

Alzheimer’s and Parkinson’s diseases are neurodegenerative disorders pathologically classified by the accumulation of amyloidogenic proteins into insoluble inclusions within the brain. Specifically, amyloid plaques in the brains of Alzheimer’s disease patients are comprised of amyloid-β (Aβ) peptide, the product of sequential cleavage of the amyloid precursor protein by β- and γ-secretases. Similarly, α-synuclein is a major component of Lewy bodies associated with Parkinson’s disease. Though both diseases increase progressively with disease and age, the soluble oligomeric forms associated with each polypeptide are, arguably, the most toxic species. Both Aβ and α-synuclein form porous oligomers that permeabilize membranes (Di Scala et al., 2016). Intracellular α-synuclein obstructs endoplasmic reticulum-Golgi traffic and following internalization, Aβ rapidly aggregates in endosomes and lysosomes. Using transgenic Caenorhabditis elegans models of neurodegeneration, Griffin et al. (2018) show that the homotypic fusion protein sorting complex, HOPS, lies at an intersection between the two pathogenic proteins to functionally protect against neurodegeneration (Figure 1).
Figure 1.
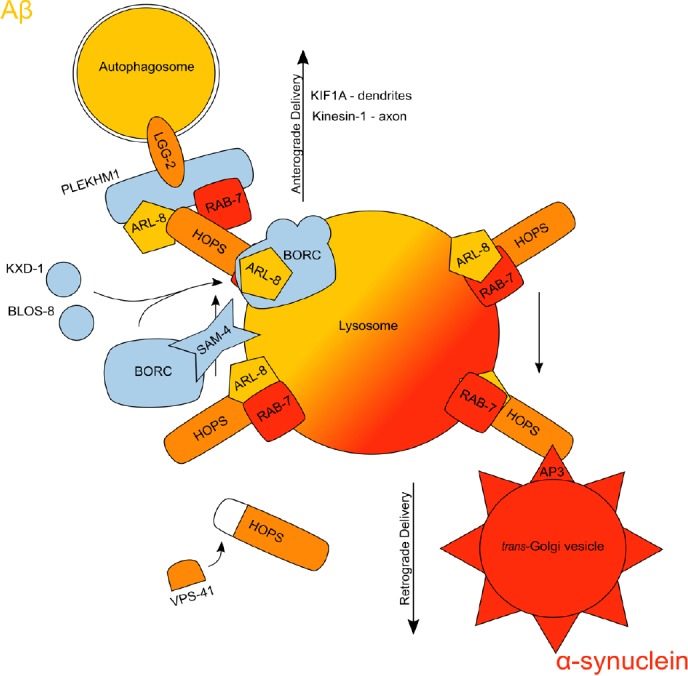
An illustration of the interactions discussed in Griffin et al. (2018).
The homotypic fusion and vacuole protein sorting (HOPS) complex (orange), which contains vacuolar protein sorting protein 41 (VPS-41), lies at the intersection between mechanisms involved in α-synuclein (red) and amyloid-β (Aβ) (yellow) toxicity. Other components of lysosomal trafficking (blue) facilitate HOPS, but it is unclear if these also participate in Aβ toxicity. As a component of the BLOC-one-related complex (BORC), the Caenorhabditis elegans orthologue of human LOH12CR1 (SAM-4) stimulates guanosine exchange within the arf-like guanosine-5′-triphosphate (GTP)ase, ARL-8. The BORC subunits, KxDL motif containing 1 (KXD-1) and biogenesis of lysosome-related organelles complex 8 (BLOS-8), are necessary for lysosomal positioning, but dispensable for synaptic vesicle trafficking. Both the kinesin family member 1A (KIF1A) and Kinesin-1 kinesins associate with BORC to differentiate dendritic targeting from axonal positioning, respectively. The pleckstrin homology and RUN domain containing M1 (PLEKHM1) protein is a scaffold for the interactions between ARL-8, the microtubule-associated proteins light chain 3 (LC3) orthologue (LGG-2), and the Ras-related protein 7 (RAB-7), however, it is unknown whether it facilitates peripheral autophagosomal clearance of Aβ. RAB-7 and HOPS on the lysosome surface coordinate to mediate tethering and fusion of trans-Golgi vesicles coated with adapter protein 3 (AP3).
In a genetic screen to identify modifiers of α-synuclein toxicity, the vacuolar protein sorting protein 41 (vps-41), which is part of the HOPS complex, increased misfolding of α-synuclein when depleted and later reduced neurodegeneration upon over-expression. This protective effect was conserved in transgenic nematodes expressing human VPS41 and was dependent on its WD40 and clathrin heavy chain repeat domains, in addition to the adaptor protein, AP3. Structure-function analysis revealed that truncation of the protein to include just WD40 and clathrin heavy chain repeat produced the smallest protective cDNA encoding hVps41. With the vps-41 gene product at the hub of endo-lysosomal interactions, Griffin et al. (2018) hypothesized that it might protect against Aβ-mediated endo-lysosomal disruptions. Overexpression of hVps41 attenuated Aβ-induced neurodegeneration, while conditional neuron-specific RNA interference of vps-41 increased neurodegeneration (Figure 1). However, RNA interference depletion of either AP3 or casein kinase, csnk-1, that modulates AP3-VPS-41 interactions in the α-synuclein model, did not increase Aβ toxicity, suggesting differential roles of hVps41 in protecting from neurodegenerative disease.
Considering the disparity between the two neurodegenerative models, the authors performed a small systematic screen of genes that define the endosomal pathway. Neuronal depletion of rab-5, required for early endosome function, increased neurodegeneration. Additionally, depletion of vps-39 or vps-41, both components of the HOPS complex, increased neurodegeneration. Depletion of sand-1, required for RAB exchange in the maturation of the early endosome into the late endosome, also increased neurodegeneration. These show that disruption of the early endosomal system exacerbates Aβ as has similarly been observed in other models (Li et al., 2012). Notably, while transgenic over-expression of the human phosphatidyl-inositol binding clathrin assembly protein (PICALM) reduced Aβ toxicity in yeast and C. elegans models (Griffin et al., 2017), RNA interference of the C. elegans PICALM increased neurodegeneration. Though single nucleotide polymorphisms in PICALM are associated with Alzheimer’s disease, it is unclear how altered PICALM specifically affects Aβ. Decreased PICALM expression exacerbates toxicity of Alzheimer’s disease-associated tau protein by decreasing SNARE protein availability for autophagy (Moreau et al., 2014), but it is unknown whether PICALM contributes to Aβ toxicity through this phenomenon. However, the results from Griffin et al. (2018) suggest that exploring the relationship between Alzheimer’s disease-associated single nucleotide polymorphisms in PICALM, autophagy, and Aβ toxicity may shed light on disease progression towards therapeutic targets.
The interaction between Rab7 and Vps41 for the tethering and fusion of endosomes with lysosomes is well-established within the literature, from yeast to mammalian cell culture. It was therefore unexpected that loss of rab-7 would increase neurodegeneration by α-synuclein, but not Aβ (Figure 1). Searching for alternative endolysosomal effectors that interact with HOPS, Griffin et al. (2018) identified a genetic relationship between hVps41/vps-41 and microtubule-associated proteins light chain 3 (LC3)/lgg-2 that was modulated by the Arf-like guanosine-5′-triphosphate (GTP)ase, arl-8 (Figure 1). While ARL-8 regulates anterograde positioning, RAB-7 is required for retrograde trafficking. A previous study found that over-expression of Arl8 increased toxicity of A53T α-synuclein (Korolchuk et al., 2011). Using wild-type α-synuclein, Griffin et al. (2018) observed no effect of arl-8 over-expression on α-synuclein-mediated neurodegeneration. Neurodegeneration by wild-type α-synuclein increased only when arl-8 was mutated into either GTP- or guanosine diphosphate (GDP)-locked states. In contrast, GTP-locked arl-8 reduced Aβ-mediated neurodegeneration, while GDP-locked arl-8 had no effect on Aβ toxicity. While differential regulation of arl-8-associated processes between neuronal subtypes is a concern, the formation of toxic oligomeric α-synuclein species is largely dependent on dopamine (Mor et al., 2017). Therefore, the effect of arl-8 might be related to the milieu of the dopaminergic neuron. However, it is unclear whether the acceleration of Aβ toxicity is similarly dependent on cell type. Due to the propensity of A53T α-synuclein towards unsolicited oligomerization, this raises the question of how hVps41 and arl-8 participate with A53T toxicity within dopaminergic neurons and other neuronal subtypes.
Considering Rab7 and Arl8 regulate opposing retrograde and anterograde trafficking, respectively, over-expression of arl-8 would theoretically recapitulate loss of rab-7 and vice versa. In this respect, with loss of rab-7 increasing α-synuclein-mediated neurodegeneration, one would expect GDP-locked arl-8 to reduce α-synuclein-mediated neurodegeneration. On the contrary, both the GTP- and GDP-locked states of arl-8 increased neurodegeneration. Because Arl8 was required for localization of Vps41 and Rab7 in human cell culture (Khatter et al., 2015), the GDP-locked state of ARL-8 might hinder proper recruitment of Rab7 in mitigating α-synuclein toxicity. Co-localization of hVps41 with ARL-8 is also affected by a single nucleotide polymorphism, T146P, in hVps41 (Khatter et al., 2015). This single nucleotide polymorphism failed to provide a protective effect in either the α-synuclein or Aβ backgrounds. Clearly, the effect of ARL-8 states on HOPS recruitment remains to be resolved, especially in the context of α-synuclein toxicity.
Previous work suggests the GTP exchange factor of ARL-8 is SAM-4 (Jia et al., 2017). The SAM-4 protein is part of a multi-subunit complex, the BLOS-related complex (BORC), that alters the recruitment and activity of kinesin through the GTP-bound state of ARL-8. Though Griffin et al. (2018) showed that the GDP-locked arl-8 was not sufficient to provide protection in the Aβ background, this does not answer the question of whether the GTPase activity or guanosine exchange in ARL-8 is necessary to confer protection. Additionally, the interaction between BORC, HOPS, and LC3 is facilitated by the Pleckstrin Homology and RUN-domain containing M1 protein (PLEKHM1/CUP-14) (Jia et al., 2017), which directly interacts with Arl8 for tethering and fusion of autophagosomes with lysosomes, mediated by the HOPS complex (Marwaha et al., 2017). It is unclear whether interactions with PLEKHM1/CUP-14 are necessary to attenuate Aβ, but over-expression of PLEKHM1/cup-14 might recapitulate the protective phenotype of hVps41 over-expression. These interactions between autophagy and lysosomes with PLEKHM1/CUP-14 are mediated through domains that interact directly with LC3, Rab7, or Arl8. Though loss of rab-7 had no effect on Aβ-mediated neurodegeneration, it is unknown whether both RAB-7 and ARL-8 are required for interaction with PLEKHM1. Thus, expression of PLEKHM1/CUP-14, and respective truncates, would reveal how these protein interactions contribute to neuroprotection.
Considering autophagy appears to play a major role in secretion (Nilsson et al., 2013), it is possible that this machinery might function to reduce the intracellular Aβ burden by engulfment and expulsion of aggregates. Extracellular deposition of Aβ is a hallmark of Alzheimer’s disease that has been observed as secretion via extracellular vesicles or the release of endolysosomal contents (Melentijevic et al., 2017). One study found that the cytochrome P450 reductase, emb-8, and the coronin-like polarity and osmotic sensitivity protein, pod-1, are necessary for the ejection of protein aggregates, including huntingtin and Aβ, from C. elegans neurons (Melentijevic et al., 2017). However, it is not known how emb-8 and pod-1 coordinate this phenotype. Components of BORC can be exchanged to specify whether it mediates the anterograde trafficking of either synaptic vesicles or lysosomes. Probing two specific components of BORC, kxd-1 and blos-8, would provide insight, since they are necessary for anterograde transport of lysosomes, but dispensable for transport of synaptic vesicle precursors (Niwa et al., 2017). As Griffin et al. (2018) have shown, there is a relationship between autophagy, hVps41/vps-41, and arl-8, but whether autophagy participates with vps-41 and arl-8 for autophagic secretion or lysosomal delivery by BORC is unclear. As previously mentioned, upregulated autophagy increases extracellular deposition of insoluble Aβ fractions in the brain, decreasing soluble intracellular oligomers (Nilsson et al., 2013). Thus, comparative western blot assay of soluble and insoluble fractions of Aβ would reveal whether over-expression of arl-8, or changes in its GTP-bound state, modifies insoluble Aβ fractions.
Additionally, the relationship between arl-8 and PLEKHM1/cup-14 in Aβ or α-synuclein toxicity remains nebulous. Since HOPS and LC3/lgg-2 directly interact with PLEKHM1/cup-14 within clearly-defined domain architecture (McEwan et al., 2015), single nucleotide polymorphisms or truncates could be tested for their functional roles in the context of α-synuclein or Aβ in vivo. Indeed, a Parkinson’s-associated single nucleotide polymorphism in a gene neighboring PLEKHM1 was also associated with altered mRNA levels of PLEKHM1. However, no study has directly investigated the effects of altered PLEKHM1 expression or function on proteotoxicity.
Alternatively, Aβ toxicity might be mitigated through lysosomal degradation at the cellular periphery. While subsequent internalization of Aβ leads to its intracellular aggregation and dysfunction of digestive compartments, defects in the endosomal machinery trap Aβ in the early endosome (Li et al., 2012), suggesting that the effects of Aβ might manifest shortly after internalization near the cell surface. Since Arl8 and BORC also recruit HOPS for peripheral autophagosomal-lysosomal fusion and positioning (Jia et al., 2017), autophagosomal envelopment and lysosomal fusion might contribute to clearance of peripheral aggregates. Delivery of lysosomes to the dendrites or axons is mediated by the activities of either KIF1A/unc-104 and Kinesin-1/unc-116, respectively, with BORC (Farías et al., 2017). Additionally, it is possible this precedes autophagy-mediated secretion of aggregates, which may be a defense mechanism to reduce the load of intracellular aggregates by jettison.
The disappointing state-of-affairs regarding unsuccessful clinical trials for diseases of neurodegeneration, and especially Alzheimer’s disease, underscore the urgent need to define new therapeutic targets and more direct mechanisms for intervention. The example set by Griffin et al. (2018), while limited to functional relationships involving hVps41-mediated protection in C. elegans, is representative of a more directed means toward accelerating the translational path through exploiting tractable in vivo models of Alzheimer’s disease and/or Parkinson’s disease-associated proteotoxicity. Though the neuroprotective mechanisms differ between models, they orbit an element shared between these two diseases, thus highlighting the therapeutic advantage of targeting commonalities underlying neurodegeneration. Nevertheless, this does not distract from the gravity of these mechanistic differences, which remain elusive in understanding the etiology of disease.
Additional file: Open peer review reports 1 (58.1KB, pdf) and 2 (58.1KB, pdf) .
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Stefano Di Santo, Inselspital Universitatsspital Bern, Switzerland; Robert L. Haining, Georgia Gwinnett College, USA.
P-Reviewers: Cho KS, Adornetto A; C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- 1.Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, Fantini J. Common molecular mechanism of amyloid pore formation by Alzheimer’s beta-amyloid peptide and alpha-synuclein. Sci Rep. 2016;6:28781. doi: 10.1038/srep28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farías GG, Guardia CM, De Pace R, Britt DJ, Bonifacino JS. BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc Natl Acad Sci U S A. 2017;114:E2955–2964. doi: 10.1073/pnas.1616363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin EF, Caldwell KA, Caldwell GA. Genetic and pharmacological discovery for alzheimer’s disease using Caenorhabditis elegans. ACS Chem Neurosci. 2017;8:2596–2606. doi: 10.1021/acschemneuro.7b00361. [DOI] [PubMed] [Google Scholar]
- 4.Griffin EF, Yan X, Caldwell KA, Caldwell GA. Distinct functional roles of Vps41-mediated neuroprotection in Alzheimer’s and Parkinson’s disease models of neurodegeneration. Hum Mol Genet. 2018;27:4176–4193. doi: 10.1093/hmg/ddy308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia R, Guardia CM, Pu J, Chen Y, Bonifacino JS. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy. 2017;13:1648–1663. doi: 10.1080/15548627.2017.1343768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khatter D, Raina VB, Dwivedi D, Sindhwani A, Bahl S, Sharma M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J Cell Sci. 2015;128:1746–1761. doi: 10.1242/jcs.162651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, O’Kane CJ, Deretic V, Rubinsztein DC. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–460. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Kanekiyo T, Shinohara M, Zhang Y, Ladu MJ, Xu H, Bu G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–44601. doi: 10.1074/jbc.M112.420224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marwaha R, Arya SB, Jagga D, Kaur H, Tuli A, Sharma M. The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J Cell Biol. 2017;216:1051–1070. doi: 10.1083/jcb.201607085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57:39–54. doi: 10.1016/j.molcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, Taub D, Parker JA, Neri C, Gabel CV, Hall DH, Driscoll M. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367–371. doi: 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mor DE, Tsika E, Mazzulli JR, Gould NS, Kim H, Daniels MJ, Doshi S, Gupta P, Grossman JL, Tan VX, Kalb RG, Caldwell KA, Caldwell GA, Wolfe JH, Ischiropoulos H. Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration. Nat Neurosci. 2017;20:1560–1568. doi: 10.1038/nn.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F, Lavau CP, Betton M, O’Kane CJ, Wechsler DS, Rubinsztein DC. PICALM modulates autophagy activity and tau accumulation. Nat Commun. 2014;5:4998. doi: 10.1038/ncomms5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 15.Niwa S, Tao L, Lu SY, Liew GM, Feng W, Nachury MV, Shen K. BORC regulates the axonal transport of synaptic vesicle precursors by activating ARL-8. Curr Biol. 2017;27:2569–2578.e4. doi: 10.1016/j.cub.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.