

Abstract
Young adult male obese Zucker rats (OZR) develop insulin resistance and hypertension with impaired baroreflex-mediated bradycardia and activation of nucleus tractus solitarius (NTS). Because type 1 diabetic rats also develop impaired baroreflex-mediated NTS activation, we hypothesized that improving glycemic control in OZR would enhance compromised baroreflexes and NTS activation. Fasting blood glucose measured by telemetry was comparable in OZR and lean Zucker rats (LZR) at 12–17 wk. However, with access to food, OZR were chronically hyperglycemic throughout this age range. By 15–17 wk of age, tail samples yielded higher glucose values than those measured by telemetry in OZR but not LZR, consistent with reports of exaggerated stress responses in OZR. Injection of glucose (1g/kg ip) produced larger rises in glucose and areas under the curve in OZR than LZR. Treatment with metformin (300 mg·kg−1·day−1) or pioglitazone (5 mg·kg−1·day−1) in drinking water for 2–3 wk normalized fed glucose levels in OZR with no effect in LZR. After metformin treatment, area under the curve for blood glucose after glucose injection was reduced in OZR and comparable to LZR. Hyperinsulinemia was slightly reduced by each treatment in OZR, but insulin was still greatly elevated compared with LZR. Neither treatment reduced hypertension in OZR, but both treatments significantly improved the blunted phenylephrine-induced bradycardia and NTS c-Fos expression in OZR with no effect in LZR. These data suggest that restoring glycemic control in OZR enhances baroreflex control of heart rate by improving the response of the NTS to raising arterial pressure, even in the presence of hyperinsulinemia and hypertension.
Keywords: baroreceptor reflex, hyperglycemia, metabolic syndrome, metformin, pioglitazone
INTRODUCTION
Excess weight gain is associated with the development of a cluster of attributes known as metabolic syndrome that foster premature cardiovascular morbidity and mortality (33, 52, 54). People with obesity are prone to hypertension with an autonomic imbalance favoring reduced cardiac vagal tone and elevated sympathetic nerve activity (SNA) to multiple cardiovascular-related targets (77, 83). In addition, obesity is linked with the development of compromised short-term control of arterial pressure (AP) by baroreflexes (67, 79, 83), which promotes reduced variability of heart rate (HR) and increased variability of AP. Diminished baroreflexes can occur independently from hypertension (28), and increased AP variability confers a significant independent risk for coronary heart disease, renal disease, stroke, and cerebrovascular-related cognitive decline (25, 71, 89). Mechanisms underlying the development of impaired baroreflexes in people with obesity are not well understood.
Obesity is also associated with the development of prediabetic attributes that impact autonomic regulation of cardiovascular function before progression into frank type 2 diabetes mellitus. Whether or not fasting hyperglycemia or hypertension are present, hyperinsulinemia and glucose intolerance coincide with impaired baroreflex-mediated control of HR (36, 45, 67, 76). Weight loss improves insulin sensitivity and baroreflexes in people with obesity (21, 27, 82), but the relationship between these deficits has not been elucidated. Insulin has the ability to raise SNA, alter baroreflexes, and increase AP variability (46, 84). However, the observation of diminished cardiac baroreflexes and HR variability in patients of normal weight with type 1 diabetes and elevated hemoglobin A1c (HbA1c) suggests that poor glucose homeostasis is a causative factor for impaired control of HR and AP (10, 18, 51).
These deleterious cardiovascular and metabolic attributes are also observed in obese rodents, allowing for more invasive study of underlying mechanisms of disease. Rats or mice made obese by a high-fat diet or by genetically driven hyperphagia of standard rodent chow, such as obese Zucker rats (OZR) and db/db mice, develop elevated SNA that drives hypertension (24, 39, 59, 81). Before the onset of hypertension, both models of rodent obesity develop impaired baroreflexes (59, 64, 75), which likely contributes to their reduced HR variability (4, 55, 65) and increased AP variability (9, 35). In obese rats, impaired baroreflexes coincide with changes in the brainstem baroreflex pathway (31, 59). Normally, an evoked rise in AP reduces HR via excitation of arterial baroreceptor afferent nerves that activate nucleus tractus solitarius (NTS) neurons in the brainstem, which, in turn, excite cholinergic neurons in nucleus ambiguus to activate cardiac parasympathetic efferent nerves and GABAergic neurons in the caudal ventrolateral medulla (CVLM) to inhibit presympathetic neurons in the rostral ventrolateral medulla (RVLM) (1). In adult male OZR and lean Zucker rats (LZR), changes in baroreceptor afferent nerve activity over a wide range of evoked changes in AP are comparable, suggesting that sensory mechanisms for detection of AP are intact (39). In contrast, direct electrical stimulation of baroreceptor afferent fibers evokes smaller decreases in SNA and mean AP in adult male OZR (39). Likewise, in rats made obese by a high-fat diet, electrical stimulation of myelinated baroreceptor afferent nerves evokes smaller decreases in HR, which is due to reduced vagal activation and sympathetic withdrawal (59). As further evidence of centrally mediated changes, acutely raising mean AP produces fewer c-Fos-positive neurons in the NTS of adult OZR (31) and less inhibition of presympathetic RVLM neuronal activity in rats made obese by a high-fat diet (37). In agreement with diminished baroreflex-mediated activation of the NTS, microinjection of glutamate into the intermediate NTS evokes smaller reductions in splanchnic SNA and mean AP in adult male OZR (31). The NTS appears to be a critical central site of impairment because glutamatergic activation of the CVLM and GABAergic inhibition of the RVLM produce equivalent decreases in SNA and AP in adult male OZR and LZR (31, 40). In contrast to adult OZR, juvenile OZR and LZR have equivalent baroreflex-mediated changes in SNA and HR that coincide with comparable physiological responses to activation of the NTS (31, 75). These data suggest that the progression of metabolic syndrome reduces baroreceptor afferent-mediated activation of the NTS to yield impaired inhibition of presympathetic RVLM neurons, SNA, and HR. Because OZR become obese with an excess intake of standard rat chow (80, 87), the development of obesity-related deficits in autonomic regulation of SNA, HR, and AP can be examined without altering diet composition.
In addition to hypertension and impaired baroreflexes, young adult male OZR have hyperinsulinemia and glucose intolerance in the presence of normal fasting glucose levels (23, 44). As reported in humans, poor glycemic control in rats is associated with a diminished short-term control of AP, even in the absence of other traits of metabolic syndrome. Streptozotocin-induced hyperglycemia in Sprague-Dawley rats produces diminished baroreceptor-mediated activation of the NTS without increasing body weight, insulin, or AP (26), and this treatment also produces impaired baroreflexes and excess AP variability that are improved by reducing blood glucose (92). Therefore, the present study examined the hypothesis that improved glycemic control after treatment with metformin or pioglitazone enhances blunted arterial baroreflex-mediated control of HR and activation of the NTS in conscious adult male OZR. In addition, we measured blood glucose continuously by telemetry to determine whether fasting glucose reflects basal glucose levels with ad libitum access to food in conscious, undisturbed rats. These experiments provide the first determination of whether improved glucose homeostasis in prediabetic male OZR enhances diminished baroreflex-mediated changes in HR and activation of the NTS.
MATERIALS AND METHODS
Animals.
Male OZR [Lepr (fa/fa)] and LZR [Lepr (+/+) and (+/fa)] were purchased from Charles River (Houston, TX) and were individually housed in centralized animal care facilities with consistent humidity (60 ± 5%), temperature (24 ± 1°C), and light cycle (lights on 7:00 AM–7:00 PM). Rats were fed a standard chow (Prolab RMH 1800, LabDiet). Experiments were performed on young adult (12–17 wk), age-matched Zucker rats. Animal experiments were conducted according to the National Institutes of Health Guide for Care and Use of Laboratory Animals and the American Physiological Society's Guiding Principles for the Care and Use of Vertebrate Animals in Research and Training. All protocols were approved by the University of North Texas Health Science Center Institutional Animal Care and Use Committee.
Implantation of glucose-sensing transmitters.
After rats were anesthetized with isoflurane, a laparotomy was performed using aseptic conditions. The tip of the transmitter catheter (HD-XG, Data Sciences International) was inserted rostrally, a laparotomy was performed to insert the tip of the transmitter catheter (HD-XG, Data Sciences International) rostrally into the abdominal aorta distal to the renal arteries. The aortic wall was sealed around the catheter with a piece of mesh and a small drop of cyanoacrylate adhesive. The transmitter was secured to the abdominal wall using a 4.0-prolene suture. After the incision was closed, rats were kept warm and monitored until fully conscious. Each cage was placed on a receiver (DSI) to continuously measure blood glucose by telemetry. Calibration of the transmitters was verified using blood samples from the tail with rats in fasted and fed states and with glucose tolerance tests (GTTs).
Glucose measures and treatment with metformin or pioglitazone.
To determine blood glucose levels from rats in fasted (18 h) and fed states, blood samples were taken from the tail between 8:00 AM and 9:00 AM with at least 3 days between the samples. To minimize disturbance to the rat during glucose measures, the tip of the tail was snipped with scissors while the rat remained in their home cage with nesting material. A drop of blood from the tail was applied to a glucose test strip that was inserted into a calibrated handheld glucometer (Accu-Chek Aviva Plus). Duplicate blood samples were taken to generate a mean value for each measurement. In fasted and fed rats, analogous baseline values were generated using telemetry by taking the average of 20-s samples every 5 min for 1 h. To determine blood glucose values over the course of one day when rats had access to food, hourly blood glucose values were generated by averaging 5-min samples spanning 60 min from each rat over a 24-h period that was at least 2 days apart from a fasting period.
The first GTT was performed 3 to 4 days after implantation of the glucose transmitter. Rats were fasted overnight for 18 h with access to water. The next morning, cages were removed from their rack and lined up on carts with their telemetry receivers, alternating lean and obese rats. A duplicate baseline blood sample was taken from the tail of each rat at ~8 AM, and then 30–60 min later, each rat was briefly lifted from their cage and injected with glucose (1 g/kg ip from a 0.5 g/ml solution). While the rat rested in its home cage, additional tail blood samples were taken at 15, 30, 45, 90, and 120 min after administration of glucose. Periodic fed glucose samples were also taken every 1–2 wk to ensure calibration of the transmitters. All tail blood sample readings were entered into the telemetry data acquisition software in duplicate for each time point as they were collected (Dataquest A.R.T. software platform, DSI).
Subsets of LZR and OZR (13−14 wk old) began treatment with metformin (300 mg·kg−1·day−1) in their drinking water (14, 22, 50) during the week after their first GTT. These rats were housed next to age-matched OZR and LZR with untreated drinking water. Fluid intake and weight were monitored every 48 h, and the concentration of metformin was adjusted to provide the correct daily dose. The metformin-treated drinking water was sweetened with an artificial sweetener (2 packets of Splenda/1 liter of water) (42), and these rats had access to HydroGel cups (clear H2O) in their cages to ensure proper hydration. Metformin was chosen for its ability to selectively normalize glucose homeostasis in the continued presence of hyperinsulinemia and hypertension (14, 50). Additionally, metformin does not impact glucose levels in LZR (22), allowing for treatment in LZR to control for other potential effects of metformin. Treatment was limited to 2–3 wk to minimize weight loss (22, 90). After 2–3 wk of drug treatment, a second GTT was performed in treated and untreated rats to ensure calibration of the transmitter and examine the efficacy of drug treatment.
To control for potential effects of weight loss with metformin, another set of rats was treated with pioglitazone (5 mg·kg−1·day−1 suspended in 0.5% methylcellulose), which tends to promote weight gain while improving glucose homeostasis. In addition, this dose produces minimal effects upon AP in OZR (19, 53). As described below, these rats were used for assessment of AP, baroreflex-mediated bradycardia, and activation of the NTS as indicated by c-Fos expression. Blood samples were taken in the fed state at ~9 AM in conscious rats to confirm effects of pioglitazone treatment upon glucose, insulin, and lipids.
Phenylephrine infusion in conscious rats to activate baroreflexes and the NTS.
After 2–3 wk of treatment, rats were anesthetized with isoflurane (5% in a ventilated box and then 2.0–2.5% through a nose cone). A catheter was implanted in the left femoral artery to record AP and HR and the left femoral vein to infuse fluids. As previously described, the free ends of the catheters were tunneled subcutaneously to exit between the scapulae (31). The rats were fitted with a button tether and dual channel swivel (Instech Laboratories) attached to a counterbalanced lever arm to allow them to move freely in a covered plexiglass cylindrical cage (MTANK/W and MTOP, Instech). The rats were allowed to recover for 24–48 h with access to food and water. On the morning of the experiment, food and water were removed, and the cages were surrounded with a cover to minimize disturbance to the rat. At ~9:00 AM a baseline blood sample (0.5 ml) was drawn through the arterial catheter, and the volume was replaced by sterile saline. Then, the arterial line was connected through the swivel to a transducer (NL108T2, Digitimer), and the venous line was connected through the swivel to an infusion pump (model A-99, Razel). After 30 min of baseline recording of AP and HR, phenylephrine was infused to raise mean AP by 40 mmHg for 90 min (13–31 µl/min of 0.5 mg/ml of phenylephrine in saline iv). The infusion rate was continuously adjusted to maintain the 40 mmHg rise in mean AP over the 90-min period. This protocol allowed for measurement of baroreflex-mediated bradycardia within the first 5 min and later activation of c-Fos expression in the brainstem. During the last 30 min of the infusion, the phenylephrine-filled syringe was replaced by a saline-filled syringe to slowly replace the phenylephrine in the line by the end of the 90-min period. Mean AP remained elevated throughout the 90-min protocol. Rats were deeply anesthetized with urethane (1.5 g/kg iv bolus) after the 90-min infusion and perfused transcardially with 250 ml of phosphate-buffered saline (pH 7.4) followed by 500 ml of 4% phosphate-buffered paraformaldehyde (Electron Microscopy Sciences). The brains were removed and stored in the same fixative for 48 h.
Measurement of plasma insulin, cholesterol, and triglycerides.
Blood drawn from the arterial catheter of conscious rats was collected into a heparinized tube and immediately centrifuged to isolate plasma. Plasma samples were stored frozen as aliquots for later analysis by ELISA. Measurements were made using a Rat Ultrasensitive Insulin ELISA kit (80-INSRTU-E01, ALPCO) for plasma insulin, a Cholesterol E kit (439-17501, Wako Diagnostics) for total plasma cholesterol, and L-Type TG M reagents for triglycerides (Color A 461-08992, Color B 461-09092 and Multi-Lipid calibrator 464-01601, Wako Diagnostics). Samples were run in duplicate to obtain an average value for each rat.
Immunohistochemistry for c-Fos.
Brainstems were sectioned with a Vibratome (30 µm, coronal plane), and sections were placed consecutively in a 24-well dish containing a cryoprotectant solution. The free-floating sections were stored at −20°C until further processing. Immunohistochemistry for c-Fos protein was performed on free-floating sections on an orbital shaker in solutions prepared in Tris-buffered saline (TBS, pH 7.4) at room temperature, unless specified otherwise. Sections from rats of different groups were run in adjacent columns within the same staining dishes to ensure comparable staining conditions. The sections were incubated with 1% hydrogen peroxide (30 min) to block endogenous peroxidases, rinsed in TBS, and blocked in 10% horse serum (45 min). Then, sections were incubated with a goat anti-c-Fos primary antibody (1:2,000, 4°C, 48 h; sc-52G, Santa Cruz Biotechnology), as previously described (31). After being rinsed in TBS, sections were incubated with a biotinylated donkey anti-goat secondary antibody (1:400, 1 h; cat. no. 705-066-147, Jackson Laboratories) followed by an avidin-biotin solution (1 h; PK-6100, Vector Laboratories). The c-Fos immunoreactivity was revealed by incubation with a nickel-intensified 3,3′diaminobenzadine solution. The reaction was closely monitored for 8–10 min and terminated with TBS rinses when staining became visible. Processed sections were mounted onto gelatin-coated slides and air-dried overnight. Slides were submerged in a series of alcohols and xylenes and then coated with DPX mounting medium (Sigma-Aldrich) to affix coverslips. The c-Fos-immunoreactive (Fos+) neurons were mapped and counted bilaterally in the NTS at four rostrocaudal levels using Neurolucida (MFB Bioscience) and blinded conditions as previously described (31).
Data acquisition and analysis.
Glucose levels were recorded by telemetry for 20 s every 5 min for 3–4 wk. For 24-h measures, hourly averages were produced from the 5-min data samples. Variability of glucose was calculated as the standard deviation of the mean using 2 h of recordings by telemetry. The AP was measured through a femoral artery catheter, and the mean AP and HR were derived from the AP pulse using a calibrated low-pass filter (N125) and a spike trigger (NL201), respectively (Neurolog System, Digitimer). Analog signals were converted to digital (Micro 1401, Cambridge Electronic Design) and viewed online using Spike2 software (Cambridge). All group data are expressed as means ± SE. Significant statistical difference was set at P < 0.05. Before treatments, baseline parameters in age-matched OZR and LZR were compared using unpaired t-tests. Values at multiple time points were compared using an ANVOA with repeated measures. After drug treatments, all measures were compared using an appropriate ANOVA, and pairwise comparisons were made using Tukey’s honestly significant difference post hoc tests. (SigmaStat, version 3.5).
RESULTS
Blood glucose values in OZR and LZR before metformin treatment.
Young adult male OZR weighed 59% more than age-matched LZR (Table 1). Morning fasted blood glucose measured by telemetry was comparable in OZR and LZR (Fig. 1A). In contrast to fasting conditions, access to food produced a rise in morning blood glucose levels in OZR but not LZR, resulting in a significantly elevated morning fed glucose level only in OZR (Fig. 1A). When rats were undisturbed in their cages with access to food, hourly averages of blood glucose over a 24-h period revealed that OZR had higher blood glucose levels than LZR at all hours of the day and night (Fig. 1B). In addition to chronically elevated blood glucose in OZR, the 24-h variability of blood glucose was higher in OZR compared with LZR (8.0 ± 0.8 vs. 4.7 ± 0.4, P < 0.05, unpaired t-test), with the blood glucose variability being highest in OZR during the night (Fig. 1C).
Table 1.
Glucose tolerance test in 12- to 14-wk-old LZR and OZR
| Group | n | Age, days | Body Weight, g | Time to Peak, min | AUC, mg/dl × 3 h |
|---|---|---|---|---|---|
| LZR | 11 | 90.5 ± 0.7 | 296.1 ± 6.4 | 15.4 ± 0.8 | 333.3 ± 40.2 |
| OZR | 12 | 91.2 ± 0.8 | 471.7 ± 10.7* | 19.2 ± 0.8* | 523.2 ± 59.5* |
Data are expressed as means± SE; n, number of rats. Age and weight were determined the morning of the glucose tolerance test (Fig. 2). Area under the curve (AUC) was measured as the difference from baseline in 5-min increments from −15 to 165 min in relation to the injection of glucose. LZR, lean Zucker rats; OZR, obese Zucker rats.
P < 0.05, unpaired t-tests.
Fig. 1.

Baseline blood glucose levels measured by telemetry in young adult (12–14 wk old), male lean Zucker rats (LZR, n = 11) and obese Zucker rats (LZR and OZR, n = 12). A: morning blood glucose levels in fasted and fed states; *P < 0.05 vs. fasted OZR and †P < 0.05 vs. fed LZR. B: hourly averages over a 24-h period in these OZR and LZR with access to food; P < 0.05, OZR vs. LZR for all hours. C: standard deviation of glucose in these LZR and OZR during peak hours of the night (9:30 PM–11:30 PM) and lowest hours of the morning (5:30 AM–7:30 AM); *P < 0.05 vs. LZR during the same time period and †P < 0.05 vs. OZR during the morning. Data were analyzed by ANOVA with repeated measures followed by Tukey’s honestly significant difference tests.
The evening before the first GTT, food was removed from the cages at ~5:00 PM. Within 8 h of fasting, blood glucose levels in OZR and LZR were equivalent to baseline measures taken the next morning, providing comparable and consistent blood glucose values for 7 h before the GTT (data not shown). Injection of glucose (1 g/kg ip) produced a higher blood glucose in OZR compared with LZR (Fig. 2) with a peak that occurred later in the OZR (20 min for 8 of 12 rats) than in LZR (15 min for 8 of 11 rats; Table 1, Fig. 2A). In addition, the return of blood glucose toward baseline levels took longer in OZR than in LZR (180 vs. 90 min, Fig. 2A), contributing to a larger area under the curve in OZR (Table 1). By 95 min, blood glucose levels were not different between OZR and LZR (Fig. 2A). Return of food to the cages 3 h after the injection of glucose produced a rise in blood glucose that peaked by 5 h in both groups (Fig. 2B). The peak blood glucose was significantly greater in OZR compared with LZR (Fig. 2B), and blood glucose levels remained higher in OZR thereafter, as seen in baseline measures with access to food (Fig. 1B).
Fig. 2.
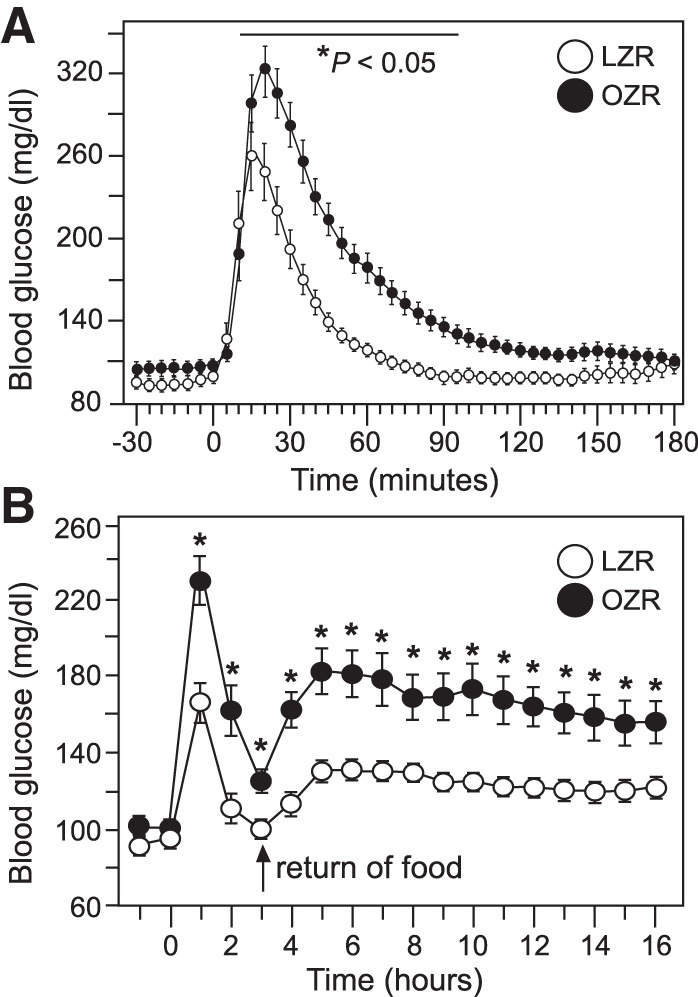
Glucose tolerance test (GTT) in obese Zucker rats (OZR) and lean Zucker rats (LZR) at 12–14 wk of age. See Fig. 1 for baseline glucose values for these rats. A: blood glucose values before and after injection of glucose (at time 0) measured by telemetry every 5 min over 210 min; *P < 0.05 vs. LZR at that time point. B: same GTT with glucose measured in hourly averages over 18 h with return of food at hour 3; *P < 0.05 vs. LZR at that time point. Pairs of groups were analyzed by ANOVA with repeated measures followed by Tukey’s honestly significant difference tests. See Table 1 for single value comparisons during the GTT.
Blood glucose values in OZR and LZR after metformin treatment.
At 15–17 wk of age, body weight was 57% higher in untreated OZR and 83% higher in metformin-treated OZR compared with like-treated, age-matched LZR (Table 2). Treatment with metformin tended to retard weight gain in both LZR and OZR, consistent with reported reductions in food intake with this dose of metformin in Zucker rats (72). In this particular set, metformin-treated LZR weighed less than untreated LZR, but no differences were observed between the OZR groups (Table 2). As seen at 12–14 wk of age, at 15−17 wk of age the fasted blood glucose measured by telemetry was comparable in untreated OZR and LZR but was higher in OZR than LZR with access to food (Fig. 3A). Treatment with metformin had no effect on fasted blood glucose levels in OZR or LZR (Fig. 3A). However, metformin treatment effectively ameliorated the elevated morning blood glucose in OZR with access to food (Fig. 3A) and dramatically reduced blood glucose levels in OZR at all hours of a 24-h period (Fig. 3B). Analysis of this 24-h period showed that metformin treatment did not alter fed blood glucose levels in LZR, and metformin-treated OZR had fed blood glucose levels equivalent to LZR (Fig. 3C). With the progression of metabolic syndrome from 12–14 wk to 15–17 wk of age, variability of blood glucose with access to food over a 24-h period was greatly increased in untreated OZR (from 8.0 ± 0.8 to 22.7 ± 2.7, P < 0.05, paired t-test), and treatment with metformin normalized blood glucose variability in OZR with no effect in LZR (Fig. 3D). Blood glucose levels were normalized in these young adult male OZR within ~1 wk with this dose and route of administration of metformin (data not shown), providing 1–2 wk of normalized blood glucose with ad libitum access to food before experiments began.
Table 2.
Glucose tolerance test in 15- to 17-wk-old LZR and OZR
| Group | n | Age, days | Body Weight, g | Time to Peak, min | AUC, mg/dl × 3 h |
|---|---|---|---|---|---|
| Untreated | |||||
| LZR | 5 | 108.2 ± 0.2 | 351.4 ± 10.5 | 21.0 ± 1.0 | 310.8 ± 80.6 |
| OZR | 5 | 108.2 ± 0.2 | 552.6 ± 20.1* | 27.0 ± 2.0* | 940.6 ± 138.5* |
| Metformin-treated | |||||
| LZR | 6 | 108.0 ± 0.0 | 273.3 ± 22.0† | 19.2 ± 0.8 | 341.6 ± 66.4 |
| OZR | 6 | 109.5 ± 1.5 | 500.0 ± 22.6* | 22.5 ± 1.1† | 547.7 ± 91.6† |
| P values | |||||
| Rat type | 0.383 | <0.001 | 0.002 | <0.001 | |
| Treatment | 0.520 | 0.005 | 0.023 | 0.074 | |
| Interaction | 0.383 | 0.536 | 0.308 | 0.040 |
Data are expressed as means ± SE; n, number of rats. Age and weight were determined the morning of the glucose tolerance test (Fig. 4). The area under the curve (AUC) was measured as the difference from baseline at 5-min increments from −15 to 165 min in relation to the injection of glucose. Each measure was analyzed by 2-way ANOVA for rat type × treatment followed by Tukey’s honestly significant difference tests. See Figs. 3 and 4 for blood glucose values in these rats. LZR, lean Zucker rat; OZR, obese Zucker rat.
P < 0.05 vs. LZR of like treatment,
P < 0.05 vs. untreated rat of same type.
Fig. 3.

Baseline blood glucose values measured by telemetry in untreated and metformin (MET)-treated lean Zucker rats (LZR) and obese Zucker rats (OZR) at 15–17 wk of age. Data are from the same rats from Figs. 1 and 2 with each group divided into untreated rats (5 LZR and 5 OZR) and metformin-treated rats (6 LZR and 6 OZR). A: morning blood glucose levels in untreated and metformin-treated rats during fasted (left) and fed (right) states. Data were analyzed separately within the same feeding status by ANOVA with Tukey’s honestly significant difference (HSD) tests; *P < 0.05 vs. untreated fed LZR and †P < 0.05 vs. untreated fed OZR. B: hourly glucose values from untreated and metformin-treated OZR over a 24-h period analyzed by ANOVA with repeated measures and Tukey’s HSD tests; P < 0.05 for untreated and metformin-treated OZR for all hours. C: hourly glucose values over a 24-h period from untreated and metformin-treated LZR and OZR (same OZR as in B). ANOVA with repeated measures for metformin-treated LZR and OZR and ANOVA for untreated and metformin-treated LZR did not yield significant differences. D: standard deviation of blood glucose values over a 24-h period with access to food. Data were analyzed by ANOVA with Tukey’s HSD tests; *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR.
At 15–17 wk of age, rats were fasted 18 h before the GTT, and at this age, 10 h were required for blood glucose levels to be equivalent in untreated OZR and LZR (data not shown). As seen at 12–14 wk of age, injection of glucose (1 g/kg ip) into fasted, untreated rats evoked a larger rise in blood glucose in OZR than LZR at 15–17 wk of age (Fig. 4A). Treatment with metformin had no effect on the response to injected glucose in LZR (Fig. 4, B and E), but metformin significantly reduced the evoked rise in blood glucose, time to peak, recovery time, and area under the curve in OZR compared with untreated OZR (Figs. 4, C and F; Table 2). Comparison of metformin-treated OZR and LZR revealed a slightly higher peak in blood glucose in the treated OZR (Fig. 4D), but time to peak and area under the curve for these metformin-treated OZR and LZR were not different (Table 2). With the return of food 3 h after glucose injection, the untreated OZR displayed a significant rise in blood glucose that was virtually absent in metformin-treated OZR and unaffected by metformin treatment in LZR (Figs. 4, E and F and 5D).
Fig. 4.

Glucose tolerance tests in untreated and metformin (MET)-treated lean Zucker rats (LZR) and obese Zucker rats (OZR) at 15−17 wk of age. See Table 2 and Fig. 3 for baseline values of these rats. A–D: blood glucose levels measured by telemetry before and after injection of glucose (at time 0), shown in pairs of groups for clarity. Each pair was analyzed by ANOVA with repeated measures with Tukey’s honestly significance difference (HSD) tests. *P < 0.05 differs at that time point. E: same glucose tolerance test in LZR from B with blood glucose measured in hourly averages and return of food at hour 3. F: same glucose tolerance test in OZR from C with blood glucose measured in hourly averages and return of food at hour 3. For E and F, the data were analyzed by ANOVA with repeated measures with Tukey’s HSD tests; *P < 0.05 vs. metformin-treated OZR at that time point.
Fig. 5.

Development of enhanced reactivity to tail sampling of blood glucose in obese Zucker rats (OZR) at 15–17 wk of age. Comparisons of fasted (A) and fed (B) blood glucose values from morning tail samples and telemetry values 1 h before the tail samples in OZR (n = 12) and lean Zucker rats (LZR) (n = 11) at 12–14 wk of age (left), in OZR (n = 6) and LZR (n = 6) at 15–17 wk of age (middle), and in OZR (n = 6) and LZR (n = 6) at 15–17 wk of age after metformin treatment (right). Each of the three sets was analyzed by ANOVA with repeated measures for sample type with Tukey’s honestly significant difference (HSD) tests. *P < 0.05 vs. tail sample in LZR and †P < 0.05 vs. tail sample in OZR. Enlargement of data shown in Fig. 4, A and D, with a tail sample taken ~1 h before injection of glucose at time 0 (C). The arrow denotes the mean time of all tail samples (51 min) with a horizontal line to denote the range of times a baseline tail sample was taken (−30 to −70 min). Like-treated rats were compared by ANOVA with repeated measures with Tukey’s HSD tests. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. metformin (MET)-treated LZR. Expansion of data shown in Fig. 4, E and F, to show 5-min averages over the time period of 95 min to 270 min (3 h) after injection of glucose (D). Like-treated rats were compared by ANOVA with repeated measures with Tukey’s HSD tests. *P < 0.05 vs. untreated LZR (circles) during that time period and †P < 0.05 vs. metformin-treated LZR (squares) during that time period.
Comparison of blood glucose values from tail samples and telemetry.
Because treatment with metformin did not completely normalize the peak in blood glucose with the GTT in OZR (Fig. 4D), but normalized the elevated basal blood glucose with access to food (Figs. 3C and 4F), we examined whether moving and handling of the rats altered blood glucose levels more in OZR than LZR. Direct comparison of baseline blood glucose levels by tail sample, which involved moving and opening cages and handling the rats’ tails, with measures of blood glucose by telemetry 1 h before any disturbance, revealed striking differences that were impacted by age, rat genotype, and feeding status. At 12–14 wk of age, fasting blood glucose values were comparable in LZR and OZR whether measured by tail sample or telemetry (Fig. 5A, left). In contrast, at 15–17 wk of age, fasted blood glucose levels measured by tail sample were significantly higher than values measured by telemetry in OZR (Fig. 5A, middle), and this difference persisted after metformin treatment (Fig. 5A, right). In contrast, comparisons of blood glucose levels by tail sample and telemetry in LZR did not differ with or without metformin treatment at 15–17 wk of age (Fig. 5A middle and right). Thus, fasted blood glucose levels were higher in 15-to 17-wk-old OZR compared with age-matched LZR when measured by tail samples, but fasted blood glucose levels were comparable in OZR and LZR when measured by telemetry (Fig. 5A).
When rats had access to food, the comparison of glucose values from tail samples and telemetry also revealed some differences that varied by age. As seen in these rats when they were fasted, at 12–14 wk of age, glucose readings by both measures were comparable in LZR and OZR (Fig. 5B, left), and with access to food, OZR had higher glucose levels than LZR whether measured by tail samples or telemetry, in agreement with Fig. 1A (Fig. 5B, left). At 15–17 wk of age, glucose levels measured by tail sample or telemetry were comparable in LZR with or without metformin treatment (Fig. 5B, middle and right). However, as seen with fasting, when OZR had access to food, their blood glucose levels were higher when measured by tail sample than with telemetry whether or not the OZR were treated with metformin (Fig. 5B, middle and right). In contrast to the fasted state, in the fed state the sampling method did not alter conclusions regarding differences between OZR and LZR. With access to food, untreated 15- to 17-wk old OZR had higher blood glucose levels than LZR, but metformin-treated OZR and LZR had comparable blood glucose levels whether measured by tail sample or telemetry (Fig. 5B, middle and right).
Similar observations regarding the impact of handling the rats upon blood glucose levels can be made by examining the 15-to 17-wk-old rats immediately before and after a GTT (using segments of data from Fig. 4). Fasting blood glucose levels were comparable in treated and untreated OZR and LZR when measured by telemetry (time span −90 to −70 min in Fig. 5C, taken from Fig. 4, A and D). However, with moving the cages and taking blood samples from the tail, blood glucose levels measured by telemetry rose in OZR but not LZR during the hour before the injection of glucose that occurred at time 0 (Fig. 5C). Although analysis of the entire time period in Fig. 4A (4 h of 5-min samples) did not detect differences observed before the injection of glucose, analysis of a shorter time span before the injection of glucose (80 min of 5-min samples) revealed clear differences between OZR and LZR (Fig. 5C). Blood glucose was higher in untreated OZR than LZR for 40 min (−50 to −10 min, Fig. 5C, from Fig. 4A), and metformin treatment in OZR and LZR reduced the duration of the difference in fasted blood glucose after tail sampling to 10 min (at −25 and −20 min; Fig. 5C, from Fig. 4D).
Differences between OZR and LZR can also be seen following the GTT when food was returned to the cages, and the cages were moved back to their rack 3 h after the injection of glucose (5-min samples starting at 180 min, Fig. 5D, after time frame shown in Fig. 4, A and D). The rise in blood glucose with the return of food is larger in untreated OZR than LZR (Fig. 5D), and this difference is also apparent in the hourly averages (Fig. 4, E and F). In metformin-treated rats, analysis of 5-min samples for 90 min after the return of food also revealed a significant rise in blood glucose in OZR compared with LZR during the time when cages were being moved, although the difference was smaller compared with untreated rats and occurred only for 10 min (Fig. 5D). Within 30 min after the return of food to their cages and moving them, blood glucose levels in metformin-treated OZR were comparable to LZR (Fig. 5D), whereas untreated OZR remained significantly elevated above metformin-treated OZR (Fig. 5D), as observed in the hourly averages (Fig. 4F).
Baseline plasma insulin and lipids in OZR and LZR after treatment with metformin.
Before the infusion of phenylephrine, a blood sample was taken from the arterial catheter in conscious, undisturbed rats after 1 day of recovery from surgery to implant indwelling femoral vascular catheters. As observed with the rats used for telemetry, metformin tended to retard weight gain. In this particular set, unlike those in Table 2, the metformin-treated LZR were comparable to untreated LZR, whereas metformin-treated OZR weighed less than untreated OZR (Table 3). As seen in Table 2, comparison of like-treated rats showed that OZR weighed significantly more than LZR (by 67% in untreated rats and 46% in metformin-treated rats; Table 3). Insulin levels were slightly reduced by metformin treatment in OZR, with no effect in LZR (Table 3). However, compared with like-treated LZR, untreated and metformin-treated OZR had insulin levels that were five times and seven times higher, respectively (Table 3). Plasma cholesterol and triglycerides were higher in metformin-treated OZR compared with LZR, and neither measure was affected by metformin treatment or differences in body weights of treated and untreated OZR (Table 3).
Table 3.
Effects of metformin treatment on plasma insulin, cholesterol, and triglycerides
| Group | n | Age, days | Body Weight, g | Insulin, ng/dl | Cholesterol, mg/dl | Triglycerides, mg/dl |
|---|---|---|---|---|---|---|
| Untreated | ||||||
| LZR | 7 | 115.3 ± 1.7 | 351.7 ± 10.5 | 3.8 ± 1.3 | 107.5 ± 7.8 | 16.3 ± 2.9 |
| OZR | 6 | 114.0 ± 1.3 | 583.5 ± 28.7* | 22.9 ± 0.4* | 227.1 ± 16.2* | 52.6 ± 16.0 |
| Metformin | ||||||
| LZR | 8 | 116.5 ± 0.3 | 338.1 ± 11.8 | 2.3 ± 0.4 | 124.4 ± 7.5 | 18.2 ± 3.0 |
| OZR | 8 | 113.0 ± 1.4 | 493.6 ± 14.2*† | 18.5 ± 2.1*† | 206.2 ± 17.8* | 68.6 ± 18.5* |
| P values | ||||||
| Rat type | 0.064 | <0.001 | <0.001 | <0.001 | 0.002 | |
| Treatment | 0.972 | 0.004 | 0.046 | 0.880 | 0.478 | |
| Interaction | 0.364 | 0.029 | 0.312 | 0.166 | 0.574 |
Values are means ± SE; n, number of rats. Blood samples were collected from arterial line at ~9:00 AM in conscious rats with access to food and water before infusion of phenylephrine. Each measure was analyzed by 2-way ANOVA for rat type × treatment followed by Tukey’s honestly significant difference tests. LZR, lean Zucker rat; OZR, obese Zucker rat.
P < 0.05 vs. LZR of like treatment,
P < 0.05 vs. untreated rat of same type.
Impact of metformin on phenylephrine-induced bradycardia and NTS c-Fos expression.
Mean AP was elevated in untreated OZR compared with LZR, and metformin treatment did not reduce mean AP in either LZR or OZR (Fig. 6A). Instead, mean AP was slightly but significantly higher in metformin-treated OZR compared with untreated OZR (Fig. 6A). There were no differences in baseline HR among the groups (data not shown), as previously reported in untreated conscious male OZR and LZR of a similar age (31). Raising mean AP by 40 mmHg with infusion of phenylephrine evoked a baroreflex-mediated bradycardia that was markedly reduced in untreated OZR compared with LZR (Fig. 6, B and C), as previously reported (31). Treatment with metformin enhanced phenylephrine-induced bradycardia in OZR with no effect in LZR (Fig. 6C). However, the bradycardia in metformin-treated OZR was still smaller compared with LZR (Fig. 6C), suggesting a partial restoration of the lower plateau of the cardiac baroreflex in the metformin-treated OZR.
Fig. 6.

Baseline mean arterial pressure (AP) and phenylephrine (PE)-induced bradycardia in untreated and metformin-treated lean Zucker rats (LZR) and obese Zucker rats (OZR). A: mean AP in untreated and metformin-treated, conscious LZR and OZR. See Table 3 for more baseline values. Data were analyzed by ANOVA with Tukey’s honestly significant difference (HSD) tests. *P < 0.05 vs. like-treated LZR and †P < 0.05 vs. untreated OZR. B: representative tracing from an untreated LZR illustrating baseline period for MAP and period of analysis for PE-induced decrease in heart rate (HR). C: PE-induced decrease in HR in untreated and metformin-treated LZR and OZR. Data were analyzed by ANOVA with Tukey’s HSD tests. *P < 0.05 vs. like-treated LZR and †P < 0.05 vs. untreated OZR.
As expected, the number of phenylephrine-induced c-Fos-expressing neurons in the caudal and intermediate NTS of OZR was reduced compared with LZR at all four rostrocaudal levels examined (Figs. 7, A–C and 8, A and B) (31). Treatment with metformin did not alter phenylephrine-induced c-Fos expression in the NTS of LZR but enhanced c-Fos expression in OZR, comparable to counts observed in LZR at all four rostrocaudal levels of the NTS examined (Figs. 7 and 8). Regions of the NTS known to receive baroreceptor inputs (13) expressed fewer c-Fos+ neurons in untreated OZR compared with LZR (Figs. 7C, left, and 8, A and B), and expression in metformin-treated OZR was restored within these regions (Figs. 7C, right, and 8D).
Fig. 7.

Phenylephrine-induced c-Fos expression in the nucleus tractus solitarius (NTS) of untreated and metformin (MET)-treated lean Zucker rats (LZR) and obese Zucker rats (OZR). A: number of c-Fos-positive neurons from bilateral counts of NTS at four rostrocaudal levels. Each bregma level was analyzed separately by ANOVA with Tukey’s honestly significant difference (HSD) tests. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR. B: total counts of c-Fos-positive neurons from all four rostrocaudal levels of the NTS. Data were analyzed by ANOVA with Tukey’s HSD tests. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR. C: representative maps of c-Fos-positive neurons at four rostrocaudal levels of the NTS in untreated (left) and metformin-treated (right) LZR and OZR. AP, area postrema; CC, central canal.
Fig. 8.

Representative bright-field photomicrographs of the left nucleus tractus solitarius at −13.8 mm caudal to bregma from an untreated lean Zucker rat (LZR) (A) and obese Zucker rat (OZR) (B) and a metformin-treated LZR (C) and OZR (D). Scale bar = 250 µm. AP, area postrema; CC, central canal; TS, tractus solitarius.
Baseline plasma insulin and lipids in OZR and LZR after treatment with pioglitazone.
Because metformin reduced weight gain in OZR and LZR, another set of rats was treated with pioglitazone and compared with untreated OZR and LZR. As expected (19), pioglitazone accelerated weight gain in OZR (Table 4). Compared with like-treated LZR, untreated and pioglitazone-treated OZR weighed 47% and 56% more, respectively (Table 4). Like metformin, treatment with pioglitazone caused a small reduction in plasma insulin levels in OZR but had no significant effect in LZR (Table 4). However, compared with like-treated LZR, insulin levels were 4.8 times higher in untreated and pioglitazone-treated OZR. Plasma cholesterol was elevated in OZR compared with LZR and was not affected by treatment with pioglitazone (Table 4). Plasma triglycerides were elevated in untreated OZR compared with LZR, and pioglitazone treatment had no significant effect in OZR or LZR (Table 4).
Table 4.
Effects of pioglitazone treatment on plasma insulin, cholesterol, and triglycerides
| Group | n | Age, days | Body Weight, g | Insulin, ng/dl | Cholesterol, mg/dl | Triglycerides, mg/dl |
|---|---|---|---|---|---|---|
| Untreated | ||||||
| LZR | 8 | 115.5 ± 1.6 | 378.4 ± 8.2 | 4.0 ± 1.0 | 83.4 ± 7.9 | 8.6 ± 1.8 |
| OZR | 6 | 113.3 ± 2.1 | 557.0 ± 12.2* | 23.2 ± 0.1* | 148.7 ± 16.2* | 30.0 ± 8.8* |
| Pioglitazone | ||||||
| LZR | 7 | 112.4 ± 0.4 | 400.1 ± 11.6 | 3.3 ± 0.4 | 58.9 ± 9.0 | 15.9 ± 2.1 |
| OZR | 8 | 111.9 ± 0.4 | 624.1 ± 24.8*† | 19.1 ± 2.0*† | 123.5 ± 11.3* | 31.1 ± 9.5 |
| P values | ||||||
| Rat type | 0.314 | <0.001 | <0.001 | <0.001 | 0.010 | |
| Treatment | 0.093 | 0.012 | 0.050 | 0.034 | 0.525 | |
| Interaction | 0.498 | 0.180 | 0.212 | 0.973 | 0.639 |
Values are means ± SE; n, number of rats. Blood samples were collected from arterial line at ~9:00 AM in conscious rats with access to food and water before infusion of phenylephrine. For triglycerides, n = 5 rats for untreated OZR because of the removal of one outlier (385.2 mg/dl). Each measure was analyzed by 2-way ANOVA for rat type × treatment followed by Tukey’s honestly significant difference tests. LZR, lean Zucker rat; OZR, obese Zucker rat.
P < 0.05 vs. LZR of like treatment,
P < 0.05 vs. untreated rat of same type.
Impact of pioglitazone on glucose and phenylephrine-induced changes in HR and NTS c-Fos expression.
Blood glucose was measured at ~9 AM in rats with access to food because these rats were not instrumented to record blood glucose by telemetry, and differences in blood glucose in untreated OZR and LZR were present only when rats had access to food (Figs. 3A and 5B). Like metformin, treatment with pioglitazone did not alter morning fed blood glucose levels in LZR (Figs. 3A and 9A). In contrast, treatment with pioglitazone greatly reduced morning fed blood glucose levels in OZR compared with those observed in LZR (Fig. 9A). Although GTTs were not performed, previous reports show normalized fed glucose in OZR coincides with restoration of glucose response to glucose challenge (19). Pioglitazone did not alter mean AP in conscious LZR or OZR, leaving the pioglitazone-treated OZR with higher mean AP than LZR (Fig. 9B). As seen with metformin, pioglitazone did not alter phenylephrine-induced bradycardia in LZR (Fig. 9C). In contrast, the phenylephrine-induced bradycardia that was blunted in untreated OZR was fully restored by treatment with pioglitazone (Fig. 9C). Treatment with pioglitazone did not alter c-Fos expression in the NTS of LZR, but the diminished c-Fos expression observed untreated OZR and was significantly enhanced by treatment with pioglitazone (Fig. 9D). However, after treatment with pioglitazone, phenylephrine-induced c-Fos expression was still slightly reduced in OZR compared with LZR (Fig. 9D).
Fig. 9.

Baseline blood glucose mean arterial pressure (AP) with phenylephrine-induced bradycardia and total counts of c-Fos-positive neurons in the nucleus tractus solitarius (NTS) of untreated and pioglitazone-treated obese Zucker rats (OZR) and lean Zucker rats (LZR). A: morning-fed glucose taken from tail samples. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR. B: baseline mean AP in conscious rats before infusion of phenylephrine. *P < 0.05 vs. untreated rat of same type. C: phenylephrine-induced decrease in heart rate (HR) using the same protocol shown in Fig. 6B. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR. D: total counts of c-Fos-positive neurons from 4 rostrocaudal levels of the NTS (−14.2, −13.8, −13.4, and −13.0 mm caudal to bregma) using the same protocol as in Fig. 7B. *P < 0.05 vs. untreated LZR and †P < 0.05 vs. untreated OZR. Each measure was analyzed by ANOVA with Tukey’s honestly significant difference tests. E: representative maps of c-Fos-positive neurons at four rostrocaudal levels of the NTS in untreated (left) and pioglitazone-treated (right) LZR and OZR. CC, central canal; AP, area postrema; PIO, pioglitazone.
DISCUSSION
Obesity is associated with the development of a constellation of attributes, known as metabolic syndrome, that promote premature morbidity and mortality (33, 52, 54, 62). Current clinical guidelines use threshold values of these attributes that may not provide adequate triggers for intervention to reduce significant health risks. In the present study, fasting blood glucose levels measured by telemetry were comparable in adult male LZR and OZR up to 17 wk of age. However, with access to food, OZR were chronically hyperglycemic with impaired glucose tolerance by 12–14 wk of age. By this age, OZR also develop hypertension and impaired baroreflexes coincident with reduced baroreflex-mediated activation of the NTS (31, 75). Treatment of OZR with metformin or pioglitazone dramatically improved glucose homeostasis and simultaneously enhanced the blunted baroreflex-mediated bradycardia and c-Fos expression in NTS with no effect in treated LZR. These attributes were restored in the persistence of hyperinsulinemia and hypertension, suggesting changes in insulin and AP did not contribute to the effects of the treatments. These data strongly suggest that impaired glucose homeostasis in prediabetic, insulin-resistant OZR contributes to reduced baroreceptor-mediated activation of the NTS and bradycardia before the onset of frank type 2 diabetes mellitus.
Metformin and pioglitazone are both highly effective for restoring glucose homeostasis without producing hypoglycemia, but their underlying primary mechanisms differ. Pioglitazone stimulates peroxisome proliferator-activated receptor gamma to increase insulin sensitivity, particularly in liver and adipose tissue (3), whereas metformin stimulates AMP-activated protein kinase to reduce hepatic gluconeogenesis and enhance glucose uptake by muscles (94). Although unidentified effects of metformin and pioglitazone may have also contributed to improvement of baroreflexes, several controls in the present study strengthen the argument that improved glucose homeostasis enhanced the brainstem’s response to baroreceptor inputs in insulin-resistant OZR. Neither treatment altered baroreflex-mediated bradycardia or activation of the NTS in the LZR, suggesting these treatments were effective by ameliorating obesity-related attributes. Metformin tended to retard weight gain in all treated rats, and weight loss can improve baroreflexes in obese subjects (21, 27, 82). However, pioglitazone accelerated weight gain while restoring glycemic control and baroreflex efficacy, providing confidence that improvement with metformin was not just a consequence of weight loss. Although plasma insulin was slightly reduced by both treatments in OZR, plasma insulin was still five to seven times higher in OZR than in LZR. Furthermore, insulin appears to act in the forebrain to alter baroreflex-mediated tachycardia without a significant effect on baroreflex-mediated bradycardia (63, 70). Hypertension is also associated with compromised baroreflexes (29), but both treatments enhanced baroreflex-mediated responses without reducing mean AP. Elevated cholesterol and triglycerides in OZR were not reduced by these treatments. Thus, the data suggest the enhanced glycemic control observed in treated OZR contributed to the improvement of baroreceptor-mediated activation of the NTS and bradycardia.
Metformin and pioglitazone both improved phenylephrine-induced bradycardia and activation of the NTS in OZR, but the degree of recovery for each measure varied by treatment. Pioglitazone was more effective than metformin for restoring baroreflex-mediated bradycardia in OZR. The partial restoration observed in metformin-treated OZR may be related to the increase in baseline mean AP or the use of a suboptimal dose, as metformin-treated OZR retained some differences in glucose compared with treated LZR. In addition, although pioglitazone-treated OZR and LZR had comparable phenylephrine-induced bradycardia, the enhanced phenylephrine-induced c-Fos expression in the NTS was still significantly less in OZR than in LZR. These data suggest that pioglitazone may also enhance baroreflexes independent of improved activation of NTS. Furthermore, a related compound, rosiglitazone, increases baroreflex gain in correlation with or independent of improved insulin sensitivity in obese rats, depending on the dose (93). Alternatively, the apparent mismatches in degree of improvement may also be related to the rudimentary assessment of baroreflex efficacy and excitation of baroreflex-related NTS neurons. Quantification of the baroreflex was limited to the maximal bradycardia evoked by a rapid rise in AP using a protocol to optimize c-Fos expression in the NTS. Bradycardia was chosen as the dependent measure because it could be readily quantified in conscious rats, and the maximal response to an evoked rise in AP is a prominent deficit observed in obese rodents (12, 59, 75). Likewise, human subjects who are overweight and insulin-resistant with normal or elevated fasting blood glucose can have diminished baroreflex-mediated bradycardia in the absence of diminished baroreflex-mediated tachycardia or changes in SNA (36). In obese rats, both sympathetic and parasympathetic contributions to baroreflex-mediated bradycardia are impaired (6, 59), but further study is needed to determine how metformin and pioglitazone improve the autonomic regulation of HR in OZR. In addition, although phenylephrine-induced c-Fos expression in NTS depends upon inputs from arterial baroreceptors (13), many activated NTS neurons are not likely to be directly involved in producing the baroreflex-mediated bradycardia. Most NTS neurons that express c-Fos after phenylephrine infusion are glutamatergic, but only a small portion of these neurons project to the region of the CVLM and nucleus ambiguus (88). Therefore, although the number of phenylephrine-induced c-Fos-expressing neurons roughly indicates a magnitude of regional activation within the NTS, future studies will be necessary to determine which neurons are altered in OZR and are affected by treatments that improve glycemic control. Nevertheless, in the present study, treatment of OZR with metformin or pioglitazone increased c-Fos expression in regions of the NTS containing neurons that project to the ventrolateral medulla (88). Despite these methodological limitations, these data show that restoration of glucose homeostasis in OZR coincides with improved baroreflex-mediated activation of the NTS and bradycardia in conscious rats.
Although the present study cannot delineate the cellular mechanisms underlying diminished baroreceptor-mediated activation of the NTS in OZR or its reversal by treatments that normalize glucose homeostasis, these changes are consistent with the impact of hyperglycemia upon the afferent limb of the baroreflex. In the absence of obesity, hypertension, or hyperinsulinemia, the presence of elevated blood glucose is accompanied by diminished phenylephrine-induced c-Fos expression in the NTS (26) and impaired baroreflexes without altering the relationship between aortic depressor nerve activity and AP (17, 20, 38). Likewise, adult male OZR develop impaired activation of the NTS with no overt changes in aortic depressor nerve activity at an age when they are chronically hyperglycemic with access to food (31, 39). The threshold AP for the onset of aortic depressor nerve activity is comparable in these OZR and LZR, and both the percent change and raw voltage of the aortic depressor nerve are equivalent over a wide range of AP. Furthermore, the modest hypertension in these OZR is accompanied by a slightly baseline higher aortic depressor nerve activity, suggesting no resetting of baroreceptor afferent nerve activity (39). In contrast, electrical stimulation of the baroreceptor afferent fibers or microinjection of glutamate into the NTS evokes smaller physiological responses in adult male obese rats (39, 93). These observations are consistent with a hyperglycemia-induced reduction in the ability of baroreceptor afferent nerves to activate the NTS.
Neurons within barosensitive regions of the NTS can be excited by raising circulating glucose within a physiological range and by local changes in glucose concentration (91). Within the NTS, glucose increases glutamate release from vagal afferent nerve terminals to enhance vagal activation of NTS neurons, and glucose produces excitatory postsynaptic effects in some NTS neurons (86, 91). In the setting of metabolic syndrome, many of these homeostatic mechanisms are disrupted or lost altogether. Just as hyperinsulinemia promotes insulin resistance, hyperglycemia fosters glucose insensitivity within the NTS. In type 1 diabetic mice, glucose fails to modulate the frequency of spontaneous excitatory postsynaptic currents in NTS neurons, suggesting that chronic hyperglycemia abrogates the glucose-enhanced glutamate release from vagal afferent nerve terminals (11). Furthermore, with streptozotocin-induced hyperglycemia, glucose-mediated augmentation of NTS neuronal excitability is lost, coincident with reduced expression and function of glucokinase within the NTS (32). Thus, acute local changes in glucose concentration play an important role in facilitating the activation of NTS neurons by afferent inputs, and the loss of glucose-mediated enhancement of neurotransmission with chronic hyperglycemia likely contributes to diminished activation of the NTS in the setting of diabetes.
Although the loss of glucose-enhanced activation of NTS neurons has been proposed to underlie impaired gastrointestinal function with diabetes (11), these changes in NTS neurotransmission with chronic hyperglycemia would also impair autonomic regulation of cardiovascular function. Whether or not the barosensitive NTS neurons are also glucose-sensitive, vagal activation of NTS neurons that regulate ingestive behaviors and digestion clearly converge to modulate the activity of barosensitive neurons in the ventrolateral medulla. Ingestion of calories stimulates duodenal release of cholecystokinin (CCK), which activates subdiaphragmatic vagal afferents that project to the NTS (66) and selectively inhibits splanchnic SNA to increase mesenteric blood flow (61, 73). In agreement, some baroactivated GABAergic CVLM neurons are also excited by CCK (61), and some baroinhibited bulbospinal RVLM neurons are inhibited by CCK with timing that corresponds to inhibition of splanchnic SNA (73). Thus, vagal activation of NTS neurons that regulate gastrointestinal function also impact autonomic control of cardiovascular targets to coordinate digestion and blood flow.
In agreement with the notion of a more widespread depression of vagal activation of NTS neurons with chronic hyperglycemia, OZR have impaired sympathoinhibitory responses to direct the stimulation of vagal afferent fibers and activation of the von Bezold-Jarisch reflex by phenylbiguanide (39). Although only a small proportion of NTS neurons activated by phenylbiguanide are also excited by increased AP (69), these reflexes converge at the ventrolateral medulla as phenylbiguanide excites baroactivated GABAergic neurons in the CVLM (74) and inhibits presympathetic RVLM neurons (85). Deficits in sympathoinhibitory reflexes initiated by vagal afferents are not specific to OZR because obese Sprague-Dawley rats on a high-fat diet have diminished CCK-induced inhibition of splanchnic SNA that coincides with preserved subdiaphragmatic vagal afferent nerve responses and reduced CCK-induced c-Fos expression in the NTS and CVLM (37). Furthermore, in these rats, bulbospinal RVLM neurons display significantly reduced barosensitivity and inhibition by CCK (37). Similarly, Wistar rats on a high-fat diet become hypertensive with impaired baroreflexes and blunted inhibition of renal SNA to volume expansion (43). Renal denervation restores these sympathoinhibitory reflexes in the obese rats (43), and this denervation improves glycemic control in insulin-resistant rats by reducing hepatic glucose production and increasing insulin sensitivity (15). Thus, poor glycemic control appears to foster a widespread impairment of autonomic reflexes that are mediated by activation of the NTS.
The long-term measurement of blood glucose by telemetry in conscious, undisturbed rats provided a more in-depth and accurate assessment of baseline values and changes in blood glucose over time. Analysis spanning 12–17 wk of age demonstrated a comparable fasting blood glucose in OZR and LZR that was chronically elevated in OZR with access to food by 12 wk of age. In addition, telemetry provided real-time changes in blood glucose that could not be readily achieved with periodic blood samples. For example, the time required for fasting to produce equivalent blood glucose levels in OZR and LZR grew longer as metabolic syndrome progressed over time, and neither group showed significant periods of hypoglycemia with fasting or drug treatments. Furthermore, within this age range, 1 wk of metformin treatment was sufficient to reduce fed glucose levels in OZR to match LZR. During GTTs, measurement of blood glucose by telemetry was essential for determining the time to reach peak glucose, which was usually longer in OZR than LZR, and an accurate determination of the peak, which would not be possible with the use of timed tail samples.
Measures of blood glucose by telemetry also revealed the development of enhanced glucose responses to mild stressors in OZR. Great care was taken to minimize disturbance to the rats during blood sampling from their tails, and LZR and OZR showed similar alerting and exploration responses with no obvious behavioral differences. At 12–14 wk of age, comparison of morning blood glucose values measured by tail and telemetry showed no differences in LZR or OZR whether they were fasted or fed, even though OZR displayed glucose intolerance when challenged. However, by 15–17 wk of age, tail sample values were much higher than telemetry values in fasted OZR but not in LZR. This discrepancy yielded striking contradictory results for fasted blood glucose levels between LZR and OZR, as OZR would be considered hyperglycemic with tail samples but not with telemetry measures. Telemetry also revealed differences in blood glucose between OZR and LZR after baseline tail samples before GTTs and return of food to cages afterward. Metformin treatment greatly reduced but did not eliminate discrepancies between tail sample and telemetry values in 15- to 17-wk-old OZR or differences in peak responses to glucose challenge between OZR and LZR. Thus, this dose of metformin was sufficient to maintain normal blood glucose levels in undisturbed OZR with access to food but was not completely effective for counteracting exaggerated rises in glucose to mild stressors. These data highlight the importance of considering that conscious rats may respond differently to the same environment and handling in healthy and disease states, and that these differences may not be detected by observing their behavior.
Adult male OZR have exaggerated sympathoexcitatory reflexes (41), but brief immobilization stress in adult male OZR evokes enhanced rises in glucose that are accompanied by normal increases in plasma norepinephrine and epinephrine in comparison with LZR (49). Instead, these hyperinsulinemic OZR have larger reductions in plasma insulin after immobilization stress (49), consistent with augmented corticosterone-mediated inhibition of insulin secretion (7). Without functional leptin receptors, OZR are not protected by leptin-mediated suppression of corticosterone release from the adrenal cortex during stressors like immobilization (34). In addition to amplifying stress responses, corticosterone can raise AP and impair glutamatergic activation of the NTS (8, 78). However, corticosterone-induced impairment of baroreflex-mediated changes in HR is related only to the gain, with no changes in the maximal phenylephrine-induced bradycardia (8). Furthermore, the dose and duration metformin used in the present study appear to have no effect on comparable morning plasma levels of corticosterone in OZR or LZR of this age (72). These data suggest that the beneficial effects of metformin and pioglitazone were not caused by reductions in corticosterone. However, enhanced corticosterone actions may contribute to the hypertension and exaggerated stress responses observed in OZR (16, 30, 34).
Perspectives and Significance
Metabolic syndrome significantly increases the risk for development of cardiovascular disease and type 2 diabetes, and the diagnosis is defined by the presence of three of five attributes with stated threshold values for central adiposity, AP, and fasting blood levels of HDL cholesterol, triglycerides, and glucose. However, mounting evidence supports the importance of dynamic measures of AP and blood glucose. Increased variability of AP is a significant, independent risk factor for coronary heart disease, renal disease, stroke, and cognitive decline, and this deficit can be detected with short-term measures, such as white coat hypertension, or long-term measures, such as visit-to-visit variability in AP (25, 48, 57, 68, 71, 89). Furthermore, the variability of AP has been shown to be a better predictor than mean AP for end-organ damage and cognitive decline (2, 47, 60). White coat hypertension is significantly associated with a higher prevalence of glucose dysregulation that can be detected by glucose challenge but not a threshold value for fasting blood glucose (56, 57). Similarly, in the absence of diabetes, a prediabetic level of fasting blood glucose (>100 mg/dl) is a better predictor of impaired HR variability and HR turbulence than any other attribute of metabolic syndrome (5). Furthermore, in patients with type 2 diabetes the variability of blood glucose has a highly significant inverse relationship with baroreflex sensitivity (58).
The present study postulates that chronic hyperglycemia with access to food is a critical link between insulin resistance and increased variability of AP because this state impairs the brainstem’s response to acute changes in AP. Furthermore, the resulting diminished baroreflex-mediated control of HR occurs independent of hypertension and hyperinsulinemia. Because insulin resistance and poor glycemic control are not typically managed until a significant increase in fasting blood glucose is observed, a prediabetic population may not be treated while hyperglycemia impairs stability of AP. The present study underscores the need for early detection of poor glycemic control in metabolic syndrome to reduce the risks of stroke, cognitive decline, and irreversible organ damage that could occur before the onset of frank type 2 diabetes.
GRANTS
This project was financially supported by National Heart, Lung, and Blood Institute Grant R01-HL-132568 and a Seed Grant from the Institute for Cardiovascular and Metabolic Disease at UNTHSC to A. M. Schreihofer. In addition, P. Chaudhary was supported by predoctoral fellowship PRE27260088 from the American Heart Association.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.C. and A.M.S performed experiments, analyzed data, interpreted results of experiments, prepared figures, drafted the manuscript, edited and revised the manuscript, and approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Steve Mifflin and Tom Cunningham for sharing resources and wisdom to improve this study. We are also grateful to Joel Little for tireless expert technical assistance regarding the surgical implantation of telemetry transmitters and use of DSI equipment and software.
REFERENCES
- 1.Aicher SA, Milner TA, Pickel VM, Reis DJ. Anatomical substrates for baroreflex sympathoinhibition in the rat. Brain Res Bull 51: 107–110, 2000. doi: 10.1016/S0361-9230(99)00233-6. [DOI] [PubMed] [Google Scholar]
- 2.Alpérovitch A, Blachier M, Soumaré A, Ritchie K, Dartigues JF, Richard-Harston S, Tzourio C. Blood pressure variability and risk of dementia in an elderly cohort, the Three-City Study. Alzheimers Dement 10, Suppl: S330–S337, 2014. doi: 10.1016/j.jalz.2013.05.1777. [DOI] [PubMed] [Google Scholar]
- 3.Anagnostis P, Siolos P, Christou K, Gkekas NK, Kosmidou N, Athyros VG, Karagiannis A. The effect of antidiabetic medications on the cardiovascular system: a critical appraisal of current data. Hormones (Athens) 17: 83–95, 2018. doi: 10.1007/s42000-018-0017-5. [DOI] [PubMed] [Google Scholar]
- 4.Apaijai N, Pintana H, Chattipakorn SC, Chattipakorn N. Effects of vildagliptin versus sitagliptin, on cardiac function, heart rate variability and mitochondrial function in obese insulin-resistant rats. Br J Pharmacol 169: 1048–1057, 2013. doi: 10.1111/bph.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balcioğlu AS, Akinci S, Çiçek D, Eldem HO, Çoner A, Bal UA, Müderrisoğlu H. Which is responsible for cardiac autonomic dysfunction in non-diabetic patients with metabolic syndrome: Prediabetes or the syndrome itself? Diabetes Metab Syndr 10, Suppl 1: S13–S20, 2016. doi: 10.1016/j.dsx.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Barringer DL, Buñag RD. Uneven blunting of chronotropic baroreflexes in obese Zucker rats. Am J Physiol 256: H417–H421, 1989. [DOI] [PubMed] [Google Scholar]
- 7.Barseghian G, Levine R. Effect of corticosterone on insulin and glucagon secretion by the isolated perfused rat pancreas. Endocrinology 106: 547–552, 1980. doi: 10.1210/endo-106-2-547. [DOI] [PubMed] [Google Scholar]
- 8.Bechtold AG, Scheuer DA. Glucocorticoids act in the dorsal hindbrain to modulate baroreflex control of heart rate. Am J Physiol Regul Integr Comp Physiol 290: R1003–R1011, 2006. doi: 10.1152/ajpregu.00345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belin de Chantemèle EJ, Ali MI, Mintz JD, Rainey WE, Tremblay ML, Fulton DJ, Stepp DW. Increasing peripheral insulin sensitivity by protein tyrosine phosphatase 1B deletion improves control of blood pressure in obesity. Hypertension 60: 1273–1279, 2012. doi: 10.1161/HYPERTENSIONAHA.112.196295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boysen A, Lewin MA, Hecker W, Leichter HE, Uhlemann F. Autonomic function testing in children and adolescents with diabetes mellitus. Pediatr Diabetes 8: 261–264, 2007. doi: 10.1111/j.1399-5448.2007.00254.x. [DOI] [PubMed] [Google Scholar]
- 11.Browning KN. Modulation of gastrointestinal vagal neurocircuits by hyperglycemia. Front Neurosci 7: 217, 2013. doi: 10.3389/fnins.2013.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buñag RD, Meyer M, Vansell N, Kerecsen L. Conscious obese rats have impaired reflex bradycardia and enhanced norepinephrine sensitivity. Am J Physiol Regul Integr Comp Physiol 271: R654–R660, 1996. doi: 10.1152/ajpregu.1996.271.3.R654. [DOI] [PubMed] [Google Scholar]
- 13.Chan RK, Sawchenko PE. Organization and transmitter specificity of medullary neurons activated by sustained hypertension: implications for understanding baroreceptor reflex circuitry. J Neurosci 18: 371–387, 1998. doi: 10.1523/JNEUROSCI.18-01-00371.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chantler PD, Shrader CD, Tabone LE, d’Audiffret AC, Huseynova K, Brooks SD, Branyan KW, Grogg KA, Frisbee JC. Cerebral cortical microvascular rarefaction in metabolic syndrome is dependent on insulin resistance and loss of nitric oxide bioavailability. Microcirculation 22: 435–445, 2015. doi: 10.1111/micc.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Chang Y, He L, Jian X, Li L, Gao L, Yang Y, Zeng M, Liu H, Zhao AZ, Yang G. Effect of renal sympathetic denervation on hepatic glucose metabolism and blood pressure in a rat model of insulin resistance. J Hypertens 34: 2465–2474, 2016. doi: 10.1097/HJH.0000000000001087. [DOI] [PubMed] [Google Scholar]
- 16.Clapham JC, Turner NC. Effects of the glucocorticoid II receptor antagonist mifepristone on hypertension in the obese Zucker rat. J Pharmacol Exp Ther 282: 1503–1508, 1997. [PubMed] [Google Scholar]
- 17.Dall’Ago P, Fernandes TG, Machado UF, Belló AA, Irigoyen MC. Baroreflex and chemoreflex dysfunction in streptozotocin-diabetic rats. Braz J Med Biol Res 30: 119–124, 1997. doi: 10.1590/S0100-879X1997000100018. [DOI] [PubMed] [Google Scholar]
- 18.de Ferranti SD, de Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, Magge SN, Marx N, McGuire DK, Orchard TJ, Zinman B, Eckel RH. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Diabetes Care 37: 2843–2863, 2014. doi: 10.2337/dc14-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Souza CJ, Eckhardt M, Gagen K, Dong M, Chen W, Laurent D, Burkey BF. Effects of pioglitazone on adipose tissue remodeling within the setting of obesity and insulin resistance. Diabetes 50: 1863–1871, 2001. doi: 10.2337/diabetes.50.8.1863. [DOI] [PubMed] [Google Scholar]
- 20.do Carmo JM, Huber DA, Castania JA, Fazan VP, Fazan R Jr, Salgado HC. Aortic depressor nerve function examined in diabetic rats by means of two different approaches. J Neurosci Methods 161: 17–22, 2007. doi: 10.1016/j.jneumeth.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Emdin M, Gastaldelli A, Muscelli E, Macerata A, Natali A, Camastra S, Ferrannini E. Hyperinsulinemia and autonomic nervous system dysfunction in obesity: effects of weight loss. Circulation 103: 513–519, 2001. doi: 10.1161/01.CIR.103.4.513. [DOI] [PubMed] [Google Scholar]
- 22.Farrar NS, Chambers NJ, Carlsson AR, Denyer G, Johnston GA. Effect of a series of novel sulphonylthioureas on glucose tolerance in the obese fa/fa Zucker rat. Clin Exp Pharmacol Physiol 28: 386–391, 2001. doi: 10.1046/j.1440-1681.2001.03466.x. [DOI] [PubMed] [Google Scholar]
- 23.Goodwill AG, Frisbee SJ, Stapleton PA, James ME, Frisbee JC. Impact of chronic anticholesterol therapy on development of microvascular rarefaction in the metabolic syndrome. Microcirculation 16: 667–684, 2009. doi: 10.3109/10739680903133722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goncalves AC, Tank J, Diedrich A, Hilzendeger A, Plehm R, Bader M, Luft FC, Jordan J, Gross V. Diabetic hypertensive leptin receptor-deficient db/db mice develop cardioregulatory autonomic dysfunction. Hypertension 53: 387–392, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124776. [DOI] [PubMed] [Google Scholar]
- 25.Gosmanova EO, Mikkelsen MK, Molnar MZ, Lu JL, Yessayan LT, Kalantar-Zadeh K, Kovesdy CP. Association of systolic blood pressure variability with mortality, coronary heart disease, stroke, and renal disease. J Am Coll Cardiol 68: 1375–1386, 2016. doi: 10.1016/j.jacc.2016.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gouty S, Regalia J, Cai F, Helke CJ. Alpha-lipoic acid treatment prevents the diabetes-induced attenuation of the afferent limb of the baroreceptor reflex in rats. Auton Neurosci 108: 32–44, 2003. doi: 10.1016/j.autneu.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Grassi G, Seravalle G, Colombo M, Bolla G, Cattaneo BM, Cavagnini F, Mancia G. Body weight reduction, sympathetic nerve traffic, and arterial baroreflex in obese normotensive humans. Circulation 97: 2037–2042, 1998. doi: 10.1161/01.CIR.97.20.2037. [DOI] [PubMed] [Google Scholar]
- 28.Grassi G, Seravalle G, Dell’Oro R, Turri C, Bolla GB, Mancia G. Adrenergic and reflex abnormalities in obesity-related hypertension. Hypertension 36: 538–542, 2000. doi: 10.1161/01.HYP.36.4.538. [DOI] [PubMed] [Google Scholar]
- 29.Grassi G, Trevano FQ, Seravalle G, Scopelliti F, Mancia G. Baroreflex function in hypertension: consequences for antihypertensive therapy. Prog Cardiovasc Dis 48: 407–415, 2006. doi: 10.1016/j.pcad.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Guillaume-Gentil C, Rohner-Jeanrenaud F, Abramo F, Bestetti GE, Rossi GL, Jeanrenaud B. Abnormal regulation of the hypothalamo-pituitary-adrenal axis in the genetically obese fa/fa rat. Endocrinology 126: 1873–1879, 1990. doi: 10.1210/endo-126-4-1873. [DOI] [PubMed] [Google Scholar]
- 31.Guimaraes PS, Huber DA, Campagnole-Santos MJ, Schreihofer AM. Development of attenuated baroreflexes in obese Zucker rats coincides with impaired activation of nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 306: R681–R692, 2014. doi: 10.1152/ajpregu.00537.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halmos KC, Gyarmati P, Xu H, Maimaiti S, Jancsó G, Benedek G, Smith BN. Molecular and functional changes in glucokinase expression in the brainstem dorsal vagal complex in a murine model of type 1 diabetes. Neuroscience 306: 115–122, 2015. doi: 10.1016/j.neuroscience.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han TS, Lean ME. A clinical perspective of obesity, metabolic syndrome and cardiovascular disease. JRSM Cardiovasc Dis 5: 2048004016633371, 2016. doi: 10.1177/2048004016633371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haque Z, Akbar N, Yasmin F, Haleem MA, Haleem DJ. Inhibition of immobilization stress-induced anorexia, behavioral deficits, and plasma corticosterone secretion by injected leptin in rats. Stress 16: 353–362, 2013. doi: 10.3109/10253890.2012.736047. [DOI] [PubMed] [Google Scholar]
- 35.Harris LE, Morgan DG, Balthasar N. Growth hormone secretagogue receptor deficiency in mice protects against obesity-induced hypertension. Physiol Rep 2: e00240, 2014. doi: 10.1002/phy2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holwerda SW, Vianna LC, Restaino RM, Chaudhary K, Young CN, Fadel PJ. Arterial baroreflex control of sympathetic nerve activity and heart rate in patients with type 2 diabetes. Am J Physiol Heart Circ Physiol 311: H1170–H1179, 2016. doi: 10.1152/ajpheart.00384.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.How JM, Wardak SA, Ameer SI, Davey RA, Sartor DM. Blunted sympathoinhibitory responses in obesity-related hypertension are due to aberrant central but not peripheral signalling mechanisms. J Physiol 592: 1705–1720, 2014. doi: 10.1113/jphysiol.2013.269670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber DA, Carmo JM, Castania JA, Fazan R Jr, Salgado HC. Does acute hyperglycemia alter rat aortic depressor nerve function? Braz J Med Biol Res 40: 1567–1576, 2007. doi: 10.1590/S0100-879X2007001100017. [DOI] [PubMed] [Google Scholar]
- 39.Huber DA, Schreihofer AM. Attenuated baroreflex control of sympathetic nerve activity in obese Zucker rats by central mechanisms. J Physiol 588: 1515–1525, 2010. doi: 10.1113/jphysiol.2009.186387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huber DA, Schreihofer AM. Altered regulation of the rostral ventrolateral medulla in hypertensive obese Zucker rats. Am J Physiol Heart Circ Physiol 301: H230–H240, 2011. doi: 10.1152/ajpheart.00075.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huber DA, Schreihofer AM. Exaggerated sympathoexcitatory reflexes develop with changes in the rostral ventrolateral medulla in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 311: R243–R253, 2016. doi: 10.1152/ajpregu.00085.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly-Cobbs A, Elgebaly MM, Li W, Ergul A. Pressure-independent cerebrovascular remodelling and changes in myogenic reactivity in diabetic Goto-Kakizaki rat in response to glycaemic control. Acta Physiol (Oxf) 203: 245–251, 2011. doi: 10.1111/j.1748-1716.2010.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan SA, Sattar MZ, Abdullah NA, Rathore HA, Abdulla MH, Ahmad A, Johns EJ. Obesity depresses baroreflex control of renal sympathetic nerve activity and heart rate in Sprague Dawley rats: role of the renal innervation. Acta Physiol (Oxf) 214: 390–401, 2015. doi: 10.1111/apha.12499. [DOI] [PubMed] [Google Scholar]
- 44.Laight DW, Desai KM, Gopaul NK, Anggård EE, Carrier MJ. Pro-oxidant challenge in vivo provokes the onset of NIDDM in the insulin resistant obese Zucker rat. Br J Pharmacol 128: 269–271, 1999. doi: 10.1038/sj.bjp.0702801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lambert EA, Rice T, Eikelis N, Straznicky NE, Lambert GW, Head GA, Hensman C, Schlaich MP, Dixon JB. Sympathetic activity and markers of cardiovascular risk in nondiabetic severely obese patients: the effect of the initial 10% weight loss. Am J Hypertens 27: 1308–1315, 2014. doi: 10.1093/ajh/hpu050. [DOI] [PubMed] [Google Scholar]
- 46.Landsberg L. Insulin-mediated sympathetic stimulation: role in the pathogenesis of obesity-related hypertension (or, how insulin affects blood pressure, and why). J Hypertens 19, Suppl: 523–528, 2001. doi: 10.1097/00004872-200103001-00001. [DOI] [PubMed] [Google Scholar]
- 47.Lattanzi S, Brigo F, Vernieri F, Silvestrini M. Visit-to-visit variability in blood pressure and Alzheimer’s disease. J Clin Hypertens (Greenwich) 20: 918–924, 2018. doi: 10.1111/jch.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lattanzi S, Viticchi G, Falsetti L, Buratti L, Luzzi S, Provinciali L, Silvestrini M. Visit-to-visit blood pressure variability in Alzheimer disease. Alzheimer Dis Assoc Disord 28: 347–351, 2014. doi: 10.1097/WAD.0000000000000040. [DOI] [PubMed] [Google Scholar]
- 49.Levin BE, Comai K, Sullivan AC. Metabolic and sympatho-adrenal abnormalities in the obese Zucker rat: effect of chronic phenoxybenzamine treatment. Pharmacol Biochem Behav 14: 517–525, 1981. doi: 10.1016/0091-3057(81)90311-7. [DOI] [PubMed] [Google Scholar]
- 50.Liepinsh E, Skapare E, Svalbe B, Makrecka M, Cirule H, Dambrova M. Anti-diabetic effects of mildronate alone or in combination with metformin in obese Zucker rats. Eur J Pharmacol 658: 277–283, 2011. doi: 10.1016/j.ejphar.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 51.Limberg JK, Farni KE, Taylor JL, Dube S, Basu A, Basu R, Wehrwein EA, Joyner MJ. Autonomic control during acute hypoglycemia in type 1 diabetes mellitus. Clin Auton Res 24: 275–283, 2014. doi: 10.1007/s10286-014-0253-y. [DOI] [PubMed] [Google Scholar]
- 52.Ma C, Avenell A, Bolland M, Hudson J, Stewart F, Robertson C, Sharma P, Fraser C, MacLennan G. Effects of weight loss interventions for adults who are obese on mortality, cardiovascular disease, and cancer: systematic review and meta-analysis. BMJ 359: j4849, 2017. doi: 10.1136/bmj.j4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mamnoor PK, Hegde P, Datla SR, Damarla RK, Rajagopalan R, Chakrabarti R. Antihypertensive effect of ragaglitazar: a novel PPARalpha and gamma dual activator. Pharmacol Res 54: 129–135, 2006. doi: 10.1016/j.phrs.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 54.Mancia G, Bousquet P, Elghozi JL, Esler M, Grassi G, Julius S, Reid J, Van Zwieten PA. The sympathetic nervous system and the metabolic syndrome. J Hypertens 25: 909–920, 2007. doi: 10.1097/HJH.0b013e328048d004. [DOI] [PubMed] [Google Scholar]
- 55.Marques-Neto SR, Castiglione RC, Pontes A, Oliveira DF, Ferraz EB, Nascimento JH, Bouskela E. Effects of incretin-based therapies on neuro-cardiovascular dynamic changes induced by high fat diet in rats. PLoS One 11: e0148402, 2016. doi: 10.1371/journal.pone.0148402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin CA, Cameron JD, Chen SS, McGrath BP. Two hour glucose post loading: a biomarker of cardiovascular risk in isolated clinic hypertension. J Hypertens 29: 749–757, 2011. doi: 10.1097/HJH.0b013e328342eeeb. [DOI] [PubMed] [Google Scholar]
- 57.Martin CA, McGrath BP. White-coat hypertension. Clin Exp Pharmacol Physiol 41: 22–29, 2014. doi: 10.1111/1440-1681.12114. [DOI] [PubMed] [Google Scholar]
- 58.Matsutani D, Sakamoto M, Iuchi H, Minato S, Suzuki H, Kayama Y, Takeda N, Horiuchi R, Utsunomiya K. Glycemic variability in continuous glucose monitoring is inversely associated with baroreflex sensitivity in type 2 diabetes: a preliminary report. Cardiovasc Diabetol 17: 36, 2018. doi: 10.1186/s12933-018-0683-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCully BH, Brooks VL, Andresen MC. Diet-induced obesity severely impairs myelinated aortic baroreceptor reflex responses. Am J Physiol Heart Circ Physiol 302: H2083–H2091, 2012. doi: 10.1152/ajpheart.01200.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miao CY, Xie HH, Zhan LS, Su DF. Blood pressure variability is more important than blood pressure level in determination of end-organ damage in rats. J Hypertens 24: 1125–1135, 2006. doi: 10.1097/01.hjh.0000226203.57818.88. [DOI] [PubMed] [Google Scholar]
- 61.Mobley SC, Mandel DA, Schreihofer AM. Systemic cholecystokinin differentially affects baro-activated GABAergic neurons in rat caudal ventrolateral medulla. J Neurophysiol 96: 2760–2768, 2006. doi: 10.1152/jn.00526.2006. [DOI] [PubMed] [Google Scholar]
- 62.Mulnier HE, Seaman HE, Raleigh VS, Soedamah-Muthu SS, Colhoun HM, Lawrenson RA, de Vries CS. Risk of myocardial infarction in men and women with type 2 diabetes in the UK: a cohort study using the General Practice Research Database. Diabetologia 51: 1639–1645, 2008. doi: 10.1007/s00125-008-1076-y. [DOI] [PubMed] [Google Scholar]
- 63.Okada M, Buñag RD. Insulin acts centrally to enhance reflex tachycardia in conscious rats. Am J Physiol 266: R481–R486, 1994. doi: 10.1152/ajpregu.1994.266.2.R481. [DOI] [PubMed] [Google Scholar]
- 64.Osmond JM, Mintz JD, Dalton B, Stepp DW. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension 53: 381–386, 2009. doi: 10.1161/HYPERTENSIONAHA.108.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Overton JM, Williams TD, Chambers JB, Rashotte ME. Cardiovascular and metabolic responses to fasting and thermoneutrality are conserved in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 280: R1007–R1015, 2001. doi: 10.1152/ajpregu.2001.280.4.R1007. [DOI] [PubMed] [Google Scholar]
- 66.Owyang C, Heldsinger A. Vagal control of satiety and hormonal regulation of appetite. J Neurogastroenterol Motil 17: 338–348, 2011. doi: 10.5056/jnm.2011.17.4.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paleczny B, Siennicka A, Zacharski M, Jankowska EA, Ponikowska B, Ponikowski P. Increased body fat is associated with potentiation of blood pressure response to hypoxia in healthy men: relations with insulin and leptin. Clin Auton Res 26: 107–116, 2016. doi: 10.1007/s10286-015-0338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parati G, Ochoa JE, Lombardi C, Bilo G. Assessment and management of blood-pressure variability. Nat Rev Cardiol 10: 143–155, 2013. [Erratum in Nat Rev Cardiol 11: 314, 2014.] doi: 10.1038/nrcardio.2013.1. [DOI] [PubMed] [Google Scholar]
- 69.Paton JF. Pattern of cardiorespiratory afferent convergence to solitary tract neurons driven by pulmonary vagal C-fiber stimulation in the mouse. J Neurophysiol 79: 2365–2373, 1998. doi: 10.1152/jn.1998.79.5.2365. [DOI] [PubMed] [Google Scholar]
- 70.Pricher MP, Freeman KL, Brooks VL. Insulin in the brain increases gain of baroreflex control of heart rate and lumbar sympathetic nerve activity. Hypertension 51: 514–520, 2008. doi: 10.1161/HYPERTENSIONAHA.107.102608. [DOI] [PubMed] [Google Scholar]
- 71.Rothwell PM. Limitations of the usual blood-pressure hypothesis and importance of variability, instability, and episodic hypertension. Lancet 375: 938–948, 2010. doi: 10.1016/S0140-6736(10)60309-1. [DOI] [PubMed] [Google Scholar]
- 72.Rouru J, Huupponen R, Pesonen U, Koulu M. Subchronic treatment with metformin produces anorectic effect and reduces hyperinsulinemia in genetically obese Zucker rats. Life Sci 50: 1813–1820, 1992. doi: 10.1016/0024-3205(92)90066-X. [DOI] [PubMed] [Google Scholar]
- 73.Sartor DM, Verberne AJ. Cholecystokinin selectively affects presympathetic vasomotor neurons and sympathetic vasomotor outflow. Am J Physiol Regul Integr Comp Physiol 282: R1174–R1184, 2002. doi: 10.1152/ajpregu.00500.2001. [DOI] [PubMed] [Google Scholar]
- 74.Schreihofer AM, Guyenet PG. Baro-activated neurons with pulse-modulated activity in the rat caudal ventrolateral medulla express GAD67 mRNA. J Neurophysiol 89: 1265–1277, 2003. doi: 10.1152/jn.00737.2002. [DOI] [PubMed] [Google Scholar]
- 75.Schreihofer AM, Mandel DA, Mobley SC, Stepp DW. Impairment of sympathetic baroreceptor reflexes in obese Zucker rats. Am J Physiol Heart Circ Physiol 293: H2543–H2549, 2007. doi: 10.1152/ajpheart.01201.2006. [DOI] [PubMed] [Google Scholar]
- 76.Seravalle G, Lonati L, Buzzi S, Cairo M, Quarti Trevano F, Dell’Oro R, Facchetti R, Mancia G, Grassi G. Sympathetic nerve traffic and baroreflex function in optimal, normal, and high-normal blood pressure states. J Hypertens 33: 1411–1417, 2015. doi: 10.1097/HJH.0000000000000567. [DOI] [PubMed] [Google Scholar]
- 77.Seravalle G, Grassi G. Obesity and hypertension. Pharmacol Res 122: 1–7, 2017. [Erratum in Pharmacol Res 124: 156, 2017.] doi: 10.1016/j.phrs.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 78.Shank SS, Scheuer DA. Glucocorticoids reduce responses to AMPA receptor activation and blockade in nucleus tractus solitarius. Am J Physiol Heart Circ Physiol 284: H1751–H1761, 2003. doi: 10.1152/ajpheart.01033.2002. [DOI] [PubMed] [Google Scholar]
- 79.Skrapari I, Tentolouris N, Perrea D, Bakoyiannis C, Papazafiropoulou A, Katsilambros N. Baroreflex sensitivity in obesity: relationship with cardiac autonomic nervous system activity. Obesity (Silver Spring) 15: 1685–1693, 2007. doi: 10.1038/oby.2007.201. [DOI] [PubMed] [Google Scholar]
- 80.Stern JS, Johnson PR. Spontaneous activity and adipose cellularity in the genetically obese Zucker rat (fafa). Metabolism 26: 371–380, 1977. doi: 10.1016/0026-0495(77)90104-4. [DOI] [PubMed] [Google Scholar]
- 81.Stocker SD, Meador R, Adams JM. Neurons of the rostral ventrolateral medulla contribute to obesity-induced hypertension in rats. Hypertension 49: 640–646, 2007. doi: 10.1161/01.HYP.0000254828.71253.dc. [DOI] [PubMed] [Google Scholar]
- 82.Straznicky NE, Lambert EA, Lambert GW, Masuo K, Esler MD, Nestel PJ. Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. J Clin Endocrinol Metab 90: 5998–6005, 2005. doi: 10.1210/jc.2005-0961. [DOI] [PubMed] [Google Scholar]
- 83.Straznicky NE, Eikelis N, Lambert EA, Esler MD. Mediators of sympathetic activation in metabolic syndrome obesity. Curr Hypertens Rep 10: 440–447, 2008. doi: 10.1007/s11906-008-0083-1. [DOI] [PubMed] [Google Scholar]
- 84.Sucharita S, Tinku T, Raj T, Kurpad AV, Vaz M. Cardiovascular autonomic responses to hyperinsulinemia in young adult males of normal and low body mass index. Auton Neurosci 161: 121–125, 2011. doi: 10.1016/j.autneu.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 85.Verberne AJ, Guyenet PG. Medullary pathway of the Bezold-Jarisch reflex in the rat. Am J Physiol Regul Integr Comp Physiol 263: R1195–R1202, 1992. doi: 10.1152/ajpregu.1992.263.6.R1195. [DOI] [PubMed] [Google Scholar]
- 86.Wan S, Browning KN. d-Glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol 294: G757–G763, 2008. doi: 10.1152/ajpgi.00576.2007. [DOI] [PubMed] [Google Scholar]
- 87.Wang Q, Dryden S, Frankish HM, Bing C, Pickavance L, Hopkins D, Buckingham R, Williams G. Increased feeding in fatty Zucker rats by the thiazolidinedione BRL 49653 (rosiglitazone) and the possible involvement of leptin and hypothalamic neuropeptide Y. Br J Pharmacol 122: 1405–1410, 1997. doi: 10.1038/sj.bjp.0701535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weston M, Wang H, Stornetta RL, Sevigny CP, Guyenet PG. Fos expression by glutamatergic neurons of the solitary tract nucleus after phenylephrine-induced hypertension in rats. J Comp Neurol 460: 525–541, 2003. doi: 10.1002/cne.10663. [DOI] [PubMed] [Google Scholar]
- 89.Yamaguchi Y, Wada M, Sato H, Nagasawa H, Koyama S, Takahashi Y, Kawanami T, Kato T. Impact of ambulatory blood pressure variability on cerebral small vessel disease progression and cognitive decline in community-based elderly Japanese. Am J Hypertens 27: 1257–1267, 2014. doi: 10.1093/ajh/hpu045. [DOI] [PubMed] [Google Scholar]
- 90.Yasuda N, Inoue T, Nagakura T, Yamazaki K, Kira K, Saeki T, Tanaka I. Metformin causes reduction of food intake and body weight gain and improvement of glucose intolerance in combination with dipeptidyl peptidase IV inhibitor in Zucker fa/fa rats. J Pharmacol Exp Ther 310: 614–619, 2004. doi: 10.1124/jpet.103.064964. [DOI] [PubMed] [Google Scholar]
- 91.Yettefti K, Orsini J-C, Perrin J. Characteristics of glycemia-sensitive neurons in the nucleus tractus solitarii: possible involvement in nutritional regulation. Physiol Behav 61: 93–100, 1997. doi: 10.1016/S0031-9384(96)00358-7. [DOI] [PubMed] [Google Scholar]
- 92.Yoshikawa T, Kishi T, Shinohara K, Takesue K, Shibata R, Sonoda N, Inoguchi T, Sunagawa K, Tsutsui H, Hirooka Y. Arterial pressure lability is improved by sodium-glucose cotransporter 2 inhibitor in streptozotocin-induced diabetic rats. Hypertens Res 40: 646–651, 2017. doi: 10.1038/hr.2017.14. [DOI] [PubMed] [Google Scholar]
- 93.Zhao D, McCully BH, Brooks VL. Rosiglitazone improves insulin sensitivity and baroreflex gain in rats with diet-induced obesity. J Pharmacol Exp Ther 343: 206–213, 2012. doi: 10.1124/jpet.112.194738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]