

Abstract
Adverse intrauterine conditions cause fetal growth restriction and increase the risk of adult cardiovascular disease. We hypothesize that intrauterine hypoxia impairs fetal heart function, is sustained after birth, and manifests as both cardiac and mitochondrial dysfunction in offspring guinea pigs (GPs). Pregnant GPs were exposed to 10.5% O2 (HPX) at 50 days of gestation (full term = 65 days) or normoxia (NMX) for the duration of the pregnancy. Pups were allowed to deliver vaginally and raised in a NMX environment. At 90 days of age, mean arterial pressure (MAP) was measured in anesthetized GPs. NMX and prenatally HPX offspring underwent echocardiographic imaging for in vivo measurement of left ventricular cardiac morphology and function, and O2 consumption rates and complex IV enzyme activity were measured from isolated cardiomyocytes and mitochondria, respectively. Prenatal HPX increased (P < 0.01) MAP (52.3 ± 1.3 and 58.4 ± 1.1 mmHg in NMX and HPX, respectively) and decreased (P < 0.05) stroke volume (439.8 ± 54.5 and 289.4 ± 15.8 μl in NMX and HPX, respectively), cardiac output (94.4 ± 11.2 and 67.3 ± 3.8 ml/min in NMX and HPX, respectively), ejection fraction, and fractional shortening in male, but not female, GPs. HPX had no effect on left ventricular wall thickness or end-diastolic volume in either sex. HPX reduced mitochondrial maximal respiration and respiratory reserve capacity and complex IV activity rates in hearts of male, but not female, GPs. Prenatal HPX is a programming stimulus that increases MAP and decreases cardiac and mitochondrial function in male offspring. Sex-related differences in the contractile and mitochondrial responses suggest that female GPs are protected from cardiovascular programming of prenatal HPX.
Keywords: heart, hypoxia, mitochondria, programming, ventricular function
INTRODUCTION
Intrauterine hypoxia is one of the most significant complications during pregnancy, challenging normal fetal growth and development (6, 27, 33). The hypoxemic fetus adapts to its environment by redistributing its cardiac output (CO) to critical organs of need, such as the heart, brain, and adrenal glands (60, 62, 65, 66), resulting in asymmetric fetal growth restriction. Chronic exposure to intrauterine hypoxia may compromise fetal organ function, depending on the duration and severity of hypoxia and the gestational age at the time of exposure. Several studies have shown that chronic hypoxia induces asymmetric fetal growth restriction, accompanied by altered cardiovascular responses associated with aortic wall thickening, cardiac hypertrophy, and enhanced sympathetic innervation of peripheral arteries (for reviews see Refs. 32 and 37) and reduced cardiomyocyte endowment (13) in animal models. Long-term high-altitude hypoxia induces cardiac contractile dysfunction of fetal sheep associated with altered cardiac Ca2+ regulation and β-adrenergic receptor function (29). Furthermore, chronic maternal hypoxia decreases gene expression of PKCε and heat shock protein 70 and increases membrane type 1 matrix metalloproteinase in fetal rat hearts (58, 59, 79) and increases expression of inducible nitric oxide synthase, metalloproteinases, and proinflammatory cytokines in fetal guinea pig hearts (23, 26, 56, 78). Thus, increased gene expression attributed to oxidative/nitrative stress and cardiac inflammation, as well as cardiac remodeling and Ca2+ dysregulation, is implicated in several animal models as underlying causes of contractile dysfunction in the hypoxic fetal heart.
Programming effects of prenatal hypoxia on cardiac function in the offspring, addressing the developmental origins of health and disease (DOHaD) hypothesis that intrauterine stress impacts fetal growth and organ function and has lasting functional consequences in the offspring (11, 34), have also been reported (32, 55). Several studies support programming effects of prenatal hypoxia in offspring mammalian hearts, demonstrating increased risk for ischemia-reperfusion injury (44, 45, 84, 85), reduced cardiac contractile function (31, 68), reduced cardiac metabolism (69), and increased ventricular arrhythmias (54).
We recently focused on the effects of prenatal hypoxia on mitochondrial function of fetal and offspring hearts. We reported that prenatal hypoxia decreases cytochrome c oxidase activity in fetal (4) and offspring (5) guinea pig heart ventricles as evidence of prenatal hypoxia as a stimulus that impairs cardiac mitochondrial function. Attenuation of mitochondrial function is well documented to increase risk of heart dysfunction in the adult (16, 48). We propose that prenatal hypoxia inhibits mitochondrial respiratory activity and contributes to reduced ventricular performance postnatally. To test this idea, we measured contractile function using noninvasive echocardiography and indexes of mitochondrial respiration of heart cells of offspring exposed to prenatal hypoxia. The results of this study link the effects of intrauterine hypoxia in the fetal heart to mitochondrial and cardiac programming in the offspring.
METHODS
Animal model.
All animal procedures were approved by the University of Maryland Institutional Animal Care and Use Committee in accordance with Association for Assessment and Accreditation of Laboratory Animal Care-accredited procedures (Animal Welfare Assurance No. A3200-01). The pregnant guinea pig is an excellent animal model of human pregnancy for study of the programming of intrauterine stress. The similarities of the pregnant guinea pig to human pregnancy include placental morphology, such as a hemomonochorial placenta with deep invasion, a luteoplacental shift and elevated progesterone levels at full term, fetal growth characteristics of similar fat content and epicardial depots, and timing of myogenesis, as well as prenatal maturation of fetal cardiovascular and nervous systems (22, 25, 35, 51, 52). This provides an excellent model for the study of cardiovascular programming of the offspring to intrauterine hypoxia because of the relative maturity of the full-term fetus and its adaptive responses to intrauterine stress.
Virgin female guinea pigs were mated in groups of three with a single male following visual evidence of vaginal membrane opening. Time-mated pregnant guinea pigs were randomly assigned to normoxia (NMX, room air) or hypoxia (HPX, 10.5% O2) treatment and exposed to NMX during the entire gestation or to HPX during the last 14 days of pregnancy, a period of rapid fetal growth and increased O2 utilization by the fetus. Chronic hypoxia does not reduce either maternal food or water intake and, therefore, represents a hypoxic model without nutrient restriction (80). Pregnant sows were allowed to deliver vaginally, and pups born into a hypoxic environment were removed to normal room air within 1 day of delivery. Newborn pups were housed in room air with their mothers until weaning (30 days) and then placed in individual cages. To generate offspring for the four experimental series (i.e., blood pressure measurement, echocardiography, cell isolation for extracellular flux analysis, and mitochondrial isolation for enzymatic assay), 89 sows were used to generate 135 offspring. A single male and a single female were selected from the same litter, if both were present, for comparison of sex differences. Litter size was selected based on two to four offspring per litter to avoid the influence of litter numbers on fetal body weight. Offspring body weight and food and water intake rates were monitored at 33 days of age and in 3-day intervals until 90 days of age, a time of established reproductive maturity and a steady growth profile. All measurements of blood pressure, left ventricular (LV) morphology and function, and indexes of mitochondrial function were acquired from 90-day-old guinea pig offspring. Food and water intake rates were obtained from animals being measured for arterial blood pressure.
Measurement of arterial blood pressure.
For measurement of arterial blood pressure, NMX or HPX male (n = 15) and female (n = 16) guinea pig offspring were anesthetized with ketamine (80 mg/kg sc)-xylazine (10 mg/kg sc). After lidocaine injection and a skin incision, the right brachial artery was cannulated with polyethylene tubing filled with heparinized saline. Systolic, diastolic, and mean arterial blood pressures were recorded (Power Laboratory 800, ADInstruments, Colorado Springs, CO) over a 5-min period and analyzed with Chart v. 4.2 software (ADInstruments).
Echocardiography.
LV morphology and function of NMX or HPX male (n = 18) and female (n = 18) offspring were measured by echocardiography. Animals were anesthetized with 1–5% isoflurane in balance with O2 and studied using a Vevo 2100 imaging system (FUJIFILM-VisualSonics, Toronto, ON, Canada) equipped with a 20-MHz scan head. LV dimension and wall thickness at the level of the papillary muscle were measured using two-dimensional guided M-mode imaging via parasternal short-axis view, which was also used for calculation of LV fractional shortening [FS = (LVEDD − LVESD)/LVEDD × 100, where LVEDD is LV end-diastolic diameter and LVESD is LV end-systolic diameter], ejection fraction [EF = (EDV − SV)/EDV × 100, where EDV is end-diastolic volume and SV is stroke volume], and LV mass. Two-dimensional guided M-mode imaging of parasternal long- and short-axis views was performed for measurements of aortic diameter and LV chamber size/wall thickness, respectively, and an apical four-chamber view (a “long”-axis view in nature) was used for measurement of the E/A ratio [an index of diastolic function assessed by the velocity waveform during passive (E wave peak velocity) and active (A wave peak velocity during atrial contraction) filling of the LV during diastole] from the mitral inflow (42, 53). Under guidance of color Doppler imaging, aortic flow and mitral inflow were measured using pulse-wave Doppler imaging via suprasternal notch view and apical four-chamber view, respectively. Data were calculated according to the generally accepted formulas, as previously reported (17, 18).
Isolation of mitochondria.
Tissue was ground with a mortar and pestle in liquid N2 to isolate mitochondria from frozen heart LV of NMX or HPX male and female offspring. Frozen tissue was homogenized in 1× homogenization buffer (0.25 M sucrose, 5 mM HEPES, and 1 mM EDTA, pH 7.2) with zirconium oxide beads in a VWR multitube vortexer at high speed for 10 min at 4°C. Samples were centrifuged twice at 600 g for 10 min at 4°C to remove cellular debris. The supernatant was carefully transferred to a clean 1.5-ml tube, and the mitochondria were pelleted by centrifugation at 12,500 g for 10 min at 4°C. The mitochondrial pellet was resuspended in 100 μl of homogenization buffer with 0.1 mM N-dodecyl β-d-maltoside (Sigma, St. Louis, MO). Mitochondrial protein of each sample was determined by the Bio-Rad protein assay. Purity of the mitochondrial-enriched fraction of the heart tissue is ∼100% (98.2–99.6%), similar to that reported for placenta mitochondria (75). This percent contamination is based on low contamination from nuclear (1.8%) or cytosolic (0.4%) proteins by targeting specific nuclear (lamin A/C), cytoplasmic (α-tubulin), and mitochondrial [voltage-dependent anion channel (VDAC)] proteins with primary antibodies using Western blot analysis. Percent contamination was quantified by normalization of band density to VDAC in the mitochondrial fraction.
Mitochondrial complex activity assay.
Respiratory complex IV contains cytochrome c oxidase, which reduces O2 to H2O in the respiratory chain. Enzyme activity was measured in isolated, enriched cardiac mitochondrial fractions of frozen LV of NMX or HPX male (n = 16) and female (n = 16) offspring. Complex IV activity is measured colorimetrically by monitoring the oxidation rate of cytochrome c. Briefly, 3 μg of mitochondrial protein were added to each well of a 96-well plate containing the assay buffer [10 mM Tris·HCl (pH 7.0) and 120 mM KCl + reduced cytochrome c] (76). Optical density was measured at 550 nm in a 96-well plate reader (BioTek, Winooski, VT) for 30 min at 10-s intervals. Our assay was optimized using a range of protein amounts (2–5 μg) that generated a minimum and a maximum rate of oxidation; a 3-μg amount was selected based on the kinetics profile for oxidation that fell between these curves and was used as the standardized amount of total mitochondrial protein for determination of the slope in the linear range.
The rate of the reaction was calculated in the linear range according to the following formula: cytochrome c oxidase activity (units/mg mitochondrial protein) = (ΔOD/Δt) ÷ [ε * protein (mg)], where OD is optical density, t is time, and ε is the extinction coefficient (7.04 mM−1·cm−1).
Isolation of cardiomyocytes of male and female offspring hearts.
After thoracotomy, hearts were excised from anesthetized and heparinized NMX or HPX male (n = 19) and female (n = 17) guinea pigs for isolation of cardiac cells. Hearts were immediately placed in iced physiological buffer solution (PBS) and mounted via the aorta onto a glass cannula of a modified Langendorff heart perfusion apparatus. Guinea pig cardiomyocytes were isolated using a modified procedure reported for isolation of fetal sheep cardiac cells (38). Hearts were retrograde-perfused at 37°C with a low-Ca2+ (no Ca2+ added) Tyrode solution (in mM: 140 NaCl, 5 KCl, 10 HEPES, 10 glucose, and 1 MgCl2, pH 7.35) without enzymes for 5 min and then with Tyrode solution containing enzymes [collagenase (80 U/ml), protease (0.59 U/ml), and albumin (1 mg/ml)] for 12 min. Hearts were then perfused with Kraft-Brühe (KB) buffer (in mM: 30 KCl, 10 HEPES, 10 glucose, 74 potassium glutamate, 20 taurine, 1.5 MgSO4·7H2O, 0.5 EGTA, and 30 KH2PO4, pH 7.37) for 5 min to wash out the enzymes. Hearts were removed from the apparatus and placed in a beaker containing warmed KB buffer for gentle mincing to release cells from the heart. Cells were filtered through 150-μm nylon mesh and flushed into a beaker containing 37°C KB buffer. Cells were pelleted twice by centrifugation (200–250 g) and resuspended in KB buffer, and the supernatant was discarded. The final cell pellet was resuspended in DMEM containing 1× penicillin (100 U)-streptomycin (100 μg) (PenStrep, catalog no. 15140-122, Life Technologies) and then transferred to a 50-ml tube containing ~10–15 ml of DMEM with d-glucose (1 g/l). After isolation, cells were counted on an Invitrogen Countess automated cell counter, and percent cell viability (~95%) was determined using Trypan blue. An aliquot volume was calculated for transfer of 20,000 cells/100 μl to laminin-coated 96-well microplates and incubated for 2–4 h at 37°C and 5% CO2 in a humidified incubator.
Mitochondrial respiration studies of isolated cardiomyocytes.
Mitochondrial respiration of isolated heart cells was measured using an extracellular flux analyzer (Seahorse XF24, Agilent Technologies, Santa Clara, CA), as previously described (64) and modified for guinea pig cardiac cells. In a separate series of experiments, conditions were optimized (20,000 cells/well) for cardiac cell density by seed density experiments (15,000–25,000 cells/well) and drug concentrations of oligomycin (1 µg/ml, an ATP synthase inhibitor), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone [FCCP, 1 μM, a protonophore that uncouples respiration, inducing maximal O2 consumption rate (OCR)], and antimycin A (10 μM, an inhibitor of complex III for determination of cellular nonmitochondrial O2 consumption). OCRs of cardiac cells obtained from a single guinea pig heart were measured at baseline and following separate injections of oligomycin, FCCP, and antimycin A. Maximal OCR is the difference between OCR measured after FCCP and nonmitochondrial OCR measured following antimycin A. Reserve capacity is the difference between maximal OCR and baseline OCR. Stock concentrations of oligomycin (catalog no. AB674, Sigma), FCCP (catalog no. C2920, Sigma), and antimycin A (catalog no. 75351, Sigma) were prepared in ethanol.
Statistics.
Values are means ± SE. Body weight and food and water intake rates were compared between NMX and prenatally HPX groups by three-way repeated-measures analysis of variance, with days, sex, and treatment as independent variables. Post hoc tests were not performed if there were no significant differences between the groups. Mitochondrial indexes (e.g., OCR and complex IV activities) and cardiac function were compared between NMX and prenatally HPX groups by two-way analysis of variance, with treatment and sex as variables. P < 0.05 was considered significantly different. Statistical analyses were carried out using Systat software (San Jose, CA).
RESULTS
Body weight and food and water intake rates.
Figure 1 illustrates the temporal profile of body weight and food and water intake rates of male (n = 8 NMX and 8 HPX) and female (n = 7 NMX and 8 HPX) guinea pig offspring. Birth weights of neonates exposed to prenatal hypoxia were significantly (P < 0.05) decreased at 1 day of age [male: 91.3 ± 5.5 g (NMX) and 80.7 ± 3.5 g (HPX); female: 97.9 ± 5.2 g (NMX) and 84.9 ± 2.4 g (HPX)] but were not different at 90 days of age (Fig. 1). Prenatal hypoxia did not affect food or water intake rates over the 90-day period. Furthermore, there were no differences in heart, liver, or kidney weight in HPX offspring compared with their NMX controls, regardless of sex. Exposure to prenatal hypoxia reduced (P < 0.05) birth weights in male and female offspring.
Fig. 1.

Effect of prenatal hypoxia on body weight (BW) and food and water intake rates of guinea pig offspring. Guinea pig offspring were exposed to prenatal normoxia (NMX) or hypoxia (HPX, 10.5% O2 for 14 days) and raised in a normoxic environment until 90 days of age. Values are means ± SE; n = 8 NMX and 8 HPX males and 7 NMX and 8 HPX females.
Mean arterial blood pressure.
Arterial blood pressures were measured in male (n = 8 NMX and 7 HPX) and female (n = 8 NMX and 8 HPX) NMX and HPX guinea pig offspring. There were no differences in arterial blood pressure between males and females born to NMX mothers. In contrast, prenatal HPX significantly increased (P < 0.01) arterial blood pressure by 12% in male (50.7 ± 1.7 and 57.4 ± 1.6 mmHg in NMX and HPX, respectively), but not female (50.5 ± 2.5 and 52.0 ± 2.0 mmHg in NMX and HPX, respectively), offspring, with no effect on heart rate (male: 161 ± 4 and 171 ± 6 beats/min in NMX and HPX, respectively; female: 169 ± 5 and 163 ± 6 beats/min in NMX and HPX, respectively). The HPX-induced increase in mean arterial blood pressure in males was accompanied by a significant increase (P < 0.01) in systolic (60.3 ± 2.1 vs. 68.3 ± 2.3 mmHg in NMX and HPX, respectively) and diastolic (41.4 ± 1.3 and 46.9 ± 1.1 mmHg in NMX and HPX, respectively) blood pressures, with no difference in pulse pressure (18.8 ± 1.0 and 21.4 ± 1.5 mmHg in NMX and HPX, respectively). In females, systolic pressure (59.7 ± 2.9 and 61.8 ± 2.4 mmHg in NMX and HPX, respectively), diastolic pressure (41.2 ± 2.1 and 42.4 ± 1.7 mmHg in NMX and HPX, respectively), pulse pressure (18.5 ± 1.1 and 19.4 ± 0.9 mmHg in NMX and HPX, respectively), and heart rate (169.1 ± 5.4 vs 163.2 ± 6.1 beats/min in NMX and HPX, respectively) were similar between NMX and HPX offspring.
Cardiac morphology and function.
LV morphology and function were measured by echocardiography from 90-day-old male (n = 9 NMX and 9 HPX) and female (n = 9 NMX and 9 HPX) offspring exposed to NMX and prenatal HPX (Fig. 2). No significant differences between NMX and HPX groups were found in males or females for LV dimension, LV wall thickness, or interventricular septal wall thickness at systole or diastole. In addition, there were no differences between NMX and HPX offspring in total heart mass (male: 1.94 ± 0.11 and 1.90 ± 0.05 g in NMX and HPX, respectively; female: 1.94 ± 0.11 and 1.90 ± 0.05 g in NMX and HPX, respectively), LV weight (male: 1.50 ± 0.10 and 1.41 ± 0.04 g in NMX and HPX, respectively; female: 1.50 ± 0.10 and 1.41 ± 0.04 g in NMX and HPX, respectively), and LV weight-to-total weight ratio (male: 0.76 ± 0.01 and 0.74 ± 0.01 in NMX and HPX, respectively, female: 0.75 ± 0.01 and 0.75 ± 0.01 in NMX and HPX, respectively). Thus there were no signs of LV hypertrophy in this model, despite the increase in mean arterial pressure.
Fig. 2.

Effect of prenatal hypoxia on morphological indexes of guinea pig offspring. Guinea pig offspring were exposed to prenatal normoxia (NMX) or hypoxia (HPX, 10.5% O2 for 14 days) and raised in a normoxic environment until 90 days of age. Morphological indexes of left ventricular (LV) diameter, LV wall thickness, and interventricular septal (IVS) thickness were measured using M-mode imaging (Vevo 2100) Doppler ultrasound biomicroscopy during systole and diastole of the cardiac cycle. Values are means ± SE; n = 9 NMX and 9 HPX males and 9 NMX and 9 HPX females.
Male HPX offspring demonstrated compromised LV global performance (SV and CO) and systolic function (EF and FS) (Fig. 3). Prenatal HPX significantly (P < 0.05) decreased SV, CO, and cardiac index (CO/body weight) but had no effect on heart rate (250 ± 14 and 237 ± 11 beats/min in NMX and HPX, respectively) in male offspring. In females, there was no effect of HPX on heart rate (222 ± 2 and 222 ± 6 beats/min in NMX and HPX, respectively), SV, CO, or cardiac index compared with NMX controls. Furthermore, prenatal HPX decreased LV EF and FS in male, not female, hearts. Prenatal HPX had no effect on the E/A ratio of the 90-day-old male offspring (1.65 ± 0.09 and 1.72 ± 0.07 in NMX and HPX, respectively) but decreased (P < 0.05) the E/A ratio in female offspring (1.88 ± 0.14 and 1.43 ± 0.08 in NMX and HPX, respectively), which was attributed to a decrease in the maximal peak velocity of the E wave.
Fig. 3.

Effect of prenatal hypoxia on functional indexes of guinea pig offspring. Guinea pig offspring were exposed to prenatal normoxia (NMX) or hypoxia (HPX, 10.5% O2 for 14 days) and raised in a normoxic environment until 90 days of age. Functional indexes of stroke volume, cardiac output, ejection fraction, and fractional shortening of left ventricular diameter (LVD) were measured using pulse-wave Doppler ultrasound. Values are means ± SE; n = 9 NMX and 9 HPX males and 9 NMX and 9 HPX females. *P < 0.05 vs. NMX.
Mitochondrial respiratory function and complex IV activity.
OCRs were measured in isolated cardiomyocytes derived from single hearts of male (n = 9 NMX and 10 HPX) and female (n = 9 NMX and 8 HPX) offspring exposed to NMX or prenatal HPX. OCRs of baseline, in association with ATP synthesis, electron leak, maximal rate, and reserve capacity, were measured as absolute OCR and OCR normalized to baseline (Fig. 4). Baseline OCR and leak OCR were similar between cardiomyocytes of NMX and HPX hearts, regardless of sex. There was a significant increase (P < 0.05) in OCR associated with ATP synthesis in male hearts exposed to prenatal HPX but no difference when OCR was normalized to baseline (Fig. 5). In contrast, HPX significantly reduced (P < 0.05) both maximal OCR and reserve capacity (percent baseline) in males, but not females, compared with their respective NMX controls. Exposure to prenatal HPX reduced complex IV activity in hearts of males (n = 8 NMX and 8 HPX), but not females (n = 8 NMX and 8 HPX), compared with NMX controls (Fig. 6).
Fig. 4.
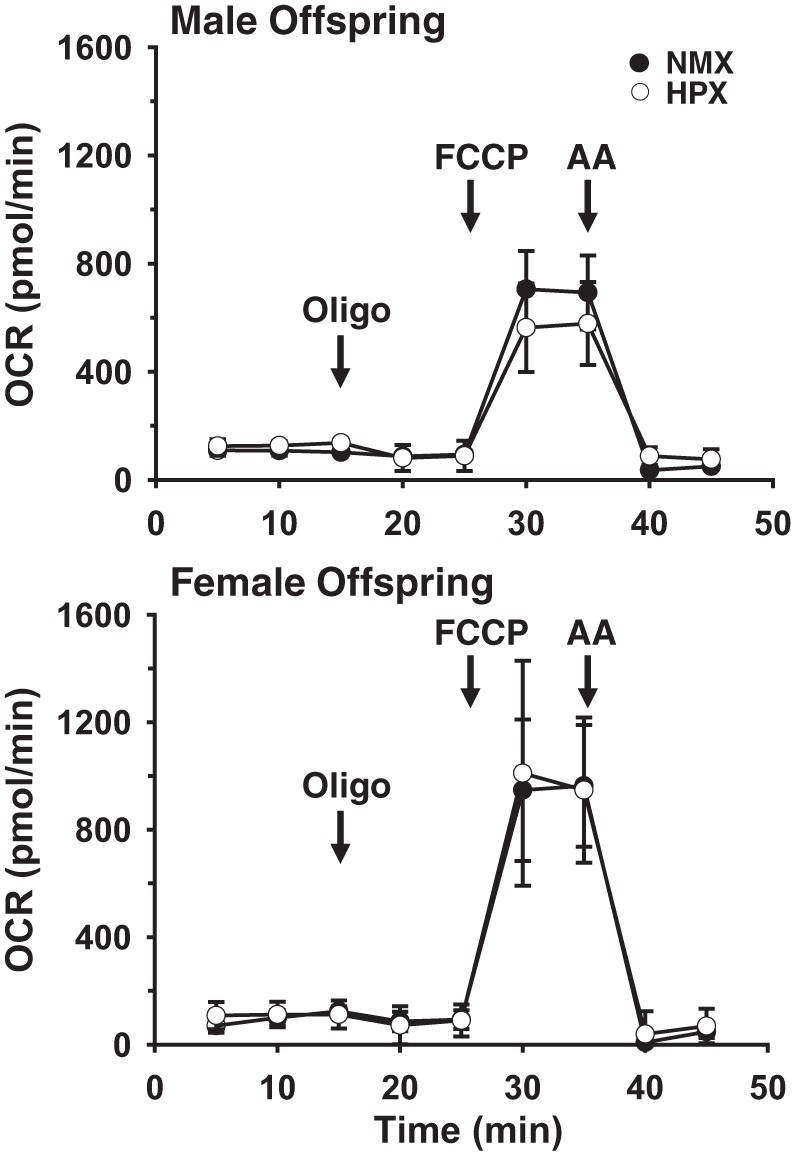
Representative traces of O2 consumption rates (OCR) measured in freshly isolated guinea pig cardiomyocytes. Cells were obtained at 90 days of age from single hearts of male or female offspring exposed to normoxia (NMX) or prenatal hypoxia (HPX). Baseline OCR was measured at 3 consecutive time points followed by addition of the ATPase inhibitor oligomycin (Oligo, 1 μg/ml). Maximal OCR was obtained in the presence of 1 μM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP). Antimycin A (AA, 10 μM) was added to inhibit mitochondrial OCR, allowing subtraction of nonmitochondrial OCR. Values are means (SD) of 3 identically treated wells.
Fig. 5.

Effect of prenatal hypoxia on O2 consumption rates (OCR) of isolated cardiac cells from guinea pig offspring. Hearts were obtained from 90-day-old guinea pig offspring exposed to prenatal normoxia (open bars) or hypoxia (hatched bars, 10.5% O2 for 14 days). Mitochondrial respiration (OCR) was measured (pmol·min−1·20,000 cells−1 or percentage of baseline OCR) at baseline (Base), following oligomycin-induced inhibition of ATP synthesis (ATP), and following carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP)-induced maximal OCR (Max), and respiratory reserve capacity (ResCap) was calculated. Values are means ± SE; n = 9 NMX and 10 HPX males and 9 NMX and 8 HPX females. *P < 0.05 vs. NMX.
Fig. 6.
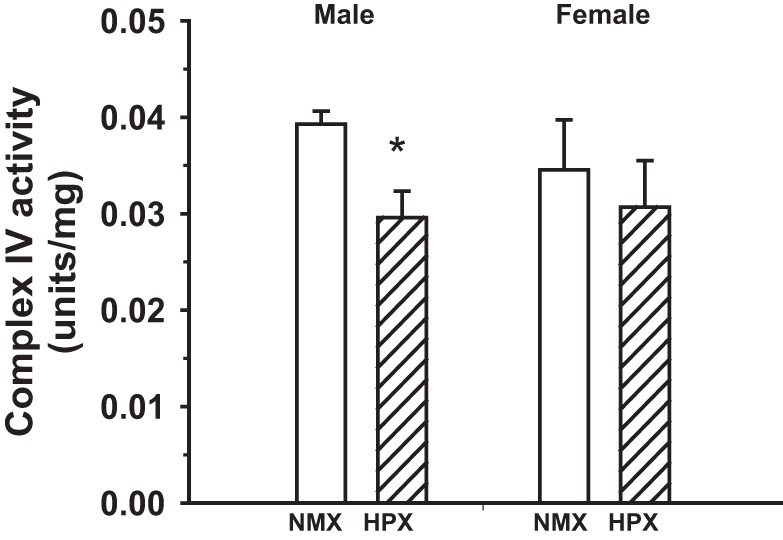
Effect of prenatal hypoxia on complex IV activity of heart ventricles of guinea pig offspring. Hearts were obtained from 90-day-old guinea pig offspring exposed to prenatal normoxia (open bars) or hypoxia (hatched bars, 10.5% O2 for 14 days). Values are means ± SE; n = 8 NMX and 8 HPX males and 8 NMX and 8 HPX females. *P < 0.05 vs. NMX.
DISCUSSION
This study showed that exposure to prenatal HPX increases mean arterial blood pressure and decreases cardiac contractile and mitochondrial function in male compared with female offspring. Prenatal HPX caused fetal growth restriction in both sexes, although differences in body weight were absent at 90 days of age. There was no effect of prenatal HPX on food or water intake rates between treatment groups to account for any nutrient intake differences after birth.
Prenatal HPX increased mean arterial blood pressure in a sex-related manner in this guinea pig model of fetal growth restriction. Previous studies showed that prenatal HPX or placental insufficiency increased arterial blood pressure in the offspring rat (1, 3, 14, 20, 43), mouse (83), and guinea pig (60), while other studies showed no effect of gestational hypoxia on blood pressure in adult rats (49, 61, 70) or chickens (71). Fetal programming of hypertension varies depending on the etiology of fetal growth restriction, with possible causes including maternal nutrient and protein restriction (24, 77), reduced uterine blood flow (2, 20), and gestational hypoxia (33). Depending on the animal model, systemic hypertension may be mediated by reduced nephrogenesis, increased sympathetic hyperinnervation of blood vessels, endothelial dysfunction, altered regulation of the renin-angiotensin system, and/or increased angiotensin II responsiveness (2, 20, 24, 32, 67). Since CO was reduced with HPX, we attribute the hypertension in the offspring to increased systemic vascular resistance, likely mediated by programming of vascular mechanisms in utero (2, 20, 32, 67).
Blood pressure values in the guinea pig are similar to those reported in other studies for guinea pigs. For example, mean arterial blood pressures of ketamine-anesthetized nonpregnant guinea pigs were 54 mmHg (73) and 47.1 ± 6.8 mmHg (15) compared with 50.7 ± 1.7 mmHg in male guinea pigs in the current study. In awake guinea pigs, blood pressures varied from 53 to 70 mmHg [59 ± 4 mmHg (72), 43 mmHg (63), 70 ± 2 mmHg (73), 53.1 ± 4.2 mmHg (15), 64 ± 1.38 mmHg (28), and 59.7 ± 7.2 mmHg (81)]; yet, despite a well-known cardiodepressive effect of anesthesia, our values fell within this range. However, heart rates of our ketamine-exposed animals were significantly reduced (161–171 beats/min) compared with those in awake guinea pigs (205–267 beats/min) (15, 28, 63, 72, 73, 81), although heart rates measured during echocardiography in urethane-exposed animals were similar (222–250 beats/min).
Previous studies using ex vivo techniques to assess cardiac function in offspring exposed to prenatal HPX report a decrease in recovery following ischemia-reperfusion injury (32, 58), cardiac β-adrenergic receptor signaling (46), and diastolic function (7). In addition, gestational hypoxia has variable effects on ventricular wall mass (32), causing increased hypertrophic growth or no change, depending on the animal model. In the present study, prenatal HPX had no effect on cardiac morphology (i.e., relative heart weight, cardiac wall thickness, and chamber diameter) in either sex, despite inhibition of functional indexes of SV, CO, EF, and FS in male guinea pigs only. Furthermore, there was no LV hypertrophy or increased heart mass, despite a significant increase in mean arterial blood pressure in male offspring exposed to prenatal HPX. This may be related to an increased afterload that is below threshold for inducing cardiac hypertrophy.
The decreased CO may be mediated by several factors, including decreased preload and contractility, increased afterload. While preload was not measured, E/A ratios provide an index of diastolic function of passive and active filling of the ventricle. Prenatal HPX had no effect on the E/A ratio in male offspring, whereas the E/A ratio was decreased in females. This was attributed to a decrease in E wave velocity and a decreased passive filling during diastole. Yet, no other functional indexes in female hearts were altered by HPX, indicating an unlikely significant effect on cardiac performance. The decreases in EF and FS in males are consistent with decreased contractility. In fetal sheep studies of high-altitude hypoxia, reduced CO was attributed to reduced Ca2+ release mechanisms associated with sarcoplasmic reticulum (29) and reduced myofibrillar Mg2+-activated ATPase (30). In the current study, a role for altered Ca2+ handling by cardiomyocytes (21, 48) cannot be ruled out. Rather, we propose mitochondrial dysfunction as an additional mechanism inhibiting LV contractility in prenatal HPX-exposed male offspring by altering respiratory efficiency and ATP availability.
Mitochondria account for ~95% of the O2 consumed in cardiac cells, which rely predominantly on oxidative phosphorylation for generation of the ATP supply (47). In fetal and adult hearts, HPX reduces respiratory enzyme activities (4, 5, 36), which reduces mitochondrial function and may contribute to cardiomyopathies and cardiac failure (16, 48, 49). Prenatal HPX had no effect on baseline OCR or normalized OCR associated with ATP synthesis in isolated heart cells of male or female offspring, although H+ leak was slightly decreased in males. Yet, maximal OCR and the respiratory reserve capacity were reduced by prenatal HPX in cardiomyocytes of male, but not female, hearts. The lack of effect of prenatal HPX on baseline OCR may be attributed to plated cardiomyocytes at rest. However, the HPX-mediated decreases in maximal OCR and respiratory reserve capacity measured ex vivo suggest a limited capacity of cardiomyocytes to increase their energy supply in response to energy demand. The HPX-induced reduction of complex IV activity may contribute to reduced electron flux along the respiratory chain and impair mitochondrial efficiency. Thus the mitochondrial deficiencies may confer an underlying risk to secondary cardiac challenges, such as systemic hypertension, exercise, or diet, when a higher level of respiration is required.
Previous studies provide insight into several possible mechanisms by which intrauterine hypoxia may mediate decreased mitochondrial and contractile function in the heart of offspring: 1) epigenetic programming of hypermethylation of specific promoters associated with contractile protein function (58, 59, 85), 2) altered cardiac cell remodeling of the fetal heart in response to intrauterine stress (12, 13, 50), 3) transcriptional regulation of mitochondrial biogenesis and cardiac cell proteins (5, 13), and 4) indirect effects of systemic hypertension and afterload on heart remodeling in the offspring (32). First, PKCε plays an important role in cardioprotection through its interaction with both contractile proteins and targeting of mitochondria (74). Previous studies identified hypermethylation of the PKCε promoter and reduced PKCε mRNA/protein levels in hearts from prenatal HPX-exposed male rat offspring (58, 59, 85), reducing the cardioprotective effect of contractile function against ischemia-reperfusion injury. Additionally, PKCε plays an important role in cardioprotection at the mitochondria via translocation of PKCε to the inner mitochondrial membrane and phosphorylation of targeted proteins such as cytochrome c oxidase, VDAC, adenine nucleotide transporter, ATP synthase, and the mitochondrial ATP-sensitive K+ channel (9, 74). The interaction of PKCε with mitochondrial proteins has been demonstrated to be integral to metabolic function associated with glycolysis, the tricarboxylic acid cycle, β-oxidation, and ion transport signaling pathways (74). Thus, reduced PKCε expression and/or translocation to the mitochondria by intrauterine hypoxia may inhibit cardiac function via contractile and metabolic pathways. Second, decreased gene expression of mitochondrial respiratory proteins, such as cytochrome c oxidase (4) and metabolic enzymes associated with fatty acid oxidation and transport (13), in hearts of prenatal HPX-exposed guinea pig offspring has been reported, indicating a role for gene repression by epigenetic mechanisms. Third, chronic hypoxia reduces the number of cardiomyocytes in both fetal (12) and offspring guinea pig hearts in a sex-related manner that favors females (13), suggesting a decrease in cardiac cell endowment that is sustained into adulthood and may limit the heart’s functional capacity against secondary challenges such as increased afterload (32). These studies indicate that intrauterine hypoxia may impart lasting perturbations in heart function of the offspring by altering mechanisms associated with contractile and metabolic function.
The sex-related differences in contractile and mitochondrial function uncovered by the current study may indicate a functional advantage in female compared with male hearts. An extensive literature has identified cardioprotection in females over males attributed to differences in estrogen levels (40, 41, 57), longevity in aging (41), and increased cardiac tolerance to ischemic injury (57). Sex-related differences in mitochondrial function favoring females are attributed to an enhanced mitochondrial antioxidant capacity (10), contributing to reduced reactive oxygen species and increased levels of estrogen, which regulate gene expression of mitochondrial proteins via activation of estrogen receptor-α/β (41, 82) in reproductively mature offspring. In addition, mitochondrial morphology of 6-wk-old male mice exhibited increased fragmented, circular and smaller mitochondria compared with age-matched female mitochondria (40), indicating a reduced biogenesis and an altered dynamic process favoring fission over fusion in males (40). Other sex-related differences include lower mitochondrial Ca2+ uptake (8), mitochondrial content (19), and efficiency, as measured by H2O2 generation (19), in females than males. There are also sex differences in mechanisms associated with Ca2+-handling and -release mechanisms (i.e., L-type Ca2+ channels, Na+/Ca2+ exchangers, and ryanodine Ca2+-release channels) of the cardiomyocyte favoring females, with a higher Ca2+ efflux from cells (reviewed in Ref. 57). Thus, while there are advantages in females over males in terms of cardioprotective mechanisms, our understanding of sex-related differences in the adaptive responses to programming requires further evaluation. For example, our study does not attribute sex differences to mitochondrial or contractile function under control conditions but exhibits a sex difference in response to hypoxia-induced programming.
In conclusion, this is the first study to identify a sexual dimorphism linking decreased cardiac contractility and mitochondrial deficits of respiratory enzyme activity and cellular respiration in hearts of guinea pig offspring. Investigation of the specific sex differences in mitochondrial mechanisms associated with biogenesis, electron transport, or dynamics is important in identifying the programming response of heart function to intrauterine hypoxia. We propose that prenatal HPX may increase the vulnerability to cardiac and mitochondrial dysfunction later in life in a sex-dependent manner, increasing the risk of heart disease and/or failure. Given the sex differences in heart failure, with the incidence lower in women than men (20), further study is needed to understand the underlying contribution of mitochondrial and contractile programming by prenatal HPX to heart dysfunction.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.P.T., L.C., and B.M.P. conceived and designed the research; L.P.T., L.C., B.M.P., G.P., and H.S. analyzed data; L.P.T., L.C., B.M.P., and H.S. interpreted results of experiments; L.P.T., B.M.P., and H.S. prepared figures; L.P.T. drafted manuscript; L.P.T., L.C., B.M.P., G.P., and H.S. edited and revised manuscript; L.P.T., L.C., B.M.P., G.P., and H.S. approved final version of manuscript; L.C., B.M.P., G.P., and H.S. performed experiments.
REFERENCES
- 1.Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 41: 457–462, 2003. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- 2.Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol 290: R1–R10, 2006. doi: 10.1152/ajpregu.00417.2005. [DOI] [PubMed] [Google Scholar]
- 3.Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr Physiol 5: 997–1025, 2015. doi: 10.1002/cphy.c140036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Hasan YM, Evans LC, Pinkas GA, Dabkowski ER, Stanley WC, Thompson LP. Chronic hypoxia impairs cytochrome oxidase activity via oxidative stress in selected fetal guinea pig organs. Reprod Sci 20: 299–307, 2013. doi: 10.1177/1933719112453509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Hasan YM, Pinkas GA, Thompson LP. Prenatal hypoxia reduces mitochondrial protein levels and cytochrome c oxidase activity in offspring guinea pig hearts. Reprod Sci 21: 883–891, 2014. doi: 10.1177/1933719113518981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aljunaidy MM, Morton JS, Cooke CM, Davidge ST. Prenatal hypoxia and placental oxidative stress: linkages to developmental origins of cardiovascular disease. Am J Physiol Regul Integr Comp Physiol 313: R395–R399, 2017. doi: 10.1152/ajpregu.00245.2017. [DOI] [PubMed] [Google Scholar]
- 7.Aljunaidy MM, Morton JS, Kirschenman R, Phillips T, Case CP, Cooke CM, Davidge ST. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol Res 134: 332–342, 2018. doi: 10.1016/j.phrs.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Arieli Y, Gursahani H, Eaton MM, Hernandez LA, Schaefer S. Gender modulation of Ca2+ uptake in cardiac mitochondria. J Mol Cell Cardiol 37: 507–513, 2004. doi: 10.1016/j.yjmcc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 9.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cε interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92: 873–880, 2003. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baños G, Medina-Campos ON, Maldonado PD, Zamora J, Pérez I, Pavón N, Pedraza-Chaverrí J. Antioxidant enzymes in hypertensive and hypertriglyceridemic rats: effect of gender. Clin Exp Hypertens 27: 45–57, 2005. doi: 10.1081/CEH-200044255. [DOI] [PubMed] [Google Scholar]
- 11.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet 341: 938–941, 1993. doi: 10.1016/0140-6736(93)91224-A. [DOI] [PubMed] [Google Scholar]
- 12.Botting KJ, McMillen IC, Forbes H, Nyengaard JR, Morrison JL. Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia-responsive genes. J Am Heart Assoc 3: e000531, 2014. doi: 10.1161/JAHA.113.000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Botting KJ, Loke XY, Zhang S, Andersen JB, Nyengaard JR, Morrison JL. IUGR decreases cardiomyocyte endowment and alters cardiac metabolism in a sex- and cause-of-IUGR-specific manner. Am J Physiol Regul Integr Comp Physiol 315: R48–R67, 2018. doi: 10.1152/ajpregu.00180.2017. [DOI] [PubMed] [Google Scholar]
- 14.Bourque SL, Gragasin FS, Quon AL, Mansour Y, Morton JS, Davidge ST. Prenatal hypoxia causes long-term alterations in vascular endothelin-1 function in aged male, but not female, offspring. Hypertension 62: 753–758, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01516. [DOI] [PubMed] [Google Scholar]
- 15.Brown JN, Thorne PR, Nuttall AL. Blood pressure and other physiological responses in awake and anesthetized guinea pigs. Lab Anim Sci 39: 142–148, 1989. [PubMed] [Google Scholar]
- 16.Casademont J, Miró O. Electron transport chain defects in heart failure. Heart Fail Rev 7: 131–139, 2002. doi: 10.1023/A:1015372407647. [DOI] [PubMed] [Google Scholar]
- 17.Chen L, Zhang J, Hu X, Philipson KD, Scharf SM. The Na+/Ca2+ exchanger-1 mediates left ventricular dysfunction in mice with chronic intermittent hypoxia. J Appl Physiol (1985) 109: 1675–1685, 2010. doi: 10.1152/japplphysiol.01372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Zadi ZH, Zhang J, Scharf SM, Pae EK. Intermittent hypoxia in utero damages postnatal growth and cardiovascular function in rats. J Appl Physiol (1985) 124: 821–830, 2018. doi: 10.1152/japplphysiol.01066.2016. [DOI] [PubMed] [Google Scholar]
- 19.Colom B, Oliver J, Roca P, Garcia-Palmer FJ. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc Res 74: 456–465, 2007. doi: 10.1016/j.cardiores.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Dasinger JH, Davis GK, Newsome AD, Alexander BT. Developmental programming of hypertension: physiological mechanisms. Hypertension 68: 826–831, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies CH, Harding SE, Poole-Wilson PA. Cellular mechanisms of contractile dysfunction in human heart failure. Eur Heart J 17: 189–198, 1996. doi: 10.1093/oxfordjournals.eurheartj.a014834. [DOI] [PubMed] [Google Scholar]
- 22.Dickinson H, Moss TJ, Gatford KL, Moritz KM, Akison L, Fullston T, Hryciw DH, Maloney CA, Morris MJ, Wooldridge AL, Schjenken JE, Robertson SA, Waddell BJ, Mark PJ, Wyrwoll CS, Ellery SJ, Thornburg KL, Muhlhausler BS, Morrison JL. A review of fundamental principles for animal models of DOHaD research: an Australian perspective. J Dev Orig Health Dis 7: 449–472, 2016. doi: 10.1017/S2040174416000477. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y, Thompson LP. Differential expression of endothelial nitric oxide synthase in coronary and cardiac tissue in hypoxic fetal guinea pig hearts. J Soc Gynecol Investig 13: 483–490, 2006. doi: 10.1016/j.jsgi.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Edwards LJ, McMillen IC. Maternal undernutrition increases arterial blood pressure in the sheep fetus during late gestation. J Physiol 533: 561–570, 2001. doi: 10.1111/j.1469-7793.2001.0561a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elias AA, Maki Y, Matushewski B, Nygard K, Regnault TRH, Richardson BS. Maternal nutrient restriction in guinea pigs leads to fetal growth restriction with evidence for chronic hypoxia. Pediatr Res 82: 141–147, 2017. doi: 10.1038/pr.2017.92. [DOI] [PubMed] [Google Scholar]
- 26.Evans LC, Liu H, Pinkas GA, Thompson LP. Chronic hypoxia increases peroxynitrite, MMP9 expression, and collagen accumulation in fetal guinea pig hearts. Pediatr Res 71: 25–31, 2012. doi: 10.1038/pr.2011.10. [DOI] [PubMed] [Google Scholar]
- 27.Fajersztajn L, Veras MM. Hypoxia: from placental development to fetal programming. Birth Defects Res 109: 1377–1385, 2017. doi: 10.1002/bdr2.1142. [DOI] [PubMed] [Google Scholar]
- 28.Flynn AJ, Dengerink HA, Wright JW. Blood pressure in resting, anesthetized and noise-exposed guinea pigs. Hear Res 34: 201–205, 1988. doi: 10.1016/0378-5955(88)90108-6. [DOI] [PubMed] [Google Scholar]
- 29.Gilbert RD. Fetal myocardial responses to long-term hypoxemia. Comp Biochem Physiol A Mol Integr Physiol 119: 669–674, 1998. doi: 10.1016/S1095-6433(98)01003-4. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert RD, Pearce WJ, Longo LD. Fetal cardiac and cerebrovascular acclimatization responses to high altitude, long-term hypoxia. High Alt Med Biol 4: 203–213, 2003. doi: 10.1089/152702903322022802. [DOI] [PubMed] [Google Scholar]
- 31.Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS, Hansell JA, Kane AD, Wooding FB, Cross CM, Herrera EA. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One 7: e31017, 2012. doi: 10.1371/journal.pone.0031017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giussani DA, Davidge ST. Developmental programming of cardiovascular disease by prenatal hypoxia. J Dev Orig Health Dis 4: 328–337, 2013. doi: 10.1017/S204017441300010X. [DOI] [PubMed] [Google Scholar]
- 33.Giussani DA, Niu Y, Herrera EA, Richter HG, Camm EJ, Thakor AS, Kane AD, Hansell JA, Brain KL, Skeffington KL, Itani N, Wooding FB, Cross CM, Allison BJ. Heart disease link to fetal hypoxia and oxidative stress. Adv Exp Med Biol 814: 77–87, 2014. doi: 10.1007/978-1-4939-1031-1_7. [DOI] [PubMed] [Google Scholar]
- 34.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73, 2008. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harvey TJ, Murphy RM, Morrison JL, Posterino GS. Maternal nutrient restriction alters Ca2+ handling properties and contractile function of isolated left ventricle bundles in male but not female juvenile rats. PLoS One 10: e0138388, 2015. doi: 10.1371/journal.pone.0138388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heather LC, Cole MA, Tan JJ, Ambrose LJ, Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ, Clarke K. Metabolic adaptation to chronic hypoxia in cardiac mitochondria. Basic Res Cardiol 107: 268, 2012. doi: 10.1007/s00395-012-0268-2. [DOI] [PubMed] [Google Scholar]
- 37.Itani N, Salinas CE, Villena M, Skeffington KL, Beck C, Villamor E, Blanco CE, Giussani DA. The highs and lows of programmed cardiovascular disease by developmental hypoxia: studies in the chicken embryo. J Physiol 596: 2991–3006, 2018. doi: 10.1113/JP274111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am J Physiol Regul Integr Comp Physiol 292: R913–R919, 2007. doi: 10.1152/ajpregu.00484.2006. [DOI] [PubMed] [Google Scholar]
- 39.Kane AD, Herrera EA, Camm EJ, Giussani DA. Vitamin C prevents intrauterine programming of in vivo cardiovascular dysfunction in the rat. Circ J 77: 2604–2611, 2013. doi: 10.1253/circj.CJ-13-0311. [DOI] [PubMed] [Google Scholar]
- 40.Khalifa ARM, Abdel-Rahman EA, Mahmoud AM, Ali MH, Noureldin M, Saber SH, Mohsen M, Ali SS. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol Rep 5: e13125, 2017. doi: 10.14814/phy2.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klinge CM. Estrogens regulate life and death in mitochondria. J Bioenerg Biomembr 49: 307–324, 2017. doi: 10.1007/s10863-017-9704-1. [DOI] [PubMed] [Google Scholar]
- 42.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging 16: 233–271, 2015. doi: 10.1093/ehjci/jev014. [DOI] [PubMed] [Google Scholar]
- 43.Langley-Evans SC, Welham SJ, Jackson AA. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci 64: 965–974, 1999. doi: 10.1016/S0024-3205(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 44.Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig 10: 265–274, 2003. doi: 10.1016/S1071-55760300074-1. [DOI] [PubMed] [Google Scholar]
- 45.Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am J Physiol Heart Circ Physiol 286: H1712–H1719, 2004. doi: 10.1152/ajpheart.00898.2003. [DOI] [PubMed] [Google Scholar]
- 46.Lindgren I, Altimiras J. Prenatal hypoxia programs changes in β-adrenergic signaling and postnatal cardiac contractile dysfunction. Am J Physiol Regul Integr Comp Physiol 305: R1093–R1101, 2013. doi: 10.1152/ajpregu.00320.2013. [DOI] [PubMed] [Google Scholar]
- 47.Lopaschuk GD. Metabolic modulators in heart disease: past, present, and future. Can J Cardiol 33: 838–849, 2017. doi: 10.1016/j.cjca.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 48.Marin-Garcia J, Goldenthal MJ, Moe GW. Mitochondrial pathology in cardiac failure. Cardiovasc Res 49: 17–26, 2001. doi: 10.1016/S0008-6363(00)00241-8. [DOI] [PubMed] [Google Scholar]
- 49.Mittmann C, Eschenhagen T, Scholz H. Cellular and molecular aspects of contractile dysfunction in heart failure. Cardiovasc Res 39: 267–275, 1998. doi: 10.1016/S0008-6363(98)00139-4. [DOI] [PubMed] [Google Scholar]
- 50.Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 293: R306–R313, 2007. doi: 10.1152/ajpregu.00798.2006. [DOI] [PubMed] [Google Scholar]
- 51.Morrison JL. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35: 730–743, 2008. doi: 10.1111/j.1440-1681.2008.04975.x. [DOI] [PubMed] [Google Scholar]
- 52.Morrison JL, Botting KJ, Darby JRT, David AL, Dyson RM, Gatford KL, Gray C, Herrera EA, Hirst JJ, Kim B, Kind KL, Krause BJ, Matthews SG, Palliser HK, Regnault TRH, Richardson BS, Sasaki A, Thompson LP, Berry MJ. Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J Physiol. In press. doi: 10.1113/JP274948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagueh SF, Smiseth OA, Appleton CP, Byrd BF III, Dokainish H, Edvardsen T, Flachskampf FA, Gillebert TC, Klein AL, Lancellotti P, Marino P, Oh JK, Popescu BA, Waggoner AD. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 29: 277–314, 2016. doi: 10.1016/j.echo.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Netuka I, Szarszoi O, Maly J, Besik J, Neckar J, Kolar F, Ostadalova I, Pirk J, Ostadal B. Effect of perinatal hypoxia on cardiac tolerance to acute ischaemia in adult male and female rats. Clin Exp Pharmacol Physiol 33: 714–719, 2006. doi: 10.1111/j.1440-1681.2006.04423.x. [DOI] [PubMed] [Google Scholar]
- 55.Nuyt AM, Alexander BT. Developmental programming and hypertension. Curr Opin Nephrol Hypertens 18: 144–152, 2009. doi: 10.1097/MNH.0b013e328326092c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh C, Dong Y, Liu H, Thompson LP. Intrauterine hypoxia upregulates proinflammatory cytokines and matrix metalloproteinases in fetal guinea pig hearts. Am J Obstet Gynecol 199: 78.e1–78.e6, 2008. doi: 10.1016/j.ajog.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Ostadal B, Netuka I, Maly J, Besik J, Ostadalova I. Gender differences in cardiac ischemic injury and protection—experimental aspects. Exp Biol Med (Maywood) 234: 1011–1019, 2009. doi: 10.3181/0812-MR-362. [DOI] [PubMed] [Google Scholar]
- 58.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ Res 107: 365–373, 2010. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCε gene in foetal rat hearts. Cardiovasc Res 93: 302–310, 2012. doi: 10.1093/cvr/cvr322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Persson E, Jansson T. Low birth weight is associated with elevated adult blood pressure in the chronically catheterized guinea-pig. Acta Physiol Scand 145: 195–196, 1992. doi: 10.1111/j.1748-1716.1992.tb09356.x. [DOI] [PubMed] [Google Scholar]
- 61.Peyronnet J, Dalmaz Y, Ehrström M, Mamet J, Roux JC, Pequignot JM, Thorén HP, Lagercrantz H. Long-lasting adverse effects of prenatal hypoxia on developing autonomic nervous system and cardiovascular parameters in rats. Pflügers Arch 443: 858–865, 2002. doi: 10.1007/s00424-001-0766-9. [DOI] [PubMed] [Google Scholar]
- 62.Poudel R, McMillen IC, Dunn SL, Zhang S, Morrison JL. Impact of chronic hypoxemia on blood flow to the brain, heart, and adrenal gland in the late-gestation IUGR sheep fetus. Am J Physiol Regul Integr Comp Physiol 308: R151–R162, 2015. doi: 10.1152/ajpregu.00036.2014. [DOI] [PubMed] [Google Scholar]
- 63.Provan G, Stanton A, Sutton A, Rankin-Burkart A, Laycock SK. Development of a surgical approach for telemetering guinea pigs as a model for screening QT interval effects. J Pharmacol Toxicol Methods 52: 223–228, 2005. doi: 10.1016/j.vascn.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 64.Readnower RD, Brainard RE, Hill BG, Jones SP. Standardized bioenergetic profiling of adult mouse cardiomyocytes. Physiol Genomics 44: 1208–1213, 2012. doi: 10.1152/physiolgenomics.00129.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reuss ML, Rudolph AM. Distribution and recirculation of umbilical and systemic venous blood flow in fetal lambs during hypoxia. J Dev Physiol 2: 71–84, 1980. [PubMed] [Google Scholar]
- 66.Richardson BS, Bocking AD. Metabolic and circulatory adaptations to chronic hypoxia in the fetus. Comp Biochem Physiol A Mol Integr Physiol 119: 717–723, 1998. doi: 10.1016/S1095-6433(98)01010-1. [DOI] [PubMed] [Google Scholar]
- 67.Rook W, Johnson CD, Coney AM, Marshall JM. Prenatal hypoxia leads to increased muscle sympathetic nerve activity, sympathetic hyperinnervation, premature blunting of neuropeptide Y signaling, and hypertension in adult life. Hypertension 64: 1321–1327, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04374. [DOI] [PubMed] [Google Scholar]
- 68.Rueda-Clausen CF, Morton JS, Davidge ST. Effects of hypoxia-induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc Res 81: 713–722, 2009. doi: 10.1093/cvr/cvn341. [DOI] [PubMed] [Google Scholar]
- 69.Rueda-Clausen CF, Morton JS, Lopaschuk GD, Davidge ST. Long-term effects of intrauterine growth restriction on cardiac metabolism and susceptibility to ischaemia/reperfusion. Cardiovasc Res 90: 285–294, 2011. doi: 10.1093/cvr/cvq363. [DOI] [PubMed] [Google Scholar]
- 70.Rueda-Clausen CF, Morton JS, Oudit GY, Kassiri Z, Jiang Y, Davidge ST. Effects of hypoxia-induced intrauterine growth restriction on cardiac siderosis and oxidative stress. J Dev Orig Health Dis 3: 350–357, 2012. doi: 10.1017/S2040174412000219. [DOI] [PubMed] [Google Scholar]
- 71.Ruijtenbeek K, Kessels CG, Janssen BJ, Bitsch NJ, Fazzi GE, Janssen GM, De Mey J, Blanco CE. Chronic moderate hypoxia during in ovo development alters arterial reactivity in chickens. Pflügers Arch 447: 158–167, 2003. doi: 10.1007/s00424-003-1170-4. [DOI] [PubMed] [Google Scholar]
- 72.Schmitz S, Henke J, Tacke S, Guth B. Successful implantation of an abdominal aortic blood pressure transducer and radio-telemetry transmitter in guinea pigs: anaesthesia, analgesic management and surgical methods, and their influence on hemodynamic parameters and body temperature. J Pharmacol Toxicol Methods 80: 9–18, 2016. doi: 10.1016/j.vascn.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 73.Schwenke DO, Cragg PA. Comparison of the depressive effects of four anesthetic regimens on ventilatory and cardiovascular variables in the guinea pig. Comp Med 54: 77–85, 2004. [PubMed] [Google Scholar]
- 74.Scruggs SB, Wang D, Ping P. PRKCE gene encoding protein kinase Cε—dual roles at sarcomeres and mitochondria in cardiomyocytes. Gene 590: 90–96, 2016. doi: 10.1016/j.gene.2016.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song H, Telugu BP, Thompson LP. Sexual dimorphism of mitochondrial function in the hypoxic guinea pig placenta. Biol Reprod. In press. doi: 10.1093/biolre/ioy167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 7: 1235–1246, 2012. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 77.Swanson AM, David AL. Animal models of fetal growth restriction: considerations for translational medicine. Placenta 36: 623–630, 2015. doi: 10.1016/j.placenta.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 78.Thompson L, Dong Y, Evans L. Chronic hypoxia increases inducible NOS-derived nitric oxide in fetal guinea pig hearts. Pediatr Res 65: 188–192, 2009. doi: 10.1203/PDR.0b013e31818d6ad0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tong W, Xue Q, Li Y, Zhang L. Maternal hypoxia alters matrix metalloproteinase expression patterns and causes cardiac remodeling in fetal and neonatal rats. Am J Physiol Heart Circ Physiol 301: H2113–H2121, 2011. doi: 10.1152/ajpheart.00356.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Turan S, Aberdeen GW, Thompson LP. Chronic hypoxia alters maternal uterine and fetal hemodynamics in the full-term pregnant guinea pig. Am J Physiol Regul Integr Comp Physiol 313: R330–R339, 2017. doi: 10.1152/ajpregu.00056.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verkeste CM, Peeters LL. The effect of labetalol on maternal haemodynamics and placental perfusion in awake near term guinea pigs. Eur J Obstet Gynecol Reprod Biol 58: 177–181, 1995. doi: 10.1016/0028-2243(95)80020-S. [DOI] [PubMed] [Google Scholar]
- 82.Viña J, Borrás C, Gambini J, Sastre J, Pallardó FV. Why females live longer than males: control of longevity by sex hormones. Sci SAGE KE 2005: pe17, 2005. doi: 10.1126/sageke.2005.23.pe17. [DOI] [PubMed] [Google Scholar]
- 83.Walton SL, Bielefeldt-Ohmann H, Singh RR, Li J, Paravicini TM, Little MH, Moritz KM. Prenatal hypoxia leads to hypertension, renal renin-angiotensin system activation and exacerbates salt-induced pathology in a sex-specific manner. Sci Rep 7: 8241, 2017. doi: 10.1038/s41598-017-08365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu Y, Williams SJ, O’Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J 20: 1251–1253, 2006. doi: 10.1096/fj.05-4917fje. [DOI] [PubMed] [Google Scholar]
- 85.Xue Q, Zhang L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase Cε. J Pharmacol Exp Ther 330: 624–632, 2009. doi: 10.1124/jpet.109.153239. [DOI] [PMC free article] [PubMed] [Google Scholar]