BMPR2 (8) or are predicted to bind through in silico analysis in PBS, BLM, and BLM-exposed IL6-deficient mice on day 33 of BLM or PBS exposure (Fig. 9). Our data show increased expression of miR-7, miR-21, miR-130a, miR-181, and miR-26a, but not miR-23a, miR-23b, or miR-130b (Fig. 9, A–H) in BLM-exposed mice compared with PBS treatment. Interestingly, statistically significantly reduced levels of miR-7, miR-21, miR-23a, miR-23b miR-130a, miR-181, and miR-26a were observed in BLM+IL6−/− compared with BLM alone (Fig. 9, A–H).
Fig. 9.

Differential microRNA (miR) expression profile in IL6-deficient mice treated with BLM. Expression levels of miR-7 (A), miR-21 (B), miR-23a (C), miR-23b (D), miR-130 (E), miR-130b (F), miR-181 (G), and miR-26 (H), using u6 as a control from isolated lung tissue from PBS, BLM, or BLM+IL6−/− treatment groups. *P ≤ 0.05 **0.001 < P < 0.01, ANOVA comparisons between PBS and BLM groups. ##0.001 < P < 0.01, #P < 0.05, ANOVA comparisons between BLM and BLM+IL6−/− treatment groups.
Increased expression of miRs that are predicted to bind to BMPR2 in IPF.
Based on our experiments using the BLM model of lung fibrosis, we next examined the expression of our miR family in human lung tissue samples from normal controls, IPF patients, and patients diagnosed with IPF+PH. Our data reveal increased expression of miR-7, miR-21, and miR-181 (Fig. 10, A, B, and G) in both IPF and IPF+PH groups compared with normal controls. We also report significantly increased expression of miR-23a and miR-130b in IPF+PH patients but not IPF patients compared with normal controls and significantly elevated levels of miR-23b and miR130a in IPF but not IPF+PH patients compared with normal controls (Fig. 10, B–F). No significant changes were observed for miR-26 (Fig. 10H).
Fig. 10.

Differential microRNA (miR) expression profile in patients with idiopathic pulmonary fibrosis (IPF). Expression levels of miR-7 (A), miR-21 (B), miR-23a (C), miR-23b (D), miR-130 (E), miR-130b (F), miR-181 (G), and miR-26 (H), using u6 as a control from normal, IPF, and IPF+PH lung tissue samples. *P ≤ 0.05, **0.001 < P < 0.01, ANOVA comparisons between normal and IPF or IPF+PH groups.
IL6 or the IL6 + IL6R complex depletes BMPR2 expression in isolated macrophages.
There is increasing recognition that macrophages, in particular alternatively activated macrophages (AAM), are critical modulators of fibrosis (1, 7, 20, 23, 28, 45). Classically activated macrophages (M1) or the loss of the AAM phenotype has been shown to be essential for the resolution of fibrosis (7, 20, 28, 49, 62, 71). However, the mechanisms that maintain macrophages in a perpetual alternative activated state and prevent phenotype switch to an M1 profile are not fully understood. In the context of PH, very limited studies have examined the role of macrophages or AAM in Group III PH. Our group demonstrated that deletion of the adenosine A2B receptor from myeloid cells (macrophage progenitor cells) was associated with reduced markers of AAM, IL6, and hyaluronan, contributing to attenuated fibrosis and PH (28). Based on this body of information our next aim was to determine whether stimuli with IL6 or IL6 trans-signaling using the IL6 + IL6R complex was able to deplete BMPR2 from isolated BMDM from mice exposed to PBS or BLM. Surprisingly, our data show that stimulus of BLM-exposed BMDMs with IL6 or the IL6 + IL6R complex necessary for trans-signaling leads to a depletion of BMPR2 in line with elevated PSTAT3 signals (Fig. 11A). Interestingly, no changes were seen in BMDM from PBS-exposed mice (data not shown) and no differences ARG-1 or CD206 expression were observed (Fig. 11A). Remarkably, trans-signaling but not IL6 alone resulted in increased macrophage transcripts for Col1A1, Col1A2, and IL6 (Fig. 11, B–D). These findings are consistent with IHC showing reduced BMPR2 signals in CD206-positive macrophages from BLM-exposed mice compared with PBS treatment (Fig. 11E). Similarly, IHC from human lung sections revealed loss of BMPR2 in CD206-positive macrophages from patients with IPF or IPF+PH but not in normal lungs (Fig. 11F).
Fig. 11.
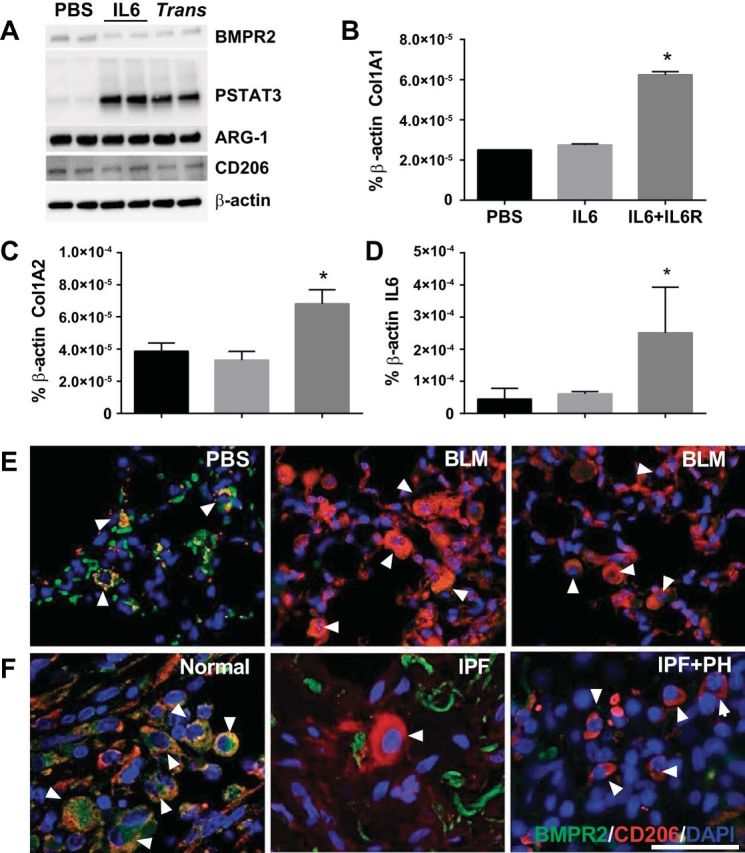
Depletion of BMPR2 from macrophages in fibrosis. Isolated bone-marrow-derived macrophages (BMDMs) from BLM-exposed mice treated with PBS, IL6 or IL6+IL6R to induce trans-signaling (trans) and subsequent immunoblots for BMPR2, PSTAT3, ARG-1, CD206, and β-actin expression levels (A); or transcript levels for Col1A1 (B), Col1A2 (C), and IL6 (D). *P ≤ 0.05, ANOVA comparisons between PBS and IL6 or IL6+ILR groups. Immunohistochemistry for BMPR2 (green signals), CD206 (red signals), merge in yellow and counterstain with DAPI from lung sections from mice exposed to PBS or BLM (E) or from human normal, IPF, and IPF+PH lung sections (F). White arrowheads denote macrophages; scale bar = 50 μm.
BMPR2 isoforms in IPF.
BMPR2 can undergo alternative splicing resulting in the expression of several protein variants (40, 41). BMPR2 isoform A is the most common and functional protein receptor, yet up to three more protein isoforms (B–D) have been identified (2, 42). Interestingly, increased levels of BMPR2 isoform B relative to the functional protein (BMPR2 isoform A) has been linked with the presence of hereditable PH in patients carrying a loss-of-function mutation of BMPR2 (13). These observations led us to determine the expression levels of the BMPR2 isoforms A and B in addition to determining the ratio of isoform A/B in our IPF cohort. Consistent with our data showing reduced BMPR2 protein expression, our results show reduced levels of BMPR2 isoform A in IPF and IPF+PH lung samples relative to normal lungs (Fig. 12A). No significant differences were seen for BMPR2 isoform B; thus elevated isoform B relative to A levels were detected in IPF and IPF+PH patients (Fig. 12, B and C). Strikingly, linear regression and Spearman correlation analysis revealed a significant association between reduced BMPR2 isoform A levels and heightened mPAP values (Fig. 12D). Linear regression and Spearman correlation analysis for BMPR2 isoform B did not reveal any significant differences; however, elevated BMPR2 isoform B relative to isoform A correlated with increased mPAP values (Fig. 12, E and F). Interestingly, further analysis of BMPR2 expression in cultured lymphocytes from patients with IPF showed increased isoform B levels relative to A that appeared to be due to increased expression of BMPR2 isoform B (Fig. 12, G–I). Taken together our data demonstrate that elevated isoform B relative to A is increased in IPF and correlates with higher mPAP values. In addition, our data suggest that the elevated expression of BMPR2 isoform B could be as a result of degradation of BMPR2 isoform A or as demonstrated in cultured lymphocytes due to increased expression of BMPR2 isoform B.
Fig. 12.

Expression levels of BMPR2 isoforms in patients with IPF. Expression levels of BMPR2 isoform A (A), BMPR2 isoform B (B), or calculated ratio of isoform A relative to isoform B (B/A ratio; C), from lung samples from normal controls, IPF, or IPF+PH patients. *P ≤ 0.05, ANOVA comparisons between normal and IPF or IPF+PH groups. Linear regression and Pearson's correlation for BMPR2 isoform A (D), BMPR2 isoform B (E), or calculated ratio of isoform A relative to isoform B (F) against mean pulmonary arterial pressure (mPAP) from patients with IPF and IPF+PH. Expression levels of BMPR2 isoform A (G), BMPR2 isoform B (H), or calculated ratio of isoform A relative to isoform B (I), from cultured lymphocytes from normal controls or IPF patients. *P ≤ 0.05, **0.001 < P < 0.01, t-test comparisons between controls and IPF groups.
BMPR2 isoforms in experimental lung injury.
We next examined levels of BMPR2 isoform A and B during the development of lung injury following BLM exposure. In these studies we observed a reduction in BMPR2 isoform A expression on day 21 of BLM exposure that remained attenuated throughout the remainder of the study (Fig. 13A). Remarkably, in our mouse model of lung fibrosis and PH, BMPR2 isoform B levels were also significantly reduced as early as day 14 of BLM exposure. Levels of BMPR2 isoform B remained reduced throughout the duration of the experiment (Fig. 13B). In line with these findings we report a reduction in BMPR2 isoform B relative to A on days 14 and 25 of BLM exposure compared with PBS exposure (Fig. 13C). These observations suggest that, in the mouse, depletion of both BMPR2 isoforms is present during the development of lung fibrosis and vascular remodeling.
Fig. 13.

Temporal expression levels of BMPR2 isoforms following BLM exposure in mice. Expression levels of BMPR2 isoform A (A), BMPR2 isoform B (B), or calculated ratio of isoform A relative to isoform B (C), from lung lysates from PBS- (day 33) or BLM-exposed mice on days 14, 21, and 25. **0.001 < P < 0.01, ***P ≤ 0.01, ANOVA comparisons between PBS and BLM treatment groups.
DISCUSSION
A major finding of our study was that reduced levels of BMPR2 were observed in patients with IPF with and without Group III PH. These observations also correlated with reduced evidence of BMPR2 signaling highlighted by depleted P-SMAD 1/5/8 but elevated P-SMAD 2/3 levels (a marker for TGF-β signaling). These results were in line with heightened IL6 expression data and P-STAT3 in patients with IPF and increased expression of a family of miRs that can target BMPR2. Our studies in our BLM-model of chronic lung injury demonstrated increased levels of IL6 and P-STAT-3 that correlated with BMPR2 depletion and the development of lung fibrosis and PH. Experiments using pharmacological inhibition or genetic deletion of IL6 demonstrated that exposure to BLM in these mice did not deplete BMPR2 levels and features of lung fibrosis and PH were significantly reduced. In these experiments we demonstrated that miRs associated with BMPR2 depletion were attenuated compared with control BLM-exposed mice. Taken together, our results point at the loss of function of BMPR2 as an important mechanism in the development of lung fibrosis and vascular remodeling. We show that BMPR2 levels are depleted following increased IL6 and activation of STAT3 that evoked a collection of miRs that downregulated BMPR2. These studies are the first to show that BMPR2 depletion is common in patients with IPF and suggest that strategies aimed at reestablishing BMPR2 signaling as a novel therapeutic target for IPF and Group III PH.
The processes that govern the progression and development of fibroproliferative lesions in IPF are not fully understood (31, 77). Mutations of BMPR2 were first described in patients with a heritable form of PH termed pulmonary arterial hypertension (PAH) where 70% or more of patients were found to be affected (14, 34). In PAH, only the vasculature is affected, with no evidence of damage or injury to the lung parenchyma or the conducting airways (57, 59, 75). BMPR2 is a ubiquitously expressed member of the TGF-β superfamily (53) and the majority of the mutations identified lead to loss of function or reduced BMPR2 expression (2, 42). Although patients with BMPR2 mutations typically do not develop lung fibrosis, at least in one case study PH preceded IPF in a BMPR2-mutation-positive patient (60). In line with this, mice with mutated BMPR2 also appear to develop worse PH when exposed to BLM (9). These studies highlight how a perturbed balance of BMPR2 and TGF-β signaling can have critical implications to lung regenerative events and lead to pathogenic processes in IPF. Recently, several studies have linked loss of function of BMPR2 as a pathological mechanism leading to increased cell survival, proliferation, and metastasis in a variety of cancer models (30, 33, 55). However, its role in promoting lung fibrosis has not been extensively studied despite the fact that loss of function of BMPR2 leads to enhanced phosphorylation of SMAD 2/3 that is activated by TGF-β (40), a molecule intrinsically linked with the pathogenesis of lung fibrosis (18, 43, 76, 77). Decreased BMPR2 signaling due to gremlin (an endogenous BMPR2 inhibitor) was shown to be elevated in asbestos-induced fibrosis (50) and in pancreatic fibrosis (68), demonstrating the importance of the BMPR2/TGF-β signaling balance in preventing and promoting lung injury. Our data derived from human lung explants from IPF patients show a marked reduction of BMPR2 (of up to 95%), P-SMAD1/5/8 and enhanced P-SMAD 2/3 in patients with IPF that is aggravated in patients with Group III PH consistent with the role that BMPR2 loss of function has on vascular remodeling and tone in PAH (14, 34). These observations are replicated in our experimental model of lung fibrosis and PH, where reduced levels of BMPR2 correlate with evidence of fibrotic deposition in the lungs and hallmarks of PH (Fig. 2). Our time-course experiments indeed show that BMPR2 depletion and alteration in BMP and TGF-β signaling track with the development of lung injury. Taken together these results clearly show that BMPR2 depletion is an important hallmark of IPF, playing a central role in fibroproliferative and vascular lesions.
To understand the mechanisms responsible for the depletion of BMPR2 in IPF and in our experimental model of lung injury, we focused on IL6, a cytokine that has been shown to be elevated in many chronic lung diseases including lung fibrosis (15, 16, 32, 35) and to play a role in the development of PH (69). Here we report elevated levels of IL6 in IPF+PH patients vs. IPF patients alone; we also demonstrate that increased IL6 expression levels correlate with elevated mPAP values, suggesting a pathogenic role for IL6 in aggravating vascular injury in IPF. Normally, IL6 binds to its membrane receptor (IL6Rα) and then associates with GP130 and activates STAT3 (24, 47, 72). However, not all cells express IL6Rα (3, 12). In cells devoid of IL6Rα, trans-signaling can occur whereby IL6 first binds to its soluble receptor sIL6Rα; the resulting complex is then able to reach the target cell membrane and associate to membrane-bound GP130, leading to activation of STAT3 (3, 12, 65). Studies from our group have shown that trans-signaling is associated with increased fibroblast proliferation and extracellular matrix deposition, demonstrating an important role for IL6 signaling in the fibroproliferative process (35). Soluble GP130 can also be used pharmacologically to remove IL6/IL6Rα complexes (12, 27, 35) that have been shown to inhibit fibrotic deposition in the lungs (35). Consistent with this, our data show that pharmacological (following sGP130 treatment) or genetic depletion of IL6 is able to significantly reduce the extent of vascular remodeling induced by BLM exposure demonstrated by morphometric analysis, RVSP, and RVH measurements (Fig. 4). In addition to these novel findings, we also show that depleting cells from IL6 is able to attenuate lung fibrosis and myofibroblasts and improve arterial oxygenation in line with our previous observations (2). Surprisingly, in line with these results we also report increased expression of BMPR2 in IL6-deficient mice compared with BLM-exposed mice, consistent with reduced P-STAT3 and elevated signals for ID1. These results suggest that IL6 and subsequent activation of STAT3 are able to modulate expression of BMPR2. Indeed activation of STAT3 by IL6 has also been associated with the increased expression of a large collection of miRs (8, 10, 78). This observation is significant given the exceptionally large 3′UTR of BMPR2 (∼7,000 bp) (13) that enables it to be targeted by a plethora of miRs. Our results in experimental lung injury show increased expression of several miRs that are known to target BMPR2 during the development of lung injury that track with reduced expression of BMPR2. Moreover, in our experiments using genetic depletion of IL6, we show that this collection of miRs is attenuated in IL6-deficient mice exposed to BLM. Furthermore, in experiments using human lung tissue, we demonstrate increased expression of this collection of miRs in patients with IPF or IPF+PH demonstrating a plausible mechanism leading to BMPR2 depletion. These studies demonstrate that IL6-mediated miR expression is an important mechanism in the regulation of BMPR2 expression. They also demonstrate that strategies aimed at targeting single miRs may not always be effective therapeutically, particularly in situations when a protein can be targeted by multiple miRs that are enhanced in disease, as is the case with BMPR2.
Furthermore, in experiments using BMDM from BLM-exposed mice, we successfully show that treatment with IL6 or the IL6+ILR is able to induce STAT3 activation and lead to depletion of BMPR2 signals concomitant with upregulation of the miR family associated with BMPR2 depletion. These results demonstrate that both conventional and trans IL6 signaling can deplete BMPR2 macrophage expression. AAM have been implicated in the pathogenesis of lung fibrosis (1, 7, 20, 23, 28, 45). Loss of AAM markers or the increased levels of M1 macrophages have been shown to be important in the resolution of lung fibrosis (7, 20, 28, 49, 62, 71). Interestingly, our results show that depletion of BMPR2 from macrophages confers these cells a greater expression of the profibrotic mediators Col1A1, Col1A2, and FN despite no changes in Arg-1 and CD206 expression. These are consistent with IHC from both BLM-exposed mice and lung sections from patients with IPF or IPF+PH showing reduced BMPR2 signals in CD206-positive macrophages. These results are consistent with our previous findings showing that CD206 expressing AAM play a role in Group III PH (28) and suggest a novel role for the depletion of BMPR2 in AAM as mechanism contributing to the development of lung fibrosis.
In both humans and animals, BMPR2 has 13 exons and is alternatively spliced to produce two primary transcripts. Isoform A is the full-length BMPR2 gene product that contains all 13 exons while isoform B is a much scarcer transcript that lacks exon 12 (5, 41, 63). Several studies have shown that exon 12 is required for the normal function of the BMPR2 protein and deletion of this exon is observed in many patients with familial PAH, where it disrupts function in a dominant negative fashion (64). A striking observation in familial PAH (FPAH) is the fact that ∼82% of families with FPAH carry mutations associated with loss of function of BMPR2; however, only 20% of carriers develop disease (53). Recently BMPR2 isoform B, a product of alternative splicing, was found to be elevated in cultured lymphocytes from FPAH patients compared with family members with BMPR2 mutations but no evidence of disease (13). In these studies, the balance between the functional isoform A and the nonfunctional isoform B was shifted to favor increased isoform B expression over A, resulting in an increased ratio of BMPR2 isoform B relative to A that was associated with the onset of disease (13). These studies suggest that loss of function of BMPR2 may require downregulation of the functional protein and enhanced levels of the nonfunctional isoform to lead to pathological consequences in humans. However, this concept has not been evaluated in IPF. Interestingly, our results show that isolated lung tissue from patients with IPF have reduced expression levels of BMPR2 isoform A (the functional receptor) and an increased ratio of BMPR2 isoform B vs. isoform A, albeit no differences in BMPR2 isoform B expression. Interestingly, reduced expression of BMPR2 isoform A correlated significantly with elevated mPAP, and elevated BMPR2 isoform B ratio to A also significantly correlated with elevated mPAP. Remarkably, in studies using cultured lymphocytes from patients with familial pulmonary fibrosis or IPF, we report enhanced isoform B expression tilting the balance toward increased expression of the nonfunctional isoform relative to the functional isoform A in conditions where lung fibrosis is apparent. These results clearly demonstrate that BMPR2 loss of function in IPF can be induced by either depletion of the functional and main BMPR2 isoform (isoform A) or through enhanced expression of the nonfunctional BMPR2 isoform B in cells such as lymphocytes. In contrast, in mice, we report depletion of both isoform A and isoform B following BLM exposure, suggesting that depletion of the functional BMPR2 isoform as a central role in the pathogenesis in murine lung fibrosis and vascular remodeling. These results also demonstrate that loss of function of BMPR2 in IPF is a complex process that involves miR-mediated depletion of the functional isoform of BMPR2 and alternative splicing leading to enhanced expression of the nonfunctional BMPR2 isoform B.
In conclusion, our studies show that depletion of BMPR2 is a common feature of IPF that plays a role in promoting fibroproliferative and vascular lesions through enhanced TGF-β signaling. We also show that miRs that are predicted to target BMPR2 are upregulated in IPF and in experimental models of lung injury and that the generation of these miRs is attenuated in IL6-deficient mice. This is in line with the maintenance of BMPR2 expression and its signaling pathway that correlates with improved outcomes in fibrosis and PH. Finally, our studies in alternative splicing show that depletion of the functional variant of BMPR2 (isoform A) but not increased expression of the nonfunctional BMPR2 variant (isoform B) is linked with the development of disease in IPF and in experimental lung injury. However, in inflammatory cells such as cultured lymphocytes, BMPR2 loss of function appears to be mediated by increased expression of the nonfunctional BMPR2 isoform B. These observations are significant since BMPR2-A can be targeted by a plethora of miRs, yet the 3′UTR of BMPR2-B is significantly smaller and largely devoid of regulatory elements (13), allowing this transcript to escape degradation resulting in elevated levels of this nonfunctional variant. In summary, our results point at rescuing BMPR2 expression and signaling as a novel therapy for the treatment of IPF that can target both the fibroproliferative and vascular remodeling components.
GRANTS
This study was supported by the American Heart Association (AHA): H. Karmouty-Quintana 14SDG18550039 and by Actelion Entelligence Young Investigator Award: H. Karmouty-Quintana 2013.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.-Y.C., S.D.C., F.L., T.T.L., A.M.H., K.P., J.M., Y.C., J.A.-G., O.A.J., R.R.B., B.A.B., R.H., and H.K.-Q. performed experiments; N.-Y.C., S.D.C., T.W., T.T.L., K.A.Y., R.H., J.D., and H.K.-Q. interpreted results of experiments; N.-Y.C., S.D.C., F.L., T.W., T.T.L., A.M.H., K.P., J.M., L.J.G.-M., Y.C., T.C.K., J.A.-G., O.A.J., R.R.B., K.A.Y., B.A.B., R.H., J.D., N.S., and H.K.-Q. approved final version of manuscript; S.D.C., F.L., T.W., T.T.L., K.P., Y.C., J.A.-G., K.A.Y., R.H., J.D., N.S., and H.K.-Q. analyzed data; S.D.C. and H.K.-Q. prepared figures; S.D.C., J.A.-G., and H.K.-Q. edited and revised manuscript; T.W. and H.K.-Q. conception and design of research; H.K.-Q. drafted manuscript.
REFERENCES
- 1.Alber A, Howie SE, Wallace WA, Hirani N. The role of macrophages in healing the wounded lung. Int J Exp Pathol : 243–251, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation : 1672–1678, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Bauer J, Bauer TM, Kalb T, Taga T, Lengyel G, Hirano T, Kishimoto T, Acs G, Mayer L, Gerok W. Regulation of interleukin 6 receptor expression in human monocytes and monocyte-derived macrophages. Comparison with the expression in human hepatocytes. J Exp Med : 1537–1549, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Behr J, Ryu JH. Pulmonary hypertension in interstitial lung disease. Eur Respir J : 1357–1367, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Beppu H, Minowa O, Miyazono K, Kawabata M. cDNA cloning and genomic organization of the mouse BMP type II receptor. Biochem Biophys Res Commun : 499–504, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, Bar-Joseph Z, Bitterman P, Blackburn MR, Bradford W, Brown KK, Chapman HA, Collard HR, Cosgrove GP, Deterding R, Doyle R, Flaherty KR, Garcia CK, Hagood JS, Henke CA, Herzog E, Hogaboam CM, Horowitz JC, King TE Jr, Loyd JE, Lawson WE, Marsh CB, Noble PW, Noth I, Sheppard D, Olsson J, Ortiz LA, O′Riordan TG, Oury TD, Raghu G, Roman J, Sime PJ, Sisson TH, Tschumperlin D, Violette SM, Weaver TE, Wells RG, White ES, Kaminski N, Martinez FJ, Wynn TA, Thannickal VJ, Eu JP. Future directions in idiopathic pulmonary fibrosis research. An NHLBI workshop report. Am J Respir Crit Care Med : 214–222, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boorsma CE, Draijer C, Melgert BN. Macrophage heterogeneity in respiratory diseases. Mediators Inflamm : 769214, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res : 1184–1191, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Bryant AJ, Robinson LJ, Moore CS, Blackwell TR, Gladson S, Penner NL, Burman A, McClellan LJ, Polosukhin VV, Tanjore H, McConaha ME, Gleaves LA, Talati MA, Hemnes AR, Fessel JP, Lawson WE, Blackwell TS, West JD. Expression of mutant bone morphogenetic protein receptor II worsens pulmonary hypertension secondary to pulmonary fibrosis. Pulm Circ : 681–690, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai Y, Chen H, Mo X, Tang Y, Xu X, Zhang A, Lun Z, Lu F, Wang Y, Shen J. Toxoplasma gondii inhibits apoptosis via a novel STAT3-miR-17-92-Bim pathway in macrophages. Cell Signal : 1204–1212, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu X, Shen Y, Huang TT. Interplay between microRNAs and the STAT3 signaling pathway in human cancers. Physiol Genomics : 1206–1214, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol : 484–494, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Cogan J, Austin E, Hedges L, Womack B, West J, Loyd J, Hamid R. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation : 1907–1916, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet : 737–744, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egwuagu CE. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine : 149–156, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell : 176–185, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farkas L, Gauldie J, Voelkel NF, Kolb M. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol : 1–15, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez IE, Eickelberg O. The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc : 111–116, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Morales LJ, Chen NY, Weng T, Luo F, Davies J, Philip K, Volcik KA, Melicoff E, Amione-Guerra J, Bunge RR, Bruckner BA, Loebe M, Eltzschig HK, Pandit LM, Blackburn MR, Karmouty-Quintana H. Altered hypoxic-adenosine axis and metabolism in group III pulmonary hypertension. Am J Respir Cell Mol Biol : 574–583, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med : 569–581, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell : 1023–1035, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haghikia A, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res : 281–289, 2014. [DOI] [PubMed] [Google Scholar]
- 23.He C, Ryan AJ, Murthy S, Carter AB. Accelerated development of pulmonary fibrosis via Cu,Zn-superoxide dismutase-induced alternative activation of macrophages. J Biol Chem : 20745–20757, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J : 297–314, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubner RH, Gitter W, El Mokhtari NE, Mathiak M, Both M, Bolte H, Freitag-Wolf S, Bewig B. Standardized quantification of pulmonary fibrosis in histological samples. BioTechniques : 507–511, 514–507, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Jarvinen PM, Laiho M. LIM-domain proteins in transforming growth factor beta-induced epithelial-to-mesenchymal transition and myofibroblast differentiation. Cell Signal : 819–825, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest : 3375–3383, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karmouty-Quintana H, Philip K, Acero LF, Chen NY, Weng T, Molina JG, Luo F, Davies J, Le NB, Bunge I, Volcik KA, Le TT, Johnston RA, Xia Y, Eltzschig HK, Blackburn MR. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J : 50–60, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, Hemnes A, Grenz A, Eltzschig HK, Blackwell TS, Xia Y, Johnston RA, Zeng D, Belardinelli L, Blackburn MR. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J : 2546–2557, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim IY, Lee DH, Lee DK, Kim BC, Kim HT, Leach FS, Linehan WM, Morton RA, Kim SJ. Decreased expression of bone morphogenetic protein (BMP) receptor type II correlates with insensitivity to BMP-6 in human renal cell carcinoma cells. Clin Cancer Res : 6046–6051, 2003. [PubMed] [Google Scholar]
- 31.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet : 1949–1961, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Knight D, Mutsaers SE, Prele CM. STAT3 in tissue fibrosis: is there a role in the lung? Pulm Pharmacol Ther : 193–198, 2011. [DOI] [PubMed] [Google Scholar]
- 33.Kodach LL, Wiercinska E, de Miranda NF, Bleuming SA, Musler AR, Peppelenbosch MP, Dekker E, van den Brink GR, van Noesel CJ, Morreau H, Hommes DW, Ten Dijke P, Offerhaus GJ, Hardwick JC. The bone morphogenetic protein pathway is inactivated in the majority of sporadic colorectal cancers. Gastroenterology : 1332–1341, 2008. [DOI] [PubMed] [Google Scholar]
- 34.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet : 81–84, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY, Pedroza M, Zhou Y, Davies J, Philip K, Molina J, Luo F, George AT, Garcia-Morales LJ, Bunge RR, Bruckner BA, Loebe M, Seethamraju H, Agarwal SK, Blackburn MR. Blockade of IL-6 trans-signaling attenuates pulmonary fibrosis. J Immunol : 3755–3768, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee CG, Kang HR, Homer RJ, Chupp G, Elias JA. Transgenic modeling of transforming growth factor-beta(1): role of apoptosis in fibrosis and alveolar remodeling. Proc Am Thorac Soc : 418–423, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesina M, Wormann SM, Neuhofer P, Song L, Algul H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin Immunol : 80–87, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest : 746–752, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol : 483–492, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Dunmore BJ, Morrell NW. Bone morphogenetic protein type II receptor mutations causing protein misfolding in heritable pulmonary arterial hypertension. Proc Am Thorac Soc : 395–398, 2010. [DOI] [PubMed] [Google Scholar]
- 41.Liu F, Ventura F, Doody J, Massague J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol Cell Biol : 3479–3486, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet : 92–102, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maher TM. Idiopathic pulmonary fibrosis: pathobiology of novel approaches to treatment. Clin Chest Med : 69–83, 2012. [DOI] [PubMed] [Google Scholar]
- 44.Michael DR, Salter RC, Ramji DP. TGF-beta inhibits the uptake of modified low density lipoprotein by human macrophages through a Smad-dependent pathway: a dominant role for Smad-2. Biochim Biophys Acta : 1608–1616, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mora AL, Torres-Gonzalez E, Rojas M, Corredor C, Ritzenthaler J, Xu J, Roman J, Brigham K, Stecenko A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol : 466–473, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morrell NW. Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc : 680–686, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science : 1808–1810, 1993. [DOI] [PubMed] [Google Scholar]
- 48.Murata S, Maruyama T, Nowatari T, Takahashi K, Ohkohchi N. Signal transduction of platelet-induced liver regeneration and decrease of liver fibrosis. Int J Mol Sci : 5412–5425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol : 723–737, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myllarniemi M, Lindholm P, Ryynanen MJ, Kliment CR, Salmenkivi K, Keski-Oja J, Kinnula VL, Oury TD, Koli K. Gremlin-mediated decrease in bone morphogenetic protein signaling promotes pulmonary fibrosis. Am J Respir Crit Care Med : 321–329, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nathan SD, King CS. Treatment of pulmonary hypertension in idiopathic pulmonary fibrosis: shortfall in efficacy or trial design? Drug Des Devel Ther : 875–885, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nathan SD, Shlobin OA, Ahmad S, Urbanek S, Barnett SD. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest : 657–663, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA 3rd, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med : 319–324, 2001. [DOI] [PubMed] [Google Scholar]
- 54.Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest : 998–1006, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Pickup MW, Hover LD, Polikowsky ER, Chytil A, Gorska AE, Novitskiy SV, Moses HL, Owens P. BMPR2 loss in fibroblasts promotes mammary carcinoma metastasis via increased inflammation. Mol Oncol : 179–191, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pitsiou G, Papakosta D, Bouros D. Pulmonary hypertension in idiopathic pulmonary fibrosis: a review. Respiration : 294–304, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Poor HD, Girgis R, Studer SM. World Health Organization Group III pulmonary hypertension. Prog Cardiovasc Dis : 119–127, 2012. [DOI] [PubMed] [Google Scholar]
- 58.Prele CM, Yao E, O′Donoghue RJ, Mutsaers SE, Knight DA. STAT3: a central mediator of pulmonary fibrosis? Proc Am Thorac Soc : 177–182, 2012. [DOI] [PubMed] [Google Scholar]
- 59.Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol : L229–L252, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raamsteeboers AJ, Bogaard HJ, Vonk Noordegraaf A. Pulmonary arterial hypertension preceding idiopathic pulmonary fibrosis in a BMPR2 mutation positive patient. Eur Respir Rev : 147–149, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med : 788–824, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW. Tumor necrosis factor-alpha accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol : 825–837, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, Heldin CH, Miyazono K. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci USA : 7632–7636, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet : 1517–1525, 2002. [DOI] [PubMed] [Google Scholar]
- 65.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta : 878–888, 2011. [DOI] [PubMed] [Google Scholar]
- 66.Sheppard D. Epithelial-mesenchymal interactions in fibrosis and repair. Transforming growth factor-beta activation by epithelial cells and fibroblasts. Ann Am Thorac Soc , Suppl 1: S21–S23, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J : 715–721, 2007. [DOI] [PubMed] [Google Scholar]
- 68.Staloch D, Gao X, Liu K, Xu M, Feng X, Aronson JF, Falzon M, Greeley GH, Rastellini C, Chao C, Hellmich MR, Cao Y, Ko TC. Gremlin is a key pro-fibrogenic factor in chronic pancreatitis. J Mol Med : 1085–1093, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res : 236–244, 228p following 244, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol : L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 71.Strieter RM. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proc Am Thorac Soc : 305–310, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell : 573–581, 1989. [DOI] [PubMed] [Google Scholar]
- 73.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res : 209–217, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Upton PD, Morrell NW. The transforming growth factor-beta-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp Physiol : 1262–1266, 2013. [DOI] [PubMed] [Google Scholar]
- 75.Ventetuolo CE, Klinger JR. WHO Group 1 pulmonary arterial hypertension: current and investigative therapies. Prog Cardiovasc Dis : 89–103, 2012. [DOI] [PubMed] [Google Scholar]
- 76.Willis BC, Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol : L525–L534, 2007. [DOI] [PubMed] [Google Scholar]
- 77.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol : 157–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu H, Han C, Lu D, Wu T. miR-17-92 cluster promotes cholangiocarcinoma growth: evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am J Pathol : 2828–2839, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]