

Abstract
Yin Yang 1 (YY1) is a multi-functional transcription factor that regulates gene expression in a range of cell types, including neurons. It controls neuronal differentiation, as well as neuronal specification and migration during the development of the mammalian nervous system. Besides, YY1 also mediates the transcription of genes that are required for neuronal survival. An impairment of the transcriptional function of YY1 causes neuronal death. This review summarizes recent research findings that unveil the dysfunction of YY1 in multiple neurodegenerative disorders. The expression of disease proteins perturbs the function of YY1 via distinct molecular mechanisms, including recruitment to protein aggregates, protein degradation and aberrant nuclear/cytoplasmic shuttling. Understanding the pathogenic roles of YY1 will further broaden our knowledge of the disease mechanisms in distinct neurodegenerative disorders.
Keywords: Alzheimer's disease, amyotrophic lateral sclerosis, neurodegeneration, protein aggregates recruitment, protein degradation, subcellular localization, transcriptional regulation, Yin Yang 1
Historical Perspective of Yin Yang 1 and Its Normal Function in the Nervous System
Yin Yang 1 (YY1) was first identified in 1991 as a multi-functional transcription factor (Shi et al., 1991). The name YY1 is adopted from Chinese, and alludes to its dual transcriptional activities in both repressing (‘Yin’) and activating (‘Yang’) gene expression (Shi et al., 1991). The genes whose expression is modulated by YY1 have widespread implications for diverse signalling pathways, leading to the involvement of YY1 in multiple cellular processes including cell differentiation, proliferation and apoptosis. The YY1 protein is ubiquitous, exerting its transcriptional activity in a range of cell types, including neurons. It is now known that YY1 supports the normal development of the mammalian nervous system by controlling the expression of two categories of developmental genes, one of which encodes transcription factors involved in brain tissue patterning, and the other encoding proteins that regulate neuronal cell specification and migration (He and Casaccia-Bonnefil, 2008). When YY1 is genetically ablated, mutant embryos display severe developmental retardation, including growth and neurulation defects (Morgan et al., 2004). Furthermore, Knauss et al. (2018) demonstrated that YY1 binds to the promoter region of a pluripotency gene, sex determining region Y-box 2, to repress its transcription in mouse cerebellar cortical neural progenitor cells. Sex determining region Y-box 2 promotes the proliferation of cerebellar cortical neural progenitor cells, and its expression is downregulated when neural progenitor cells differentiate into neurons. The YY1-mediated inhibition of sex determining region Y-box 2 results in the suppression of neural progenitor cells proliferation, while accelerating neuronal differentiation. This finding therefore suggests a role of YY1 in governing neuronal differentiation in the developing cerebellum (Knauss et al., 2018). In addition to regulating neuronal development and differentiation, YY1 also controls neuronal viability. This review highlights recent advances in studying YY1 function in neuronal survival, and demonstrates several mechanisms that lead to neuronal death due to the dysfunction of the YY1 protein. The background information in this review was collected and discussed based on a Pubmed literature search of articles published from October 1991 to September 2018.
Yin Yang 1 Maintains Neuronal Survival
Several lines of evidence have demonstrated a correlation between YY1 and neuronal survival. In a recent study, Chen et al. (2018) reported that YY1 functions as a transcription repressor in regulating the expression of a pro-apoptotic gene, Fuz. When YY1 was overexpressed, Fuz promoter was hypermethylated, and Fuz expression was downregulated. Conversely, when YY1 expression was diminished, Fuz promoter was hypomethylated and Fuz transcription was induced. Fuz is a major player in planar cell polarity signalling, a pathway that directs the polarised cell migration along the tissue plane. Dominant mutations in the Fuz gene have been uncovered in patients with neural tube defects, and a functional study revealed that Fuz mutants impair the directional cell movement and cell fusion, leading to the perturbation of neural tube closure (Seo et al., 2011). This finding highlights an important developmental role of Fuz in the human nervous system. Interestingly, Chen et al. (2018) found that when Fuz expression exceeded 2.5 times the normal level, an upregulation of neuronal apoptosis was observed, indicating a novel pro-apoptotic function of Fuz. The authors further demonstrated a Dishevelled-Ras-related C3 botulinum toxin substrate 1-mitogen-activated protein kinase-caspase signalling cascade that was exploited by Fuz to trigger neuronal apoptosis (Chen et al., 2018). In normal neurons, YY1 was found to associate with Fuz promoter to ensure sufficient DNA methylation, implying that the YY1-Fuz promoter interaction restricts the expression of Fuz to prevent the induction of neuronal apoptosis (Chen et al., 2018).
In addition to suppressing pro-apoptotic gene expression in normal neurons, YY1 also functions to promote pro-survival gene transcription. For example, YY1 was reported to bind directly to the promoter of glucose-6-phosphate dehydrogenase and stimulate its transcription. The induction of glucose-6-phosphate dehydrogenase expression subsequently leads to the activation of the pentose phosphate pathway in support of cell survival (Wu et al., 2018). It is noteworthy that in aged mice, upregulation of the brain glucose-6-phosphate dehydrogenase level alleviates neurodegeneration, suggesting a beneficial role of glucose-6-phosphate dehydrogenase in protecting against neuronal loss (Jeng et al., 2013). Taken together, YY1 maintains neuronal survival via inhibiting pro-apoptotic gene expression and simultaneously activating pro-survival genes (Chen et al., 2018; Wu et al., 2018). Any impairment of YY1’s transcriptional function would result in an imbalance of pro-apoptotic and pro-survival gene expression, and compromise neuronal survival.
Yin Yang 1 Function Is Impaired via Distinct Mechanisms in Neurodegenerative Disease Models
In 1997, Korhonen et al. (1997) first reported the impairment of functional YY1 in neuronal degeneration. The authors demonstrated that when cerebellar granule cells received excessive glutamate, a well-known excitotoxic agent that triggers neuronal death, a de-association of the YY1-DNA transcriptional complex was observed (Korhonen et al., 1997). This observation suggests a perturbation of functional YY1 during neurodegeneration. Because YY1 governs the expression of pro-apoptotic and pro-survival genes, it is possible to hypothesise that treatment with excitotoxic agents perturbs YY1’s transcriptional function, which in turn causes an imbalance of cell apoptotic and survival events, leading to the stimulation of neuronal apoptosis and contributing to neurodegeneration.
However, the ultimate cause of the deterioration of YY1 transcriptional activity in neurodegeneration, especially in neurodegenerative diseases, remains elusive. We highlight some recent research findings that unveil the impairment of YY1 via distinct mechanisms in multiple neurodegenerative diseases (Chen et al., 2018; Yin et al., 2018). More importantly, such impairment causes YY1-mediated gene dysregulation, which contributes to various pathologies.
The recruitment of Yin Yang 1 to protein aggregates
Polyglutamine (polyQ) diseases comprise a group of neurodegenerative disorders, including Huntington’s disease and several types of spinocerebellar ataxia such as spinocerebellar ataxia type 3 (SCA3). These diseases are caused by the expansion of glutamine-coding CAG trinucleotide repeats within the open reading frames of the disease genes (Lieberman et al., 2018). The formation of insoluble protein aggregates is one of the pathogenic features of polyQ diseases. Several essential cellular components, such as transcription factors, molecular chaperones and proteasomal subunits, are sequestered to polyQ protein aggregates, causing biological dysfunctions including transcriptional dysregulation, protein misfolding and perturbation of the ubiquitin proteasome system. These dysfunctions trigger the downstream cell death pathways, leading to neuronal deterioration, which contributes to the neurodegeneration in polyQ diseases (Lieberman et al., 2018).
In a recent study, Chen et al. (2018) described a novel pathogenic pathway that illustrates the relationship between polyQ protein aggregates and apoptosis induction in SCA3. This pathway involves two important partners, the transcriptional regulator YY1 and the planar cell polarity gene Fuz. The authors further showed that the level of soluble YY1 was downregulated due to the recruitment of YY1 protein to expanded SCA3-polyQ protein aggregates. Such recruitment depressed YY1 function, leading to Fuz promoter DNA hypomethylation followed by Fuz induction. The accumulation of Fuz protein consequently caused neuronal apoptosis in SCA3 disease models (Chen et al., 2018).
Similar to polyQ diseases, disease protein aggregates are observed in other neurological disorders as well. In Alzheimer’s disease, the formation of protein aggregates is caused by the expression of toxic Aβ1–42 peptide (Davis et al., 2018). Fuz induction has also been detected in Aβ1–42 neurons. The authors further demonstrated that this upregulation also resulted from the decreased DNA methylation level of Fuz promoter accompanied by the recruitment of YY1 protein to Aβ1–42 aggregates (Chen et al., 2018). These findings thus suggest a pathogenic mechanism that involves the recruitment of YY1 to disease protein aggregates, leading to the transcriptional dysregulation of a pro-apoptotic gene, Fuz, which in turn triggers apoptosis in polyQ disease and Alzheimer’s disease models.
Proteolytic cleavage of Yin Yang 1 protein
Krippner-Heidenreich et al. (2005) identified two caspase- recognised cleavage sites within the full-length YY1 protein sequence. They further provided experimental evidence that when caspases were activated, full-length YY1 was cleaved into shorter fragments, leading to the degradation of YY1 protein (Krippner-Heidenreich et al., 2005). The cleaved YY1 fragments retain the DNA-binding activity but lose the ability to regulate gene transcription, therefore behaving as a dominant negative product in occupying YY1-binding sites, in turn preventing the interaction between full-length YY1 and DNA sequences (Krippner-Heidenreich et al., 2005).
Apart from in the SCA3 and Aβ1–42 neuronal models, Fuz upregulation has further been detected in tau-expressing neurons (Chen et al., 2018). Tau protein likewise aggregates when expressed in neurons. The formation of these pathological protein aggregates compromises the neuronal functions, causing a class of neurodegenerative disorders named tauopathy (Davis et al., 2018). Interestingly, YY1 was not recruited to tau protein aggregates, suggesting that an alternative mechanism may contribute to tau-mediated Fuz induction (Chen et al., 2018). The authors were then inspired to examine whether the YY1 protein level was attenuated in tau-expressing cells. Interestingly, as demonstrated by immunoblotting results, YY1 protein was found to undergo protein degradation, resulting in an accumulation of truncated YY1 fragments accompanied by a reduction of the full-length functional YY1 level (Figure 1). Based on this observation, we hypothesise that both the reduction of full-length YY1 and accumulation of truncated fragments contribute to the impairment of YY1’s normal function, causing Fuz upregulation in tau-expressing neurons. More importantly, YY1 protein degradation was detected in the brains of Alzheimer’s disease patients (Aubry et al., 2015), further suggesting a clinical implication of YY1 degradation in neurodegenerative disorders.
Figure 1.
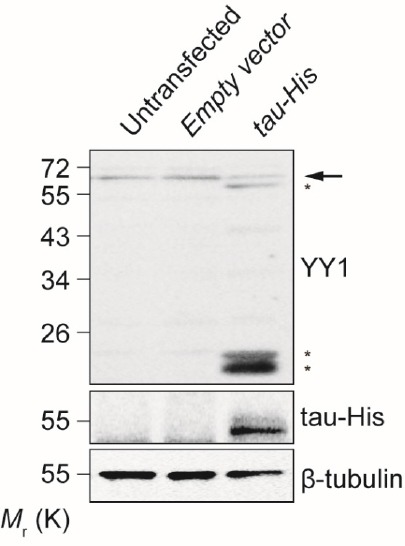
Overexpression of tau promotes Yin Yang 1 (YY1) protein degradation.
Human embryonic kidney 293 cells were transfected with a tau-overexpressing DNA construct (Chen et al., 2018). The pcDNA3.1(+) DNA construct was used as an empty vector control. The expression of YY1, tau-His and β-tubulin proteins was determined using anti-YY1 antibody (1:1000, ab109237, Abcam, Cambridge, MA, USA), anti-His antibody (1:1000, 27-4710-01, Sigma-Aldrich, St. Louis, MO, USA) and anti-β-tubulin antibody (1:2000, ab6046, Abcam), respectively. In tau-overexpressing cells, full-length YY1 protein (arrow) was found to undergo protein degradation, resulting in the accumulation of truncated YY1 protein fragments (asterisks). Beta-tubulin was used as a loading control. Experiments were independently repeated three times. Only one representative blot is shown.
Subcellular localisation of Yin Yang 1 protein
YY1 protein localises predominantly in the cell nucleus, which coincides with the fact that nuclear YY1 functions as a transcription factor to mediate gene transcription. In astrocytes derived from a SOD1G93A amyotrophic lateral sclerosis mouse model, an aberrant nuclear/cytoplasmic ratio of YY1 protein was reported. YY1 protein was found to be further enriched in the cell nuclei of diseased astrocytes (Yin et al., 2018). The accumulation of nuclear YY1 in turn caused downregulation of excitatory amino acid transporter-2, whose gene product is responsible for re-translocating glutamate into neurons to eliminate excessive extracellular glutamate. The impairment of excitatory amino acid transporter-2 expression was accompanied by impaired glutamate clearance ability, and accumulation of toxic glutamate further caused neuronal cell death in SOD1G93A astrocytes (Yin et al., 2018). In addition to protein-aggregate recruitment and degradation, another contributor to YY1 dysfunction in neurodegenerative disease is aberrant nuclear/cytoplasmic shuttling.
Concluding Remarks and Future Perspectives
Taken together, in multiple neurodegenerative disorders, the mutant disease proteins perturb the function of YY1 via distinct mechanisms, including recruitment to protein aggregates, promotion of protein degradation and alteration of nuclear/cytoplasmic distribution (Figure 2). The dysfunction of YY1 in turn causes the transcriptional dysregulation of downstream target genes (Figure 2). A future objective in YY1 research would be to uncover other potential mechanisms that lead to YY1 dysfunction in neurodegenerative diseases, such as aberrant post-translational modifications (He and Casaccia-Bonnefil, 2008).
Figure 2.
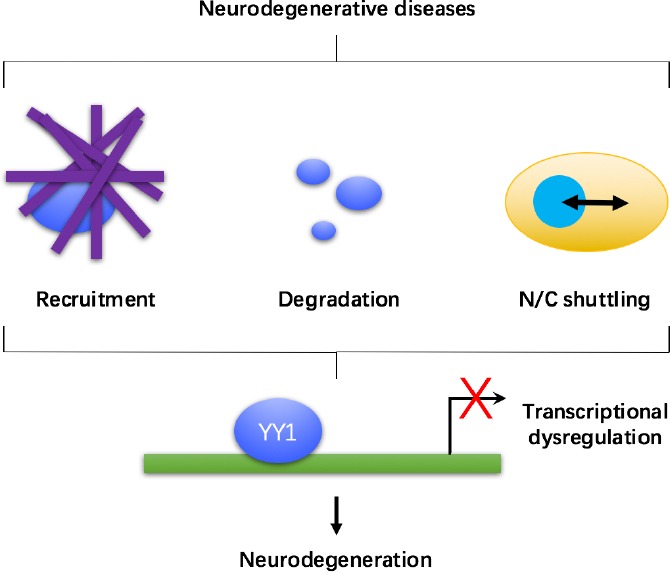
Summarised diagram illustrating Yin Yang 1 (YY1) dysfunction and transcriptional dysregulation in neurodegenerative diseases.
In multiple neurodegenerative diseases, the function of YY1 is perturbed via distinct mechanisms, including recruitment to protein aggregates, protein degradation and aberrant nuclear/cytoplasmic (N/C) shuttling. These impairments hinder the transcriptional activity of YY1, leading to gene transcriptional dysregulation, which contributes to the disease pathologies.
The central role of YY1 in governing diverse downstream genes/pathways highlights this protein as an essential therapeutic target in neurodegenerative disorders. In SCA3, when the functional level of YY1 is restored, Fuz upregulation is diminished, accompanied by the relief of neuronal apoptosis (Chen et al., 2018). Therefore, identification of potential activators of YY1, or the delivery of ectopic YY1, will help replenish YY1’s transcriptional function and correct the dysregulation of downstream genes/pathways. The restoration of the gene transcription network will thus provide therapeutic benefits against the neuronal toxicity of neurodegenerative disorders.
Other than neurons, the function of YY1 is also impaired in glial cells of neurodegenerative diseases. In astrocytes derived from SOD1G93A mouse brain, aberrant nuclear/cytoplasmic distribution of YY1 causes dysregulation of excitatory amino acid transporter-2, which in turn leads to the accumulation of glutamate that hinders neuronal viability (Yin et al., 2018). Furthermore, YY1 controls expression of excitatory amino acid transporter-1 (EAAT1), another astrocytic glutamate transporter gene (Karki et al., 2015). The dysregulation of EAAT1 is reported in multiple neurological disorders, including Alzheimer’s disease and prion diseases (Scott et al., 2002; Chretien et al., 2004). Therefore, it would be interesting to examine whether the dysfunction of YY1 leads to EAAT1 dysregulation, and contributes to disease pathologies.
Intriguingly, in addition to astrocytes, YY1 also modulates gene transcription in other types of glial cells, such as oligodendrocytes (He et al., 2007) and microglia (Zhang et al., 2018). Moreover, oligodendrocytes and microglia have been implicated in neurodegenerative disease pathogenesis (Nasrabady et al., 2018; Spiller et al., 2018). Another interesting perspective would be to investigate whether YY1’s function is dysregulated in disease oligodendrocytes and microglia, and how such dysregulation contributes to the disease pathologies. Unravelling these novel mechanisms will broaden our knowledge of YY1’s pathogenic role in various neurodegenerative diseases.
Additional file: Open peer review report 1 (55.7KB, pdf) .
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by the General Research Fund (14100714) of the Hong Kong Research Grants Council (to HYEC); The Chinese University of Hong Kong Vice-Chancellor’s One-Off Discretionary Fund (VCF2014011) (to HYEC); The Chinese University of Hong Kong One-of Funding for Joint Lab/Research Collaboration (3132980) (to HYEC); The Chinese University of Hong Kong Faculty of Science Strategic Development Fund (FACULTY-P17173) (to HYEC); The Chinese University of Hong Kong Gerald Choa Neuroscience Centre (7105306) (to HYEC); and donations from Chow Tai Fook Charity Foundation (6903898) (to HYEC) and Hong Kong Spinocerebellar Ataxia Association (6903291) (to HYEC). Zhefan Stephen Chen is supported by the Postdoctoral Fellowship in Clinical Neurosciences of The Chinese University of Hong Kong, China.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewers: Sergei Fedorovich, Institute of Biophysics and Cell Engineering of National Academy of Sciences of Belarus, Belarus; Hans-Gert Bernstein, Otto-von-Guericke University Magdeburg, Germany.
Funding: This work was supported by the General Research Fund (14100714) of the Hong Kong Research Grants Council (to HYEC); The Chinese University of Hong Kong Vice-Chancellor’s One-Off Discretionary Fund (VCF2014011) (to HYEC); The Chinese University of Hong Kong One-of Funding for Joint Lab/Research Collaboration (3132980) (to HYEC); The Chinese University of Hong Kong Faculty of Science Strategic Development Fund (FACULTY-P17173) (to HYEC); The Chinese University of Hong Kong Gerald Choa Neuroscience Centre (7105306) (to HYEC); and donations from Chow Tai Fook Charity Foundation (6903898) (to HYEC) and Hong Kong Spinocerebellar Ataxia Association (6903291) (to HYEC). Zhefan Stephen Chen is supported by the Postdoctoral Fellowship in Clinical Neurosciences of The Chinese University of Hong Kong, China.
P-Reviewers: Fedorovich S, Bernstein HG; C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- 1.Aubry S, Shin W, Crary JF, Lefort R, Qureshi YH, Lefebvre C, Califano A, Shelanski ML. Assembly and interrogation of Alzheimer’s disease genetic networks reveal novel regulators of progression. PLoS One. 2015;10:e0120352. doi: 10.1371/journal.pone.0120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen ZS, Li L, Peng S, Chen FM, Zhang Q, An Y, Lin X, Li W, Koon AC, Chan TF, Lau KF, Ngo JCK, Wong WT, Kwan KM, Chan HYE. Planar cell polarity gene Fuz triggers apoptosis in neurodegenerative disease models. EMBO Rep. 2018;19:e45409. doi: 10.15252/embr.201745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chretien F, Le Pavec G, Vallat-Decouvelaere AV, Delisle MB, Uro-Coste E, Ironside JW, Gambetti P, Parchi P, Creminon C, Dormont D, Mikol J, Gray F, Gras G. Expression of excitatory amino acid transporter-1 (EAAT-1) in brain macrophages and microglia of patients with prion diseases. J Neuropathol Exp Neurol. 2004;63:1058–1071. doi: 10.1093/jnen/63.10.1058. [DOI] [PubMed] [Google Scholar]
- 4.Davis AA, Leyns CEG, Holtzman DM. Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol. 2018;34:545–568. doi: 10.1146/annurev-cellbio-100617-062636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He Y, Casaccia-Bonnefil P. The Yin and Yang of YY1 in the nervous system. J Neurochem. 2008;106:1493–1502. doi: 10.1111/j.1471-4159.2008.05486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeng W, Loniewska MM, Wells PG. Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem Neurosci. 2013;4:1123–1132. doi: 10.1021/cn400079y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karki P, Kim C, Smith K, Son DS, Aschner M, Lee E. Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappaB and Yin Yang 1 (YY1) J Biol Chem. 2015;290:23725–23737. doi: 10.1074/jbc.M115.649327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knauss JL, Miao N, Kim SN, Nie Y, Shi Y, Wu T, Pinto HB, Donohoe ME, Sun T. Long noncoding RNA Sox2ot and transcription factor YY1 co-regulate the differentiation of cortical neural progenitors by repressing Sox2. Cell Death Dis. 2018;9:799. doi: 10.1038/s41419-018-0840-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korhonen P, Huotari V, Soininen H, Salminen A. Glutamate-induced changes in the DNA-binding complexes of transcription factor YY1 in cultured hippocampal and cerebellar granule cells. Brain Res Mol Brain Res. 1997;52:330–333. doi: 10.1016/s0169-328x(97)00310-0. [DOI] [PubMed] [Google Scholar]
- 11.Krippner-Heidenreich A, Walsemann G, Beyrouthy MJ, Speckgens S, Kraft R, Thole H, Talanian RV, Hurt MM, Luscher B. Caspase-dependent regulation and subcellular redistribution of the transcriptional modulator YY1 during apoptosis. Mol Cell Biol. 2005;25:3704–3714. doi: 10.1128/MCB.25.9.3704-3714.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieberman AP, Shakkottai VG, Albin RL. Polyglutamine repeats in neurodegenerative diseases. Annu Rev Pathol. 2018 doi: 10.1146/annurev-pathmechdis-012418-012857. doi: 10.1146/annurev-pathmechdis-012418-012857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan MJ, Woltering JM, In der Rieden PM, Durston AJ, Thiery JP. YY1 regulates the neural crest-associated slug gene in Xenopus laevis. J Biol Chem. 2004;279:46826–46834. doi: 10.1074/jbc.M406140200. [DOI] [PubMed] [Google Scholar]
- 14.Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6:22. doi: 10.1186/s40478-018-0515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott HL, Pow DV, Tannenberg AE, Dodd PR. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer’s disease. J Neurosci. 2002;22:RC206. doi: 10.1523/JNEUROSCI.22-03-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo JH, Zilber Y, Babayeva S, Liu J, Kyriakopoulos P, De Marco P, Merello E, Capra V, Gros P, Torban E. Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet. 2011;20:4324–4333. doi: 10.1093/hmg/ddr359. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Seto E, Chang LS, Shenk T. Transcriptional repression by YY1, a human GLI-Kruppel-related protein, and relief of repression by adenovirus E1A protein. Cell. 1991;67:377–388. doi: 10.1016/0092-8674(91)90189-6. [DOI] [PubMed] [Google Scholar]
- 18.Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG, Roberts CJ, Miller KR, Ransohoff RM, Trojanowski JQ, Lee VM. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci. 2018;21:329–340. doi: 10.1038/s41593-018-0083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Wang H, Li Y, Xie Y, Huang C, Zhao H, Miyagishi M, Kasim V. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 2018;78:4549–4562. doi: 10.1158/0008-5472.CAN-17-4047. [DOI] [PubMed] [Google Scholar]
- 20.Yin X, Wang S, Qi Y, Wang X, Jiang H, Wang T, Yang Y, Wang Y, Zhang C, Feng H. Astrocyte elevated gene-1 is a novel regulator of astrogliosis and excitatory amino acid transporter-2 via interplaying with nuclear factor-kappaB signaling in astrocytes from amyotrophic lateral sclerosis mouse model with hSOD1(G93A) mutation. Mol Cell Neurosci. 2018;90:1–11. doi: 10.1016/j.mcn.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XC, Liang HF, Luo XD, Wang HJ, Gu AP, Zheng CY, Su QZ, Cai J. YY1 promotes IL-6 expression in LPS-stimulated BV2 microglial cells by interacting with p65 to promote transcriptional activation of IL-6. Biochem Biophys Res Commun. 2018;502:269–275. doi: 10.1016/j.bbrc.2018.05.159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.