

Abstract
Aneurysmal subarachnoid hemorrhage remains serious hemorrhagic stroke with high morbidities and mortalities. Aneurysm rupture causes arterial bleeding-induced mechanical brain tissue injuries and elevated intracranial pressure, followed by global cerebral ischemia. Post-subarachnoid hemorrhage ischemia, tissue injuries as well as extravasated blood components and the breakdown products activate microglia, astrocytes and Toll-like receptor 4, and disrupt blood-brain barrier associated with the induction of many inflammatory and other cascades. Once blood-brain barrier is disrupted, brain tissues are directly exposed to harmful blood contents and immune cells, which aggravate brain injuries furthermore. Blood-brain barrier disruption after subarachnoid hemorrhage may be developed by a variety of mechanisms including endothelial cell apoptosis and disruption of tight junction proteins. Many molecules and pathways have been reported to disrupt the blood-brain barrier after subarachnoid hemorrhage, but the exact mechanisms remain unclear. Multiple independent and/or interconnected signaling pathways may be involved in blood-brain barrier disruption after subarachnoid hemorrhage. This review provides recent understandings of the mechanisms and the potential therapeutic targets of blood-brain barrier disruption after subarachnoid hemorrhage.
Keywords: blood-brain barrier, early brain injury, endothelial cell, subarachnoid hemorrhage, tight junction, inflammation, matricellular protein, Toll-like receptor 4, TLR4
Introduction
The rupture of cerebral aneurysms extravasates and spreads arterial blood into the subarachnoid space, causing subarachnoid hemorrhage, which remains a serious condition with high morbidities and mortalities (Suarez et al., 2006; Suzuki et al., 2018b). The aggregate worldwide incidence of aneurysmal subarachnoid hemorrhage is about 2–5% of all strokes and about 10.5 cases per 100,000 person-years, but the increasing incidences are known in specific regions (especially in Finland and Japan), older ages (mean age, 55 years), women, blacks, cases with a family history of first-degree relatives with subarachnoid hemorrhage and some heritable connective-tissue disorders (Suarez et al., 2006; Connolly et al., 2012). The modifiable risk factors include hypertension, smoking, alcohol abuse, and cocaine use; and surgical treatment of unruptured aneurysms having high risks for rupture (larger size, specific locations and shapes) is recommend for preventing subarachnoid hemorrhage (Connolly et al., 2012).
The most important determinant of poor outcome of aneurysmal subarachnoid hemorrhage is early brain injury, which begins within minutes after the initial bleeding by increased intracranial pressure and subsequent global cerebral ischemia as well as extravasated blood (Suzuki et al., 2018a). Therefore, early aneurysmal obliteration by surgical clipping or endovascular coiling to prevent rebleeding is the most important to improve the outcome (Connolly et al., 2012). However, even if ruptured aneurysm is successfully treated, intensive medical care is required to manage delayed cerebral ischemia with and without cerebral vasospasm, and other various medical complications (Connolly et al., 2012). Major clinical factors associated with poor outcome are advanced age and surrogate markers of early brain injury (initial clinical severity, increased amount of subarachnoid hemorrhage, and global cerebral edema), the latter of which is also a predictor of delayed cerebral ischemia (Suarez et al., 2006; Suzuki et al., 2017, 2018a; Nakatsuka et al., 2018; Nishikawa et al., 2018b). Blood-brain barrier disruption is an important component of both early brain injury and delayed cerebral ischemia, and we have performed a PubMed literature search of articles published in the period January 2014–October 2018 on brain-blood barrier disruption after subarachnoid hemorrhage, focusing on the mechanisms in this paper.
Characteristics of Blood-Brain Barrier Disruption after Subarachnoid Hemorrhage
Blood-brain barrier disruption is a prominent pathological characteristic of both ischemic and hemorrhagic strokes, and is usually associated with poor outcome (Keep et al., 2014; Jiang et al., 2018; Suzuki et al., 2018a). Disruption of blood-brain barrier after intracerebral hemorrhage is mainly caused by extravasated blood components (Keep et al., 2014), while focal ischemia itself plays a large role in blood-brain barrier disruption after ischemic stroke (Jiang et al., 2018). However, blood-brain barrier disruption after aneurysmal subarachnoid hemorrhage is largely different from that after cerebral infarction or intracerebral hemorrhage: it may be caused by the combination of transient global cerebral ischemia developing immediately after aneurysmal rupture, delayed cerebral ischemia and extravasated components at days 4 to 14 after onset (Suzuki et al., 2018a). Thus, in aneurysmal subarachnoid hemorrhage, global cerebral edema is observed in 8–67% of patients at admission, and 12% of patients develop delayed onset of cerebral edema within two weeks of onset (Suzuki et al., 2018a). Especially, global cerebral edema or increased blood-brain barrier permeability at an acute stage of aneurysmal subarachnoid hemorrhage has been correlated with unfavorable outcomes, and is an important manifestation of pathophysiologies called early brain injury by basic scientists of experimental subarachnoid hemorrhage (Ivanidze et al., 2018; Suzuki et al., 2018a). In addition, relatively young people (mean age, 55 years) suffer aneurysmal subarachnoid hemorrhage compared with other types of strokes (Suarez et al., 2006), and different comorbidities may occur between patients with subarachnoid hemorrhage and those with other types of strokes. Comorbidities also influence blood-brain barrier disruption and outcome after any type of stroke (Jiang et al., 2018; Suzuki and Nakano, 2018). Thus, blood-brain barrier disruption after subarachnoid hemorrhage is unique, and therefore different therapeutic approach should be needed.
Because global cerebral edema is a prognostic factor, its timely and effective treatment may have a chance to improve outcomes after subarachnoid hemorrhage (Suzuki et al., 2018a). However, therapies such as hyperosmolar agents, hypertonic saline, therapeutic hypothermia, barbiturates, and decompressive craniectomy have been tried to reduce brain edema and/or to control intracranial pressure for poor-grade subarachnoid hemorrhage patients, but the efficacy has not been established well (Komotar et al., 2009; de Oliveira Manoel et al., 2016). Although similar players have been reported to be involved in both early brain injury and cerebral vasospasm, drugs that prevent vasospasm have failed to improve outcome (Suzuki and Nakano, 2018): this could be explained by a limited therapeutic window of early brain injury or acute blood-brain barrier disruption. That is, as early brain injury or acute blood-brain barrier disruption begins within minutes after the initial bleeding, early administration of a drug even within 24–72 hours of the ictus might prevent cerebral vasospasm, but not early brain injury (Suzuki and Nakano, 2018). Or more specific treatments against blood-brain barrier disruption may be needed to modulate it. Anyway, the mechanisms of blood-brain barrier disruption after subarachnoid hemorrhage should be clarified furthermore to develop new therapies against it.
Animal Models Used in Subarachnoid Hemorrhage Studies
Subarachnoid hemorrhage-induced brain injuries are very complex, and more than 72 kinds of animal models using various species have been proposed with some modifications dependent on the research question (Suzuki and Nakano, 2018). However, there remains a lack of guidance in the performance and modeling of experimental subarachnoid hemorrhage including differences in outcome measures and methodological shortcomings, causing the failure in translation of preclinical findings into clinical trials (Marbacher et al., 2018). Preclinical researchers also know that many factors including genetic background, strain-related differences, age, sex, comorbidities, and anesthetics strongly influence outcome measures (Marbacher et al., 2018; Suzuki and Nakano, 2018). Nowadays, acute subarachnoid hemorrhage models in mice or rats produced by endovascular perforation are increasingly used to study early brain injury (Marbacher et al., 2018). In an endovascular perforation model, acute physiological and metabolic changes such as intracranial pressure elevation, subarachnoid blood distribution and reactions by the blood including high mortalities are similar to clinical findings in aneurysmal subarachnoid hemorrhage (Schwartz et al., 2000). Thus, the endovascular perforation animal model becomes the most popular one for studies of blood-brain barrier disruption after subarachnoid hemorrhage (Suzuki and Nakano, 2018), and most of findings described in this paper have been obtained from the model.
Mechanisms Underlying Blood-Brain Barrier Disruption After Subarachnoid Hemorrhage
Blood-brain barrier consists of a continuous endothelial cell layer, astrocyte end feet and pericytes known as a neurovascular unit. To maintain barrier functions or unique properties of blood-brain barrier, the adjacent endothelial cells strictly bind via tight junctions, thus preventing the extravasation of intravascular contents and the invasion of immune cells. Under normal conditions, blood-brain barrier selectively passes specific substances such as water, ions and small molecular weight particles, and regulates brain homeostasis (Chow and Gu, 2015). However, when blood-brain barrier is disrupted, harmful blood contents (thrombin, fibrinogen, and so on) enter brain parenchyma and are directly exposed to brain tissues (Keep et al., 2014). In addition, dysfunction of blood-brain barrier allows the abnormal transmigration and infiltration of leukocytes into brain parenchyma (Fujimoto et al., 2018), causing increased releasing of various cytokines, chemokines, reactive oxygen species and proteases (Figure 1A). Blood-brain barrier disruption further aggravates brain tissue damages, worsens cerebral edema formation, and elevates intracranial pressure (Chen et al., 2014). Thus, blood-brain barrier disruption is considered as an important therapeutic target to treat early brain injury and to improve outcomes of aneurysmal subarachnoid hemorrhage.
Figure 1.
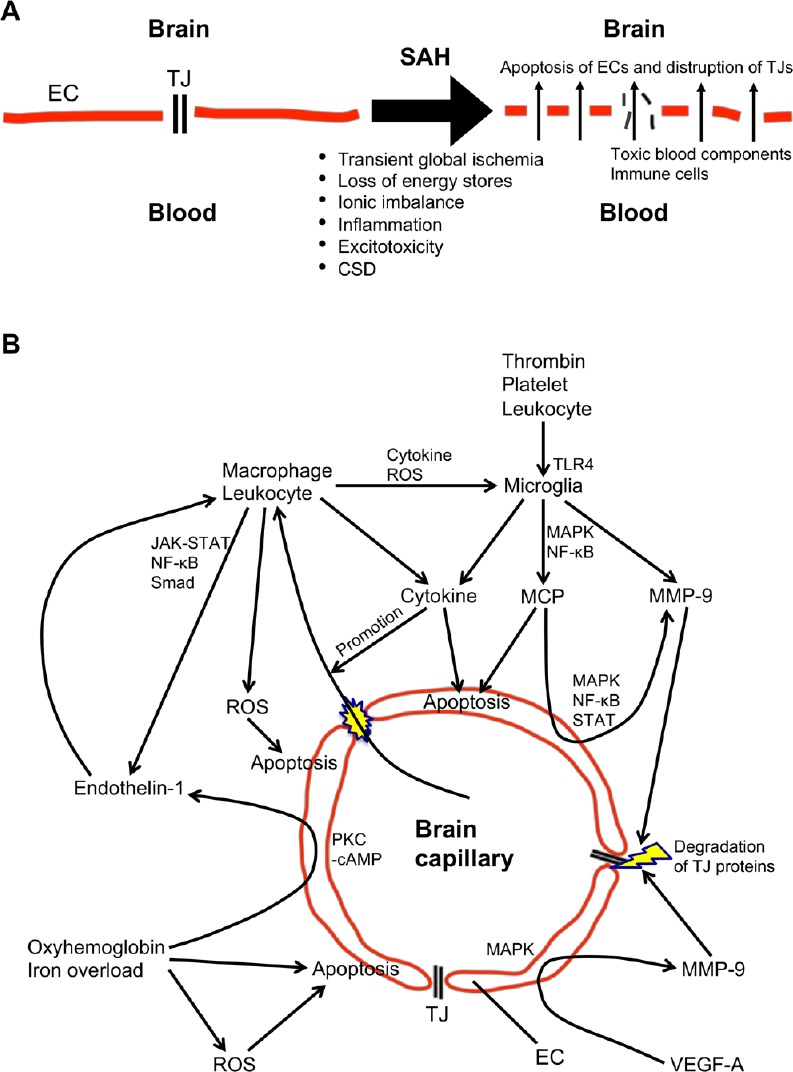
Schema of potential mechanisms (A) and signaling pathways (B) of blood-brain barrier disruption after subarachnoid hemorrhage (SAH).
CSD: Cortical spreading depolarization; EC: endothelial cell; JAK: Janus kinase; MAPK: mitogen-activated protein kinase; MCP: matricellular protein; MMP-9: matrix metalloproteinase-9; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; PKC-cAMP: protein kinase C-cyclic adenosine monophosphate; ROS: reactive oxygen species; STAT: signal transducer and activator of transcription; TJ: tight junction; TLR4: Toll-like receptor 4; VEGF-A: vascular endothelial growth factor-A.
Blood-brain barrier disruption after aneurysmal subarachnoid hemorrhage may be developed by multiple mechanisms including endothelial cell apoptosis and disruption of tight junctions (Peeyush Kumar et al., 2018). After a rupture of cerebral aneurysm, extravasated blood under arterial pressure causes mechanical damages of the surrounding brain tissues as well as elevated intracranial pressure, followed by global cerebral ischemia. Moreover, various blood components and the breakdown products are spread in the subarachnoid space. These hemorrhage, ischemia and brain tissue damage are all contributors to blood-brain barrier disruption with inducing many cascades such as inflammatory reactions. This review focuses on the mechanisms and the potential therapeutic targets of blood-brain barrier disruption after subarachnoid hemorrhage (Figure 1B).
Endothelial Cell Dysfunction or Apoptosis as a Potential Target
Normal brain capillary endothelial cells maintain the function of blood-brain barrier, but aneurysmal rupture triggers endothelial cell dysfunction and apoptosis, leading to blood-brain barrier disruption. After aneurysmal subarachnoid hemorrhage, multiple factors including oxidative stress, oxyhemoglobin, and iron overload can induce endothelial cell apoptosis within 10 minutes to 24 hours of onset (Friedrich et al., 2012; Peeyush Kumar et al., 2018). Oxyhemoglobin a representative of subarachnoid blood component directly exerts cytotoxic effects on brain endothelial cells via caspases-3, -8 or -9, elevation of intracellular Ca2+, activation of matrix metalloproteinase (MMP)-9, and generation of free radicals (Peeyush Kumar et al., 2018). There are several sources for the excessive generation of free radicals after subarachnoid hemorrhage, such as disrupted mitochondrial respiration, extracellular hemoglobin following erythrolysis and the subsequent iron overload, and endothelial cells are known to be susceptible to oxidative stress (Ayer and Zhang, 2008). Reactive oxygen species can activate apoptotic signals including p53, caspases-3 and -9 to promote apoptotic cell death, causing blood-brain barrier disruption (Chen et al., 2014).
There are many other players possibly involved in brain endothelial cell injuries. Post-subarachnoid hemorrhage ischemia, tissue injuries and subarachnoid blood components including heme, thrombin, fibrinogen, platelets and leukocytes can activate microglia as well as Toll-like receptor 4, which recognizes damage-associated molecular patterns and initiates inflammatory cascades via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and/or activator protein-1 that is mainly mediated by mitogen-activated protein kinases (MAPKs) (Okada and Suzuki, 2017; Suzuki et al., 2018a). Activation of Toll-like receptor 4 produces pro-inflammatory cytokines and mediators such as tumor necrosis factor-α, interleukins-1β, -6, -8, and -12, and MMP-9 (Okada and Suzuki, 2017). Pro-inflammatory cytokines cause caspase-dependent endothelial cell apoptosis and blood-brain barrier dysfunction, and also upregulate specific cell adhesion molecules on endothelial cells, which consequently allow macrophages and neutrophils to migrate into the subarachnoid space and brain tissues and to release a multitude of inflammatory factors including endothelins and oxidative radicals through activation of Janus kinase-2 and signal transducer and activator of transcription-3, NF-κB, and Smad signaling pathways (Chen et al., 2009; Peeyush Kumar et al., 2018). Oxyhemoglobin also directly induces endothelin-1 production in endothelial cells and vascular smooth muscle cells via protein kinase C-cyclic adenosine monophosphate signaling, and the binding of endothelin-1 to endothelin-A receptors activates macrophages, increases neutrophil-vessel wall interactions, and induces free radical production, all of which lead to endothelial cell dysfunction in myocardial ischemia (Singhal et al., 2010; Peeyush Kumar et al., 2018). However, clazosentan an endothelin receptor antagonist decreased large-artery vasospasm, but did not suppress oxidative stress, endothelial nitric oxide synthase dysfunction, and microthromboembolism after experimental subarachnoid hemorrhage in mice (Sabri et al., 2011).
A derangement in neurotransmitter release and inhibition of the reuptake leading to excitotoxicity, cortical spreading depolarization, and loss of energy stores causing ionic imbalance also occur after subarachnoid hemorrhage, and contribute to subarachnoid hemorrhage-induced endothelial cell death (Chen et al., 2014). Glutamate, a major excitatory transmitter, is synthesized by activated astrocytes and microglia, and mediates toxic effects via excessive activation of N-methyl D-aspartate receptors, leading to massive Ca2+ influx and subsequent apoptotic cell death and necrosis (Chen et al., 2014).
Disruption of Tight Junctions
The contraction of endothelial cells and disassembly of tight junctions increase the permeability of blood-brain barrier and cause blood-brain barrier disruption, leading to brain edema formation after subarachnoid hemorrhage (Peeyush Kumar et al., 2018). Two main routes exist for the transport of materials across the blood-brain barrier: the paracellular (via tight junctions between cells) and the transcellular (caveolin- and clathrin-mediated endocytosis and macropinocytosis) pathways. Tight junction proteins include occludin, zonula occludens-1 and claudin-5, which are phosphoproteins and whose interaction, redistribution and transmembrane protein localization are regulated by changes in the phosphorylation state (Li et al., 2015). Caveolin-1 is the main component of caveolae, the specific microstructures on plasma membrane surfaces, and mediates transcellular transport pathways (Deng et al., 2012). In a study investigating changes in the expression of blood-brain barrier-related proteins, expressions of caveolin-1 and claudin-5 in the basement membrane were not changed, but tight junction proteins occludin and zonula occludens-1 were downregulated in an acute stage of subarachnoid hemorrhage (Li et al., 2015). Thus, it is considered that increased permeability or disruption of blood-brain barrier after subarachnoid hemorrhage may be caused by the disturbance of paracellular pathways.
A role for MMP-9 in the early disruption of blood-brain barrier has been repeatedly reported after subarachnoid hemorrhage (Peeyush Kumar et al., 2018; Suzuki et al., 2018a). MMP-9 is induced by inflammatory cytokines and reactive oxygen species, and degrades the extracellular matrix of cerebral microvessel basal lamina such as collagen IV, laminin, fibronectin, and inter-endothelial tight junction proteins such as zonula occludens-1, causing blood-brain barrier disruption after subarachnoid hemorrhage (Guo et al., 2010; Peeyush Kumar et al., 2018). The activation of NF-κB is known to directly regulate the transcription of MMP-9 and tissue inhibitor of MMP-1 in addition to orchestrating the inflammatory cascade, and the balanced interaction between MMP-9 and tissue inhibitor of MMP-1 may determine the severity of blood-brain barrier disruption after subarachnoid hemorrhage (Suzuki et al., 2010a). In addition, several reports have shown that blood-brain barrier disruption after subarachnoid hemorrhage is caused by MAPK-mediated MMP-9 activation (Okada et al., 2018). Tenascin-C and periostin, matricellular proteins, were respectively induced and caused blood-brain barrier disruption after subarachnoid hemorrhage via MAPK-mediated activation of MMP-9 and subsequent zonula occludens-1 degradation in brain capillary endothelial cells in mice (Fujimoto et al., 2016; Liu et al., 2017; Nishikawa and Suzuki, 2017; Shiba and Suzuki, 2019). Tenascin-C and periostin may form a positive feedback mechanism and upregulate each other to aggravate blood-brain barrier disruption after subarachnoid hemorrhage (Liu et al., 2017). Another matricellular protein galectin-3 also activated endothelial cell MMP-9 and caused blood-brain barrier disruption via MAPK and signal transducer and activator of transcription pathways (Nishikawa and Suzuki, 2018; Nishikawa et al., 2018a), while another matricellular protein osteopontin was induced in reactive astrocytes and capillary endothelial cells in a delayed fashion, and prevented MMP-9 activation and blood-brain barrier disruption by inactivating MAPK and NF-κB (Suzuki et al., 2010b, 2018c). Post-subarachnoid hemorrhage Toll-like receptor 4 activation can activate both NF-κB and MAPKs, and upregulates pro-inflammatory cytokines and mediators as well as matricellular proteins (Okada et al., 2017). Actually, selective blockage of Toll-like receptor 4 inhibited post-subarachnoid hemorrhage upregulation of these molecules and activation of MAPKs and MMP-9, resulting in maintaining the blood-brain barrier function (Okada et al., 2018).
Accumulated evidence suggests that many molecules, for example, vascular endothelial growth factor (VEGF)-A, VEGF-B, angiopoietin (Ang)-1, Ang-2, MAPKs, and MAPK phosphatase-1, are involved acting simultaneously or at different stages during blood-brain barrier disruption (Suzuki et al., 2010b). VEGF-A is a potent inducer of blood-brain barrier disruption, while VEGF-B and Ang-1 block effects of VEGF-A and have a potent anti-permeability property possibly by regulating MAPK activation: MAPKs are upstream and downstream of VEGF-A, and MAPK phosphatase-1 is an endogenous MAPK inhibitor (Nag et al., 2009; Suzuki et al., 2010b). Moreover, Ang-2 is a naturally occurring inhibitor of Ang-1 (Nag et al., 2009). Blood-brain barrier disruption after subarachnoid hemorrhage was associated with downregulation of Ang-1 and MAPK phosphatase-1, activation of MAPKs, and upregulation of VEGF-A, whereas the restoration of the damaged blood-brain barrier was associated with MAPK phosphatase-1 induction, MAPK inactivation, and VEGF-A downregulation (Suzuki et al., 2010b). However, expressions of VEGF-B and Ang-2 were unchanged after subarachnoid hemorrhage (Suzuki et al., 2010b). The remarkable thing is that recombinant osteopontin could regulate theses molecules toward preserving and restoring the blood-brain barrier (Suzuki et al., 2010b).
In addition to osteopontin, VEGF-B, Ang-1 and MAPK phosphatase-1, activation of β-catenin transcription may be a promising therapeutic target for maintaining blood-brain barrier integrity as a kind of endogenous protective mechanisms against blood-brain barrier disruption. β-catenin is an adherens junction protein linking to the actin cytoskeleton, a part of blood-brain barrier, and is a Wnt pathway signal transducer in endothelial cells. Disruption of β-catenin leads to downregulation of tight junction proteins, blood-brain barrier disruption, and neuroinflammation, which are attenuated by activation of Wnt-β-catenin signaling (Tran et al., 2016). In fact, some interventions to stabilize β-catenin or promote β-catenin nuclear translocation were reported to protect blood-brain barrier integrity by increasing the expression of tight junction proteins (claudin-3, claudin-5, occludin, and zonula occludens-1) and an adherens junction protein vascular endothelial-cadherin in experimental subarachnoid hemorrhage animal models (Zuo et al., 2017). However, aberrant activation of Wnt-β-catenin signaling may accumulate β-catenin in the nucleus and promote the transcription of many oncogenes, contributing to tumorigenesis and tumor progression of several cancers (Shang et al., 2017). Although it is unknown whether β-catenin as a part of blood-brain barrier provides tumorigenesis, further studies are needed.
Perspective
Many pathways have been reported to disrupt the blood-brain barrier after subarachnoid hemorrhage, but the exact mechanisms remain unclear. In addition, lots of therapies to prevent endothelial cell apoptosis and to preserve tight junction proteins have been proposed in experimental subarachnoid hemorrhage studies, but not decisive in subarachnoid hemorrhage in a clinical setting. Possibly, multiple independent and/or interconnected signaling pathways may be involved in blood-brain barrier disruption after subarachnoid hemorrhage with complex pathogeneses, and therefore it may be difficult to block only one specific molecule and then to prevent or treat blood-brain barrier disruption after subarachnoid hemorrhage in vivo. Thus, further discovery of novel therapeutic targets against blood-brain barrier disruption would contribute and lead to improving outcomes in patients with subarachnoid hemorrhage. Further strict experimental as well as clinical studies are needed to prove that treatments against blood-brain barrier disruption improve outcome of subarachnoid hemorrhage patients (Suzuki and Nakano, 2018).
Additional file: Open peer review report 1 (56.8KB, pdf) .
Footnotes
Conflicts of interest: None declared.
Financial support: This work was supported by a grant-in-aid for Scientific Research from Japan Society for the Promotion of Science (grant number: 17K10825) to HS.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Nathan K. Evanson, Cincinnati Children’s Hospital Medical Center, USA.
Funding: This work was supported by a grant-in-aid for Scientific Research from Japan Society for the Promotion of Science (grant number: 17K10825) to HS.
P-Reviewer: Evanson NK; C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- 1.Ayer RE, Zhang JH. Oxidative stress in subarachnoid haemorrhage: Significance in acute brain injury and vasospasm. Acta Neurochir Suppl. 2008;104:33–41. doi: 10.1007/978-3-211-75718-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen G, Zhang S, Shi J, Ai J, Hang C. Effects of recombinant human erythropoietin (rhEPO) on JAK2/STAT3 pathway and endothelial apoptosis in the rabbit basilar artery after subarachnoid hemorrhage. Cytokine. 2009;45:162–168. doi: 10.1016/j.cyto.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Feng H, Sherchan P, Klebe D, Zhao G, Sun X, Zhang J, Tang J, Zhang JH. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol. 2014;115:64–91. doi: 10.1016/j.pneurobio.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow BW, Gu C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015;38:598–608. doi: 10.1016/j.tins.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connolly ES, Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- 6.de Oliveira Manoel AL, Goffi A, Marotta TR, Schweizer TA, Abrahamson S, Macdonald RL. The critical care management of poor-grade subarachnoid haemorrhage. Crit Care. 2016;20:21. doi: 10.1186/s13054-016-1193-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng J, Huang Q, Wang F, Liu Y, Wang Z, Wang Z, Zhang Q, Lei B, Cheng Y. The role of caveolin-1 in blood-brain barrier disruption induced by focused ultrasound combined with microbubbles. J Mol Neurosci. 2012;46:677–687. doi: 10.1007/s12031-011-9629-9. [DOI] [PubMed] [Google Scholar]
- 8.Friedrich V, Flores R, Sehba FA. Cell death starts early after subarachnoid hemorrhage. Neurosci Lett. 2012;512:6–11. doi: 10.1016/j.neulet.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg. 2016;124:1693–1702. doi: 10.3171/2015.4.JNS15484. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H. Effects of tenascin-C knockout on cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol. 2018;55:1951–1958. doi: 10.1007/s12035-017-0466-x. [DOI] [PubMed] [Google Scholar]
- 11.Guo Z, Sun X, He Z, Jiang Y, Zhang X, Zhang JH. Matrix metalloproteinase-9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res. 2010;32:715–720. doi: 10.1179/016164109X12478302362491. [DOI] [PubMed] [Google Scholar]
- 12.Ivanidze J, Ferraro RA, Giambrone AE, Segal AZ, Gupta A, Sanelli PC. Blood-brain barrier permeability in aneurysmal subarachnoid hemorrhage: correlation with clinical outcomes. AJR Am J Roentgenol. 2018;211:891–895. doi: 10.2214/AJR.17.18237. [DOI] [PubMed] [Google Scholar]
- 13.Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF, Shi Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018;163-164:144–171. doi: 10.1016/j.pneurobio.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keep RF, Zhou N, Xiang J, Andjelkovic AV, Hua Y, Xi G. Vascular disruption and blood-brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS. 2014;11:18. doi: 10.1186/2045-8118-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komotar RJ, Schmidt JM, Starke RM, Claassen J, Wartenberg KE, Lee K, Badjatia N, Connolly ES, Jr, Mayer SA. Resuscitation and critical care of poor-grade subarachnoid hemorrhage. Neurosurgery. 2009;64:397–411. doi: 10.1227/01.NEU.0000338946.42939.C7. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Liang G, Ma T, Li J, Wang P, Liu L, Yu B, Liu Y, Xue Y. Blood-brain barrier permeability change and regulation mechanism after subarachnoid hemorrhage. Metab Brain Dis. 2015;30:597–603. doi: 10.1007/s11011-014-9609-1. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Kawakita F, Fujimoto M, Nakano F, Imanaka-Yoshida K, Yoshida T, Suzuki H. Role of periostin in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2017;48:1108–1111. doi: 10.1161/STROKEAHA.117.016629. [DOI] [PubMed] [Google Scholar]
- 18.Marbacher S, Grüter B, Schöpf S, Croci D, Nevzati E, D’Alonzo D, Lattmann J, Roth T, Bircher B, Wolfert C, Muroi C, Dutilh G, Widmer HR, Fandino J. Systematic review of in vivo animal models of subarachnoid hemorrhage: species, standard parameters, and outcomes. Transl Stroke Res. 2018 doi: 10.1007/s12975-018-0657-4. doi: 101007/s12975-018-0657-4. [DOI] [PubMed] [Google Scholar]
- 19.Nag S, Manias JL, Stewart DJ. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol. 2009;118:197–217. doi: 10.1007/s00401-009-0541-0. [DOI] [PubMed] [Google Scholar]
- 20.Nakatsuka Y, Shiba M, Nishikawa H, Terashima M, Kawakita F, Fujimoto M, Suzuki H pSEED group. Acute-phase plasma osteopontin as an independent predictor for poor outcome after aneurysmal subarachnoid hemorrhage. Mol Neurobiol. 2018;55:6841–6849. doi: 10.1007/s12035-018-0893-3. [DOI] [PubMed] [Google Scholar]
- 21.Nishikawa H, Liu L, Nakano F, Kawakita F, Kanamaru H, Nakatsuka Y, Okada T, Suzuki H. Modified citrus pectin prevents blood-brain barrier disruption in mouse subarachnoid hemorrhage by inhibiting galectin-3. Stroke. 2018a;49:2743–2751. doi: 10.1161/STROKEAHA.118.021757. [DOI] [PubMed] [Google Scholar]
- 22.Nishikawa H, Nakatsuka Y, Shiba M, Kawakita F, Fujimoto M, Suzuki H pSEED group. Increased plasma galectin-3 preceding the development of delayed cerebral infarction and eventual poor outcome in non-severe aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2018b;9:110–119. doi: 10.1007/s12975-017-0564-0. [DOI] [PubMed] [Google Scholar]
- 23.Nishikawa H, Suzuki H. Implications of periostin in the development of subarachnoid hemorrhage-induced brain injuries. Neural Regen Res. 2017;12:1982–1984. doi: 10.4103/1673-5374.221150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishikawa H, Suzuki H. Possible role of inflammation and galectin-3 in brain injury after subarachnoid hemorrhage. Brain Sci. 2018;8:E30. doi: 10.3390/brainsci8020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okada T, Kawakita F, Nishikawa H, Nakano F, Liu L, Suzuki H. Selective Toll-like receptor 4 antagonists prevent acute blood-brain barrier disruption after subarachnoid hemorrhage in mice. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1145-2. doi: 101007/s12035-018-1145-2. [DOI] [PubMed] [Google Scholar]
- 26.Okada T, Suzuki H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regen Res. 2017;12:193–196. doi: 10.4103/1673-5374.200795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peeyush Kumar T, McBride DW, Dash PK, Matsumura K, Rubi A, Blackburn SL. Endothelial cell dysfunction and injury in subarachnoid hemorrhage. Mol Neurobiol. 2018 doi: 10.1007/s12035-018-1213-7. doi: 10.1007/s12035-018-1213-7. [DOI] [PubMed] [Google Scholar]
- 28.Sabri M, Ai J, Macdonald RL. Dissociation of vasospasm and secondary effects of experimental subarachnoid hemorrhage by clazosentan. Stroke. 2011;42:1454–1460. doi: 10.1161/STROKEAHA.110.604728. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz AY, Masago A, Sehba FA, Bederson JB. Experimental models of subarachnoid hemorrhage in the rat: a refinement of the endovascular filament model. J Neurosci Methods. 2000;96:161–167. doi: 10.1016/s0165-0270(00)00156-4. [DOI] [PubMed] [Google Scholar]
- 30.Shang S, Hua F, Hu ZW. The regulation of beta-catenin activity and function in cancer: therapeutic opportunities. Oncotarget. 2017;8:33972–33989. doi: 10.18632/oncotarget.15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shiba M, Suzuki H. Lessons from tenascin-C knockout mice and potential clinical application to subarachnoid hemorrhage. Neural Regen Res. 2019;14:262–264. doi: 10.4103/1673-5374.244789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singhal AK, Symons JD, Boudina S, Jaishy B, Shiu YE. Role of endothelial cells in myocardial ischemia-reperfusion injury. Vasc Dis Prev. 2010;7:1–14. doi: 10.2174/1874120701007010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. N Engl J Med. 2006;354:387–396. doi: 10.1056/NEJMra052732. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki H, Ayer R, Sugawara T, Chen W, Sozen T, Hasegawa Y, Kanamaru K, Zhang JH. Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit Care Med. 2010a;38:612–618. doi: 10.1097/CCM.0b013e3181c027ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010b;41:1783–1790. doi: 10.1161/STROKEAHA.110.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki H, Fujimoto M, Kawakita F, Liu L, Nakatsuka Y, Nakano F, Nishikawa H, Okada T, Kanamaru H, Imanaka-Yoshida K, Yoshida T, Shiba M. Tenascin-C in brain injuries and edema after subarachnoid hemorrhage: Findings from basic and clinical studies. J Neurosci Res. 2018a doi: 10.1002/jnr.24330. doi: 10.1002/jnr.24330. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki H, Nakano F. To improve translational research in subarachnoid hemorrhage. Transl Stroke Res. 2018;9:1–3. doi: 10.1007/s12975-017-0546-2. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki H, Nakatsuka Y, Yasuda R, Shiba M, Miura Y, Terashima M, Suzuki Y, Hakozaki K, Goto F, Toma N. Dose-dependent inhibitory effects of cilostazol on delayed cerebral infarction after aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2018b doi: 10.1007/s12975-018-0650-y. doi: 10.1007/s12975-018-0650-y. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki H, Nishikawa H, Kawakita F. Matricellular proteins as possible biomarkers for early brain injury after aneurysmal subarachnoid hemorrhage. Neural Regen Res. 2018c;13:1175–1178. doi: 10.4103/1673-5374.235022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki H, Shiba M, Nakatsuka Y, Nakano F, Nishikawa H. Higher cerebrospinal fluid pH may contribute to the development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Transl Stroke Res. 2017;8:165–173. doi: 10.1007/s12975-016-0500-8. [DOI] [PubMed] [Google Scholar]
- 41.Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Gothert JR, Malik AB, Valyi-Nagy T, Zhao YY. Endothelial β-catenin signaling is required for maintaining adult blood-brain barrier integrity and central nervous system homeostasis. Circulation. 2016;133:177–186. doi: 10.1161/CIRCULATIONAHA.115.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zuo S, Ge H, Li Q, Zhang X, Hu R, Hu S, Liu X, Zhang JH, Chen Y, Feng H. Artesunate protected blood-brain barrier via sphingosine 1 phosphate receptor 1/phosphatidylinositol 3 kinase pathway after subarachnoid hemorrhage in rats. Mol Neurobiol. 2017;54:1213–1228. doi: 10.1007/s12035-016-9732-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.