

Abstract
The Plant Homeodomain 6 gene (PHF6) is frequently mutated in human T-cell acute lymphoblastic leukemia (T-ALL), however its specific functional role in leukemia development remains to be established. Here we show that loss of PHF6 is an early mutational event in leukemia transformation. Mechanistically, genetic inactivation of Phf6 in the hematopoietic system enhances hematopoietic stem cell (HSC) long-term self-renewal and hematopoietic recovery after chemotherapy by rendering Phf6 knockout HSCs more quiescent and less prone to stress-induced activation. Consistent with a leukemia-initiating tumor suppressor role, inactivation of Phf6 in hematopoietic progenitors lowers the threshold for the development of NOTCH1-induced T-ALL. Moreover, loss of Phf6 in leukemia lymphoblasts activates a leukemia stem cell transcriptional program and drives enhanced T-ALL leukemia-initiating cell activity. These results implicate Phf6 in the control of HSC homeostasis and long-term self-renewal and support a role for PHF6 loss as a driver of leukemia-initiating cell activity in T-ALL.
Keywords: PHF6, T-ALL, T-cell acute lymphoblastic leukemia, hematopoietic stem cell, leukemia initiating cell, stem cell self-renewal, cancer stem cell
Introduction
Somatic loss-of-function mutations in the plant homeodomain factor 6 (PHF6) are highly prevalent in T-ALL (1) and are also present in acute myeloid leukemia (AML) (2,3). In addition, PHF6 is the causative gene mutated in Börjeson-Forssman-Lehman syndrome (BFLS; MIM#301900), a rare X-linked Mendelian disease characterized by mental retardation, obesity, hypogonadism, gynecomastia, digit abnormalities, large ears, and coarse and characteristic facial features (4).
Isolation of protein complexes has shown the interaction of PHF6 with the Nucleosome Remodeling Deacetylase (NuRD) complex, a major chromatin regulator controlling nucleosome positioning and transcription with important roles in development, genome integrity and cell cycle progression (5,6). In addition, PHF6 localizes to the nucleolus and interacts with the PAF1 transcription elongation complex (7) implicated in the control of RNA Polymerase I activity and ribosomal DNA (rDNA) transcription (8), and with UBF (7,9), a transcriptional activator in the RNA Pol I pre-initiation complex, supporting a role for PHF6 in the control of ribosome biogenesis.
PHF6 mutations seem restricted to hematologic tumors, are most frequently found in tumors from male patients (1,2) and are typically nonsense and frameshift truncating alleles resulting in complete loss of protein expression (1–3,10,11). In all, genetic loss of PHF6 as a result of deletions or mutations is present in about 20% of T-ALLs, in 20-25% of mixed phenotype acute leukemias (MPAL) with Early T cell Precursor (ETP) and T/myeloid characteristics and in 3% of AML cases (1–3,10,11). Interestingly, the development of pediatric T-ALL in a male BFSL patient harboring a germline PHF6 nonsense mutation (12) and the presence of PHF6 mutations in pre-leukemic clonal hematopoiesis (13,14) support a role for this tumor suppressor in leukemia initiation and HSC self-renewal, respectively.
Results
PHF6 mutations are early events in leukemia transformation and drive enhanced HSC self-renewal
To evaluate the potential role of PHF6 loss as a leukemia initiating event we analyzed the timing of somatically acquired mutations in T-ALL using Integrated Sequential Network (ISN) (15) analysis of clonal evolution and mutation dynamics using whole exome sequencing data from diagnostic and relapse leukemias. This analysis revealed that somatic mutations in PHF6 occur as early lesions in the natural history of T-ALL (P = 0.03) (Fig. 1A), prompting us to evaluate a mechanistic link between the loss of Phf6, HSC function and transformation leading to leukemia initiation. Towards this goal, we generated conditional Phf6 knockout mice (Supplementary Fig. S1A-C) and crossed them with a Vav-iCre line to inactivate Phf6 in fetal HSCs. Analysis of 8-week-old Phf6−/Y Vav-iCre animals revealed an expansion of total immature hematopoietic LSK progenitors (Fig. 1B-D) resulting from increased numbers of multipotent MPP2 and MPP3 populations compared with controls (Phf6+/Y Vav-iCre) (Fig. 1B-G) and no differences in more mature myeloid (MEP, CMP, GMP) and uncommitted lymphoid (CLP) bone marrow progenitors (Supplementary Fig. S1D-G). Phf6 knockout mice showed no significant differences in bone marrow B-cell precursors (Supplementary Fig. S2A and B), and analysis of thymic populations revealed only a modest but significant reduction of double negative DN2 and DN3 thymic progenitors (Supplementary Fig. S2C-G).
Figure 1.
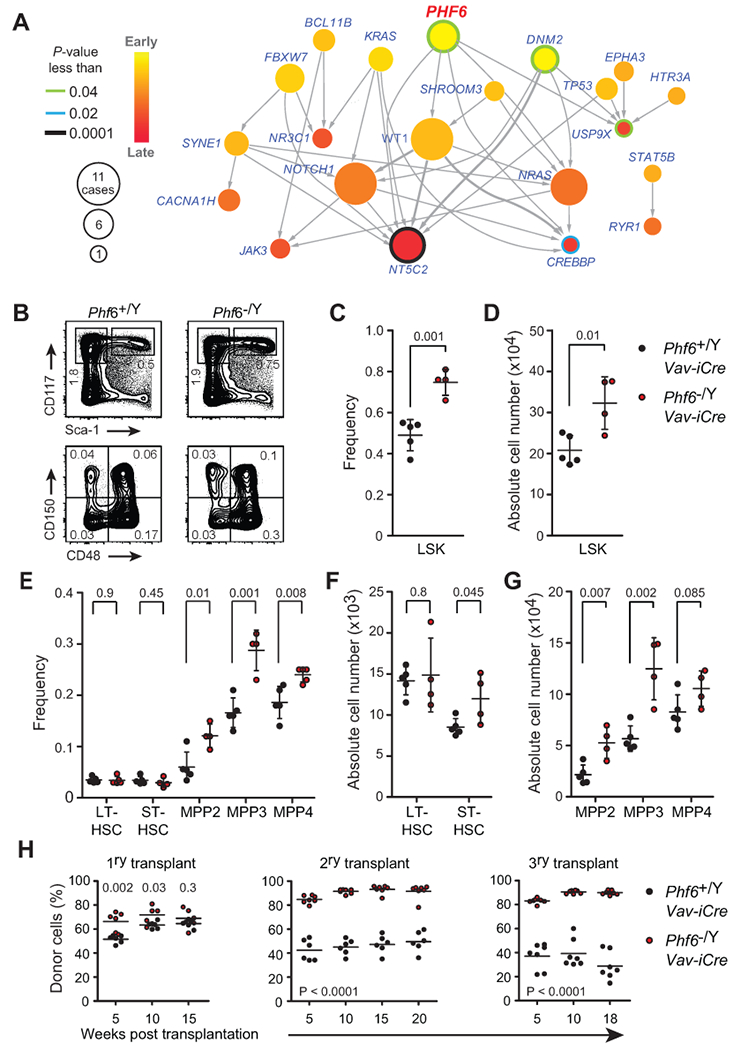
Phf6 mutations are early events in T-ALL and loss of Phf6 expands the hematopoietic stem compartment. A, Integrated Sequential Network (ISN) illustrating the sequential order of mutations (nodes) in diagnosis and relapse ALL samples (n = 37) by pooling evolutionary paths (arrows) across patients. B, FACS plots at the top show representative analysis of total myeloid progenitor cells (MyP: Lin− CD117+ Sca1−) and total hematopoietic stem and progenitor cells (LSK: Lin− CD117+ Sca1+) from Phf6 wild-type (Phf6+/Y Vav-iCre) and Phf6 knockout (Phf6−/Y Vav-iCre) littermates at 8 weeks of age. FACS plots at the bottom show representative analysis of LSK subpopulations: long-term HSCs (LT-HSCs: Lin− CD117+ Sca1+ CD150+ CD48−), short-term HSCs (ST-HSCs: Lin− CD117+ Sca1+ CD150− CD48−), MPP2 (Lin− CD117+ Sca1+ CD150+ CD48+) and MPP3 (Lin− CD117+ Sca1+ CD150− CD48+). For each representative population, frequencies reflect the mean percentage gated on total live cells. C, Frequency of LSK cells derived from Phf6 wild-type (n = 5) and Phf6 knockout (n = 4) littermates at 8 weeks of age. D, Quantification of total LSK cell numbers of populations depicted in B and C. E, The frequency of LT-HSCs, ST-HSCs, MPP2, MPP3 and lymphoid-restricted MPP4 (Lin− CD117+ Sca1+ CD135+ CD150−) progenitors derived from Phf6 wild-type (n = 5) and Phf6 knockout (n = 4) littermates. F, Absolute number of LT-HSCs and ST-HSCs as in B and E. G, Quantification of total cell numbers in multipotent (MPP2, and MPP3) and lymphoid restricted (MPP4) progenitor cell populations as in B and E. H, Total donor-derived cell frequencies in peripheral blood after primary, secondary and tertiary competitive transplantation of bone marrow cells from Phf6 wild-type (Phf6+/Y Vav-iCre) or Phf6 knockout (Phf6−/Y Vav-iCre) control mice (n = 6 - 7 recipients per group). Plots of frequency and absolute cell number show individual mice; bars represent mean ± standard deviation. P values were calculated using two-tailed Student’s t-test.
The observed expansion of LSK cells in hematopoietic-specific Phf6 knockout mice further supports a potential role for Phf6 in the control of HSC activity. To test this possibility, we evaluated HSC function in mixed bone marrow chimera serial transplantation assays. Primary transplants of Phf6 knockout (Phf6−/Y Vav-iCre) cells showed improved early hematopoietic reconstitution 5 and 10 weeks post-transplant, yet equivalent long-term total lymphoid and myeloid regeneration capacity 15 weeks post-transplant compared with Phf6+/Y Vav-iCre wild-type controls (Fig. 1H and Supplementary Fig. S3A and B). In secondary transplants, Phf6 knockout (Phf6−/Y Vav-iCre) cells showed increased total hematopoietic reconstitution and pronounced improvement in both lymphoid and myeloid lineage repopulation compared with controls (Fig. 1H and Supplementary Fig. S3A). This phenotype was further accentuated in tertiary transplants, in which Phf6 knockout cells markedly outcompeted their Phf6 wild-type counterparts (Fig. 1H). In these experiments, Phf6 wild-type serially transplanted HSCs showed a myeloid bias and decreased ability to generate B cells (Supplementary Fig. S3A and C), features characteristically associated with HSC aging and functional exhaustion (16,17). In contrast, HSCs from hematopoietic specific Phf6 knockout mice effectively contributed to both lymphoid and myeloid reconstitution in tertiary transplants, demonstrating preservation of lymphoid-lineage differentiation potential (Supplementary Fig. S3A and C).
Most somatically acquired mutations in T-ALL presumably occur postnatally in the adult hematopoietic system. To specifically evaluate the effects of Phf6 deletion in adult HSC function, we analyzed the hematopoietic system of recipient mice engrafted with bone marrow cells from control wild-type (tamoxifen-treated Phf6f/Y Rosa26+/+; referred to simply as Phf6f/Y Rosa26+/+) and adult Phf6 knockout mice (tamoxifen-treated Phf6−/Y Rosa26+/Cre-ERT2; hereafter referred to as Phf6−/Y Rosa26+/Cre-ERT2). In this setting, loss of Phf6 in adult HSCs resulted in expansion of immature hematopoietic LSK progenitors, resulting from increased numbers of MPP2 and MPP3 multipotent progenitors, as well as lymphoid committed MPP4 and CLP populations (Fig. 2A and B, and Supplementary Fig. S4A and B). Furthermore, we also observed a modest increase in CMP committed myeloid bone marrow progenitors, with no changes in MEP and GMP populations (Supplementary Fig. S4C and D). We observed no differences in in cell cycle distribution between wild-type and Phf6 knockout-derived HSCs (LSK CD48−) and MPP cells (LSK CD48+) (Supplementary Fig. 4E and F). Animals transplanted with Phf6−/Y Rosa26+/Cre-ERT2 bone marrow showed largely preserved B cell development, with only a modest decrease in pre-B and immature B-precursor cell populations (Supplementary Fig. S5A and B). In addition, and as noted in Phf6−/Y Vav-iCre knockout mice, analysis of the thymus in mice reconstituted with Phf6−/Y Rosa26+/Cre-ERT2 cells revealed a modest but significant reduction in DN2 and DN3 thymic progenitor cells (Supplementary Fig. S5C-G). Consistently, mixed bone marrow chimera serial transplantation assays showed that Phf6−/Y Rosa26+/Cre-ERT2 adult HSCs display improved competitive multi-lineage reconstitution, increased serial repopulation capacity (Fig. 2C), and successively enhanced retention of lymphoid potential in secondary and tertiary transplants compared with Phf6f/Y Rosa26+/+ controls (Supplementary Fig. S6A-C). Moreover, as a result of exhaustion of HSC function in these experiments, we noted a progressive decrease in the number of LSK cells with each consecutive transplantation of Phf6f/Y Rosa26+/+ wild-type bone marrow (Fig. 2D), which was accompanied by an increase in the relative frequency of LT-HSCs as a result of decreased hematopoietic output (Fig. 2E-H).
Figure 2.
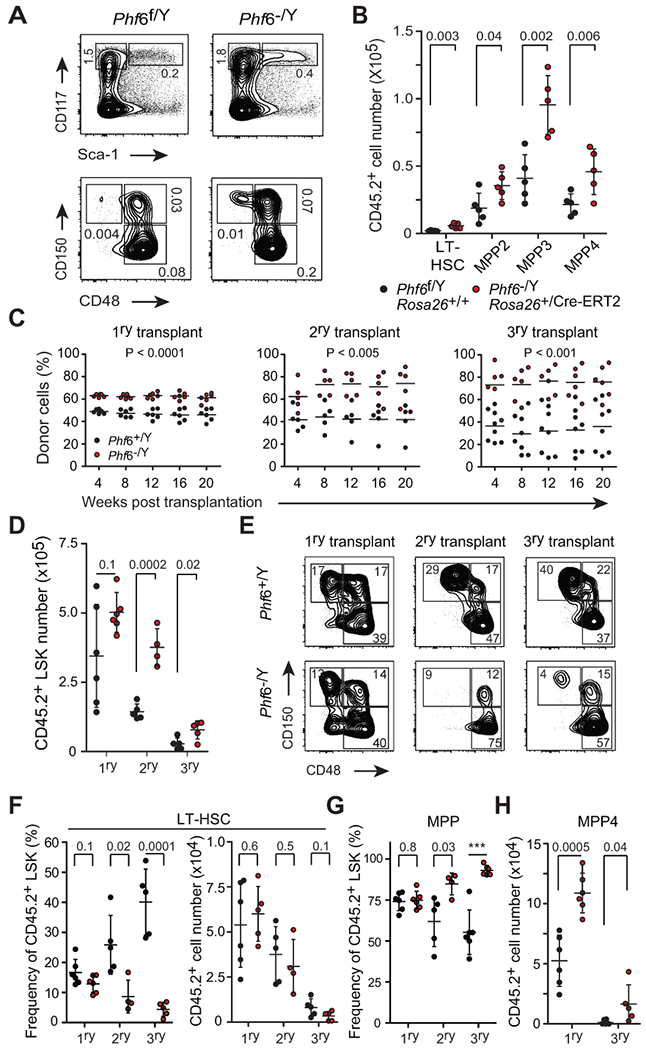
Phf6 deletion in adult hematopoiesis increases hematopoietic stem cell self-renewal following long-term competitive serial transplantation. A, Representative FACS plots of total donor-derived myeloid progenitor cells (MyP: Lin− CD117+ Sca1−) and total hematopoietic stem and progenitor cells (LSK: Lin− CD117+ Sca1+) 8-10 weeks post transplantation of lineage negative cells isolated from Phf6 wild-type (Phf6f/Y Rosa26+/+) or Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) mice. For each representative population, frequencies reflect the mean percentage gated on total live cells (n = 5 per group). B, Absolute number of donor-derived LT-HSC, MPP2, MPP3 and MPP4 cells in recipients of Phf6 wild-type or Phf6 knockout donor bone marrow cells. C, Total donor-derived cell reconstitution in peripheral blood after primary, secondary and tertiary competitive transplantation of bone marrow cells from Phf6 wild-type and Phf6 knockout mice (n = 5–7 recipients per group). D, Total number of donor-derived wild-type (Phf6f/Y Rosa26+/+) and knockout (Phf6−/Y Rosa26+/Cre-ERT2) LSK cells in the bone marrow of recipient mice 24 weeks after primary (n = 6 per group), secondary (n = 5 and 4 respectively) and tertiary (n = 5 per group) competitive mixed bone-marrow transplantation. E, Representative FACS plots of total donor-derived stem and progenitor cells in mice after primary, secondary and tertiary competitive mixed bone marrow transplantation with Phf6 wild-type or Phf6 knockout bone marrow cells. For each population, the mean percentage gated on total LSK cells is indicated. F, Frequency and absolute cell number of LT-HSCs in primary, secondary and tertiary competitive mixed bone marrow recipients as in D and E. G, Frequency of total multipotent progenitor cells (MPP) in primary, secondary and tertiary competitive mixed bone marrow recipients as in D and E. H, Absolute numbers of lymphoid-restricted MPP4 progenitor cells in primary and tertiary competitive mixed bone marrow recipients as in D and E. Plots of both frequency and absolute cell number show individual mice; bars represent mean ± standard deviation. P values were calculated using two-tailed Student’s t-test. *** indicates P < 0.0001.
In comparison, secondary and tertiary recipients of Phf6−/Y Rosa26+/Cre-ERT2 knockout cells had a less severe and delayed decline in LSK numbers (Fig. 2D), showed preservation of hematopoietic function, improved MPP output, and lacked the characteristic increase in relative frequency of LT-HSCs observed in mice serially transplanted with wild-type cells (Fig. 2E-H and Supplementary Fig. S6D and E). In addition, in-depth analysis of bone marrow MPP progenitor populations revealed preserved numbers of Phf6−/Y knockout-derived lymphoid restricted MPP4 cells in tertiary- relative to primary recipients of adult-deleted Phf6 knockout bone marrow chimeras, a finding consistent with their distinctive long-term retention of lymphoid-lineage differentiation potential (Fig. 2G and H, and Supplementary Fig. S6).
Next, to further and directly test the role of Phf6 in HSC self-renewal, we compared the reconstitution capacity of purified LT-HSCs from Phf6−/Y Rosa26+/Cre-ERT2 and wild-type Phf6f/Y Rosa26+/+ control mice after serial transplantation into isogenic recipients. In this experiment, primary recipient mice engrafted with Phf6 null LT-HSCs showed accelerated early-stage hematopoietic reconstitution at 5 weeks post transplantation. Increased long-term donor-derived reconstitution derived directly from Phf6 null LT-HSCs was maintained at 15 weeks post transplantation (Fig. 3A). Of note, equal numbers of primary transplanted Phf6 null and wild-type LT-HSCs gave rise to equivalent numbers of LT-HSCs in the bone marrow of recipient mice 16 weeks post-transplant (Fig. 3B). Strikingly, Phf6 null LT-HSCs gave rise to increased numbers of Phf6 null-derived down-stream progenitor cells compared to Phf6 wild-type LT-HSCs; increased numbers of Phf6 null donor-derived cells included ST-HSCs, MPP2-4 progenitor cells and myeloid-committed MEP, CMP and GMP progenitors (Fig. 3A and B). This phenotype was further augmented in recipients of secondary transplants performed with sorted LT-HSCs isolated 16 weeks post primary transplantation (Fig. 3C). Of note, and as observed in competitive bone marrow chimeras, Phf6 adult-deleted knockout LT-HSCs showed improved retention of B lymphoid-cell reconstitution potential compared with wild-type controls, which showed preferential myeloid-lineage differentiation (Fig. 3D). In all, these experiments indicate that loss of Phf6 results in increased LT-HSC self-renewal and hematopoietic reconstitution capacity with maintenance of long-term lymphoid-lineage commitment.
Figure 3.
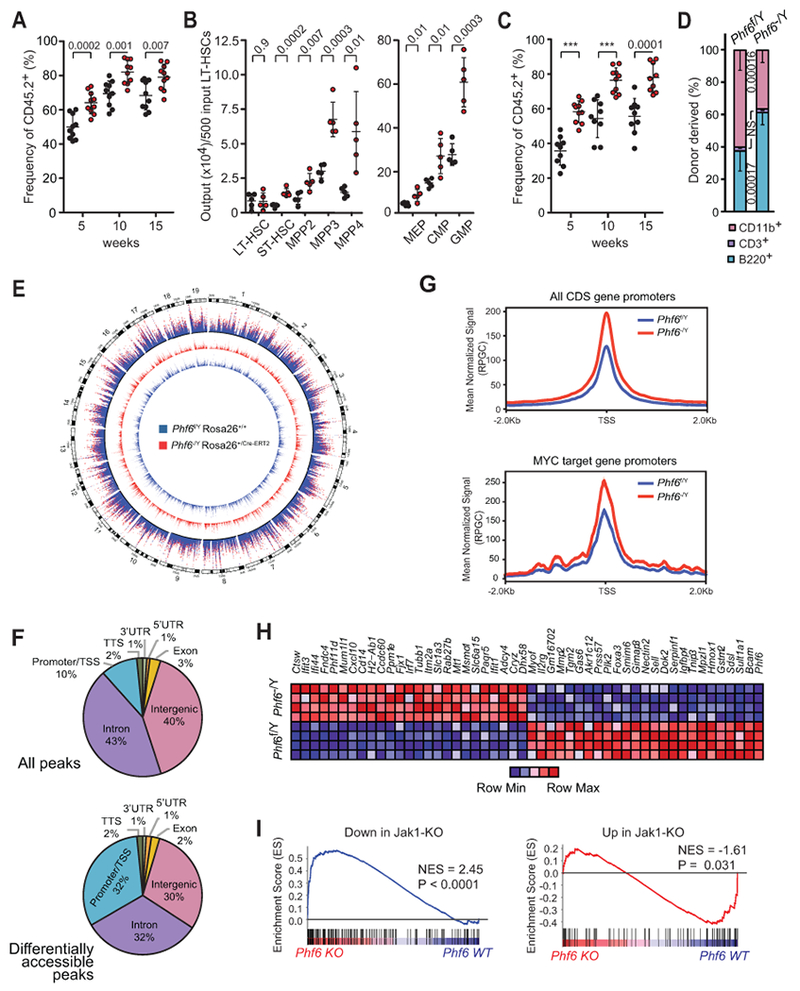
Phf6 deletion in adult LT-HSCs increases hematopoietic repopulation capacity associated with increased chromatin accessibility and transcriptional upregulation of JAK-STAT signature genes. A, Frequency of total donor-derived cells in peripheral blood 5, 10 and 15 weeks after transplantation of 500 LT-HSCs sorted from Phf6 wild-type (Phf6f/Y Rosa26+/+) or Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) donor mice (n = 10 recipients per group). B, The absolute number of hematopoietic stem and progenitor cells (LT- and ST-HSCs, MPP2-4) and myeloid-restricted progenitor cells (MEP, CMP, GMP) derived from 500 Phf6 wild-type or Phf6 knockout sorted LT-HSCs in the bone marrow of recipient mice16 weeks after transplantations (n = 5 per group), as in A. C, Frequency of total donor-derived reconstitution in peripheral blood after secondary transplantation of 1000 re-sorted Phf6 wild-type or Phf6 knockout LT-HSCs (n = 10 per group). D, Relative distribution of B cell (B220+), T cell (CD3+) and myeloid (CD11b+) populations in peripheral blood 15 weeks post-transplant, as in C. Values are shown for individual mice. Graphs represent mean ± standard deviation. P values were calculated using two-tailed Student’s t-test. *** indicates P < 0.0001. E, Genome-wide distribution of normalized chromatin accessibility in Phf6 knockout LT-HSCs (red track) and wild-type (blue track) LT-HSCs shown separately and in overlay (outer track). F, Distribution of ATACseq peaks, and of significantly differentially accessible peaks in Phf6 knockout compared to wild-type LT-HSCs. G, Normalized average ATAC-Seq signal across all protein-coding transcript promoters (top) and MYC target genes (DANG_MYC_TARGETS_UP, C2 MSIG M6506) centered on the TSS (Transcription Start Site). H, Heatmap representation of top differentially expressed genes in Phf6 knockout compared with Phf6 wild-type LT-HSCs. I, Gene set enrichment plots from GSEA of a Jak1-loss signature comprised by genes differentially expressed in LT-HSCs of Jak1 knockout mice tested in Phf6 knockout LT-HSCs. Genes that display decreased expression in Jak1 knockout LT-HSCs were tested separately (left) from those that show increased expression (right). Normalized enrichment score (NES) and the FEWER p-value are shown.
To explore the mechanisms mediating the effects of Phf6 in in LT-HSCs we performed genome-wide chromatin accessibility profiling by Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq). This analysis identified 140,485 transposase accessible peaks in mouse LT-HSCs of which 2,707 were significantly differentially accessible (adjusted P value: Padj < 0.01; fold change > 4) in Phf6 knockout versus wild-type LT-HSCs. Of these significantly differentially accessible peaks, 2,334 (86%) became more accessible in Phf6 knockout LT-HSCs and 373 (14%) became less accessible, indicating a global increase in chromatin accessibility in the Phf6 knockout setting (Fig. 3E). Notably, while only 10% of the total ATAC-seq peaks identified in LT-HSCs fell within proximal promoter regions, 32% of the significantly differentially accessible peaks in Phf6 knockout LT-HSCs were located at the proximal promoter, indicating a preferential chromatin opening around transcription start sites (TSS) (Fig. 3F and G). Functional enrichment analysis of promoter/TSS peaks with increased accessibility in Phf6 knockout LT-HSCs revealed significant enrichment of GO Term categories potentially related to HSC homeostasis including cell cycle (GO:0007049, P = 9.67 · 10−12); translation (GO:0006412, P = 4.75 · 10−12) and ribosome biogenesis (GO:0042254, P = 2.49 · 10−7); MYC targets (MSIGDB:M15774, P = 5.3 · 10−9; MSIGDB:M17753, P = 1.807 · 10−10), and cellular response to stress (GO:0033554, P = 2.28 · 10−7). Consistently, we observed increased promoter accessibility at MYC target gene promoters in Phf6 knockout LT-HSCs (Fig. 3G). In addition, gene expression profiling by RNA-seq of sorted Phf6 wild-type and knockout LT-HSCs revealed modest changes in gene expression with 306 differentially expressed genes (P < 0.01; log2 fold change > 0.3) of which 150 were up-regulated and 156 were down-regulated in Phf6 knockout LT-HSCs, respectively (Fig. 3H). Notably, this transcriptional program was significantly enriched in genes controlled by Jak1 in HSCs (18) (Fig. 3I), supporting a potential convergent transcriptional mechanism between genetic inactivation of Phf6 and JAK-STAT signaling in HSC self-renewal.
Loss of Phf6 increases HSC reconstitution capacity and quiescence after insult
Under homeostatic conditions LT-HSCs are predominantly quiescent, a property characteristically required to maintain their self-renewal capacity and functionally protective under stress (19). However, following hematopoietic challenges such as genotoxic insult by radiation or chemotherapy, HSCs transiently proliferate to ensure efficient replenishment of blood and immune cellularity (19). Similarly, inflammatory antiviral responses and interferon signaling also induce activation and proliferation of LT-HSCs, and their return to quiescence is essential to avoid accumulation of DNA damage and apoptosis (20). Loss of homeostatic equilibrium in the hematopoietic system in the context of chemotherapy or immune challenge has been implicated in the expansion of HSCs harboring clonal hematopoiesis-associated mutations, and in leukemia development (21). In this context, we explored the capacity of Phf6 null HSCs to withstand stress-induced activation by examining the kinetics of recovery after myeloablative treatment with 5-fluorouracil (5-FU) compared to wild-type controls (22). In these experiments, mice reconstituted with Phf6−/Y Rosa26+/Cre-ERT2 knockout cells showed accelerated and improved expansion of LSK cells and total myeloid progenitors (MyP: Lin− CD117+ Sca1−) on day 9 and 12 post treatment compared with wild-type Phf6f/Y Rosa26+/+ controls (Fig. 4A and 4B, and Supplementary Fig. S7). In addition, recipients of Phf6 knockout bone marrow showed improved recovery of total bone marrow cellularity, yet lower levels of activation-induced expansion in the HSC compartment (LSK CD48−), following genotoxic ablation of hematopoietic progenitors with cyclophosphamide and rescue with G-CSF cytokine stimulation (23) (Fig. 4C-E). To more directly explore a potential cell-intrinsic role of Phf6 loss in HSC quiescence, we tested the effects of interferon stimulation on HSC activation by polyinosinic:polycytidylic acid (poly (I:C)) treatment of mice reconstituted with control Phf6 wild-type (Phf6f/Y Rosa26+/+) and Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) bone marrow 8 weeks post-transplant, a timeframe in which the hematopoietic system has returned to homeostatic equilibrium. Consistently, wild-type and Phf6 knockout-derived HSCs (LSK CD48−) and MPP cells (LSK CD48+) showed no differences in cell cycle distribution in basal conditions (Supplementary Fig. 4E and F). In contrast, and most notably, 24 hours post poly (I:C) administration, Phf6 knockout HSCs (LSK CD48−) showed a more limited proliferative response and improved retention of the quiescent G0 cell-cycle state compared with wild-type controls (Fig. 4F and 4G). Conversely, unrestrained cell-cycle re-entry was observed in Phf6 knockout multipotent progenitor cells (LSK CD48+) (Fig. 4F and 4G). Taken together, these results support that the loss of Phf6 limits the proliferative response of LT-HSCs in reaction to genotoxic stress and facilitates hematopoietic reconstitution after injury.
Figure 4.
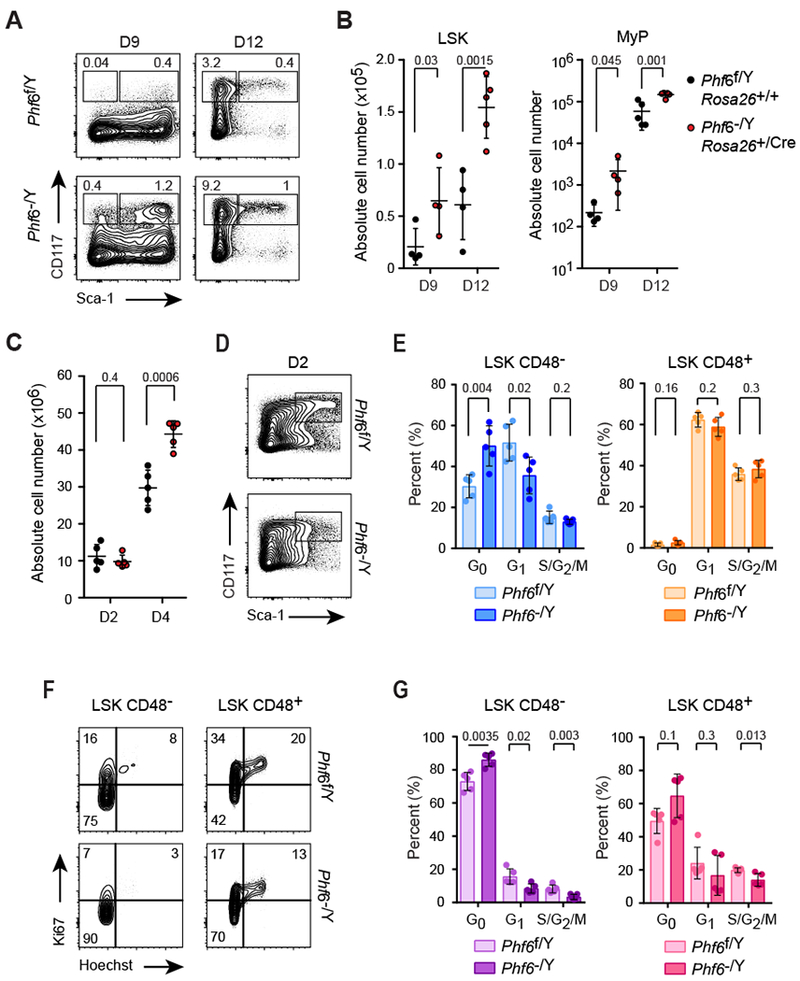
Phf6 loss facilitates hematopoietic recovery following genotoxic stress and enhances retention of cell quiescence after DNA damage and interferon challenge. A, Representative FACS plots of CD117 and Sca-1 expression in lineage negative bone marrow cells of Phf6 wild-type (Phf6f/Y Rosa26+/+) and Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) mice 9 days (n = 4 per group) and 12 days (n = 5 per group) after 5-FU treatment. Average frequencies of LSK (Lin− CD117+ Sca-1+) and total myeloid progenitor cells (MyP: Lin− CD117+ Sca-1−) are indicated. B, Absolute quantification of LSK and MyP populations as in A. Graphs show values for individual mice, bars indicate mean and standard deviation. C, Absolute bone marrow cell numbers of Phf6 wild-type (Rosa26+/+ Phf6f/Y) and Phf6 knockout (Rosa26+/Cre-ERT2 Phf6−/Y) mice following cyclophosphamide treatment (Cy) and 2 (D2) or 4 (D4) consecutive doses of G-CSF. D, Representative FACS plots of CD117 and Sca-1 expression in lineage negative bone marrow cells of Phf6 wild-type (Rosa26+/+ Phf6f/Y) and Phf6 knockout (Rosa26+/Cre-ERT2 Phf6−/Y) mice following cyclophosphamide treatment (Cy) and 2 (D2) consecutive doses of G-CSF. E, Cell cycle distribution of Phf6 wild-type (n = 5) and Phf6 knockout (n = 5) CD48− LSK and CD48+ LSK cells following cyclophosphamide treatment (Cy) and 2 (D2) consecutive doses of G-CSF. F, Representative FACS plots of Ki67 expression and DNA content (Hoechst) in bone marrow LSK CD48− stem and LSK CD48+ progenitor cells of Phf6 wild-type (Rosa26+/+ Phf6f/Y) and Phf6 knockout (Rosa26+/Cre-ERT2 Phf6−/Y) mice 24 hours after p (I:C) treatment. G, Cell cycle distribution in bone marrow LSK CD48− stem and LSK CD48+ progenitor populations of Phf6 wild-type (n = 5) and Phf6 knockout (n = 4) mice 24 hours after p(I:C) treatment. P values were calculated using two-tailed Student’s t-test. Bar graphs show values for individual mice ± standard deviation.
Deletion of Phf6 sensitizes hematopoietic progenitors to oncogenic NOTCH1-induced transformation
To formally and directly test the role of Phf6 loss as a leukemia-initiating event in T-ALL transformation we infected hematopoietic progenitor cells from Phf6 knockout (Phf6−/Y Vav-iCre) and wild-type (Phf6+/Y Vav-iCre) controls with bicistronic GFP retroviruses driving expression of a constitutively active T-ALL mutant form of the NOTCH1 receptor (NOTCH1-L1601P-ΔPEST) (24,25). Of note, activating mutations in NOTCH1 are highly prevalent in T-ALL (24) and frequently co-occur with mutational loss of PHF6 (1). In this model, forced expression of mutant NOTCH1 typically results in a wave of extrathymic T-cell lymphopoiesis 3 weeks after transplantation, which manifests as a transient appearance of GFP+ preleukemic CD4+ CD8+ double-positive (DP) T cells in peripheral blood, and is followed by full T-ALL leukemia transformation 10-15 weeks later (25). Mice transplanted with Phf6 knockout NOTCH1-L1601P-ΔPEST-expressing progenitors showed a consistent wave of GFP+ circulating cells in peripheral blood 3 weeks post-transplant (Fig. 5A). Nonetheless, analysis of the thymic compartment in this setting revealed increased numbers of GFP+ cells in mice transplanted with Phf6 knockout NOTCH1-L1601P-ΔPEST-transduced bone marrow progenitor cells (Fig. 5B). Moreover, analysis of circulating tumor cells 5 weeks and 11 weeks post-transplant revealed accelerated leukemia development in mice transplanted with Phf6 knockout NOTCH1-L1601P-ΔPEST-expressing progenitors (Fig. 5C), which ultimately translated to increased leukemia penetrance and markedly accelerated leukemia-associated mortality in this group (Phf6 wild-type NOTCH1-L1601P-ΔPEST median survival = 23.6 weeks; Phf6 knockout NOTCH1-L1601P-ΔPEST median survival = 13.3 weeks; Log-rank test P = 0.042) (Fig. 5D). Interestingly, flow cytometry analysis of hematopoietic tissues at endpoint showed increased GFP+ leukemia lymphoblast infiltration in the thymus of animals bearing Phf6 knockout leukemia, despite nearly equivalent tumor burden in the bone marrow and spleen (Fig. 5E and 5F). Moreover, analysis of leukemia lymphoblasts recovered from leukemia infiltrated tissues showed expression of T cell surface markers with increased prevalence of a CD4+ CD8+ double-positive immunophenotype in Phf6 knockout tumors compared with Phf6 wild-type controls (Fig. 5G). Western blot analysis confirmed complete loss of Phf6 expression in Phf6 knockout tumor cells (Fig. 5H).
Figure 5.
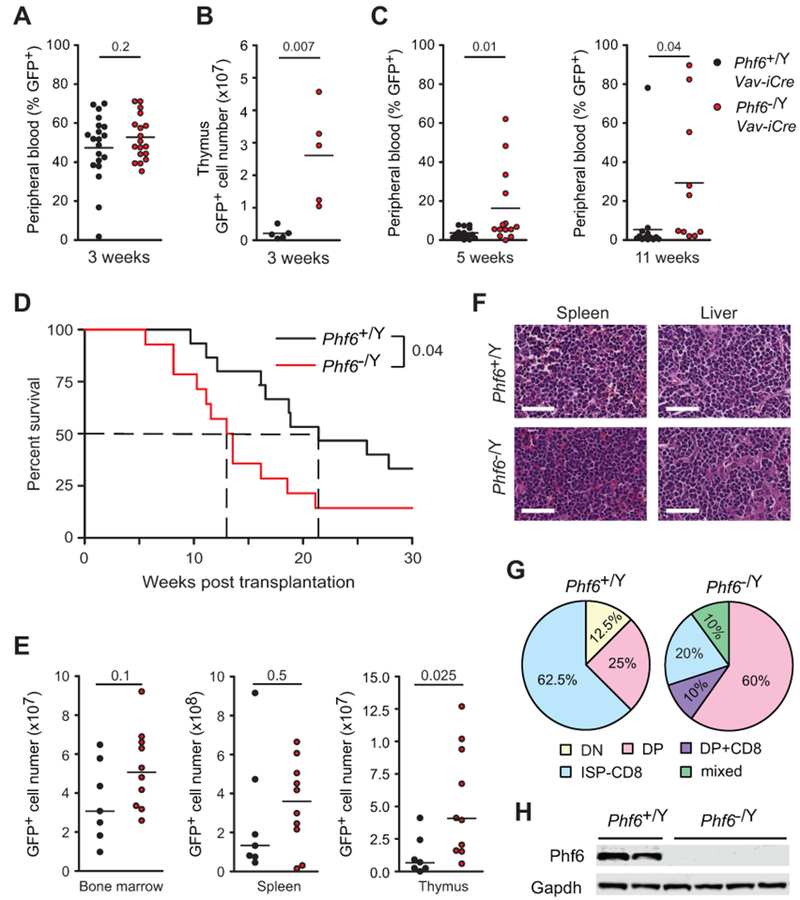
Loss of Phf6 enhances NOTCH1-induced T-ALL development. A, Peripheral blood quantification of GFP+ cells in mice transplanted with NOTCH1-L1601P-ΔPEST-transduced Phf6 wild-type (Phf6+/Y Vav-iCre) (n = 20) and Phf6 knockout (Phf6−/Y Vav-iCre) (n = 19) bone marrow progenitors showing transient NOTCH1-driven preleukemic T-cell expansion 3 weeks post-transplant. B, Quantification of GFP+ preleukemia cell infiltration in the thymus of mice 3 weeks after transplantation with NOTCH1 L1601P-ΔPEST-transduced Phf6 wild-type and Phf6 knockout (n = 5 per group) bone marrow progenitors. C, Peripheral blood analysis of early (5 weeks) and late (11 weeks) leukemia development detected by circulating GFP+ leukemia cells in mice transplanted with NOTCH1-L1601P-ΔPEST-transduced Phf6 wild-type and Phf6 knockout bone marrow progenitors. D, Kaplan-Meier survival curves of mice harboring Phf6 wild-type (Phf6+/Y Vav-iCre; n = 14) and Phf6 knockout (Phf6−/Y Vav-iCre; n = 14) NOTCH1-induced leukemias, as in C. E, Flow cytometry quantification of leukemia infiltration in bone marrow, spleen and thymus of Phf6 wild-type and Phf6 knockout NOTCH1-induced leukemia-bearing mice at endpoint. F, Representative micrographs of histology analysis of Phf6 wild-type and Phf6 knockout NOTCH1-induced leukemias. G, Phenotypic categorization of Phf6 wild-type and Phf6 knockout NOTCH1-induced leukemias based on CD4 and CD8 surface expression analyzed by flow cytometry. H, Western blot analysis of Phf6 expression in Phf6 wild-type and Phf6 knockout NOTCH1-induced leukemia lymphoblasts. P values in A-C and E were calculated using two-tailed Student’s t-test. P values in D were calculated using the log-rank test. Scale bars in F, 50 μm.
Phf6 loss drives leukemia initiating cell activity in T-ALL
To better explore the specific role of Phf6 in the pathogenesis of T-ALL without the confounding effect of genetic and immunophenotypic heterogeneity of this model, we developed isogenic tumors with loss of Phf6. Towards this goal we generated NOTCH1-L1601P-ΔPEST-induced leukemias as described above using hematopoietic progenitors from tamoxifen-inducible conditional Phf6 knockout (Phf6f/Y Rosa26+/CreERT2) mice. The resulting Phf6 conditional knockout leukemias were transplanted into secondary recipients and treated with vehicle only or tamoxifen to generate matched Phf6 wild-type and knockout tumors, respectively (Supplementary Fig. S8A-C). When transplanted into isogenic recipients, Phf6 knockout leukemia cells showed modestly accelerated tumor progression compared with Phf6 wild-type isogenic controls as documented by increased GFP+ cell counts in peripheral blood (Fig. 6A) and accelerated mortality (Log-rank test P = 0.013) (Fig. 6B). However, and most notably, limiting dilution transplantation assays demonstrated a marked increase in leukemia initiating cell activity in Phf6 knockout tumors compared with wild-type isogenic controls, supporting a role for loss of Phf6 in promoting leukemia blast self-renewal (Fig. 6C and 6D, and Supplementary Fig. S8D).
Figure 6.
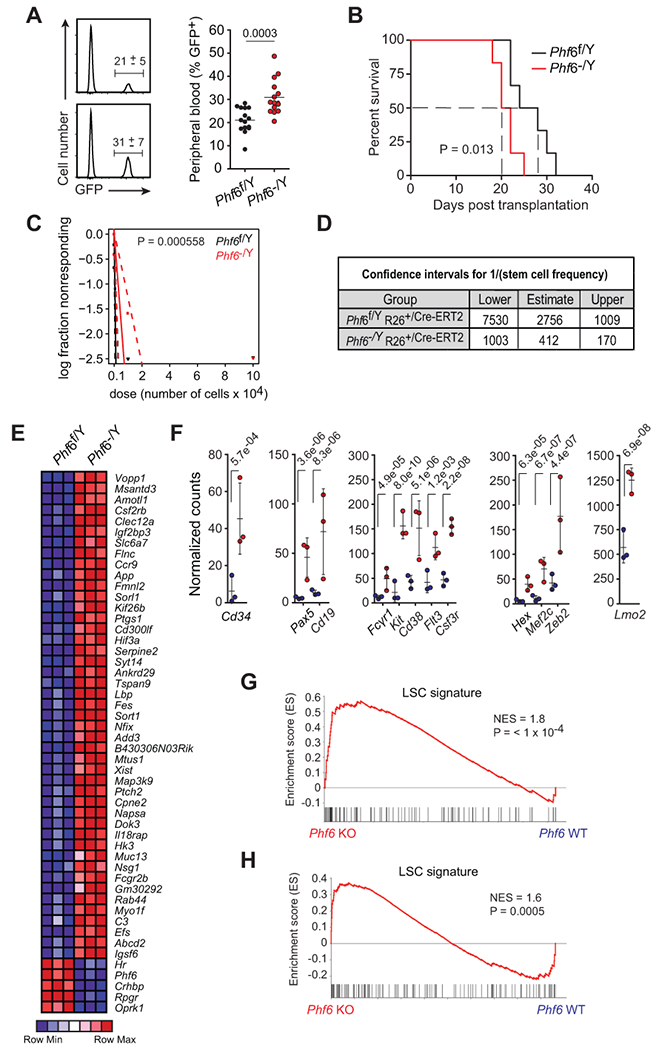
Loss of Phf6 activates an LIC program and increases LIC frequency in T-ALL. A, Quantification of GFP+ leukemia cells in peripheral blood of mice engrafted with Phf6f/Y Rosa26+/Cre-ERT2 wild-type and isogenic Phf6−/Y Rosa26+/Cre-ERT2 knockout leukemia cells on day 15 after transplantation (n = 15 per group). B, Kaplan-Meier survival curves of mice harboring Phf6 wild-type and isogenic Phf6 knockout NOTCH1-induced leukemias (n = 10 per group), as in A. C, Leukemia-initiating cell (LIC) analysis in mice transplanted with Phf6f/Y Rosa26+/Cre-ERT2 wild-type and isogenic Phf6−/Y Rosa26+/Cre-ERT2 knockout leukemia cells (n = 6 per group). D, Confidence intervals showing 1/ (stem cell frequency) based on C. E, Heatmap representation of relative gene expression for top 50 ranking genes differentially expressed between Phf6f/Y Rosa26+/Cre-ERT2 wild-type and isogenic Phf6−/Y Rosa26+/Cre-ERT2 knockout NOTCH1-induced leukemia cells. F, Normalized gene expression of selected genes differentially expressed between isogenic wild-type and Phf6 knockout NOTCH1-induced leukemia cells. G, GSEA analysis depicting enrichment of LSC-signature genes in the expression signature associated with Phf6−/Y Rosa26+/Cre-ERT2 knockout NOTCH1-induced leukemia lymphoblast cells compared with Phf6f/Y Rosa26+/Cre-ERT2 wild-type isogenic controls. H, GSEA analysis depicting enrichment of LSC-signature genes in the expression signature associated with human PHF6 mutant T-ALL compared with matched PHF6 wild-type controls. Graphs in A depict measurements from individual mice with mean and standard deviation. P values were calculated using two-tailed Student’s t-test in A, the log-rank test in C, and using the Chi-square test in D.
Analysis of transcriptional programs associated with Phf6 loss in matched isogenic Phf6 wild-type and Phf6 null leukemia samples revealed a distinct gene expression signature dominated by upregulated transcripts (608 genes up-regulated, 67 genes down-regulated; P < 0.001, fold change > 2) (Fig. 6E). Genes upregulated in Phf6 null leukemia lymphoblasts included the early hematopoietic marker Cd34, as well as genes associated with B cell (Cd19, Pax5) and myeloid (Kit, Cd14, Cd38, Flt3, Csf3r, Fcgr1) differentiation, even though Phf6 wild-type and Phf6 knockout isogenic tumors showed no apparent differences in their immunophenotype (Fig. 6F). Notably, suppression of Phf6 in mouse B-cell lymphoma cells has been described to increase lineage plasticity, in this case resulting in decreased expression of B-cell lineage genes and upregulation of T-cell markers (26), and PHF6 mutations are prevalent in ETP ALLs and myeloid/T MPALs, which are characterized by mixed expression programs and lineage plasticity (1–3,10,11). In addition, genes upregulated upon Phf6 knockout in our NOTCH1 leukemia model include bona fide T-ALL oncogenes such as Lmo2, Mef2c, Zeb2 and Hex (27) (Fig. 6F), which are characteristically expressed at higher levels in early immature ETP ALL leukemias (28,29). In addition, and in agreement with an increase in leukemia initiating cell activity, gene set enrichment analysis showed preferential activation of a gene expression program associated with human leukemia stem cell activity (30) in Phf6 knockout NOTCH1-induced T-ALL cells compared with isogenic wild-type controls (Fig. 6G). Finally, and most notably, analysis of this leukemia-initiating cell signature in human T-ALLs validated and extended these results showing a marked enrichment of leukemia stem cell genes (31) in human PHF6 mutant leukemias compared with matched PHF6 wild-type controls (Fig. 6H).
Discussion
Chemoresistant leukemia-initiating cells with expanded self-renewal potential are major drivers of leukemia relapse and are associated with poor prognosis (32). Self-renewal potential and intrinsic resistance to therapeutic agents are characteristic properties of HSCs, and transcriptional profiling of human leukemia-initiating cell populations has established a link between the transcriptional programs operating in HSCs and those present in leukemia stem cells (33). These observations are in agreement with data that support HSCs representing the cell-of-origin of many human leukemias and further support an overlap between the circuitries governing HSC self-renewal and those operating in leukemia initiating cell populations.
PHF6 regulation of HSC activity
Here we identify PHF6 as a new leukemia-initiating tumor suppressor gene implicated in the control of long-term self-renewal in HSCs and as a driver of leukemia-initiating cell activity in T-ALL. Mutations that increase the self-renewal capacity of hematopoietic progenitors are postulated to play important roles in leukemia initiation and as potential drivers of leukemia-initiating cell activity (21,34). In this context, DMT3A and TET2, two epigenetic factors governing HSC self-renewal have emerged as prominent tumor suppressors frequently mutated in clonal hematopoiesis, and as early events in the pathogenesis of acute myeloid leukemia and myelodysplastic syndromes (21,34). As in the case of DNMT3A and TET2, PHF6 mutations can be found in clonal hematopoiesis associated with aging (14) and following recovery from aplastic anemia (13), and Phf6 inactivation increases the long-term self-renewal capacity of HSCs. However, while DMT3A and TET2 both regulate DNA methylation, PHF6 is a nucleolar protein implicated in transcriptional control, ribosome biogenesis and the regulation of nucleosome positioning via interaction with the NuRD nucleosome remodeling complex (35). Notably analysis of Phf6 wild-type and knockout mice showed increased chromatin accessibility in Phf6 null LT-HSCs preferentially involving MYC targets and genes implicated in cell cycle, translation and response to stress, in support of a chromatin remodeling function of Phf6 in HSC homeostasis. In addition, gene expression profiling showed inverse effects of Phf6 inactivation and Jak1 knock-out in LT-HSCs potentially indicating a convergent mechanism of action between Phf6 inactivation and JAK-STAT signaling in promoting HSC self-renewal.
PHF6 in leukemia development
Clonal evolution mapping analysis supports that PHF6 mutations may occur early in T-ALL transformation and we demonstrate experimentally that Phf6 loss can enhance the oncogenic activity of constitutively-active mutant NOTCH1. Increased penetrance of NOTCH1-induced leukemia from Phf6 null cells may result from a priming effect resulting from increased chromatin accessibility in MYC target genes and in genes involved in cell growth and protein biosynthesis, programs that are tightly linked to T-ALL transformation and directly controlled by NOTCH1 (25,36,37). In addition, and most notably, secondary deletion of Phf6 in NOTCH1-induced leukemia cells increased leukemia initiating cell activity, a phenotype linked to the effects of PHF6 inactivation in human leukemia by the enrichment in both mouse Phf6 null tumors and in PHF6-mutant human T-ALLs of a human leukemia-initiating cell transcriptional program. Based on these observations we propose that loss of PHF6 favors leukemia initiation by enhancing the self-renewal capacity of HSCs and lowering the threshold for oncogenic transformation in hematopoietic progenitors, and postulate a role for PHF6 mutations as early drivers of leukemia initiating cell activity in T-ALL.
Open questions
Although the specific molecular functions of the PHF6 protein remain to be fully established (35), the association of Phf6 loss with increased chromatin accessibility warrants further evaluation of the role of PHF6 regulation of NuRD activity in HSCs and leukemia. Further studies will also characterize the interaction of Phf6 inactivation with leukemia oncogenes other than NOTCH1, and evaluate the role and mechanisms of Phf6 mutations in myeloid and mixed lineage leukemias. Finally, analysis of Phf6 loss in the hematopoietic system of female mice will help clarify the mechanisms responsible for the observed male predominance of PHF6 mutations in human leukemia.
Methods
Patient samples.
DNA from leukemic ALL blasts at diagnosis and relapse and matched remission samples were provided by the Hemato-Oncology Laboratory at University of Padua, Italy; the Children’s Oncology Group, and the Eastern Cooperative Oncology Group (ECOG). Samples were collected under the supervision of local Institutional Review Boards for participating institutions and analyzed under the supervision of the Columbia University Medical Center Institutional Review Board (Protocol Number: IRB-AAAB3250). All samples were obtained with written informed consent at study entry.
Mice and animal procedures.
All animals were housed in specific pathogen-free facilities at the Irving Cancer Research Center at the Columbia University Medical Center in accordance with NIH guidelines for the Care and Use of Laboratory Animals. All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC). To generate Phf6 conditional knockout mice, we used homologous recombination in C57BL/6 embryonic stem cells to introduce a LoxP site upstream of exon 4 in intron 3-4, and the LoxP/FRT-flanked Neo selection cassette downstream of exon 5 in intron 5-6. We generated chimeras in C57BL/6 albino blastocysts using three independent targeted embryonic stem cell clones identified by PCR analysis and verified by Southern blot. We verified germline transmission in the offspring of highly chimeric male mice crossed with C57BL/6 females. To remove the neomycin selection cassette, we crossed mice harboring the targeting construct with a Flp germline deleter line (Gt(ROSA)26Sortm1(FLP1)Dym/RainJ, the Jackson Laboratory), and crossed the resulting mice with C57BL/6 to breed out the Flp allele. To generate conditional Phf6 knockout mice, we bred animals harboring the conditional targeted allele with Vav-iCre+/− animals (B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J, the Jackson Laboratory) (38) expressing the Cre recombinase under control of the pan-haematopoietic Vav1 oncogene promoter. To generate conditional inducible Phf6 knockout mice, we bred animals with Rosa26+/Cre-ERT2 mice (Gt(ROSA)26Sortm1(cre/ERT2)Thl), which express a tamoxifen-inducible form of the Cre recombinase from the ubiquitous Rosa26 locus (39). Rosa26+/Cre-ERT2 mice were a kind gift from T. Ludwig. Primers used for genotyping are provided in the Supplementary Methods section (Supplementary Table 1).
Phf6−/Y Vav-iCre and Phf6+/Y Vav-iCre male mice were used in experiments at 7 - 9 weeks of age. Phf6 inactivation in age-matched Phf6f/Y Rosa26+/Cre-ERT2 and Phf6f/Y Rosa26+/+ control mice was induced at 5 – 6-weeks of age by intraperitoneal administration of 1 mg/day tamoxifen for 3 consecutive days. Mice were used in experiments a minimum of 8 weeks after tamoxifen treatment.
Male C57BL/6J mice (6-week-old) were purchased from the Jackson Laboratory (Cat. # 000664) and used as recipients in all mouse leukemia-model experiments.
Male CD45.1 mice (B6.SJL-Ptprca/BoyAiTac) were purchased from Taconic Farms (Cat. # 4007) and were used as recipients in non-competitive transplantation experiments, and as both bone marrow donors and congenic recipients in competitive transplantation experiments.
Bone marrow reconstitution experiments.
In non-competitive bone marrow transplantation experiments, 0.5 million lineage negative bone marrow cells isolated from Phf6−/Y Rosa26+/Cre-ERT2 and Phf6+/Y Rosa26+/Cre-ERT2 bone marrow donor mice (CD45.2; 6 - 8-week-old) 2 weeks after tamoxifen treatment, were transplanted into the retro-orbital sinus of lethally irradiated (990 Rads) congenic CD45.1 recipients. All experimental procedures were performed 8-10 weeks after bone marrow transplantation. Competitive transplants were performed using 0.5 million lineage negative Phf6−/Y Rosa26+/Cre-ERT2 and Phf6+/Y Rosa26+/Cre-ERT2 -derived bone marrow cells 2 weeks post tamoxifen treatment (as above) combined with 0.5 million lineage negative bone marrow cells derived from age-matched CD45.1 congenic mice. Lethally irradiated (990 Rads) 7–8-week-old congenic CD45.1 male mice served as primary mixed bone marrow recipients. Serial transplantation was performed after 24 weeks by transplanting 3 million total bone marrow cells from primary mixed bone marrow chimeras into secondary recipients, and from secondary mixed bone marrow chimeras into tertiary recipients. Donor frequency and lineage commitment were assessed every 4 weeks by flow cytometric analysis of peripheral blood.
Generation of NOTCH1-induced leukemia.
To generate NOTCH1-induced T-ALL tumors in mice, we performed retroviral transduction of lineage negative-enriched bone marrow cells (Lineage Cell Depletion kit, Miltenyi Biotec, Cat. # 130-090-858) with activated forms of the NOTCH1 oncogene (NOTCH1-L1601P-ΔPEST) tagged with the green fluorescent protein (GFP) and transplanted them into the lethally irradiated (990 Rads) isogenic recipients (40). T-ALL tumor development was assessed by monitoring CD4+ CD8+ GFP+ cells in peripheral blood by flow cytometry.
To generate isogenic Phf6 wild-type and Phf6 knockout tumors, we generated NOTCH1-L1601P-ΔPEST-induced leukemia using Phf6f/Y Rosa26+/CreERT2 donor cells as before (40). In all subsequent in vivo studies, 1 million Phf6f/Y Rosa26+/Cre-ERT2 T-ALL tumor cells were transplanted into primary sub-lethally irradiated (450 Rads) congenic C57BL/6 recipients by retro-orbital injection. After 48 hours, mice received either 3 mg tamoxifen or an equivalent volume of corn oil by intraperitoneal injection. Resulting isogenic Phf6 wild-type (Phf6f/Y Rosa26+/Cre-ERT2 and Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) tumor cells recovered from primary recipient mice were transplanted via retro-orbital injection into isogenic secondary recipients for analysis of tumor progression (10,000 tumor cells/recipient; n = 10-15 animals per group) and leukemia initiating cell activity (10, 000 – 1 tumor cells/recipient; n = 6 animals per group).
Drug treatments.
Tamoxifen (Cat. # T5648) and 5-fluorouracil (5-FU; Cat. # F6627) were purchased from Sigma-Aldrich. Tamoxifen was dissolved in corn oil at 55°C to a stock concentration of 30 mg ml−1 and stored at −20°C. In all leukemia model experiments, a single 3 mg dose of tamoxifen in 100 μl was administered by intraperitoneal injection. Vehicle treated animals received 100 μl of corn-oil. 5-FU was dissolved in Phosphate Buffer Saline (PBS) at 65 °C to a final concentration of 15 mg ml−1 and administered by intraperitoneal injection at a dose of 150 mg kg−1. A 1 mg ml−1 working solution of polyinosine-polycytidylic acid high molecular weight (poly (I:C); Invivogen, Cat. # 11C21-MM) was prepared in physiological saline solution (NaCL 0.9%) and was administered at a dose of 5 μg g−1 body weight by intraperitoneal injection.
Western blotting.
Western blot analysis was performed using standard protocols. Rabbit antibody against PHF6 (Sigma-Aldrich, Cat. # HPA001023) was used at a 1:1000 dilution, and rabbit anti-GAPDH antibody (Cell Signaling, Cat. # 2118) was used at a 1:10, 000 dilution.
Cell isolation and flow cytometry analysis.
Single-cell suspensions of total bone marrow cells were prepared by crushing the long bones and passing cells through a 40 μm filter. For experiments involving HSC sorting, bone marrow cells were additionally isolated from the illia and spinal vertebrae. Single-cell suspensions of soft tissues were obtained by pressing the tissue through a 40 μm filter. All cell suspensions were prepared in PBS supplemented with 2% FCS, 1 mM Hepes and 0.5 μM EDTA. Erythrocytes were eliminated using ACK lysing buffer. Absolute quantification of cell number was performed using CountBright™ absolute Counting Beads (Invitrogen, Cat. # MP36950). B-cell progenitor populations were defined as follows: pre-pro B (B220+ IgD− CD19−), pro-B (B220+ IgD− CD19− IgM− CD25−), pre-B (B220+ IgD− CD19− IgM− CD25+), immature B (B220+ IgD− CD19+ IgM+ CD25−), total B cells (B220+ IgD−). A full list of fluorochrome-conjugated anti-mouse antibodies is included in the Supplementary Methods section (Supplementary Table 2).
Flow cytometry analysis was performed on a FACSCantoII, LSRII and FortessaLSRII (BD Biosciences) and analyzed using Flowjo Treestar software (FlowJo LLC). Flourescence activated cell sorting was performed on a BD Influx cell sorter.
Cell cycle analysis.
After staining of surface antigens, bone marrow cells were fixed using the Cytofix Fixation/Permeabilization Kit (BD Biosciences, Cat. # 554714) according to the manufacturer’s instructions. Cells were stained with anti-Ki67 antibody (1:30 dilution) overnight on ice. 1 hour prior to analysis, cells were incubated with 1.62 μM Hoechst 33342 (Invitrogen, Cat. # H3570) at room temperature. Acquisition was performed using a BD LSRII™ (BD Biosciences) flow cytometry analyzer.
Mobilization Protocol.
Mice were injected intraperitoneally with 200 mg kg−1 of cyclophosphamide (Cy; Calbiochem, Cat #239785) and then on succeeding days with 5 μg day−1 recombinant human G-SCF (Amgen, Cat. # 55513-924-10) administered by subcutaneous injection. The day of cyclophosphamide treatment was considered day −1 and the first day of G-CSF treatment was counted as day 0 (for example, mice sacrificed on day 4 of the mobilization protocol were sacrificed on the day after the fourth G-CSF injection, designated as D4).
Cell sorting.
LT-HSCs (defined here as Lin− Sca1+ CD117+ CD135− CD150+ CD48−) were sorted from lineage-depleted bone marrow cells isolated from wild-type (Phf6f/Y Rosa26+/+) and Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) mice 8 weeks after tamoxifen treatment. Cell sorting was performed using a BD Influx cell sorter BD Biosciences).
ATAC Sequencing.
ATAC sequencing was performed as previously described (41) in 20,000 LT-HSCs sorted from three individual Phf6 knockout (Phf6−/Y Rosa26+/Cre-ERT2) and Phf6 wild-type (Phf6f/Y Rosa26+/+) mice 8 weeks post tamoxifen treatment. Nuclei were isolated and tagmented using Nextera Tn5 Transposase with single indexed, library-specific sequencing adapters. ATACseq libraries were quantified based on the average fragment size according to Bioanalyzer results, pooled at equimolar concentrations and sequenced as a pool to a total read depth target of approximately 7 · 107, paired-end, 75 nucleotide-long reads per sample on an Illumina NextSeq 500. BCL files were demultiplexed and FASTQ files generated on the BaseSpace platform (Illumina). Reads were trimmed of contaminating adapter sequences using cutadapt and aligned to the GRCM38 build mouse genome using Bowtie2 in very-sensitive mode. Peaks of transposase accessible chromatin were called using MACS2 v2.1.1 (42) in SPMR mode on each sample independently and were subsequently centered by symmetrically extending peak summits in the 5’ and 3’ direction by 200 nucleotides. Overlapping peaks were merged into single regions and the set of all non-overlapping peaks was used for further analysis. We used the featureCounts method from the Rsubread v1.32.1 package to quantify the accessibility of each peak in all samples. Statistical analysis of differentially accessible regions was performed using total library size normalization and a negative binomial model in R using the DESeq2 1.22.1 package (43). We annotated ATAC-seq accessible peaks according to their nearest genomic feature using the annotatePeaks script from the HOMER package v4.10. Gene ontology analysis was also performed for all regions with significantly increased accessibility using HOMER. Enrichment of gene signatures was calculated using the hypergeometric distribution. Genome-wide distribution of chromatin accessibility was plotted using the Circlize v0.4.4 package in R. Global proximal promoter accessibility was plotted with deeptools, 2Kb 5’ and 3’ to (and centered at) the transcription start site (TSS) of all protein-coding transcripts annotated using the RPGC normalized signal for all protein coding genes in the GRCm38.p6 comprehensive gene annotation from Gencode.
RNA Sequencing and Gene Expression Profiling.
For RNA-seq analysis of isogenic Phf6 wild-type and knockout mouse T-ALL cells, RNA was extracted from 106 cells (n = 3 per group) using the RNeasy Mini Kit (Qiagen, Cat. # 74106). cDNA synthesis and subsequent library synthesis were performed using NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs; Cat. # E7530). For RNA-seq analysis of Phf6 wild-type and knockout LT-HSCs, cells were sorted directly into buffer RLT Plus and total RNA was isolated using the RNeasy Plus Micro Kit (Qiagen, Cat. # 74034). First-strand cDNA synthesis and full-length ds cDNA amplification were performed using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Clontech Laboratories, Cat. # 634888). Sequencing-ready libraries were generated using the Low Input Library Prep Kit v2 (Clontech Laboratories, Cat. # 634899). The Agencourt AMPure XP kit was used to purify cDNA (Beckman Coulter, Cat. # A63880). Libraries were quantified using the Qubit dsDNA HS Assay Kit (Invitrogen; Cat. # Q32854) and by quantitative PCR using KAPA Library Quantification Kit (Kapa Biosystems, Cat. # KR0405). Libraries were pooled in equimolar concentrations and sequenced on an Illumina NextSeq 500 to a total target depth of 10 million 150bp paired-end reads per sample. All protocols were performed following the manufacturer’s instructions.
Raw reads were de-multiplexed and FASTQ files were generated using bcl2fastq v2.20.0.422 (Illumina). Reads were aligned to a hybrid mouse (GRCm38.p6)-ERCC genome using STAR v2.3.5.3a and gene-count matrices were computed using featureCounts v1.6.0 (Rsubread). The Removal of Unwanted Variance (RUV) method (44) was used to normalize based on the synthetic spike-in controls (Ambion, Cat. # 4456740) in RUVseq v1.14.0. Differential expression analysis was performed using the negative binomial distribution to estimate the variance-mean dependence and to test for statistical significance in DESeq2 v1.20.0 (43).
GSEA.
Gene set enrichment analysis (45) was performed using regularized log transformed normalized count data in the javaGSEA program (Broad Institute). We used the Jak1 mouse knockout hematopietic stem cell signature gene set from (18) and the human 128 leukemia initiating cell signature gene set from (31). For analysis of mouse tumors human gene identifiers were converted to mouse in R using the biomaRt bioconductor package. Analysis of human T-ALL tested the enrichment of the leukemia initiating signature in PHF6 wild-type (n=5) vs. PHF6 mutant (n=5) leukemias (46). Analysis was restricted to tumors in the TLX1 and TLX3 group to avoid the confounding effect of TLX1/TLX3 association with PHF6 mutations (1). Enrichment scores were computed and 10,000 gene permutations were used to estimate P-values.
Data availability.
RNA-seq and ATAC-seq datasets generated during this study, cited RNA-seq datasets and microarray datasets are available in Gene Expression Omnibus (GEO) under accession GSE117165, GSE85745 and GSE42328, respectively.
Integrated Sequential Network (ISN) of T-ALL.
We illustrated the sequential order of somatic mutations in T-ALL using the Integrated Sequential Network (ISN) which pools evolutionary paths across all patients. We selected recurrently mutated genes that were previously defined as drivers of pediatric T-ALL (27) and relapse-specific genes. Synonymous single nucleotide variants were not included in the analysis. For each patient, we generated a sequential network that defined early events as mutations observed in both primary tumor and relapsed tumor. Each node represents a gene, and each arrow points from a gene with an early event to a gene with a late event. The ISN then pooled sequential networks across all patients. To test whether a gene within the ISN was significantly early or late, we used the binomial test based on in-degree and out-degree of each node. Specifically, the PHF6 P value = 0.03 corresponds to the binomial cumulative distribution function for x=0 arrows going into PHF6, n=5 total trials, and probability P = 0.5 that any arrow will point towards PHF6 [binocdf(x=0, N=4, p=0.5)].
Statistical analyses.
Statistical analysis was performed using GraphPad Prism software v5.0 (GraphPad Software, La Jolla, CA, USA). We assumed normality and equal distribution of variance between groups, and we considered results with Student’s t test P < 0.05 as statistically significant. Kaplan-Meier curves were used to represent survival, where significance was calculated with the log-rank test. We analyzed serial limited dilution leukemia-initiating cell data using the ELDA online resource software (47).
Supplementary Material
Significance.
Phf6 controls HSC homeostasis, leukemia initiation, and T-ALL leukemia initiating cell self-renewal. These results substantiate a role for PHF6 mutations as early events and drivers of leukemia stem cell activity in the pathogenesis of T-ALL.
Acknowledgements
National cancer institute
We are grateful to T. Ludwig (The Ohio State University Comprehensive Cancer Center) for the Rosa26+/Cre-ERT2 mouse. We thank E. Passegué for insightful suggestions in experimental design and interpretation of our results.
Grant support
This work was supported by the National Institute of Health grants R35 CA210065 (AF), R01 CA155743 (AF), CA180827 (EP), CA196172 (EP), U54 CA193313 (RR), R01 CA185486 (RR), U54 CA209997 (RR), CA180820 (ECOG-ACRIN), CA189859 (ECOG-ACRIN), CA14958 (ECOG-ACRIN), CA180791 (ECOG-ACRIN), CA17145 (ECOG-ACRIN), U10 CA180827 (ECOG-ACRIN), U10 CA98543 (JMG, MLL), the Human Specimen Banking Grant U24 CA114766 (JMG) and P30 CA013696 (in support of the Herbert Irving comprehensive Cancer Center Genomics, Flow Cytometry and Transgenic Mouse Shared Resources). AAW was supported by a Rally Foundation fellowship. PvV was supported by the Fund for Scientific Research (FWO) Flanders (postdoctoral fellowship and Odysseus type 2 grant). MR was supported by a Damon-Runyon Sohn Pediatric Cancer fellowship.
The authors declare no financial conflicts of interest. Funding for these studies was provided by: National Institute of Health grants R35 CA210065 (AF), R01 CA155743 (AF), CA180827 (EP), CA196172 (EP), U54 CA193313 (RR), R01 CA185486 (RR), U54 CA209997 (RR), CA180820 (ECOG-ACRIN), CA189859 (ECOG-ACRIN), CA14958 (ECOG-ACRIN), CA180791 (ECOG-ACRIN), CA17145 (ECOG-ACRIN), U10 CA180827 (ECOG-ACRIN), U10 CA98543 (JMG, MLL), the Human Specimen Banking Grant U24 CA114766 (JMG) and P30 CA013696 (in support of the Herbert Irving comprehensive Cancer Center Genomics, Flow Cytometry and Transgenic Mouse Shared Resources). AAW was supported by a Rally Foundation fellowship. PvV was supported by the Fund for Scientific Research (FWO) Flanders (postdoctoral fellowship and Odysseus type 2 grant). MR was supported by a Damon-Runyon Sohn Pediatric Cancer fellowship.
Footnotes
Disclosure of Potential Conflicts of Interest
No conflicts of interest have been disclosed by any of the authors.
References
- 1.Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nature genetics 2010;42(4):338–42 doi 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia 2011;25(1):130–4 doi 10.1038/leu.2010.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England journal of medicine 2012;366(12):1079–89 doi 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nature genetics 2002;32(4):661–5 doi 10.1038/ng1040. [DOI] [PubMed] [Google Scholar]
- 5.Todd MA, Picketts DJ. PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. Journal of proteome research 2012;11(8):4326–37 doi 10.1021/pr3004369. [DOI] [PubMed] [Google Scholar]
- 6.Torchy MP, Hamiche A, Klaholz BP. Structure and function insights into the NuRD chromatin remodeling complex. Cellular and molecular life sciences : CMLS 2015;72(13):2491–507 doi 10.1007/s00018-015-1880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, et al. The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron 2013;78(6):986–93 doi 10.1016/j.neuron.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Smith ADt, Renfrow MB, Schneider DA. The RNA polymerase-associated factor 1 complex (Paf1C) directly increases the elongation rate of RNA polymerase I and is required for efficient regulation of rRNA synthesis. The Journal of biological chemistry 2010;285(19):14152–9 doi 10.1074/jbc.M110.115220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Leung JW, Gong Z, Feng L, Shi X, Chen J. PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. The Journal of biological chemistry 2013;288(5):3174–83 doi 10.1074/jbc.M112.414839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018;562(7727):373–9 doi 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481(7380):157–63 doi 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao MM, Todd MA, Kontny U, Neas K, Sullivan MJ, Hunter AG, et al. T-cell acute lymphoblastic leukemia in association with Borjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatric blood & cancer 2010;55(4):722–4 doi 10.1002/pbc.22574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. The New England journal of medicine 2015;373(1):35–47 doi 10.1056/NEJMoa1414799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018;559(7714):400–4 doi 10.1038/s41586-018-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer cell 2014;26(6):813–25 doi 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang WW, Schrier SL, Weissman IL. Age-associated changes in human hematopoietic stem cells. Seminars in hematology 2017;54(1):39–42 doi 10.1053/j.seminhematol.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Elias HK, Bryder D, Park CY. Molecular mechanisms underlying lineage bias in aging hematopoiesis. Seminars in hematology 2017;54(1):4–11 doi 10.1053/j.seminhematol.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Kleppe M, Spitzer MH, Li S, Hill CE, Dong L, Papalexi E, et al. Jak1 Integrates Cytokine Sensing to Regulate Hematopoietic Stem Cell Function and Stress Hematopoiesis. Cell stem cell 2018;22(2):277 doi 10.1016/j.stem.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015;520(7548):549–52 doi 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 20.Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. The Journal of experimental medicine 2014;211(2):245–62 doi 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrando AA, Lopez-Otin C. Clonal evolution in leukemia. Nature medicine 2017;23(10):1135–45 doi 10.1038/nm.4410. [DOI] [PubMed] [Google Scholar]
- 22.Randall TD, Weissman IL. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood 1997;89(10):3596–606. [PubMed] [Google Scholar]
- 23.Morrison SJ, Wright DE, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proceedings of the National Academy of Sciences of the United States of America 1997;94(5):1908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (New York, NY) 2004;306(5694):269–71 doi 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 25.Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature medicine 2014;20(10):1130–7 doi 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soto-Feliciano YM, Bartlebaugh JME, Liu Y, Sanchez-Rivera FJ, Bhutkar A, Weintraub AS, et al. PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes & development 2017;31(10):973–89 doi 10.1101/gad.295857.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nature reviews Cancer 2016;16(8):494–507 doi 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- 28.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. The Lancet Oncology 2009;10(2):147–56 doi 10.1016/s1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, Haydu JE, Rigo I, Hadler M, et al. ETV6 mutations in early immature human T cell leukemias. The Journal of experimental medicine 2011;208(13):2571–9 doi 10.1084/jem.20112239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016;540(7633):433–7 doi 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 31.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nature medicine 2011;17(9):1086–93 doi 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 32.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends in cell biology 2005;15(9):494–501 doi 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 33.Vedi A, Santoro A, Dunant CF, Dick JE, Laurenti E. Molecular landscapes of human hematopoietic stem cells in health and leukemia. Annals of the New York Academy of Sciences 2016;1370(1):5–14 doi 10.1111/nyas.12981. [DOI] [PubMed] [Google Scholar]
- 34.Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell stem cell 2018;22(2):157–70 doi 10.1016/j.stem.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd MA, Ivanochko D, Picketts DJ. PHF6 Degrees of Separation: The Multifaceted Roles of a Chromatin Adaptor Protein. Genes 2015;6(2):325–52 doi 10.3390/genes6020325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proceedings of the National Academy of Sciences of the United States of America 2006;103(48):18261–6 doi 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margolin AA, Palomero T, Sumazin P, Califano A, Ferrando AA, Stolovitzky G. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proceedings of the National Academy of Sciences of the United States of America 2009;106(1):244–9 doi 10.1073/pnas.0806445106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. European journal of immunology 2003;33(2):314–25 doi 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 39.Guo K, McMinn JE, Ludwig T, Yu YH, Yang G, Chen L, et al. Disruption of peripheral leptin signaling in mice results in hyperleptinemia without associated metabolic abnormalities. Endocrinology 2007;148(8):3987–97 doi 10.1210/en.2007-0261. [DOI] [PubMed] [Google Scholar]
- 40.Herranz D, Ambesi-Impiombato A, Sudderth J, Sanchez-Martin M, Belver L, Tosello V, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. 2015;21(10):1182–9 doi 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Current protocols in molecular biology 2015;109:21.9.1–9 doi 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome biology 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nature biotechnology 2014;32(9):896–902 doi 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, et al. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood 2013;122(1):74–82 doi 10.1182/blood-2013-03-491092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods 2009;347(1-2):70–8 doi 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ATAC-seq datasets generated during this study, cited RNA-seq datasets and microarray datasets are available in Gene Expression Omnibus (GEO) under accession GSE117165, GSE85745 and GSE42328, respectively.