

Abstract
Male rats offered 30% sucrose solution in addition to chow develop leptin resistance without an increase in energy intake or body fat. This study tested whether the leptin resistance was dependent on the physical form of the sucrose. Sprague-Dawley rats were offered a sucrose-free (NS) diet, a 66.6% of energy as sucrose (HS) diet, or the NS diet + 30% sucrose solution (LS). Sucrose intake of LS rats equaled that of HS rats, but total carbohydrate intake exceeded that of HS rats. After 33 days, male and female LS rats were resistant to the inhibitory effect of peripherally administered leptin on food intake. LS rats drank small, frequent meals of sucrose during light and dark periods, whereas HS rats consumed more meals during the dark than the light period and remained responsive to leptin. Diet did not affect daily energy intake or insulin sensitivity. There was a small increase in body fat in the female rats. Leptin sensitivity was restored within 5 days of withdrawal from sucrose in male LS rats. This rapid reversal suggested that leptin resistance was associated with the metabolic impact of drinking sucrose. An experiment was carried out to test whether activity of the hexosamine biosynthetic pathway and glycation of leptin signaling proteins were increased in LS rats, but the results were equivocal. A final experiment determined that female LS rats were leptin-resistant within 18 days of access to sucrose solution and that the small, but significant, increase in body fat was associated with increased adipocyte glucose utilization and insulin responsiveness, which may have been secondary to adipocyte leptin resistance.
Keywords: body composition, food intake, meal pattern, sucrose solution
INTRODUCTION
It has been reported that male rats offered free access to a 30% solution of sucrose in addition to chow and water consume >60% of their calories as sucrose and develop leptin resistance within 4 wk, even though there is no increase in body fat mass (13). Signal transducer and activator of transcription 3 (STAT3) is essential for effective control of energy balance by leptin (3). Phosphorylation of STAT3 at Tyr705 (STAT3Y) is frequently used as a marker of leptin receptor activation (37) and is required for dimerization and translocation of the transcription factor to the nucleus. STAT3 is also phosphorylated at Ser727 (STAT3S). Little attention has been paid to the role of phosphorylated STAT3S (pSTAT3S) in leptin signaling, but it has been hypothesized that pSTAT3S is essential for the association of dimerized STAT3 to transcription sites (34). Western blot analysis of tissue blocks containing the hypothalamus or brain stem from sucrose-drinking rats indicated an increase in basal levels of pSTAT3Y but normal basal levels of pSTAT3S (13). Leptin failed to further stimulate pSTAT3S or pSTAT3Y, and immunohistochemical analysis suggested that the increase in hypothalamic pSTAT3Y was limited to the arcuate nucleus of the hypothalamus (Arc) (13).
In vitro studies with HepG2 cells incubated in medium containing high concentrations of glucose confirmed that exposure to elevated glucose levels increased basal pSTAT3Y, but not pSTAT3S, and prevented leptin stimulation of pSTAT3Y or pSTAT3S (50), suggesting that leptin resistance in sucrose-drinking rats is secondary to increased glucose utilization. The hexosamine biosynthetic pathway (HBP) has been referred to as a nutrient-sensing pathway (36), because the amount of glucose that enters the pathway is increased in the setting of high-glucose or excess-calorie intake. The product of the pathway, uridine diphosphate N-acetylglucosamine, is a substrate for β-linked N-acetylglucosamine (O-GlcNAc) modification of threonine and serine residues of hundreds of proteins (16). Although O-GlcNAcylation is critical for a multitude of regulatory cell processes, chronic activation has been associated with development of cardiovascular disease (28), cancer (25), and insulin resistance (42). Others have reported that STAT3 is subject to O-GlcNAcylation, with modification confirmed on Thr717 and other potential modification sites, including Thr714, Thr716, Thr721, and Ser727 (24). Therefore, it is possible that O-GlcNAcylation of STAT3 disrupts phosphorylation and that this is responsible for the leptin resistance in sucrose-drinking rats (13).
In previous in vivo experiments, sucrose solution was offered to male rats with access to rat chow (12, 13). Resistance to leptin was apparent as early as 17 days after initiation of the diet (12), which contrasts with the extended period of time required for high-fat diet-fed rats to become leptin-resistant (6, 47). Therefore, the present studies were carried out to test for early leptin resistance to demonstrate a specific effect of diet, independent of the development of obesity. The primary objective of the studies described here was to test whether the development of leptin resistance was selective for consumption of sucrose as a liquid, or whether consumption of a similar amount of sucrose from a dry, formulated diet would also induce leptin resistance. This was tested in male and female rats in experiment 1, which also allowed us to determine whether there was a sex difference in the response to sucrose. In addition, if leptin resistance was secondary to a metabolic response to sucrose consumption, then it would be expected that leptin resistance would be rapidly reversed if sucrose was withdrawn, and this was tested in experiment 2. The results of experiment 1 suggest that, unlike male rats, female rats gained more body fat when offered sucrose in solution than when sucrose was consumed as part of a dry diet; therefore, experiment 3 measured basal and insulin-stimulated levels of glucose and fatty acid utilization in white adipose tissue of female rats that had been consuming a sucrose-free control diet, a high-sucrose formulated diet, or a sucrose-free diet + 30% sucrose solution.
METHODS
Sprague-Dawley rats (Envigo RMS) were housed individually in hanging wire mesh cages to allow measurement of daily food intake corrected for spillage. The room was maintained at 22°C (72°F) and ~50% humidity, with lights on for 12 h/day from 6:00 AM. Each rat was given a small Nylabone for enrichment. All animal procedures were approved by the Institutional Animal Care and Use Committee at Augusta University, and animals were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (32). Rats had free access to water and food, unless specified otherwise. They were offered a sucrose-free (NS) diet (product no. D11724, Research Diets, New Brunswick, NJ), which contained 67.7% of kcal from carbohydrate (15.4% maltodextrin and 51.2% cornstarch), 11.5% of kcal from fat, and 20.8% of kcal from protein, or a high-sucrose (HS) diet (product no. D11725, Research Diets), which contained 67.7% of kcal from carbohydrate (66.6% kcal sucrose), 11.5% of kcal from fat, and 20.8% of kcal from protein. Both diets had an energy content of 3,902 kcal/kg. LS rats were offered a 30% (wt/vol) solution of sucrose in addition to the NS diet and water.
Activity of the HBP was measured by Western blotting in brain and/or peripheral tissue in all experiments. O-GlcNAc is a measure of total O-GlcNAcylation of tissue protein, glutamine:fructose-6-phosphate amidotransferase (GFAT) controls the flux of glucose into the HBP, O-GlcNAc transferase (OGT) catalyzes the addition of a single N-acetylglucosamine molecule to serine or threonine residues in cellular proteins, and 3-O-(N-acetyl-d-glucosaminyl)-l-serine/threonine N-acetylglucosaminyl hydrolase (OGA) catalyzes the removal of N-acetylglucosamine molecules from proteins.
Experiment 1: development of leptin resistance in male and female rats.
Twenty-one male rats (275–300 g body wt) and 21 female rats (175–200 g body wt) were each divided into 3 weight-matched groups of 7 animals. One group of males and one group of females were offered the NS diet, one group each of male and female rats was offered the HS diet, and the final male and female groups were offered the LS diet. Body weights and food and sucrose intakes were measured daily between 7:30 and 8:30 AM.
An insulin tolerance test was performed on the female rats on day 29 of the experiment and on the male rats on day 30. Food and sucrose were removed from the cages at 7:30 AM on the day of the test. Starting at 1:00 PM, a small blood sample was collected from the tail of each rat for measurement of blood glucose; then the rat received an intraperitoneal injection of insulin (0.5 U/kg) in a volume of 0.1 ml/100 g body wt. After measurement of blood glucose at 5, 10, 13, 20, 30, 40, and 50 min after injection, food was returned to the cage. Glucose was measured using a glucometer (EasyGluco Plus, US Diagnostics, New York, NY). Leptin responsiveness was measured on days 33 and 37, because sucrose intake of the LS rats progressively increased during the first part of the experiment and more slowly in males than females. Day 33 was 1 wk after stabilization of the intake of the male LS rats but 12 days after stabilization of the intake of the female LS rats. Food and sucrose were removed from the cages at 7:30 AM. Starting at 5:00 PM, each rat received an intraperitoneal injection of 2 mg/kg leptin (rat recombinant leptin, R & D Systems, Minneapolis, MN) or PBS in a volume of 0.2 ml/100 g body wt. Food and sucrose were returned to the cages at 6:00 PM, and intake during the next 62 h was analyzed to test for leptin response. On day 33, half of the rats in each treatment group were injected with leptin and the others with PBS. On day 37, the rats that had been injected with leptin on day 33 were injected with PBS and the rats that had been injected with PBS were injected with leptin, so that each animal served as its own control.
Starting on day 45, subgroups of rats were housed in the calorimeter (TSE LabMaster, Metabolic Research Platform, TSE Systems International, Chesterfield, MO) for 3 days, and oxygen consumption, carbon dioxide production, and total activity were measured every 39 min and food, water, and sucrose intake were measured every minute on the last of the 3 days. These data were used to determine meal size and frequency and duration of consumption of the dry food and the sucrose solution during 12 h of the light period and 12 h of the dark period. Consumption of ≥0.01 g/min was considered initiation of a meal; an intermeal interval was a ≥3-min period during which no food was consumed. Only 12 calorimetry cages were available, and three different cohorts were tested (6 rats total from each treatment group), with each treatment group represented in each cohort.
On days 53 and 54, food was removed from the cages of half of the male and half of the female rats at 7:30 AM. Starting at 10:00 AM, the rats were killed by decapitation, and trunk blood was collected for measurement of serum leptin (mouse/rat leptin Quantikine ELISA kit, R & D Systems), insulin (rat insulin RIA kit, Millipore, Burlington, MA), glucose, free fatty acids (FFA; NEFA C kit, Wako Chemicals, Richmond, VA), and triglycerides (TG; L-type TG H kit, Wako Chemicals). The brain was rapidly removed, and tissue blocks containing the hypothalamus were dissected, snap-frozen, and subsequently used to measure pSTAT3Y, pSTAT3S, O-GlcNAc, GFAT, OGA, and OGT by Western blotting, as described previously (13, 50). The dissection was carried out as described previously (10, 19). The optic chiasm was used as an anterior marker and the posterior border of the mammillary bodies as a posterior marker for the hypothalamic section. The block was cut laterally to include the lateral hypothalamus, and a dorsal cut was made through the mammillothalamic tract. This tissue block included the lateral, dorsomedial, ventromedial, arcuate, and paraventricular nuclei of the hypothalamus and the median eminence. The anterior cut for the hindbrain included the medial vestibular nucleus, and the posterior cut included the spinal vestibular nucleus. A ventral cut was made to include the area postrema and medial nucleus of the solitary tract. pSTAT3 was normalized against total STAT3, and the other proteins were normalized against actin. Primary antibodies for pSTAT3Y, pSTAT3S, STAT3, and GFAT were obtained from Cell Signaling Technology (Danvers, MA), antibody for actin from Millipore, OGA from Proteintech (Rosemont, IL), and O-GlcNAc and OGT from Abcam (Cambridge, MA). Secondary anti-rabbit and anti-mouse antibodies were obtained from Jackson Laboratories (Bar Harbor, ME). Inguinal (Ing), gonadal, retroperitoneal (RP), and mesenteric fat and the liver were dissected and weighed and then returned to the carcass. One lobe of each liver was snap-frozen for Western blot analysis of leptin signaling and HBP proteins. The carcass less the gastrointestinal tract was frozen and subsequently analyzed for composition, as described previously (11).
Experiment 2: reversal of leptin resistance in sucrose-drinking rats.
Twenty-four male rats were offered ad libitum access to the NS diet. After 4 days, they were divided into two weight-matched groups: one group of 8 continued to have access to only the dry diet (NS rats), and the second group of 16 was offered 30% sucrose solution in addition to the diet (LS rats). Food intake corrected for spillage, sucrose intake, and body weight were measured daily. Leptin responsiveness of the animals was tested twice, with each rat serving as its own control (see above). The test days were days 24 and 30 for the first test and days 37 and 41 for the second test. On day 45, sucrose solution was withdrawn from half of the LS rats (LS-Rev), and all the rats were tested for leptin responsiveness again on days 47 and 50. On day 52, food and sucrose were removed from the cages at 7:00 AM, and starting at 10:00 AM the rats were euthanized by decapitation. Fat depots and the liver were dissected and weighed. One lobe of the liver was snap-frozen for determination of liver glycogen and lipid content and for Western blot analysis of HBP and leptin signaling proteins. The brain was rapidly removed, and tissue blocks containing the hypothalamus or brain stem were snap-frozen for Western blot analysis. The remaining tissue was returned to the carcass, which was analyzed for determination of body composition. Trunk blood was collected for measurement of serum IL-6 and TNF-α by ELISA (Sigma Aldrich, St. Louis, MO).
Experiment 3: glucose and fatty acid utilization of white adipose tissue in sucrose-drinking rats.
Twenty-four female rats were individually housed and offered rat chow for 3 days and then divided into three weight-matched groups: NS, HS, and LS. Food intake, sucrose consumption, and body weight were recorded daily, and the rats were tested for leptin responsiveness on days 11, 15, and 18, with each rat receiving alternate injections of PBS or leptin. Data from days 15 and 18 indicated that the LS rats were leptin-resistant. On days 20 and 21, half of the rats from each treatment group were food- deprived for 3 h beginning at 7:00 AM. The rats were decapitated, and blood was collected for measurement of serum glucose, insulin, leptin, TG, FFA, glycerol (total glycerol kit, Sigma), IL-6, and TNF-α. White fat depots and the liver were dissected and weighed, and RP fat and one lobe of the liver were snap-frozen for Western blot analysis of pSTAT3Y and pSTAT3S, O-GlcNAc, GFAT, OGT, and OGA. One Ing pad was used for in vitro measurements of glucose and fatty acid utilization (see below), and the remaining tissues were returned to the carcass for analysis.
One Ing pad from each rat was minced with scissors and then digested in Krebs-Ringer bicarbonate buffer (10 mM HEPES, 5.0 mM glucose, 0.5 mM palmitic acid, 2% bovine serum albumin, and 2 mg/ml collagenase, pH 7.45). Each cell digest was incubated at 37°C and subjected to 150 oscillations/min for exactly 30 min. The cells were washed three times with collagenase-free medium, resuspended in ~20 ml of medium, and left to rest at 37°C for 20 min. For measurement of glucose and fatty acid utilization, adipocytes were incubated in collagenase-free Krebs-Ringer bicarbonate buffer containing 0.04 μCi/μmol [U-14C]glucose or 1.0 μCi/μmol [1-14C]palmitic acid. Glucose oxidation, de novo fatty acid synthesis, fatty acid oxidation, and fatty acid esterification were measured as described previously (14). Glucose utilization was measured in basal conditions and in the presence of 100 μU/ml insulin. Fatty acid utilization was measured only in basal conditions. A 500-μl aliquot of each cell suspension was fixed in osmium tetroxide for measurement of cell number by Coulter Counter and Channelizer (Multisizer 3, Beckman Coulter, Brea, CA). Metabolic data are expressed per 106 cells.
Statistical analysis.
Data for single time point measures were compared by one-way analysis of variance (ANOVA) and post hoc Tukey’s test. Results from leptin tests were analyzed by repeated-measures ANOVA, with diet and leptin as dependent variables, number of rats as a covariant, and time as the repeated variable. Post hoc two-way ANOVA and a subsequent Tukey’s test with number of rats as the covariate were run at each time point. Daily measures of intake or body weight were initially analyzed by repeated-measures ANOVA, with day as the repeated measure; then data from individual days were tested by one-way ANOVA and post hoc Tukey’s test. All comparisons were made using Statistica 9.0 (StatSoft, Cary, NC) and were considered significant at P < 0.05.
RESULTS
Experiment 1.
Sucrose intake of male LS rats gradually increased over the first 26 days of the experiment and then stabilized at the level of sucrose intake of male HS rats, which were consuming a diet containing 66.6% of kcal as sucrose (Fig. 1A; diet effect: P < 0.001, day effect: P < 0.001, interaction: P < 0.001). Although sucrose intake increased over time, consumption of the NS diet by the LS rats gradually declined, so that energy intake remained stable and was not different on a daily basis from that of NS and HS rats (Fig. 1B; diet effect: P < 0.001, day effect: P < 0.001, interaction: not significant). Because the NS diet contained cornstarch and maltodextrin, the percentage of calories consumed as carbohydrate was higher (~87%) in LS than NS or HS rats. Comparison over the 52 days of the experiment showed that total energy intake differed between the three dietary groups: it was lowest in HS rats and highest in LS rats (Fig. 1D). Comparison over the last 26 days of the experiment showed that total energy intake was higher for LS rats than for the two other groups (Fig. 1E). There were no differences in body weight of the male rats during the experiment (Fig. 1C; diet effect: not significant, day effect: P < 0.0001, interaction: not significant), and carcass analysis revealed no differences in body composition at the end of the experiment, although mesenteric fat was increased in LS rats (Table 1). Therefore, despite small, but significant, differences in energy intake of the dietary groups, these differences were not great enough to cause a measurable change in growth rate or body composition. Similarly, there was no effect of diet on glucose clearance during the insulin tolerance test (data not shown) or on serum hormone and metabolite levels measured at the end of the experiment (Table 1).
Fig. 1.

A–C: daily sucrose intake, total energy intake, and body weight of male and female rats in experiment 1. ITT, insulin tolerance test. Values are means ± SE for groups of 7 rats. *Significant difference between HS and LS rats. D and E: total energy intake during the 52 days of the experiment and during the last 26 days of the experiment, when sucrose intake of LS rats had stabilized. Values for energy intake for male or female rats that do not share a common superscript are significantly different (P < 0.05), as determined by a repeated-measures ANOVA followed by post hoc Tukey’s test on each day.
Table 1.
Body composition and serum hormone levels of male and female rats in experiment 1
| Males | Females | |||||
|---|---|---|---|---|---|---|
| NS | HS | LS | NS | HS | LS | |
| Carcass composition, g | ||||||
| Carcass | 398 ± 11 | 382 ± 11 | 386 ± 6 | 240 ± 5 | 228 ± 5 | 241 ± 4 |
| Fat | 37 ± 4 | 34 ± 2 | 39 ± 4 | 27 ± 3A,B | 24 ± 2A | 33 ± 2B |
| Lean | 345 ± 8 | 333 ± 10 | 331 ± 4 | 201 ± 4 | 193 ± 1 | 196 ± 4 |
| Ash | 16.0 ± 0.9 | 15.7 ± 0.5 | 15.5 ± 0.6 | 12.0 ± 0.8 | 12 ± 0.1 | 12.6 ± 0.4 |
| Fat depot wt, g | ||||||
| Inguinal | 8.2 ± 0.6 | 8.0 ± 0.4 | 9.4 ± 0.5 | 4.1 ± 0.4 | 3.3 ± 0.3 | 4.3 ± 0.3 |
| Gonadal | 5.8 ± 0.4 | 5.2 ± 0.2 | 6.0 ± 0.5 | 5.8 ± 0.8A,B | 3.9 ± 0.7A | 7.7 ± 0.7B |
| Retroperitoneal | 3.8 ± 0.5 | 3.3 ± 0.2 | 4.3 ± 0.4 | 1.8 ± 0.3A,B | 1.2 ± 0.2A | 2.0 ± 0.2B |
| Mesenteric | 3.2 ± 0.3a,b | 2.5 ± 0.1a | 4.0 ± 0.4b | 2.3 ± 0.2A,B | 1.4 ± 0.25A | 2.6 ± 0.2B |
| Liver | 13.6 ± 0.4 | 14.2 ± 0.9 | 15.2 ± 0.5 | 7.4 ± 0.3A | 7.9 ± 0.2A,B | 8.7 ± 0.3B |
| Serum hormones and metabolites | ||||||
| Leptin, ng/ml | 1.4 ± 0.2 | 1.2 ± 0.3 | 1.5 ± 0.1 | 0.8 ± 0.3 | 0.7 ± 0.3 | 1.3 ± 0.3 |
| Insulin, ng/ml | 25 ± 6 | 19 ± 3 | 33 ± 7 | 8 ± 3 | 5 ± 2 | 6 ± 3 |
| Glucose, mg/dl | 148 ± 0.6 | 163 ± 0.7 | 160 ± 7 | 129 ± 7 | 112 ± 6 | 128 ± 5 |
| FFA, mEq/l | 0.60 ± 0.03 | 0.60 ± 0.04 | 0.62 ± 0.02 | 0.66 ± 0.02 | 0.64 ± 0.03 | 0.70 ± 0.05 |
| TG, mg/dl | 108 ± 20 | 86 ± 9 | 126 ± 17 | 96 ± 10 | 132 ± 30 | 128 ± 33 |
Values are means ± SE for groups of 7 rats. Male and female Sprague-Dawley rats were offered diets of different sucrose content [sucrose-free (NS) diet, 66.6% of energy as sucrose (HS) diet, and NS diet + 30% sucrose solution (LS) diet] for 53 or 54 days. FFA, free fatty acid; TG, triglyceride. Values within male or female groups that do not share a common superscript are significantly different (P < 0.05), as determined by 1-way ANOVA and post hoc Tukey’s test.
The effect of leptin on energy intake was diet-dependent (diet effect: P < 0.001, leptin effect: P < 0.05, time effect: P < 0.001, diet × time interaction: P < 0.001, diet × leptin × time interaction: P < 0.07), with a similar pattern of response at 14 and 24 h after injection and at 38 and 62 h after injection; therefore, only results from 24 and 38 h are shown in Fig. 2. Leptin caused a significant inhibition of food intake in NS and HS rats, but at different times after injection. There was no effect of leptin on intake of LS rats at any of the times tested.
Fig. 2.

Cumulative energy intake of male and female rats at 24 and 38 h in experiment 1 after injection of leptin (2 mg/kg ip) or an equivalent volume of PBS. Values are means ± SE for groups of 7 rats. *Significantly different (P < 0.05) from PBS, as determined by 2-way ANOVA followed by post hoc Tukey’s test, with number of rats as covariate.
Similar to the male rats, consumption of sucrose solution by the female rats increased with time. By day 21, sucrose intake of LS rats was equal to that of HS rats and stabilized at this level for the rest of the experiment (diet effect: P < 0.004, day effect: P < 0.001, interaction: P < 0.001). Consumption of dry food by female LS rats decreased as sucrose intake increased, and there were no significant differences in energy intake between the dietary groups when they were compared on a daily basis (diet effect: P < 0.001, day effect: P < 0.001, interaction: not significant), although total carbohydrate intake of LS rats was higher than that of NS and HS rats. When total intakes of the groups during the 52-day experimental period were compared, energy intake of LS rats was significantly higher than that of NS and HS rats (Fig. 1D), but during the last 26 days of the experiment, when sucrose intake of LS and NS rats was the same, intake of HS rats was significantly lower than that of NS and LS rats (Fig. 1E). Female HS rats gained less weight during the experiment (diet effect: not significant, day effect: P < 0.001, interaction: P < 0.001), but post hoc analysis did not identify any day on which body weight was significantly different between groups. At the end of the experiment, carcass weight of HS rats was lower than that of LS rats, and this was associated with a significant reduction in carcass fat content and weights of RP, mesenteric, and gonadal fat depots (Table 1). By contrast, liver weight was significantly greater in LS than NS, but not HS, rats (Table 1).
Similar to male rats, the effect of leptin on energy intake of female rats was diet-dependent (diet effect: P < 0.0005, leptin effect: P < 0.07, time effect: P < 0.001, diet × time interaction: P < 0.0001). Leptin caused a significant inhibition of food intake in NS and HS rats, but at different times after injection. The effect was significant at 14 h for NS rats and at 24 h for HS rats (Fig. 2). This pattern of response was the same as that for male rats, but it ended earlier in female rats. There was no effect of leptin on energy intake of LS rats at any of the times tested.
There were no effects of diet on energy expenditure or respiratory exchange ratio during the light or dark period for male or female rats (data not shown). Meal patterns of male and female rats were similar but differed between the light and dark phase and by dietary group (Fig. 3). Rats offered the NS and HS diets ate fewer meals during the light than the dark period. LS rats ate very little of the dry diet during the light period but consumed as many meals of sucrose solution during the light period as during the dark period (phase effect: P < 0.0001, diet effect: P < 0.0001, interaction: P < 0.001). There was no difference in the duration of meals during the light or dark period for any of the rats, although caloric intake during a meal was lower for consumption of sucrose solution than for dry diet. This was significant only for female rats (phase effect: not significant, diet effect: P < 0.0001, interaction: not significant). Intermeal interval was significantly increased for consumption of dry diet by male and female LS rats during the light period (phase effect: P < 0.0001, diet effect: P < 0.0001, interaction: P < 0.0002). Total energy intake was higher during the dark than the light period for all treatment groups, but there was no effect of diet on energy intake during the light or dark period (phase effect: P < 0.0001, diet effect: not significant, interaction: not significant).
Fig. 3.

Meal patterns of male and female rats in experiment 1 during the last 24 h in the calorimeter. Initiation of a meal was defined as consumption of ≥0.01 g, and an intermeal interval was a ≥3-min period during which no food was consumed. NS, sucrose-free; HS, 66.6% of energy as sucrose; LS, NS diet + 30% sucrose solution. Values are means ± SE for groups of 6 rats. Values for a specific parameter that do not share a common superscript are significantly different (P < 0.05), as determined by 2-way ANOVA followed by post hoc Tukey’s test.
Western blot analysis of the hypothalamus for male and female rats showed a nonsignificant 13% increase in pSTAT3Y in LS compared with NS and HS rats and a similar nonsignificant increase in hypothalamic O-GlcNAc, with no changes in pSTAT3S, GFAT, OGT or OGA (data not shown). There were no effects of diet on expression of any of the proteins in liver tissue (data not shown).
Experiment 2.
Similar to experiment 1, sucrose intake of all rats that had access to sucrose solution (LS and LS-Rev) increased over time and then plateaued after 20 days (Fig. 4A). Despite the increase in sucrose intake, energy intake of the rats did not change due to a simultaneous reduction in consumption of dry diet (Fig. 4B). Leptin tests indicated that rats with access to sucrose solution were leptin-resistant on days 37 and 41. Leptin responsiveness in NS rats was represented by a significant reduction in food intake during the 36 h after leptin treatment (Fig. 5, days 37 and 41).
Fig. 4.
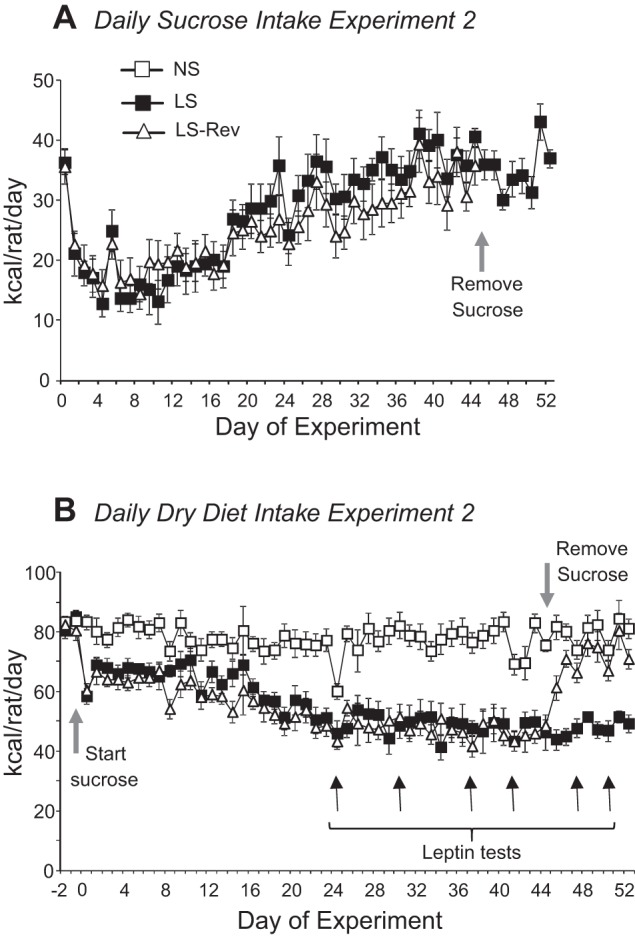
Daily sucrose (A) and dry diet (B) intake of male LS rats and half of the LS rats from which sucrose solution was withdrawn (LS-Rev) in experiment 2. Access to sucrose was removed from LS-Rev rats on day 45. Leptin responsiveness was tested on days 24, 30, 37, 41, 47, and 50. Values are means ± SE for groups of 8 rats.
Fig. 5.

A: cumulative energy intake after injection of leptin (2 mg/kg ip) or an equivalent volume of PBS in male rats in experiment 2. Data are from leptin responsiveness tests while LS-Rev rats still had access to sucrose solution (days 37 and 41) or after sucrose had been removed from the LS-Rev rats (days 47 and 50). Values within each time period that do not share a common superscript are significantly different (P < 0.05). B and C: Western blots showing expression of STAT3 phosphorylated at Tyr705 (pSTAT3Y) in hypothalamic and brain stem tissue. Values that do not share a common superscript are significantly different (P < 0.05). Values are means ± SE for groups of 8 rats. Images from representative Western blots have been adjusted for brightness and contrast.
When sucrose solution was withdrawn from LS-Rev rats, their energy intake decreased for 1 day but then returned to the same level as in NS and LS rats (data not shown). Total energy intake and weight gain (Table 2) during the 8 days after removal of sucrose were significantly lower in LS-Rev than NS and LS rats, as they did not compensate for the initial hypophagia. LS-Rev rats also were leptin-responsive when tested 2 and 5 days after removal of sucrose (Fig. 5, days 47 and 50). Because LS-Rev rats were consuming less than NS controls, food intake following a PBS injection was the same as that of the leptin-injected NS rats, and leptin inhibited this intake further in LS-Rev rats (Fig. 5A). At the end of the experiment, there were no differences in liver weight, carcass weight, fat mass, or lean body mass between the groups (Table 2). Liver weight was increased in LS compared with NS and LS-Rev rats, and this weight difference was associated with an increase in liver glycogen, but not lipid (Table 3). All the fat pads tended to be larger in LS rats, and this reached significance for Ing, RP, and mesenteric depots (Table 2).
Table 2.
Weight gain and body composition of male rats in experiment 2
| NS | LS | LS-Rev | |
|---|---|---|---|
| Body wt, g | |||
| Start | 270 ± 2 | 272 ± 2 | 270 ± 2 |
| Day 45 | 424 ± 6 | 439 ± 5 | 423 ± 9 |
| End | 444 ± 5 | 457 ± 6 | 434 ± 9 |
| Change day 45–53 | 18 ± 2A | 20 ± 1A | 11 ± 1B |
| Carcass composition | |||
| Weight, g | 402 ± 5 | 411 ± 6 | 399 ± 10 |
| Fat, g | 29 ± 1 | 32 ± 4 | 28 ± 2 |
| Lean, g | 358 ± 5 | 355 ± 10 | 365 ± 7 |
| Liver, g | 13.9 ± 0.5A | 16.1 ± 0.2B | 13.4 ± 0.7A |
| Liver glycogen, mg | 549 ± 64A | 893 ± 56B | 526 ± 52A |
| Liver lipid, mg | 578 ± 24 | 636 ± 57 | 576 ± 53 |
| Fat depot wt, g | |||
| Inguinal | 7.4 ± 0.2A | 9.1 ± 0.6B | 7.3 ± 0.3A |
| Epididymal | 5.5 ± 0.2 | 6.5 ± 0.5 | 5.6 ± 0.5 |
| Retroperitoneal | 3.4 ± 0.3A | 4.2 ± 0.4B | 2.9 ± 0.2A |
| Mesenteric | 2.3 ± 0.2A | 2.9 ± 0.2B | 2.3 ± 0.2A |
Values are means ± SE for groups of 8 rats. LS-Rev, half of the LS rats from which sucrose solution was withdrawn on day 45.Values for a specific parameter that do not share a common superscript are significantly different (P < 0.05), as determined by 1-way ANOVA and post hoc Tukey’s test.
Table 3.
Weight gain and body composition of female rats in experiment 3
| NS | HS | LS | |
|---|---|---|---|
| Start wt, g | 191 ± 2 | 191 ± 3 | 188 ± 2 |
| End wt, g | 227 ± 4 | 223 ± 2 | 228 ± 2 |
| Total energy intake, kcal·rat−1·23 days−1 | 1,286 ± 24A | 1,222 ± 21A | 1,498 ± 35B |
| Carcass composition, g | |||
| Carcass wt | 200 ± 3 | 192 ± 2 | 197 ± 1 |
| Fat | 16 ± 1A | 17 ± 1A | 21 ± 2B |
| Lean | 147 ± 2A | 140 ± 2B | 141 ± 1A,B |
| Liver | 6.8 ± 0.4 | 7.2 ± 0.9 | 7.8 ± 0.2 |
| Fat depot wt, g | |||
| Inguinal | 2.6 ± 0.1A | 2.7 ± 0.1A,B | 3.2 ± 0.2B |
| Retroperitoneal | 0.9 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.1 |
| Mesenteric | 1.2 ± 0.116A | 1.3 ± 0.1A | 1.8 ± 0.2B |
| Serum hormones and metabolites | |||
| Leptin, ng/ml | 0.64 ± 0.08A | 0.77 ± 0.11A | 1.17 ± 0.13B |
| Insulin, ng/ml | 25 ± 4 | 19 ± 2 | 15 ± 5 |
| Glucose, mg/dl | 113 ± 4 | 117 ± 7 | 115 ± 8 |
| FFA, mEq | 0.63 ± 0.03 | 0.61 ± 0.04 | 0.73 ± 0.05 |
| TG, mg/dl | 83 ± 5 | 118 ± 36 | 69 ± 7 |
| Glycerol, nmol/ml | 12.4 ± 0.7 | 12.8 ± 1.5 | 14.3 ± 0.8 |
| TNF-α, pg/ml | 92 ± 34 | 67 ± 43 | 94 ± 56 |
| IL-6, pg/ml | 16 ± 3 | 15 ± 3 | 14 ± 2 |
Values are means ± SE for groups of 8 rats. Values for a specific parameter that do not share a common superscript are significantly different (P < 0.05), as determined by 1-way ANOVA and post hoc Tukey’s test.
There was no effect of diet or removal of access to sucrose in LS rats on liver expression of pSTAT3Y or pSTAT3S or proteins associated with the HBP (data not shown). In the hypothalamus, pSTAT3Y expression was significantly higher in LS than NS, but not LS-Rev, rats, in which pSTAT3 expression was intermediate between NS and LS rats. In the brain stem, pSTAT3Y was nearly twofold higher in LS than NS or LS-Rev rats (Fig. 5). There were no differences in any of the other proteins measured in either tissue (data not shown). Circulating levels of IL-6 and TNF-α were very low and at, or below, the detectable limit of the assay for all groups of rats (data not shown).
Experiment 3.
Diet composition had no effect on weight gain of female rats in experiment 3 (Table 3). Sucrose consumption of LS rats was the same as that of HS rats by day 7 (Fig. 6; diet effect: not significant, day effect: P < 0.001, interaction: P < 0.001). Fluctuations in daily sucrose intake appeared to follow the estrous cycles of the LS rats, but the measurements necessary to confirm this were not performed. These cyclic changes in sucrose consumption were reflected in total energy intake and were more dramatic than those exhibited by HS or NS rats, so that energy intake of LS rats was significantly greater than that of the other groups on some, but not all, days of the experimental period (Fig. 6; diet effect: P < 0.0001, day effect: P < 0.0001, interaction: P < 0.03).
Fig. 6.
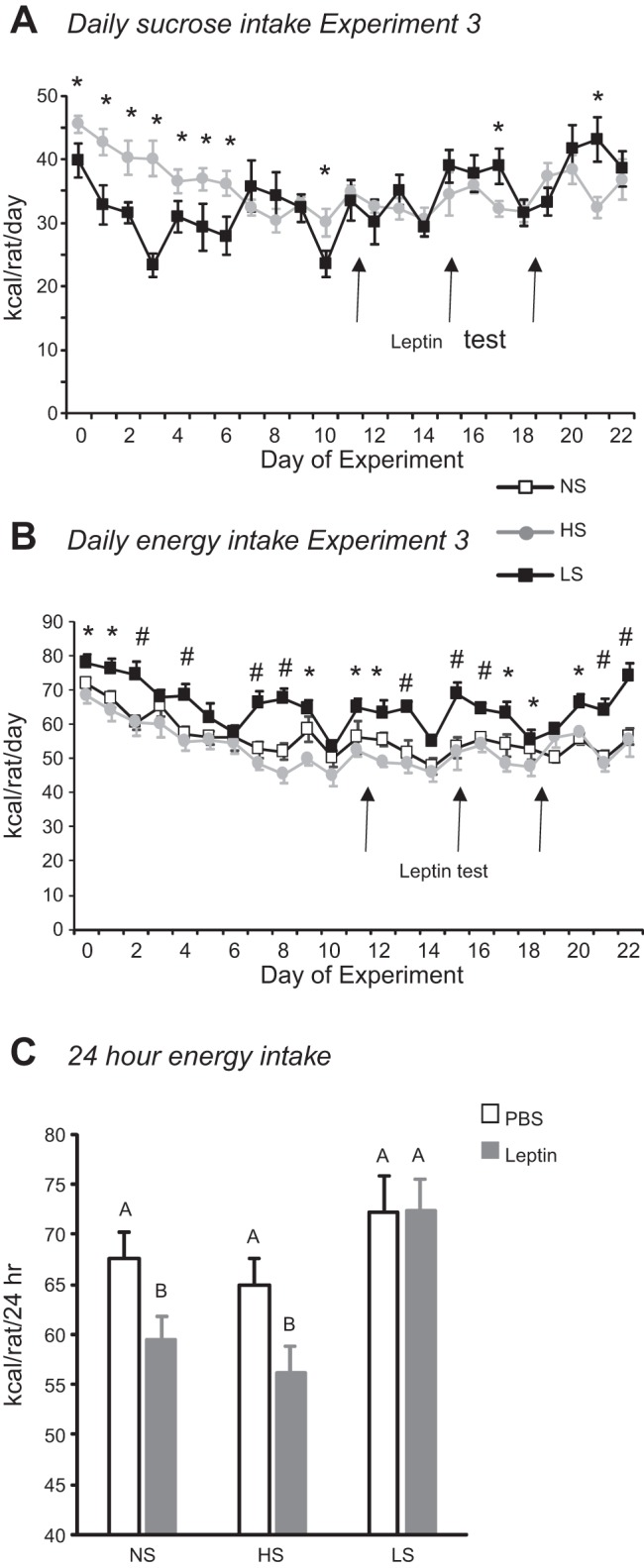
Daily sucrose (A) and energy (B) intake of female rats in experiment 3. Values are means ± SE for groups of 8 rats. *Significant difference between LS and HS rats. #Significant difference between LS and both NS and HS rats. C: cumulative energy intake 24 h after injection of leptin (2 mg/kg ip) or PBS. Values that do not share a common superscript are significantly different (P < 0.05), as determined by 2-way ANOVA followed by post hoc Tukey’s test, with number of rats as a covariate.
Leptin injections on days 15 and 18 significantly inhibited energy intake of NS and HS rats at 24 h, but the effect was reversed by 38 h (Fig. 6C; diet effect: P < 0.001, leptin effect: P < 0.02, time effect: P < 0.0001, diet × time interaction: P < 0.08). At the end of the experiment, LS rats had significantly more body fat than NS or HS rats, whereas NS rats had significantly more lean body mass than HS rats. The increase in body fat was reflected by an increase in the size of Ing and mesenteric fat depots. Serum leptin was also increased in LS rats compared with the two other groups, but there were no differences in serum glucose, insulin, FFA, glycerol, or TG (Table 3). Circulating levels of TNF-α and IL-6 were very low in all groups and at, or below, the reliable threshold for detection.
Incubation of adipocytes isolated from Ing fat showed no effect of diet on basal fatty acid oxidation or esterification, but ~100 times more fatty acid was esterified than oxidized. Insulin significantly stimulated glucose oxidation in adipocytes isolated from rats fed all the diets (diet effect: P < 0.04, insulin effect: P < 0.0001, interaction: not significant). Basal and insulin-stimulated glucose oxidation was significantly higher in adipocytes from LS than NS rats. There was no effect of diet or insulin on glucose incorporation into fatty acids. The amount of glucose incorporated into fatty acids was approximately equivalent to the amount of glucose oxidized.
Western blot analysis of liver tissue did not show an effect of diet on leptin signaling or HBP protein expression (data not shown). In RP fat, there was a significant increase in pSTAT3Y expression but no effect of diet on pSTAT3S (Fig. 7, E and F). O-GlcNAc protein was also increased in LS compared with NS rats (Fig. 7G), but there was no effect of diet on expression of GFAT, OGT, or OGA (data not shown).
Fig. 7.

A–D: glucose oxidation, glucose incorporation into esterified fatty acids, fatty acid oxidation, and fatty acid esterification in inguinal adipocytes isolated from female rats at the end of experiment 3. E–G: pSTAT3Y expression, pSTAT3S expression, and β-linked N-acetylglucosamine (O-GlcNAc) protein for retroperitoneal (RP) fat from female rats at the end of experiment 3. Values for a specific parameter that do not share a common superscript are significantly different (P < 0.05), as determined by 1-way ANOVA and post hoc Tukey’s test. Values are means ± SE for 8 rats. Images from representative Western blots have been adjusted for brightness and contrast.
DISCUSSION
The experiments described here determined whether the leptin resistance that develops rapidly in male rats or mice offered liquid sucrose in addition to chow (13, 50) is specific to consumption of sucrose in solution. The results demonstrate that male and female rats become leptin-resistant when offered sucrose in solution, but not when an equivalent amount of sucrose is consumed from a composite, dry diet (HS rats). Although female LS rats gained more fat than their controls, there was no effect of diet on body composition of male rats, indicating that leptin resistance was independent of obesity. This contrasts with rats that become leptin-resistant, but also obese, when fed a high-fat diet (6, 47). In addition, leptin resistance was independent of a change in whole body insulin sensitivity, and there was no effect on basal levels of glucose or insulin measured at the end of the experiment. This is consistent with studies in which rats were offered sucrose solution in addition to rat chow. There were no changes in basal blood glucose, but there was an increase in insulin released in response to a glucose challenge (2, 12, 20), which was not measured here. By contrast, it has been reported that mice fed a dry 50% sucrose diet for 55 wk had normal basal glucose and glucose clearance but improved insulin sensitivity (44).
Experiment 1 identified a difference in the pattern of sucrose intake between LS and HS rats that may contribute to the difference in leptin responsiveness. Both male and female LS rats consumed sucrose solution as frequent small meals across 24 h. The average intermeal interval for sucrose solution was ~40 min during the day and at night, whereas that for the HS diet was ~100 min during the day and 50 min at night. The frequent consumption of sucrose by the LS rats would result in repeated excursions in blood glucose. Although the meals were relatively small (0.6−0.8 kcal), the sucrose was consumed independent of other nutrients and was likely rapidly digested and absorbed compared with sucrose consumed in combination with other dietary components by HS rats. Harris and Apolzan found that blood glucose remains above baseline for 2 h following a glucose gavage of 1 g/kg (1.2 kcal for a 300-g rat) (12); thus the repeated consumption of sucrose solution by LS rats may have resulted in chronic stimulation of pathways that metabolize glucose, including the HBP. In the present study, blood glucose concentrations were not measured across the day, and additional studies are needed to determine how the pattern of sucrose intake influences blood glucose, glucose metabolism, and leptin responsiveness in LS and HS rats. The role of activation of the HBP in inducing leptin resistance has been studied (13, 50), and whether the repeated consumption of sucrose results in an increase in the activity of this pathway, which could ultimately modify leptin signaling, has been hypothesized.
The control HS rats were offered a diet that contained 66.6% of calories as sucrose, which is 50% glucose and 50% fructose. The NS controls consumed a diet that contained 66.6% of calories as the glucose polymers cornstarch and maltodextrin. The liver metabolizes 98% of fructose consumed, and almost half of this enters gluconeogenesis (35, 45). Thus glucose delivery to extrahepatic tissue in LS and HS rats is increased by glucose derived directly from sucrose and by increased hepatic production of glucose from fructose. Although the amount of glucose reaching the brain is tightly controlled, it has been shown that peripheral hyperglycemia increases glucose in ventricular cerebrospinal fluid, including central extracellular fluid (40, 41). Thus it is possible that consumption of large quantities of sucrose results in increased glucose flux in all tissues, including the brain. It is unlikely that fructose intake alone accounted for leptin resistance in LS rats, because HS rats consumed the same amount of fructose. The LS rats also consumed some NS diet and would have consumed a further 20% of their calories as cornstarch and maltodextrin. Therefore, they not only consumed sucrose throughout the day, but their total carbohydrate intake was greater than that of HS or NS rats, which could have influenced blood glucose levels and metabolism.
Others have reported no effect of sex on rat preference for saccharide solutions in a 24-h two-bottle test with water (9). The effect appears to be specific to carbohydrate in solution, as rats show a strong preference for sucrose solution adulterated with a bitter flavor to unadulterated sucrose powder (39). In the present experiments, the daily intake of sucrose increased with time in males and females, and although there was a sex difference and a between-experiment difference in the time required for sucrose intake to plateau, intake stabilized at 66% of caloric intake in males and females. Studies with mice have shown that intraduodenal, but not hepatic portal, infusions of glucose produce a conditioned taste preference, supporting a preabsorptive role for intestinal sweet taste receptors driving consumption of carbohydrate solutions (1). In contrast, it has been suggested that consumption of sweet solutions is determined by metabolic effects of the carbohydrate. Stimulation of sweet taste receptors in the small intestine increases glucose uptake (27) and influences the release of incretins to promote glucose clearance (43). In addition, activation of sweet taste receptors on beta cells by fructose exaggerates glucose-stimulated insulin secretion (22). These observations support the notion that consumption of sucrose as a liquid, rather than as part of a solid diet, would change blood glucose in addition to glucose and fructose metabolism.
As noted above, female LS rats increased intake of sucrose solution to a stable level faster than males. This sex effect varied between experiments, but it is not obvious why. The greatest difference between experiments 1 and 3 was that the male and female rats were housed in the same room during experiment 1, but not experiment 3. However, it is not clear why this would influence the time course of response in the females. Leptin resistance also appeared earlier in females, but this may have been secondary to the earlier increase in sucrose consumption of female than male LS rats. Although the estrous cycle was not monitored in these experiments, it has the potential to influence leptin responsiveness, because estradiol suppresses expression of hypothalamic long-form leptin receptors but increases expression of short-form receptors (4). Although it appeared that the estrous cycle influenced sucrose intake in experiment 3, it is unlikely that it influenced leptin responsiveness, because each test was carried out with 2 injection days, and not all rats would have been injected at the same stage of the cycle. In experiment 1, female HS rats had a lower energy intake and weighed less than the other groups. This resulted in less body fat in the female HS than LS rats at the end of the study. It is unclear why this would have happened, as LS and HS rats consumed high levels of fructose, and NS rats consumed a similar percentage of energy as carbohydrate. There were no differences in basal levels of blood glucose, insulin, or lipids at the end of the study, but the response to a glucose challenge was not tested.
A second difference between the sexes was that female LS rats were fatter than their controls, whereas body composition of the males was not changed by diet. This may be explained by the greater requirement to support growth in males than females. Examination of adipocyte glucose and fatty acid metabolism showed increased glucose utilization in cells from female rats consuming a high proportion of daily calories as sucrose, but the response was greater in LS than HS rats. This may have been due to the higher total carbohydrate intake of LS rats or because the form and pattern of consumption played additive roles in driving carbohydrate utilization. Although in vivo and in vitro studies show little effect of leptin on basal glucose utilization (18, 30, 49), physiological concentrations of leptin inhibit insulin-stimulated glucose metabolism (31). Therefore, the increased insulin responsiveness in adipocytes from LS rats could have been secondary to development of leptin resistance in adipose tissue. If this was the case, then adipocytes from male LS rats would also be expected to have an exaggerated insulin response, as these rats were leptin-resistant, even though their body fat remained at control levels.
In cell culture experiments, overnight exposure of HepG2 cells to 20 mM glucose or 1 mM glucosamine increased activity of the HBP, as indicated by increased O-GlcNAcylation of cell protein. Basal levels of pSTAT3Y, but not pSTAT3S, increased, and leptin did not increase phosphorylation of STAT3Y or STAT3S (50). STAT3 is expressed in many different cell types and can either promote or inhibit specific cellular functions (46). Tyr705 (STAT3Y) and Ser727 (STAT3S) are the primary phosphorylation sites on STAT3, and pSTAT3S has been reported to enhance transcriptional activity following STAT3Y phosphorylation (33) and also to inhibit activity of pSTAT3 (5). Little is known about the importance of pSTAT3S in effective leptin signaling. O’Rourke and Shepherd (34) reported that leptin stimulates phosphorylation of both STAT3Y and STAT3S, that phosphorylation of STAT3S is dependent on MAPK, and that inhibition of pSTAT3S inhibits STAT3 DNA binding. Zimmerman and Harris confirmed that leptin stimulates phosphorylation of STAT3Y and STAT3S (50), but further investigation is needed to determine its importance as a mediator of the different biological activities of leptin. Because protein O-GlcNAcylation targets serine and threonine residues, if HBP activity interferes with leptin signaling, then STAT3S is a potential site of modification. In cell culture, an inhibitor of GFAT and glucose entry into the HBP reverses these changes in STAT3 activation and leptin responsiveness, supporting the notion that activation of the HBP and protein O-GlcNAcylation are involved in the development of leptin resistance in cells exposed to high levels of glucose (50). We previously found that male mice offered 30% sucrose solution in addition to chow were fatter than their controls, were leptin-resistant and insulin-insensitive, and had increased HBP activity and increased basal levels of pSTAT3Y in the liver and hypothalamus. In a separate experiment, O-GlcNAc and pSTAT3Y in hypothalamic tissue were increased in mice infused with glucosamine for 2 h (50). Glucosamine enters the HBP independent of control by GFAT; therefore, these observations also support the idea that activation of the HBP increased pSTAT3Y.
Studies with rats provided less conclusive results. Harris and Apolzan did not find significant increases in markers of HBP activity in hypothalamic, hindbrain, liver, or adipose tissue of leptin-resistant rats offered 30% sucrose solution, but hypothalamic and hindbrain pSTAT3Y were increased (13). Western blots from the experiments described here provided similar results. There was no significant increase in any marker of HBP activity or pSTAT3Y activation in the liver from LS rats. By contrast, O-GlcNAc was increased in RP fat, and this was associated with increased levels of pSTAT3Y. The tissue-specific differences may be determined by the ability to metabolize glucose. In the liver, glucose transport is rarely saturated, glucose is stored as glycogen, and metabolism is optimized for handling large variations in glucose supply. In adipose tissue, glucose transport is insulin-dependent, and TG represent the major form of energy storage, although adipocytes do contain small stores of glycogen (23). Therefore, a chronic increase in glucose availability would change substrate flux through different pathways in different tissues. In experiment 2, pSTAT3Y was significantly increased in hypothalamic and brain stem tissue of LS rats, but no measurable increase in O-GlcNAc was found. The increase in pSTAT3Y was partially reversed 8 days after sucrose solution had been removed, with activation levels not different from either control NS or leptin-resistant LS rats, but the rats had recovered their sensitivity to leptin, supporting the notion that leptin resistance had a metabolic basis driven by consumption of sucrose. However, the maintenance of leptin responsiveness in HS rats indicates that the resistance is specific to consumption of sucrose solution, and whether this is mediated by the HBP remains unclear.
Leptin resistance in LS rats correlated with increased basal levels of pSTAT3Y and failure of leptin to stimulate phosphorylation further (50). By contrast, leptin resistance caused by feeding a high-fat diet or aging is associated with failure to fully activate STAT3 (21, 38). However, Scarpace et al. (38) found elevated basal levels of hypothalamic pSTAT3Y in rats made leptin-resistant by prolonged central overexpression of leptin. Therefore, there is precedence for leptin resistance in the presence of increased basal pSTAT3Y, but whether the resistance in LS rats is due to failure to increase pSTAT3Y or failure of leptin to phosphorylate pSTAT3S remains to be determined. Harris and Apolzan reported that the increase in pSTAT3Y in the hypothalamus is confined to the Arc (13), consistent with the leptin resistance represented as failure of leptin to inhibit food intake. It is well established that activation of leptin receptors in the Arc inhibits expression of the orexigenic neuropeptides neuropeptide Y and agouti-related hormone and increases expression of the anorexigenic neuropeptides cocaine and amphetamine-regulated transcript and the α-melanocyte-stimulating hormone precursor pro-opiomelanocortin (29). We did not confirm that leptin resistance in LS rats was associated with failure of leptin to change expression of these neuropeptides. The site-specific change in pSTAT3Y in LS rats may explain why only small changes in pSTAT3Y were detected by Western blot analysis of tissue blocks that included the whole hypothalamus.
A high-fructose diet can increase tissue oxidative stress (7), which promotes inflammation and increases risk for insulin resistance (8) and cardiovascular disease (26). Because it has also been shown that leptin can attenuate the response to an inflammatory challenge (48), whether leptin resistance in LS rats was associated with a change in inflammatory status was tested, but no significant levels of proinflammatory cytokines were found in serum from LS or HS rats. Although this does not exclude the possibility of local tissue inflammation, there was no evidence of an overall inflammatory state. Feeding a high-fat, high-sucrose diet has also been established as a model for inducing nonalcoholic fatty liver disease (17). The accumulation of liver lipid has been attributed to fructose entry into glycolysis as trioses that can be used for gluconeogenesis or for lipogenesis and TG synthesis. Normally, a majority of fructose is converted to glucose and glycogen, and this appears to be the case in our experiments, where liver glycogen content was increased but liver lipid was not changed. If the rats had been overeating and gaining weight, it is possible that liver lipid would have been increased once glycogen stores were replete.
Perspectives and Significance
In summary, these experiments indicate that chronic access to sucrose solution results in development of leptin resistance in both male and female rats independent of any substantial increase in energy intake or body fat mass. The sucrose intake of LS and HS rats was the same, but total carbohydrate intake was increased in LS rats. Therefore, the selective effect of consumption of sucrose in solution may be due to total carbohydrate intake or the pattern of sucrose consumption by LS rats that drank small volumes of sucrose solution throughout the day and night. Future experiments will determine whether this results in a sustained elevation of blood glucose when blood glucose would normally be at basal levels. We did not explore whether leptin resistance developed in response to sweet taste or as a result of postabsorptive carbohydrate metabolism, and further studies are needed to identify which attribute of sucrose solution is critical to the response. Previous studies suggested a link between activation of the HBP and development of leptin resistance. Analysis of tissues from experiments described here did not confirm or refute a direct relation between sucrose consumption, O-GlcNAc protein modification, and STAT3 phosphorylation.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-109997 and DK-053903 and support from the Diabetes Action Research and Education Foundation awarded to R. B. S. Harris.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.B.H. conceived and designed research; R.B.H. performed experiments; R.B.H. analyzed data; R.B.H. interpreted results of experiments; R.B.H. prepared figures; R.B.H. drafted manuscript; R.B.H. edited and revised manuscript; R.B.H. approved final version of manuscript.
ACKNOWLEDGMENTS
The author thanks Chanel Young, Lakeisha Grant, and Bianca Marsh for assistance with the Western blots.
REFERENCES
- 1.Ackroff K, Yiin YM, Sclafani A. Post-oral infusion sites that support glucose-conditioned flavor preferences in rats. Physiol Behav 99: 402–411, 2010. doi: 10.1016/j.physbeh.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apolzan JW, Harris RB. Differential effects of chow and purified diet on the consumption of sucrose solution and lard and the development of obesity. Physiol Behav 105: 325–331, 2012. doi: 10.1016/j.physbeh.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG Jr. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421: 856–859, 2003. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 4.Bennett PA, Lindell K, Karlsson C, Robinson IC, Carlsson LM, Carlsson B. Differential expression and regulation of leptin receptor isoforms in the rat brain: effects of fasting and oestrogen. Neuroendocrinology 67: 29–36, 1998. doi: 10.1159/000054295. [DOI] [PubMed] [Google Scholar]
- 5.Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 17: 6508–6516, 1997. doi: 10.1128/MCB.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab 301: E187–E195, 2011. doi: 10.1152/ajpendo.00056.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delbosc S, Paizanis E, Magous R, Araiz C, Dimo T, Cristol JP, Cros G, Azay J. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 179: 43–49, 2005. doi: 10.1016/j.atherosclerosis.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Faure P, Rossini E, Lafond JL, Richard MJ, Favier A, Halimi S. Vitamin E improves the free radical defense system potential and insulin sensitivity of rats fed high fructose diets. J Nutr 127: 103–107, 1997. doi: 10.1093/jn/127.1.103. [DOI] [PubMed] [Google Scholar]
- 9.Feigin MB, Sclafani A, Sunday SR. Species differences in polysaccharide and sugar taste preferences. Neurosci Biobehav Rev 11: 231–240, 1987. doi: 10.1016/S0149-7634(87)80031-3. [DOI] [PubMed] [Google Scholar]
- 10.Haring SJ, Harris RB. The relation between dietary fructose, dietary fat and leptin responsiveness in rats. Physiol Behav 104: 914–922, 2011. doi: 10.1016/j.physbeh.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris RB. Growth measurements in Sprague-Dawley rats fed diets of very low fat concentration. J Nutr 121: 1075–1080, 1991. doi: 10.1093/jn/121.7.1075. [DOI] [PubMed] [Google Scholar]
- 12.Harris RB, Apolzan JW. Changes in glucose tolerance and leptin responsiveness of rats offered a choice of lard, sucrose, and chow. Am J Physiol Regul Integr Comp Physiol 302: R1327–R1339, 2012. doi: 10.1152/ajpregu.00477.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris RB, Apolzan JW. Hexosamine biosynthetic pathway activity in leptin resistant sucrose-drinking rats. Physiol Behav 138: 208–218, 2015. doi: 10.1016/j.physbeh.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris RB, Martin RJ. Metabolic response to a specific lipid-depleting factor in parabiotic rats. Am J Physiol Regul Integr Comp Physiol 250: R276–R286, 1986. doi: 10.1152/ajpregu.1986.250.2.R276. [DOI] [PubMed] [Google Scholar]
- 16.Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446: 1017–1022, 2007. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 17.Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, MacLean PS, Diggle CP, Asipu A, Inaba S, Kosugi T, Sato W, Maruyama S, Sánchez-Lozada LG, Sautin YY, Hill JO, Bonthron DT, Johnson RJ. High-fat and high-sucrose (Western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 58: 1632–1643, 2013. doi: 10.1002/hep.26594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 389: 374–377, 1997. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 19.Kasser TR, Harris RB, Martin RJ. Level of satiety: fatty acid and glucose metabolism in three brain sites associated with feeding. Am J Physiol Regul Integr Comp Physiol 248: R447–R452, 1985. doi: 10.1152/ajpregu.1985.248.4.R447. [DOI] [PubMed] [Google Scholar]
- 20.Kendig MD, Ekayanti W, Stewart H, Boakes RA, Rooney K. Metabolic effects of access to sucrose drink in female rats and transmission of some effects to their offspring. PLoS One 10: e0131107, 2015. doi: 10.1371/journal.pone.0131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. PLoS One 5: e11376, 2010. doi: 10.1371/journal.pone.0011376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kyriazis GA, Soundarapandian MM, Tyrberg B. Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc Natl Acad Sci USA 109: E524–E532, 2012. doi: 10.1073/pnas.1115183109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence JC Jr, Larner J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J Biol Chem 253: 2104–2113, 1978. [PubMed] [Google Scholar]
- 24.Li X, Zhang Z, Li L, Gong W, Lazenby AJ, Swanson BJ, Herring LE, Asara JM, Singer JD, Wen H. Myeloid-derived cullin 3 promotes STAT3 phosphorylation by inhibiting OGT expression and protects against intestinal inflammation. J Exp Med 214: 1093–1109, 2017. doi: 10.1084/jem.20161105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Z, Vosseller K. O-GlcNAc in cancer biology. Amino Acids 45: 719–733, 2013. doi: 10.1007/s00726-013-1543-8. [DOI] [PubMed] [Google Scholar]
- 26.Maarman GJ, Mendham AE, Lamont K, George C. Review of a causal role of fructose-containing sugars in myocardial susceptibility to ischemia/reperfusion injury. Nutr Res 42: 11–19, 2017. doi: 10.1016/j.nutres.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Mace OJ, Affleck J, Patel N, Kellett GL. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J Physiol 582: 379–392, 2007. doi: 10.1113/jphysiol.2007.130906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsh SA, Chatham JC. The paradoxical world of protein O-GlcNAcylation: a novel effector of cardiovascular (dys)function. Cardiovasc Res 89: 487–488, 2011. doi: 10.1093/cvr/cvq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meister B. Control of food intake via leptin receptors in the hypothalamus. Vitam Horm 59: 265–304, 2000. doi: 10.1016/S0083-6729(00)59010-4. [DOI] [PubMed] [Google Scholar]
- 30.Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 48: 287–291, 1999. doi: 10.2337/diabetes.48.2.287. [DOI] [PubMed] [Google Scholar]
- 31.Müller G, Ertl J, Gerl M, Preibisch G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. J Biol Chem 272: 10585–10593, 1997. doi: 10.1074/jbc.272.16.10585. [DOI] [PubMed] [Google Scholar]
- 32.National Research Council Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies, 1996. [Google Scholar]
- 33.Ng J, Cantrell D. STAT3 is a serine kinase target in T lymphocytes. Interleukin 2 and T cell antigen receptor signals converge upon serine 727. J Biol Chem 272: 24542–24549, 1997. doi: 10.1074/jbc.272.39.24542. [DOI] [PubMed] [Google Scholar]
- 34.O’Rourke L, Shepherd PR. Biphasic regulation of extracellular-signal-regulated protein kinase by leptin in macrophages: role in regulating STAT3 Ser727 phosphorylation and DNA binding. Biochem J 364: 875–879, 2002. doi: 10.1042/bj20020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renold AE, Hastings AB, Nesbett FB. Studies on carbohydrate metabolism in rat liver slices. III. Utilization of glucose and fructose by liver from normal and diabetic animals. J Biol Chem 209: 687–696, 1954. [PubMed] [Google Scholar]
- 36.Rossetti L. Perspective: hexosamines and nutrient sensing. Endocrinology 141: 1922–1925, 2000. doi: 10.1210/endo.141.6.7566. [DOI] [PubMed] [Google Scholar]
- 37.Scarpace PJ, Matheny M, Tümer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience 104: 1111–1117, 2001. doi: 10.1016/S0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 38.Scarpace PJ, Matheny M, Tümer N, Cheng KY, Zhang Y. Leptin resistance exacerbates diet-induced obesity and is associated with diminished maximal leptin signalling capacity in rats. Diabetologia 48: 1075–1083, 2005. doi: 10.1007/s00125-005-1763-x. [DOI] [PubMed] [Google Scholar]
- 39.Sclafani A, Vigorito M. Effects of SOA and saccharin adulteration on Polycose preference in rats. Neurosci Biobehav Rev 11: 163–168, 1987. doi: 10.1016/S0149-7634(87)80021-0. [DOI] [PubMed] [Google Scholar]
- 40.Shram NF, Netchiporouk LI, Martelet C, Jaffrezic-Renault N, Cespuglio R. Brain glucose: voltammetric determination in normal and hyperglycaemic rats using a glucose microsensor. Neuroreport 8: 1109–1112, 1997. doi: 10.1097/00001756-199703240-00009. [DOI] [PubMed] [Google Scholar]
- 41.Silver IA, Erecińska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14: 5068–5076, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srinivasan V, Tatu U, Mohan V, Balasubramanyam M. Molecular convergence of hexosamine biosynthetic pathway and ER stress leading to insulin resistance in L6 skeletal muscle cells. Mol Cell Biochem 328: 217–224, 2009. doi: 10.1007/s11010-009-0092-7. [DOI] [PubMed] [Google Scholar]
- 43.Steinert RE, Gerspach AC, Gutmann H, Asarian L, Drewe J, Beglinger C. The functional involvement of gut-expressed sweet taste receptors in glucose-stimulated secretion of glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). Clin Nutr 30: 524–532, 2011. doi: 10.1016/j.clnu.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Sumiyoshi M, Sakanaka M, Kimura Y. Chronic intake of high-fat and high-sucrose diets differentially affects glucose intolerance in mice. J Nutr 136: 582–587, 2006. doi: 10.1093/jn/136.3.582. [DOI] [PubMed] [Google Scholar]
- 45.Sun SZ, Empie MW. Fructose metabolism in humans—what isotopic tracer studies tell us. Nutr Metab (Lond) 9: 89, 2012. doi: 10.1186/1743-7075-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel TP, Milner JD, Cooper MA. The ying and yang of STAT3 in human disease. J Clin Immunol 35: 615–623, 2015. doi: 10.1007/s10875-015-0187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilsey J, Zolotukhin S, Prima V, Scarpace PJ. Central leptin gene therapy fails to overcome leptin resistance associated with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 285: R1011–R1020, 2003. doi: 10.1152/ajpregu.00193.2003. [DOI] [PubMed] [Google Scholar]
- 48.Xiao E, Xia-Zhang L, Vulliémoz NR, Ferin M, Wardlaw SL. Leptin modulates inflammatory cytokine and neuroendocrine responses to endotoxin in the primate. Endocrinology 144: 4350–4353, 2003. doi: 10.1210/en.2003-0532. [DOI] [PubMed] [Google Scholar]
- 49.Zierath JR, Frevert EU, Ryder JW, Berggren PO, Kahn BB. Evidence against a direct effect of leptin on glucose transport in skeletal muscle and adipocytes. Diabetes 47: 1–4, 1998. doi: 10.2337/diab.47.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Zimmerman AD, Harris RB. In vivo and in vitro evidence that chronic activation of the hexosamine biosynthetic pathway interferes with leptin-dependent STAT3 phosphorylation. Am J Physiol Regul Integr Comp Physiol 308: R543–R555, 2015. doi: 10.1152/ajpregu.00347.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]