

Cell cycle regulation and brain injury: Traumatic brain injury (TBI) is one of the primary causes of morbidity and mortality in patients, and it affects more than 1.7 million Americans each year. Other than direct damage resulting from TBI, the brain injury triggers several secondary mechanisms which either individually or in combination contribute to the neurological deficits after TBI (Gupta and Sen, 2016). These secondary mechanisms include neuronal death and stimulate proliferation of astrocytes and microglial cells, which in turn triggers neuro-inflammation (Simon et al., 2017). The association between proliferation of dividing cells like astrocytes and microglia with cell cycle has already been highlighted in a recent publication (Simon et al., 2017). However, in this article, we will emphasize the importance of cell cycle in post-mitotic cells like mature neurons following brain injury.
Briefly, the cell cycle is considered as an essential cellular mechanism to determine the fate of cells and typically consists of four phases: S‐phase, during which DNA replication occurs; M‐phase, where cell division, or mitosis, takes place, and the gap phases that separate the two; G1 and G2, respectively (Herrup and Yang, 2007). However, neurons exist as a non-dividing and quiescent stage known as G0, and remain terminally differentiated in the brain. As a result, they cannot enter into the cell cycle. Under cellular stress, these mitotically inactive neurons which are in G0 phase, become wrongly activated and forced to enter the cell cycle; however, these neurons were incapable of completing the cell cycle and triggered the cell death pathways to kill themselves through apoptosis (Herrup and Yang, 2007).
The expression of the proteins involved in the cell cycle is significantly lowered in neurons compared to other dividing cells like astrocytes and glial cells in the brain. Thus, there was a concern whether the lack of cell cycle regulatory proteins in the neuron is responsible for induction of cell death in neurons. Several independent studies concluded that it was not the fact; instead, several cell cycle regulatory proteins such as cyclin D1 was aberrantly induced and forces mature neurons to enter into the cell cycle process and ultimately leads to cell death following brain trauma (Cernak et al., 2005; Byrnes and Faden, 2007). Interestingly, the activation of cyclin D1 is not exclusive to neurons. Previous studies from our group (Saha et al., 2018) and others (Kabadi et al., 2012; Skovira et al., 2016) found that cyclin D1 level was also increased in astrocytes and microglial cells. The effect of increased cyclin D1 in these cells is different from neuronal fate. Previously, it was demonstrated that proliferation of microglial and astrocytic cells is associated with the other cell cycle proteins and caspase activation in neurons following TBI (Skovira et al., 2016). As a proof-of-fact, treatment with an inhibitor of cell-cycle kinase which acts in concomitant with cyclin, reduced neuronal cell death, brain lesion volume, astroglial scar formation, and microglial activation, as well as subsequent neurological deficits (Di Giovanni et al., 2005). However, the major limitation of this study is that the underlying mechanism remains obscure. Our study fulfilled the void and elucidated the underlying mechanism how an induction of cyclin D1 affects neuronal fates following TBI. Our recent study established that an induction of cyclin D1 mediates the neurotoxicity through promoting mitochondrial dysfunction following TBI.
Mitochondrial biogenesis and TBI: Mitochondria are essential to maintaining the neuronal cell homeostasis through a balanced process of mitophagy and biogenesis. In the process of mitophagy, the damaged mitochondria which have lost their membrane potential were removed from the cell. If mitophagy is impaired, the damaged mitochondria will be accumulated inside cells and the excessive reactive oxygen species generated from the damaged mitochondria will affect other mitochondria and ultimately will lead to cell death. Thus, regulated mitophagy is required for healthy cells; however, disruption of this process during stress conditions like TBI causes toxicity. The biogenesis of mitochondria is the process to replenish the pool of mitochondria. In fact, the mitochondrial biogenesis and mitophagy have always remained in the equilibrium within the healthy cells. Thus, the proper intracellular distribution of mitochondria is assumed to be critical for normal physiology of neuronal cells (Anne Stetler et al., 2013; Wang et al., 2017).
Mitochondrial mass, by itself, represents the net balance between rates of biogenesis and degradation and mitochondrial mass can be regulated by mitochondrial DNA content which is known to be synthesized inside the nucleus through activation of several transcription factors (Lee and Wei, 2005). Mitochondrial mass is one of the critical factors to maintain the function of mitochondria including energy metabolism. The mitochondrial oxidative phosphorylation (OXPHOS) is critical for energy (ATP) production in eukaryotic cells. The OXPHOS enzymes are multimeric complexes (Anne Stetler et al., 2013), and PGC-1α is a co-transcriptional regulation factor that induces mitochondrial mass by activating different transcription factors, including NRF1, which promotes the expression of mitochondrial transcription factor A (TFAM). NRF1 is an essential contributor to the sequence of events leading to the increase in transcription of key mitochondrial enzymes, and it has been shown to regulate TFAM, which drives transcription and replication of mitochondrial DNA (Lee and Wei, 2005).
Our study suggests that activation of cyclin D1 following TBI affects mitochondrial mass through impairment of a key transcription factor, NRF1 in the nucleus. NRF1 mostly transcribes genes coding for mitochondrial proteins involved in energy production. Thus, either depletion or inactivation of NRF1 will lead to an impairment in OXPHOS which ultimately leads to mitochondrial dysfunction and oxidative stress inside cells. We have shown that NRF1 could interact and acetylated by an acetyltransferase p300/CBP and acetylation of NRF1 enhances its transcriptional activation by augmenting its DNA binding (Saha et al., 2018). TBI leads to a decrease in acetylation of NRF1 due to a reduced interaction between NRF1 and p300. An increase in the level of cyclin D1 blocks the interaction between NRF1 and p300 in the nucleus, and as a result, the transcriptional activity of NRF1 was reduced. Administration of RNAi for cyclin D1 rescues the interaction between p300 and NRF1 and recovers the transcriptional activity of NRF1 following TBI (Anne Stetler et al., 2013) (Figure 1).
Figure 1.
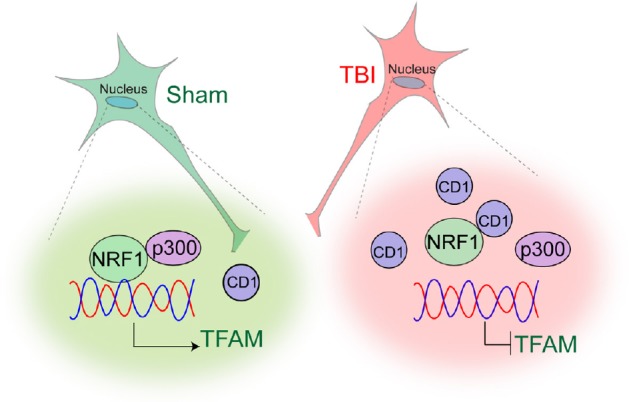
A model showing how cyclin D1 (CD1) affects mitochondrial mass following traumatic brain injury (TBI).
TBI leads to a decrease in acetylation of NRF1 due to a reduced interaction between NRF1 and p300. An increase in the level of CD1 blocks the interaction between NRF1 and p300 in the nucleus, and as a result, the transcriptional activity of NRF1 was reduced. TFAM: Mitochondrial transcription factor A.
Collectively, our study not only re-establish the importance of cyclin D1 in the neural cell death, but also uniquely discover the influence of cyclin D1 in mitochondrial function. This study provides evidence in support of the fact that augmentation in cyclin D1 can directly influence the mitochondrial mass via modulating the transcriptional activity of NRF1. TBI-induced decrease in transcriptional activation of NRF1, can explain how a loss of mitochondrial mass contributes to compromise in the mitochondrial function and induce oxidative stress. In addition, our innovative approach of rescuing the loss of mitochondrial mass by reducing the level of cyclin D1 provides a novel strategy to rescue mitochondrial function following TBI. Considering that mitochondrial dysfunction is a common mechanism of pathology associated with several neurodegenerative diseases, the identification of the role of cyclin D1 to mitochondrial mass can be extended to these diseases to refine our current understanding of the related pathology.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Masahito Kawabori, Hokkaido University, Japan.
P-Reviewer: Kawabori M; C-Editors: Zhao M, Li JY; T-Editor: Liu XL
References
- 1.Anne Stetler R, Leak RK, Gao Y, Chen J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J Cereb Blood Flow Metab. 2013;33:22–32. doi: 10.1038/jcbfm.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrnes KR, Faden AI. Role of cell cycle proteins in CNS injury. Neurochem Res. 2007;32:1799–1807. doi: 10.1007/s11064-007-9312-2. [DOI] [PubMed] [Google Scholar]
- 3.Cernak I, Stoica B, Byrnes KR, Di Giovanni S, Faden AI. Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle. 2005;4:1286–1293. doi: 10.4161/cc.4.9.1996. [DOI] [PubMed] [Google Scholar]
- 4.Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005;102:8333–8338. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta R, Sen N. Traumatic brain injury: a risk factor for neurodegenerative diseases. Rev Neurosci. 2016;27:93–100. doi: 10.1515/revneuro-2015-0017. [DOI] [PubMed] [Google Scholar]
- 6.Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- 7.Kabadi SV, Stoica BA, Loane DJ, Byrnes KR, Hanscom M, Cabatbat RM, Tan MT, Faden AI. Cyclin D1 gene ablation confers neuroprotection in traumatic brain injury. J Neurotrauma. 2012;29:813–827. doi: 10.1089/neu.2011.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Saha P, Gupta R, Sen T, Sen N. Activation of cyclin D1 affects mitochondrial mass following traumatic brain injury. Neurobiol Dis. 2018;118:108–116. doi: 10.1016/j.nbd.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171–191. doi: 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skovira JW, Wu J, Matyas JJ, Kumar A, Hanscom M, Kabadi SV, Fang R, Faden AI. Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J Neuroinflammation. 2016;13:299. doi: 10.1186/s12974-016-0769-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Figueiredo-Pereira C, Oudot C, Vieira HL, Brenner C. Mitochondrion: a common organelle for distinct cell deaths? Int Rev Cell Mol Biol. 2017;331:245–287. doi: 10.1016/bs.ircmb.2016.09.010. [DOI] [PubMed] [Google Scholar]