

Abstract
Glioblastomas are heterogeneous and invariably lethal tumours. They are characterized by genetic and epigenetic variations among tumour cells, which makes the development of therapies that eradicate all tumour cells challenging and currently impossible. An important component of glioblastoma growth is communication with and manipulation of other cells in the brain environs, which supports tumour progression and resistance to therapy. Glioblastoma cells recruit innate immune cells and change their phenotype to support tumour growth. Tumour cells also suppress adaptive immune responses, and our increasing understanding of how T cells access the brain and how the tumour thwarts the immune response offers new strategies for mobilizing an antitumour response. Tumours also subvert normal brain cells — including endothelial cells, neurons and astrocytes — to create a microenviron that favours tumour success. Overall, after glioblastoma-induced phenotypic modifications, normal cells cooperate with tumour cells to promote tumour proliferation, invasion of the brain, immune suppression and angiogenesis. This glioblastoma takeover of the brain involves multiple modes of communication, including soluble factors such as chemokines and cytokines, direct cell–cell contact, extracellular vesicles (including exosomes and microvesicles) and connecting nanotubes and microtubes. Understanding these multidimensional communications between the tumour and the cells in its environs could open new avenues for therapy.
Glioblastomas remain one of the most aggressive malignancies, with no change in the standard of care for almost 20 years and a median lifespan from time of diagnosis to death of about 15 months1. This bleak outcome has stimulated ongoing efforts to reveal new insights into these tumours and the surrounding cells to facilitate development of new treatment strategies. New studies and technologies have deepened our understanding of the factors that make these tumours so formidable but have highlighted two major challenges. First, a lack of models that can authentically reproduce the genetic and phenotypic properties of human glioblastoma (BOX 1), especially regarding the analysis of glioblastoma microenvironmental communication, is hampering progress into the development of new therapies for the condition. Second, as underlined by the 2016 WHO classification system, evidence increasingly demonstrates that glioblastoma is genetically heterogeneous (BOX 2) and thus will probably require combinatorial approaches for different subtypes of tumour cell even within a single glioblastoma tumour.
Box 1 |. Models for glioblastoma.
One strategy to improve models of glioblastoma has been to isolate cells from patient tumours and maintain them as neurospheres or organoids in serum-free medium such that they retain their genetic heterogeneity and tumour-initiating cells (also known as cancer stem cells) when reimplanted into immune-compromised mice158,159. Tumour-initiating cells represent a population of highly malignant tumour cells that lurk in different vascular and hypoxic niches within the tumour and are able to expand to reform malignant tumours after therapeutic intervention160,161. Authentic reproduction of the genetic and phenotypic properties of human glioblastoma can also be achieved in models by implanting and passaging portions of patient tumours in immune-compromised mice, referred to as patient-derived xenograft models162,163. Other currently favoured models of glioblastoma include syngeneic mouse models, in which tumours are initially induced by chemicals or viruses and are then established as cell lines that can be transplanted back into the mouse brain164. Spontaneous brain tumours can also be induced with known driver mutations in genetically engineered mouse models165. However, none of these models are a perfect representation of human glioblastoma. Neurospheroid cultures, patient-derived xenograft models and cell lines suffer from genetic instability166, and cell lines frequently have low invasiveness. Glioma-derived cells also display a different genomic methylation pattern and transcriptome in culture and in vivo167. Genetically engineered mouse models represent only a few driver mutations and thus have few neoantigens. Given the current focus of therapeutic research on alerting the immune system to glioblastoma, use of more than one type of mouse model is advisable, and research should include syngeneic mouse glioma lines and genetically engineered mouse-derived glioma cells that can be grown in immune-competent mice. Glioma cell lines that have been passaged extensively are genetically unstable168 and have immunological peculiarities169; hence, they do not represent reliable models of human glioblastoma.
Box 2 |. Genetic and epigenetic heterogeneity of glioblastoma.
Deep sequencing of the genome and transcriptome together with study of the epigenome of glioblastoma cells has revealed both genetic and epigenetic differences among tumour cells within the same glioblastoma, with many genetic drivers represented in almost every glioblastoma170,171. Evidence of the complexity of this disease can be found in the 2016 WHO classification system as well as in experimental subclassification studies. The WHO classification system for diffuse glioma now defines subtypes of intrinsic brain tumours that have a predictable prognosis; importantly, IDH1 or IDH2 mutation with chromosome 1p/19q co-deletion confers a better outcome than other genetic subtypes do172. In addition, epigenetic variation is present among tumour cells. Mutation of IDH1 or IDH2 results in altered transcriptional regulation of many other genes through interference with topologically associated domains, adding another dimension to the genetic complexity173. Experimental transcriptome classification has defined three subtypes in glioblastoma with wild-type IDH1 and IDH2: proneural, classical and mesenchymal171. Glioblastoma driver mutations have been found in up to 45 genes174, including common mutations in genes such as TP53, ATRX, TERT, NF1, PTEN and EGFR172. As such, a strategy of hitting subsets of these genes with combinatorial treatments will be difficult to achieve owing to the large number of genes175, although some pathways are more crucial than others.
In addition to this deepening understanding of the genetic and phenotypic variability within glioblastoma, the field has gained increasing awareness of the ability of these tumours to manipulate and exploit normal brain cells. Almost all cell types in the tumour environs are affected: the tumour is able to stimulate angiogenesis and co-opt existing vasculature2, suppress immune cell functions3, disarm microglia and macrophages that should recognize and fight foreign elements in the brain4, coerce astrocytes into joining tumour support5 and even change the extracellular matrix (ECM) to facilitate invasion6. Conversely, new insights into the presence of adaptive immune cells in the brain and the presence of a CNS lymphatic system7,8 might give rise to therapeutic opportunities that manipulate this system to recognize tumour neoantigens9 similarly to the strategies currently being clinically applied for some melanoma and lung cancer patients.
Glioblastoma recruits normal cells in its environs to promote growth, sustenance and encroachment of the tumour into the brain (FIG. 1). This glioblastoma ‘takeover’ of the brain involves multiple types of communication and directive exchanged between tumour cells and surrounding cells. Cell-secreted soluble factors, including transforming growth factor-β (TGFβ), IL-6, Notch, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF) and stromal cell-derived factor 1 (SDF1; also known as CXCL12), are well known to serve as signalling molecules by binding to receptors on target cells, but the importance of other routes of communication — such as gap junctions, extracellular vesicles and nanotubes — are now being recognized (FIG. 2). A distinguishing feature of glioblastomas is their ability to form a virtual nuclear and cytoplasmic continuum with neighbouring cells, whereby they can introduce not only inorganic elements but also genetic elements and proteins into normal cells to change their phenotype and rescue fellow tumour cells that are in trouble, for example, as a result of radiotherapy or chemotherapy. These newly recognized transit routes can transmit non-secretable molecules, including transcription factors, directive RNAs and DNA and even mitochondria and nuclei. Small molecules such as Ca2+, ATP, metabolites and microRNAs (miRNAs) can be transferred between adjacent cells through gap junctions10,11. Connexins, which form a structural component of these junctions, are upregulated in tumourinitiating cells12 and are associated with increased invasiveness of gliomas11. Non-secretable proteins (including transcription factors), RNA, DNA, lipids and metabolites can be transferred through extracellular vesicles released from cells via fusion of multivesicular bodies with the cell membrane (which yields exosomes), budding from the plasma membrane (giving rise to microvesicles and large oncosomes)13–15 or budding off the tips of nanotubes that extend out from the cells16,17. These tumour-derived extracellular vesicles can change the phenotype of normal cells to promote angiogenesis, immune suppression, tumour cell invasion and metabolic regulation18–20. Tumour cells can also be linked by ‘tunnelling’ nanotubes and microtubes that form gap junctions or a cytoplasmic continuum between cells to enable transport of molecules and organelles21–23. The involvement of microtubes has been indicated in the regrowth of tumours after surgery and in conferring resistance to chemotherapy24, although they are not apparent in some glioma models25.
Fig. 1 |. Glioblastoma microenvironment.
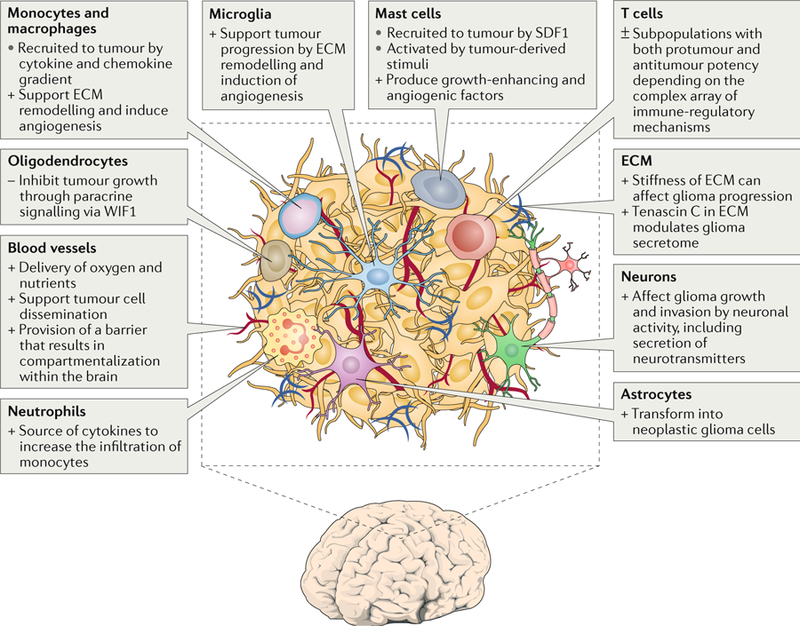
The glioblastoma environ consists of tumour cells, extracellular matrix (ECM), blood vessels, innate immune cells (monocytes, macrophages, mast cells, microglia and neutrophils), T cells and non-tumorous neurons, astrocytes and oligodendrocytes. +, protumour function; −, antitumour function; ±, mixed protumour and antitumour functions; SDF1, stromal cell-derived factor 1; WIF1, WNT inhibitory factor 1.
Fig. 2 |. Routes of communication between tumour cells and cells in their environs.
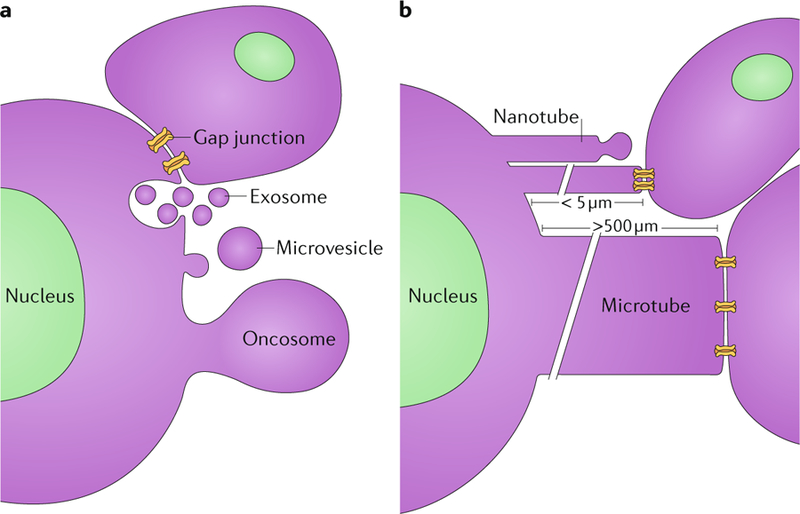
a | Gap junctions (24 nm in diameter) form across the adjacent membranes of cells that are in physical contact, enabling the passage of small molecules. Cells also release exosomes (50–100 μm) from multivesicular bodies that fuse with the plasma membrane. In addition, microvesicles (100–200 μm) and even large oncosomes (1–10 μm) bud off from the plasma membrane and can interact with and be taken up by other cells. b | Tunnelling nanotubes (50–200 nm in width and up to 5 μm in length) extend out from cells and can either bud off vesicles at their tips or form gap junctions with other cells. Microtubes extend out from tumour cells (1–2 μm in width and >500 μm in length) and can form gap junctions with other cells.
These different modes of physical support among tumour cells, and the two-way crosstalk between tumour cells and normal cells in their vicinity, together with the epigenetic flexibility of cells, enable the tumour to create a pluripotent environment that can adapt to changes and thus give the tumour many options to survive therapeutic assault. Given the presence of multiple routes of information transfer in the tumour microenvironment, it is difficult to determine which route mediates which functions and how best to disrupt this multifaceted tumour-supportive network.
This Review focuses on new findings that elucidate the complex and dynamic modes of communication between tumour cells and normal cells in the tumour microenvironment. We discuss the ways in which tumours recruit and subvert normal cells to change their phenotype to support tumour progression, suppress immune defences and defy therapeutic interventions.
Innate immune system and glioblastoma
Interaction between glioblastoma and microglia, monocytes or macrophages.
The glioblastoma microenvironment contains brain-resident microglia and infiltrating monocytes. Once monocytes have infiltrated the tumour, they can differentiate into macrophages26,27. Although often grouped together under the term tumour-associated macrophages or myeloid cells (TAMs), these cells represent distinctly different populations26. Microglia are derived from immature yolk sac progenitors during early embryonic development and maintain themselves in the brain through self-renewal28,29. In non-pathological settings, microglia are the main innate immune cells in the brain and are important in the defence against pathogens and noxious stimuli30. Glioblastoma leads to some disruption of the blood–brain barrier (BBB), which enables bone marrow haematopoietic stem cell-derived monocytes and macrophages to infiltrate the tumour26,27,31. Studies have shown that in specific cases up to 50% of the glioblastoma mass can consist of TAMs32. Chimeric and cell lineage models have shown that the exact composition of TAMs changes over time26,31. One study examined the infiltration of peripheral immune cells in a chimeric GL261 mouse glioma model that received head-protected irradiation, in which BBB disruption due to irradiation is avoided. Fluorescently tagged myeloid-derived monocytes and macrophages transplanted into these mice constituted up to 25% of TAMs in the glioblastoma tumour after 21 days, with lower percentages of myeloid-derived TAMs observed at earlier time points31. The influx of myeloid-derived monocytes in mouse glioblastoma tumours was confirmed in a haematopoietic stem cell lineage tracing model, in which >35% of TAMs were myeloid-derived26. As such, the population of glioblastoma TAMs can progress from strictly microglial in early phases to a mixture of microglia and infiltrating monocytes and macrophages in late phases of tumour progression. In mice, accurate separation of microglia and macrophages can be obtained by fluorescence-activated cell sorting using αM integrin (also known as CD11b) and receptor-type tyrosine-protein phosphatase C (also known as CD45) markers26. In humans, α4 integrin (also known as CD49D) can accurately separate these two cell types in tumours26. Here, when studies used these specific markers for separation of microglia and myeloid-derived cells we refer to the cellular subpopulation studied, otherwise the generic term ‘TAMs’ is used.
TAM recruitment.
The recruitment of TAMs to glioma is mostly mediated by cytokine and chemokine gradients released by glioblastoma cells (FIG. 3). These factors have been extensively reviewed elsewhere and include CC-chemokine ligand 2 (CCL2; also known as MCP1) and CCL7 (also known as MCP3), glial cell line-derived neurotrophic factor (GDNF), hepatocyte growth factor (HGF), SDF1, tumour necrosis factor (TNF), VEGF, ATP, macrophage colony-stimulating factor 1 (CSF1) and granulocyte–macrophage colony-stimulating factor (GM–CSF)32,33. TAMs can also be recruited to a specific subset of glioblastoma cells, such as oligodendrocyte transcription factor 2 (OLIG2)-expressing and transcription factor SOX2-expressing tumour-initiating cells, which secrete periostin to recruit TAMs34. Medical interventions can also stimulate TAM recruitment; for example, intracranial biopsies can increase infiltration of circulating monocytes into the tumour in a CCL2-dependent manner35. Microglia and macrophages themselves also secrete CCL2 to increase infiltration of CCR2+Ly6C+ monocytes, thus creating a positive feedback loop for the continued infiltration of myeloid cells36.
Fig. 3 |. Interactions between glioma and TAMs.
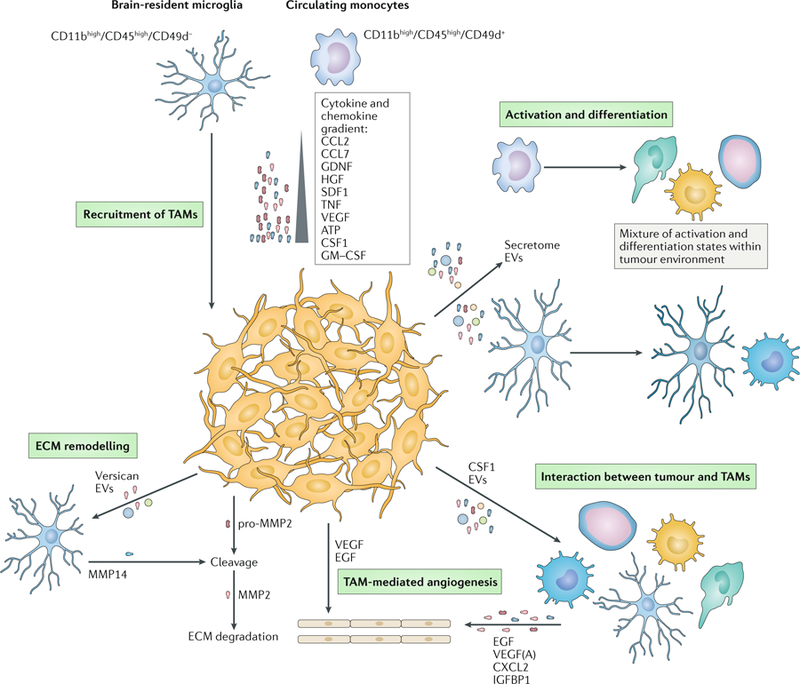
Recruitment of tumour-associated macrophages or myeloid cells (TAMs), including blood monocytes and brain-resident microglia, is based on the gradient of chemokines and cytokines released by the glioblastoma cells. Once recruited, TAMs can be activated and differentiated under the influence of the secretome and extracellular vesicles (EVs) released by the tumour. The various recruited and activated TAMs can affect tumour growth by promoting angiogenesis through secretion of epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), CXC-chemokine ligand 2 (CXCL2) and insulin-like growth factor-binding protein 1 (IGFBP1). This process is further promoted by the release of tumour-derived VEGF and EGF. Invasion and growth of the tumour are accomplished by remodelling the extracellular matrix (ECM) surrounding the tumour. For example, versican and EVs from the tumour induce the release of matrix metalloproteinase 14 (MMP14) by microglia. The release will facilitate the cleavage of tumour-derived pro-MMP2 following extracellular degradation by the active enzyme MMP2. CCL2, CC-chemokine ligand 2 (also known as MCP1); CSF1, macrophage colony-stimulating factor 1; GDNF, glial cell line-derived neurotrophic factor; HGF, hepatocyte growth factor; SDF1, stromal cell-derived factor 1; TNF, tumour necrosis factor.
TAM activation state.
Interaction between glioblastoma cells and TAMs is multifactorial and occurs both in close proximity by direct cell–cell contact and distantly by the release of soluble and membrane-encased factors. This secretome consists of a multitude of molecules, including soluble lipids, cytokines and chemokines33,37. Glioblastomas also release extracellular vesicles that contain a cargo of many types of molecules that have been shown to influence TAM status in a combinatorial way in culture and in vivo25,38. However, no techniques currently are available to suppress extracellular vesicle release from glioblastomas specifically; therefore, the overall relevance of the interaction between glioblastoma extracellular vesicles and TAMs remains to be elucidated. Ultimately, the combination and timing of all glioblastoma-released factors determine the activation state and function of TAMs.
The traditional model of the activation states of TAMs describes a binary system of either tumour-suppressive (M1) or tumour-supportive (M2) macrophages39. This model was based on stimulation of cells in culture by IFNγ, lipopolysaccharide (LPS) or IL-4 and was later extended to include M2 subtypes activated by other types of stimulation, comprising M2a (IL-4 and IL-13), M2b (immune complexes, Toll-like receptor (TLR) or IL-1R) and M2c (IL-10)40. However, RNA sequencing extended the number of stimuli to a combination of 28 known factors and revealed that a wide spectrum of activation states could be induced. The findings demonstrated that macrophage differentiation is much more complex than the binary M1– M2 model41, even when stimulated in culture. This complexity became more apparent when microglia, monocytes and macrophages were isolated from glioblastoma in vivo and analysed by RNA sequencing. The most upregulated genes were found to be shared between traditional M1, M2a, M2b and M2c transcriptomes, suggesting that the activation state in vivo is very different from that in culture26,42,43. Single-cell sequencing confirmed that activation of both M1 and M2 signatures can be observed even in individual cells in an in vivo brain trauma model44. Consequently, the M1 and M2 designations are being replaced by more precise situation-specific models39. Altogether, these findings suggest that TAMs express gene sets in vivo that are associated with stimulation by different factors, highlighting the variety of information transfer in the tumour microenvironment.
TAMs contribute to tumour proliferation.
The role of secreted molecules on TAM function and, subsequently, on tumour growth has been studied extensively32. This interplay between glioblastoma cells and TAMs is especially apparent in tissue remodelling and is necessary for glioblastoma cells to infiltrate the brain (FIG. 3). One group of proteins that is crucial in tissue remodelling is matrix metalloproteinases (MMPs)45. In glioblastoma, MMP2 has an important role in ECM degradation, which facilitates glioblastoma cell migration and invasion46. MMP2 is released in a precursor form (pro-MMP2) that is cleaved by MMP14 to an active state32. However, glioblastoma cells secrete pro-MMP2, but not MMP14. Conversely, microglia in the tumour microenvironment are a major source of MMP14. Two different glioblastoma-derived factors act to increase microglial MMP14 release38,47. First, the ECM protein versican is released from glioma and induces MMP14 release by TAMs through its upstream receptor TLR2 (REF.47). Second, studies of cell-culture models have shown that glioblastoma-derived extracellular vesicles can also induce microglial expression of MMP14 RNA, although the mechanism and in vivo relevance remain to be elucidated38.
Owing to their rapid growth, glioblastomas are in constant need of neovascularization and release angiogenic factors, such as EGF and VEGF33. Additionally, in glioblastoma, microglia and macrophages accumulate around blood vessels and also produce the pro-angiogenic chemokines VEGF and CXC-chemokine ligand 2 (CXCL2)48. Furthermore, glioblastoma cells might promote angiogenesis indirectly through microglial cells, as CSF1 secreted by glioblastoma cells in vitro induces microglia cells to release insulin-like growth factor-binding protein 1 (IGFBP1), which can induce angiogenesis49. RAGE (receptor for advanced glycation end products; also known as AGER) is thought to play a part in a number of diseases, including tumours. In tumour-bearing mice, RAGE ablation increases survival by reducing the levels of VEGFA secreted by infiltrating TAMs, which results in leaky (rather than fully developed) vasculature and disturbed tumour perfusion50. However, these effects were reported in syngeneic GL261 mouse tumours (a frequently used cellular model of glioma), which do not represent the invasive growth pattern observed in glioblastoma patients. Thus, TAMs have a crucial role in tumour angiogenesis through multiple signalling mechanisms.
Overall, the interaction between glioblastoma and TAMs is bidirectional and multifactorial. This plethora of paracrine loops can determine the ultimate effects of TAMs on tumour growth and can differ depending on local variables such as hypoxia, the extent of necrosis, TAM infiltration density and/or TAM activation state.
Neutrophils and mast cells.
In addition to macrophages and microglia, other innate immune cells, including neutrophils and mast cells, are recruited into the tumour microenvironment51,52. The release of various chemokines and cytokines, such as IL-8, TNF and CCL2, by tumour cells or stromal cells after biopsy or resection can recruit neutrophils to the tumour microenvironment35,53. Tumour-associated neutrophils secrete cytokines that further increase the infiltration of neutrophils and monocytes and thus might indirectly support tumour growth by increasing TAM density at the glioma site54.
Similarly, various chemokines produced by glioblastoma cells, including SDF1, macrophage migration inhibitory factor and plasminogen activator inhibitor 1, recruit mast cells to the tumour microenvironment52,55,56. Mast cells are bone-marrow-derived immune cells that play a part in innate and adaptive immune responses. Upon activation by glioblastoma cells, mast cells produce soluble factors, including IL-6, IL-8, VEGF and TNF, that can facilitate tumour growth and angiogenesis57. What role other forms of communication — including extracellular vesicles, nanotubes and microtubes — have in the communication between tumour cells and neutrophils and mast cells remains to be investigated.
An increasing body of evidence, therefore, suggests that multiple communication loops in the tumour microenvironment are responsible for the recruitment and activation of various cells of the innate immune system. An improved understanding of these complex interactions between glioblastoma and innate immune cells could open novel therapeutic avenues for glioblastoma via the blockade of these interactions.
Adaptive immune response to brain tumours
Beyond their direct interactions with glioma cells described above, innate immune cells also have essential roles in the activation of adaptive immune responses, which in turn can restrict glioma growth. For a long time, the historic notion that the brain is an immune-privileged organ that is both invisible to the adaptive immune system and physically excludes migratory immune cells through the BBB has discouraged the idea that CNS tumours, including glioblastoma, might be susceptible to spontaneous or therapeutically induced adaptive antitumour immune responses. This dogmatic view has largely eroded over the past few years, partly owing to recognition of phenomena that conflict with this concept, including the fact that multiple sclerosis is driven by T cells58, that non-CNS tumours can induce paraneoplastic autoimmune reactions that affect the CNS59, that restriction of T cell trafficking can elicit opportunistic CNS infections60 and that CNS antigens are detected by T cells in extracranial lymphoid tissues61. As a result, the traditional concept of passive immune privilege through physical isolation of the CNS from the adaptive immune system has given way to the concept of active immune privilege that is dynamically maintained by mechanisms of immune regulation, which can be revoked when necessary62. Efforts to understand the role of adaptive immune cells, and particularly T cells, in the glioma tumour environment have consequently intensified.
Glioblastoma neoantigens and T cell activation.
High-grade glioma ranks in the bottom half of human tumours in terms of mutational neoantigen load63, which currently is considered a crucial determinant of an effective adaptive antitumour response. Nevertheless, T cell infiltration of tumours positively correlates with clinical outcome in patients with glioblastoma64–68, suggesting — although not proving — that these cells can control tumour growth. In contrast to innate immune cells, cells of the adaptive immune system (namely, T and B cells) become effector cells that are capable of exerting antitumour effects only through a dramatic expansion of small populations of naive antigen-specific precursor cells, in conjunction with differentiation that equips them with immunological effector activities. With regards to T cells, effector differentiation enables migration to non-lymphoid and tumour tissues. The activation process occurs in secondary lymphoid organs, such as the lymph nodes and spleen, where resident and lymphborne migratory dendritic cells that originate in peripheral tissues present antigens to T cells (FIG. 4a). Cellular and molecular tracer studies revealed a functional connection between the CNS and the deep cervical lymph nodes decades ago69, but a bona fide lymphatic drainage system for the CNS was demonstrated only in rodents and humans in 2015 (REFs7,8). Mouse studies also suggest that antigen-presenting cells (APCs) traffic from the CNS to deep cervical lymph nodes and present brain tumour antigens, which induce expression of a trafficking receptor pattern by T cells that enables their homing to the CNS; by contrast, the same activity is not demonstrated by APCs that originate from skin tumours. Thus, unique features of the brain tumour environment help to educate APCs that then travel to deep cervical lymph nodes via lymphatic vessels to imprint T cells with brain tropism61.
Fig. 4 |. T lymphocytes in the glioblastoma environment.
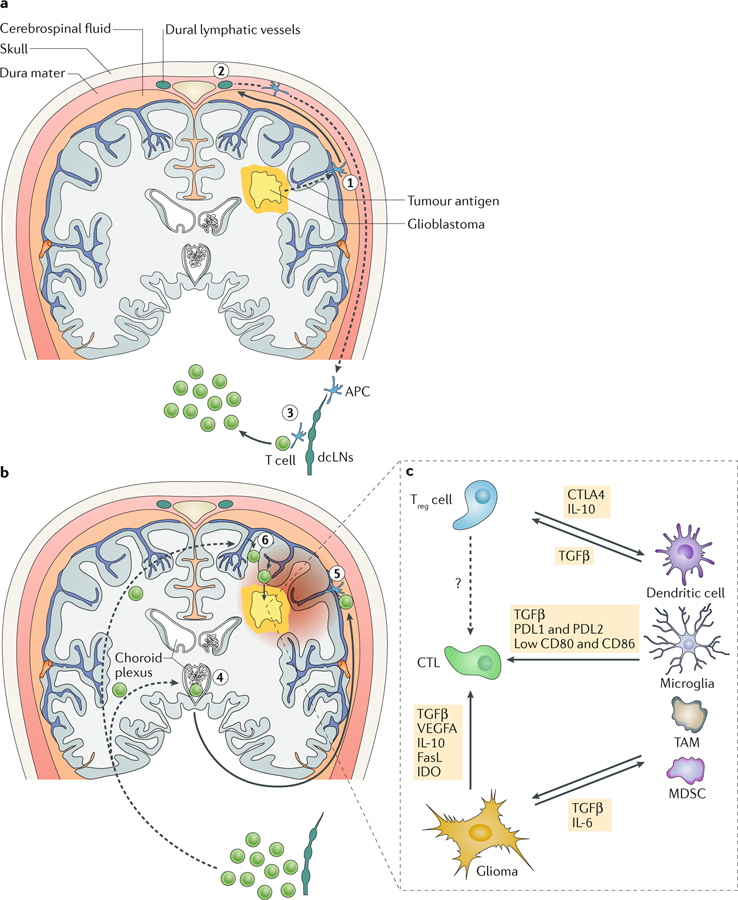
a | A hypothetical scenario for the induction of anti-glioblastoma T cell responses. Tumour antigens travel with interstitial fluid along the perivascular glymphatic system to the brain surface (step 1), where they enter the cerebrospinal fluid or are taken up by local antigen-presenting cells (APCs). A fraction of cerebrospinal fluid enters dural lymphatic vessels, potentially along with migratory APCs (step 2), and by this route tumour antigen reaches the deep cervical lymph nodes (dcLNs). There, resident or migratory APCs interact with and activate tumour antigen-specific naive T cells (step 3). b | Potential pathways by which activated or memory T cells reach the CNS and glioblastoma tumours. T cells expressing CC-chemokine receptor 6 (CCR6; and possibly other chemokine receptors) enter the cerebrospinal fluid in ventricles via the choroid plexus (step 4) and percolate along the brain surface. Upon re-encounter with their cognate tumour antigen (step 5), they initiate a local inflammatory reaction that activates the local microvasculature to recruit additional T cells in a CCR6-independent and potentially CXC-chemokine receptor 3 (CXCR3)-dependent fashion (step 6), facilitating their accumulation in the glioblastoma environment. c | In and around the glioblastoma, effector T cells, including cytotoxic T lymphocytes (CTLs), are exposed to a network of immune-regulatory mechanisms that promote their differentiation into a dysfunctional state. CTLA4, cytotoxic T lymphocyte protein 4; FasL, Fas antigen ligand; IDO, indoleamine 2,3-dioxygenase; MDSC, myeloid-derived suppressor cell; PDL1, programmed cell death 1 ligand 1; TAM, tumour-associated macrophages or myeloid cells; TGFβ, transforming growth factor-β; Treg cell, regulatory T cell; VEGFA, vascular endothelial growth factor A.
Routes of T cells into the brain.
Once primed in deep cervical lymph nodes, T cells enter the bloodstream and have two principal access routes to the brain (FIG. 4b). In the steady state, CC-chemokine receptor 6 (CCR6)-expressing T helper 17 cells can enter the cerebrospinal fluid via the choroid plexus and percolate around the brain surface, where they might re-encounter their cognate antigen on meningeal APCs. This interaction can trigger an initially inflammatory reaction that then licenses local CNS microvessels to support recruitment of additional T cells and other immune cells in a CCR6-independent way70. Whether this immune-surveillance pathway is relevant for anti-glioma responses is currently unknown. Alternatively, inflammation induced by the invasive growth of glioblastomas and by the factors these tumours produce could directly drive microvascular changes that support recruitment of T cells and innate immune cells via pre-existing brain microvessels and neo-vessels that develop in response to pro-angiogenic factors released by tumours.
Immune regulation in the brain.
Upon entry into the glioma microenvironment, tumour-reactive T cells are faced with a complex array of immune-regulatory mechanisms (FIG. 4c). Most of these mechanisms are also operative in non-CNS tumours, but it is possible that the conditions underlying active immune privilege in the healthy CNS also support a particularly immunesuppressive environment in glioblastomas. TGFβ2, which was discovered under the name glioma-derived T cell suppressor factor, was one of the earliest immunosuppressive factors found and was identified on the basis of its ability to suppress IL-2-dependent T cell survival71. TGFβ2 and TGFβ1, the predominant forms of TGFβ produced by immune cells, have remained centre stage in glioma microenvironment research, not only because of their organization of the interplay between tumour cells and myeloid cells (as described above) but also because of their direct effects on T cells. CD8+ T cells rapidly induce expression of ITGAE (encoding αE integrin), a gene for which the expression is strictly TGFβ-dependent, upon entry into the brain tumour microenvironment in mice or humans, indicating that they are locally exposed to strong TGFβ signals72. Although αE integrin expression improves retention of cytotoxic T lymphocytes in the brain, TGFβ also attenuates the ability of these cells to express crucial effector molecules, such as granzyme B and IFNγ, which probably reduces their efficacy against glioma73.
Beyond TGFβ2, glioblastomas have been reported to produce a wide range of membrane-bound, secreted and metabolic factors, as described for other tumours, including Fas antigen ligand (FasL; also known as FASLG), programmed cell death 1 ligand 1 (PDL1), VEGFA, indoleamine oxidase and others74–77. Glioblastoma can also induce TGFβ and IL-10 expression in pericytes, which routinely interact with extravasating T cells and reduce their expression of co-stimulatory molecules78. Both glioblastoma and pericyte-derived immune-regulatory factors might act on antitumour effector T cells through direct mechanisms or indirectly by promoting the recruitment and function of tolerogenic APCs and regulatory T (Treg) cells. Either way, these factors produce a local state of reduced T cell effector function commonly referred to as T cell exhaustion or dysfunction79. This cellular differentiation state is also observed in chronic viral infection and presumably serves to prevent immunopathological tissue damage due to ongoing T cell activity against a virus, whereas in the tumour environment it restricts T cell antitumour functions.
T cell dysfunction in glioblastoma.
One hallmark of T cell dysfunction is the co-expression of multiple co-inhibitory receptors, including programmed cell death protein 1 (PD1), T cell membrane protein 3 (TIM3; also known as HAVCR2), lymphocyte activation gene 3 protein (LAG3) and T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT)80. Improved understanding of the mechanisms by which expression of these receptors is induced in tumourinfiltrating T cells will be necessary, but they probably include T cell receptor activation in the absence of sufficient co-stimulation, resulting in binding of nuclear factor of activated T cell (NFAT) to exhaustion-associated genes instead of effector genes81. Treg cells can downregulate co-stimulatory molecules on tumour-associated APCs and thereby promote T cell exhaustion82. In addition, TGFβ and VEGFA, which are both produced by the glioma, amplify T cell receptor-driven induction of PD1 and other co-inhibitory receptors83,84 and might explain their high expression in tumour-infiltrating T cells.
Glioma cells and APCs in the glioma environment express co-inhibitory receptor ligands, which engage with T cells and attenuate functions triggered by concurrent T cell receptor engagement, reducing the antitumour activity of these cells. Expression of PDL1, one of the known ligands for PD1, is induced on tumour cells by IFNγ and therefore reflects ongoing antitumour immune activity85. In addition, oncogenic mutations, such as those causing a loss of PTEN function in glioma, can activate PDL1 expression in tumour cells86.
Immune checkpoint inhibition.
Use of blocking antibodies to interrupt binding of the co-inhibitory receptors PD1 and cytotoxic T lymphocyte protein 4 (CTLA4) with their ligands — so-called checkpoint blockade — has recently established itself as a powerful new modality of cancer therapy. In a notable study from 2016, blockade of the PD1 and/or CTLA4 pathways eliminated established mouse glioblastomas in the majority of treated animals and produced immune memory protective against tumour rechallenge87. However, patients with glioblastoma generally carry a lower neoantigen load than the mouse glioblastoma model used here; therefore, it remains to be determined whether CTLA4-targeted or PD1-targeted therapy will be effective in humans, given the correlation between neoantigen load and response to either of these therapies88,89. Indeed, although phase I trial data on PD1 blockade in recurrent glioblastoma indicated a transient increase in progression-free survival90, a phase III arm of the same trial failed to show an increase in overall survival91. In addition to low antigenicity, a low degree of inflammation and the overwhelming immune-regulatory activity intrinsic to the CNS might have contributed to this failure. Consequently, measures to enhance the basal immune reactivity of glioblastoma might be needed to render PD1-targeted therapy effective. However, irrespective of the question of tumour antigenicity and despite CNS immune privilege, brain tumours are, in principle, susceptible to the effects of checkpoint blockade antibodies and the enhanced antitumour T cell function they elicit. This conclusion is also supported by data from humans showing that brain metastasis of malignant melanoma is tractable to checkpoint inhibitor treatment92.
Endothelial cells and tumour vasculature
Glioblastoma growth is accompanied by angiogenesis, and tumour vasculature has several key functions enabling tumour growth, including the delivery of oxygen and nutrients, modulation of the immune response, support of tumour cell dissemination and provision of barriers for compartmentalization within the brain. Notably, tumour-initiating cells are usually located close to endothelial cells, in the so-called perivascular niche, which serves as a hub for generation of multiple cellular phenotypes and has important functions in the maintenance of tumour-initiating cells, in communication between the tumour and vascular compartments and in therapeutic resistance (reviewed in depth elsewhere2,93).
Factors promoting angiogenesis in the tumour microenvironment.
Glioma cells release many pro-angiogenic factors that shape tumour vasculature, including TGFβ, VEGF, proteolytic enzymes, ribonucleases (such as plasminogen activators and angiogenin) and chemokines. These factors are released in the forms of extracellular vesicles, soluble factors and extravesicular ribonuclear complexes. For example, extracellular vesicle-derived VEGFA has a pro-angiogenic effect, at least in culture94. In addition, glioma-derived factors in the ECM, such as tenascin, dynamically modulate the glioma secretome and might also alter endothelial cell proliferation and tubulogenesis, contributing to overall angiogenesis95. An intriguing idea that highly plastic tumour-initiating cells might themselves transdifferentiate into endothelial cells and promote angiogenesis in glioblastoma96 has also received considerable support in studies based on live-cell imaging and patient tumour histopathology97,98. Integrated transcriptomic and epigenomic analysis has revealed that activation of the WNT5a pathway can drive differentiation of tumour-initiating cells to endothelial-like cells99. In this context, understanding how the tumour microenvironment modulates this process will be important.
Vascular integrity.
Normalization of the tumour vasculature integrity, which is severely compromised in glioblastoma, represents one therapeutic strategy to improve drug delivery100. Invading glioblastoma cells cause physical dislocation of astrocytes from endothelial cells and thus disrupt gliovascular coupling of the BBB101. VEGF can also increase access of drugs to glioblastoma across the BBB102. In addition, extracellular vesicles released by glioblastoma can disrupt the endothelial barrier via delivery of semaphorin 3A, among other factors103. Interestingly, vascular integrity can also be modulated remotely by delivery of miR-132 in extracellular vesicles released from neurons, via the indirect upregulation of the adherens junction protein, VE-cadherin104. However, insight into environmental mechanisms that regulate integrity of the vasculature and BBB in glioblastoma is currently limited and would benefit from improved, orthotopic animal models and drugs that are designed to access the brain. BBB penetration by chemotherapeutic drugs and antibodies currently remains a major limitation in treating glioblastomas105.
Neurons
Although many patients with glioblastoma have epileptic seizures, substantial cognitive dysfunctions, memory impairment and personality changes, little is known about the interactions between glioblastoma and surrounding cortical neurons. The effect of the tumour on neuronal networks has been proposed to be due to mechanical pressure. However, considering the range of factors released by glioma and the complexity of inter-cellular signalling mechanisms, the effects are probably much more complicated. The most profound neurotoxicity and seizures are associated with excessive glutamate release by glioma cells106. Interestingly, expression of glutamate transporters is increased in peritumoural cells, which seems to have a neuroprotective function and provides some survival benefit in animal models107. In addition, recurrent seizures and the associated excessive glutamate release might lead to increased vascular permeability through the activation of NMDA (N-methyl-d-aspartate) receptors108. This permeability could result in increased peritumoural oedema, but it could also have a positive effect on drug delivery to the tumour.
However, glioblastomas might have developed additional mechanisms of neurotoxicity that are unrelated to glutamate release. Our analysis of the proteins secreted by tumour-initiating cells and the ribonuclear complexes released by glioblastoma cells identified multiple oncogenic and cell-cycle-promoting factors109, including a cofactor of the DNA polymerase proliferating cell nuclear antigen (PCNA) and a number of miRNAs that are oncogenic in the brain microenvironment. These miRNAs include miR-10b and miR-26, which drive cell-cycle progression via interaction with cell-cycle inhibitors (cyclin-dependent kinase inhibitor 1 and 2) and retinoblastoma-associated protein, respectively110,111. These molecules promote tumour growth, but transfer of such proliferative signals to neurons can lead to neurodegeneration112. For example, increased levels of miR-26 cause aberrant cell-cycle entry in postmitotic neurons that leads to neuronal cell death113. Intercellular transfer of such molecules from glioblastoma to neurons could affect neuronal health, postmitotic state and overall cell viability.
Although mitogenic signalling from malignant cells to adjacent normal cells is expected, the glioma-supportive signalling from neurons to glioma is, perhaps, more surprising. Several reports suggest that neurotransmitters can affect glioma growth and invasion114–116, and seminal work in the past few years from Monje and colleagues has revealed a novel and unexpected mechanism through which neuronal activity supports glioblastoma growth117,118. Using an optogenetics-based approach, these researchers found that firing activity of cortical projection neurons promotes glioma proliferation and growth in vivo via regulated secretion of the synaptic protein neuroligin 3. This postsynaptic adhesion molecule is secreted by neurons and oligodendrocyte precursor cells and is found mostly in excitatory synapses. Its actions include downstream signalling, activation of the focal adhesion kinase (FAK; also known as PTK)–phosphoinositide 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) pathway, growth factor receptors and oncogenic proteins and feedforward expression of neuroligin 3 in glioma cells. Remarkably, growth of glioblastoma xenografts from adult and paediatric patients, as well as xenografts from diffuse intrinsic pontine glioma, was strongly impaired in Nlgn3-knockout mice. Monje and colleagues propose that blockade of neuroligin 3 release, mediated largely by the membrane protease ADAM10 (disintegrin and metalloproteinase domain-containing protein 10), could represent a therapeutic approach for glioblastoma118. Additional factors regulated by neuronal activity, such as brain-derived neurotrophic factor and endoplasmic reticulum chaperone BiP (GRP78; also known as HSPA5), have also been identified that have central roles in normal synaptic functions and can act as glioma mitogens in the tumour microenvironment117, highlighting the importance of neuronal-to-glioblastoma communication. Generally, dissection of molecular mechanisms that couple the activity of neurons and synapses with the behaviour of neighbouring glioma cells, including tumour-initiating cells and invading glioma cells, could lead to a new class of selective and targeted anti-glioma therapies that spare normal neuronal functions.
Astrocytes and other glial populations
Astrocytes are the endogenous native cells of the brain that are phenotypically most similar to the bulk of glioblastoma and represent one of the cell types from which gliomas can originate. Astrocytes can be transformed to neoplastic glioma cells or tumour-initiating cells in vitro and in vivo by a variety of oncogenes, such as MYC, RAS and EGFR variant III (REFS119,120), which raises the question of whether the communication between glioblastoma and surrounding reactive astrocytes can promote the process of transformation and thereby feed the tumour with newly recruited oncogenic cells. Although the exact mechanism is unknown, media conditioned by glioma cells inhibits expression of the p53 tumour suppressor in normal astrocytes, which in turn enriches laminin and fibronectin in the ECM and promotes glioma cell survival121. Studies show that the inevitable recurrence of the tumour could arise from quiescent tumour-initiating cells, infiltrating tumour cells and/or recruitment of apparently normal cells from the peritumoural brain zone122. In support of the latter origin of cells, genes involved in growth, proliferation and motility are induced in peritumoural white matter that is free of tumour cells, whereas several tumour suppressors and genes involved in neurogenesis are downregulated relative to normal white matter123.
Indeed, co-implantation of tumour-associated glial cells (that is, brain-derived glial cells conditioned by glioblastoma xenografts) and glioblastoma cells promoted tumour growth compared with implantation of tumour cells alone or tumour cells with unconditioned glial cells124. Furthermore, resection and radiation-induced injury can cause transcriptome and secretome alterations in reactive astrocytes that further potentiate tumour aggressiveness5,125.
Signalling between glioma cells and reactive peritumoural astrocytes in vivo has yet to elucidated, largely owing to the lack of highly specific markers discriminating these cell populations, but studies of glioblastoma biology and the secretome present some intriguing possibilities. For example, malignant transformation of astrocytes requires expression of the oncogenic miR-10b, which is normally silenced in these cells126. miR-10b is highly abundant in all glioblastoma subtypes and is released in extracellular vesicles from tumour cells. Transfer of this and other pro-oncogenic regulators, such as miR-21 and miR-26, from glioblastoma to the peritumoural astrocytes could facilitate their transformation. Interestingly, a greater density of astrocytes adjacent to the tumour is predictive of a shorter lifespan in glioblastoma patients127. This finding supports the concept that the tumour-associated astrocytes are coerced into promoting tumour growth and facilitating tumour invasion128,129.
By contrast, endogenous oligodendrocytes seem to inhibit glioblastoma growth and proliferation by paracrine signalling via WNT inhibitory factor 1 (REF.130). Although less is known about interactions between glioma and oligodendrocytes than astrocytes, the presence of crosstalk between oligodendrocytes, microglia and astrocytes through multiple cytokines131 suggests a complex relationship with these endogenous cells and glioblastoma that can include both tumour-promoting and tumour-suppressing signals. Furthermore, a subtype of reactive astrocytes has been identified that is induced by activated microglia and promotes the death of neurons and oligodendrocytes132, which points to the multifaceted interactions within the glioma microenvironment.
Perspectives and therapeutic insights
The many new insights into the communication between glioblastoma cells and other cells within the tumour microenvironment, as outlined in this Review, reveal new strategies for potential therapies for glioma. These include therapies that target modes of communication between glioblastoma and surrounding cells, as well as therapies that directly or indirectly target molecules with tumour-supportive properties, including factors involved in the innate and adaptive immune system, angiogenesis and interaction with normal cells, such as neurons.
One mode of communication that is increasingly recognized as a potential method for glioblastoma cells to influence the microenvironment is tumour-derived extracellular vesicles. The contents of extracellular vesicles derived from tumour-initiating cells, for example, have been shown to include various oncogenic and cell-cycle-promoting factors, including PCNA and miRNAs109. Future studies could validate their functional effects on different cell types in the tumour microenvironment by interfering with extracellular-vesicle-mediated communication, for example, by influencing extracellular vesicle generation or uptake. Extracellular vesicle release can be reduced by GW4869 — a neutral sphingomyelinase inhibitor that prevents the ceramide-mediated inward budding of multivesicular bodies and the release of mature exosomes into the extracellular space133. GW4869 has been shown to reduce the levels of extracellular vesicles in various systems, including in a mouse model of Alzheimer disease, where it reduced the number of exosomes in both serum and brain134–136. On the other hand, uptake of extracellular vesicles by recipient cells can be reduced by heparin137 or by silencing of annexin A1 (REF.138). However, these strategies have been applied only in preclinical studies thus far and do not distinguish between tumour-specific extracellular vesicle interactions and physiological processes related to extracellular vesicles. Much work will be needed to explore their potential for specific, effective and safe routine clinical applications.
Further strategies for glioma therapy include targeting other mechanisms of communication within the tumour microenvironment, such as cytokines, gap junctions and nanotubes or microtubes. Inhibition of certain cytokines has shown promise for glioblastoma treatment in mouse models, including CCL2, which reversed the induction of migration and proliferation of tumour cells observed after biopsy35. In addition, recruitment of CCR4+ Treg cells mediated by CCL2 can be blocked using an antagonist against CCR4 and results in an improvement in the median survival of tumour-bearing animals36. The gap junctions formed in glioblastomas between astrocytes and tumour cells via connexin 43 support growth and invasion23 and can be modulated using the cyclooxygenase inhibitors meclofenamate or tonabersat138. These cyclooxygenase inhibitors have been shown to reduce established brain metastases in an experimental model139. Therapies that alter the structure of the plasma membrane — for example, focal hyperthermia (in combination with radiation therapy), which has gained clinical attention, with several trials under way — could also suppress nanotubule and microtubule formation and even extracellular vesicle release. In addition, the ECM is a crucial site for extracellular communication in the tumour environment. Cell-culture studies have shown that the stiffness of the ECM can affect glioma progression, and interfering with this substrate might provide an effective therapy beyond direct targeting of the tumour cells140. One approach would be to block ECM remodelling using inhibitors of extracellular MMPs141 and heparanases142.
Over the past few years, interactions between glioblastoma cells and cells of the immune system have gained attention as targets for novel treatment options. One interesting strategy aimed at modulating the communication between the tumour and cells of the innate immune system involves inhibition of the CSF1 receptor (CSF1R), which is expressed on TAMs. For example, this inhibition decreased the tumour-supportive capacity of TAMs and increased survival in a mouse proneural glioblastoma model143. Unfortunately, subsequent longitudinal studies have shown that resistance to CSF1R blockage is acquired as a result of macrophage-induced secretion of insulin growth factor 1 (IGF1), leading to PI3K activation in glioblastoma cells, which again promotes tumour growth144. Co-treatment with CSF1R and IGF1 antagonists in a mouse model of glioma reduced the levels of resistance substantially144. A clinical trial showed that CSF1R inhibition alone in the recurrent setting had little to no effect; however, the activation of PI3K signalling in many glioblastomas strongly suggests the effectiveness of CSF1R and IGF1 antagonists as a combination therapy145,146.
Treatments aimed at ameliorating the adaptive immune response have been successful in some tumours, but their effect in glioblastoma has been very modest, partly owing to the low neoantigen load of glioblastoma. Thus far, no clinical benefit has been demonstrated by large clinical trials on dendritic cell vaccination aiming to elicit anti-glioblastoma T cell responses; however, overall survival was improved upon vaccination with dendritic cells loaded with tumour lysate in a small subset of nine patients with glioblastoma who exhibited a dominantly mesenchymal gene signature, indicating that some forms of glioblastoma might be more susceptible to immunotherapy than others147. Although the failure of immune checkpoint therapy to improve overall survival in patients with recurrent glioblastoma has been disappointing (such as in the CheckMate-143 trial148), multiple avenues remain to explore the potential utility of this approach, especially in the setting of newly diagnosed disease. Phase III trials on PD1 blockade for newly diagnosed glioblastoma patients are continuing (CheckMate-498 (REF.149) and CheckMate-548 (REF.150)). Similar to tumours with a mesenchymal gene signature, hypermutant glioblastoma in paediatric patients with germline biallelic mismatch repair deficiency might constitute a subset of cases with enhanced responsiveness to this form of therapy151. However, in most cases, additional interventions might be needed to sensitize cells to immune checkpoint therapy. Interestingly, a preclinical study of mouse glioblastoma showed that intracranial treatment with an IL-12-expressing oncolytic herpes simplex virus gave a highly effective response when combined with anti-PD1 and anti-CTLA4 therapy152. Effectiveness might also be increased by including inflammatory agents, such as oncolytic adenovirus and herpes simplex virus vectors, with immune checkpoint inhibitors153. Overall, PD1 blockage in glioblastoma tumours in humans has not resulted in increased survival, but strategies to increase efficiency or select favourable patients might yield more encouraging results.
Treatments targeting the tumour vasculature, especially antiangiogenic therapies such as bevacizumab, have been studied extensively (reviewed in detail elsewhere154). However, these trials did not show the much hoped for undisputed improvement in overall survival. Consequently, an alternative strategy aiming to normalize tumour vasculature for improved drug delivery has received attention. Dual inhibition of the VEGF receptor and angiogenin 2 normalized tumour vasculature and prolonged survival in two glioblastoma mouse models109. In a different model, angiopoietin 1 receptor activation and angiogenin 2 inhibition normalized the vasculature, elicited a favourable tumour microenvironment (including an altered immune cell profile) and improved the delivery of chemotherapeutic agents into tumours155. The therapeutic potential of antiangiogenic drugs, therefore, has the potential to be reinvigorated; however, current means have yet to be successful.
Finally, dissection of the mechanisms linking the activity of neurons and other endogenous cells in their environs with the behaviour of glioma cells could provide new opportunities and might result in a new class of selective and targeted anti-glioma therapies. For example, release of neuroligin 3, which is cleaved from neurons and oligodendrocytes via ADAM10, promotes glioma proliferation117. Furthermore, tumour growth was markedly reduced in a paediatric glioblastoma and diffuse pontine glioma model using the ADAM10 inhibitor GI254023X118. The ADAM10 inhibitors INCB7839 and XL-784 have been tested in clinical trials for breast cancer and have been shown to penetrate the BBB156,157. Consequently, further studies are warranted to explore inhibition of this neuronal support of tumour growth.
Conclusions
Glioblastomas represent a complex set of tumour cells that differ greatly in genetic and epigenetic characteristics and are surrounded by endogenous cells that are transformed into predominantly tumour-supporting stromal cells through various modes of bidirectional communication. This rampant multimodal networking in the tumour environment offers targets for new therapies yet simultaneously highlights the challenges ahead. Microenvironmental compensatory mechanisms, such as stromal IGF1 secretion after CSF1R blockade, demonstrate the need for extended and repeated interventions to overcome dynamic changes in tumour growth incentives. These mechanisms also underscore the need to undercut the dependency by glioblastoma on its environs in the development of much needed novel therapies for this inevitably lethal disease.
Key Points.
Glioblastomas use numerous forms of communication to hijack many different cell types in the brain environs to support tumour progression.
Communication routes include secreted proteins and molecules, gap junctions between cells, extracellular vesicles, tunnelling nanotubes and microtubes.
Tumour cells co-opt microglia and infiltrating macrophages for their own benefit through the release of cytokines and extracellular vesicles.
Glioblastomas and pericytes generate a state of reduced T cell effector function that is commonly referred to as T cell exhaustion or dysfunction.
The interaction of tumour cells with normal brain cells, such as neurons, is not unidirectional, and neuronal activity is subverted to promote glioblastoma progression.
Comprehension and disruption of tumour directives in the glioblastoma microenvironment could improve therapeutic intervention for these lethal tumours.
Acknowledgements
The authors thank S. McDavitt for her skilled editorial assistance. This work was supported by U19 CA179563 by the US NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director (X.O.B., A.M.K. and T.R.M.), and the US NIH National Cancer Institute (P01 CA069246 (X.O.B.), R01 AI123349 (T.R.M.) and R21 NS098051 (A.M.K.)).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Neurology thanks W. Wick and the other, anonymous reviewers for their contribution to the peer review of this work.
References
- 1.Stupp R et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Jhaveri N, Chen TC & Hofman FM Tumor vasculature and glioma stem cells: contributions to glioma progression. Cancer Lett 380, 545–551 (2016). [DOI] [PubMed] [Google Scholar]
- 3.See AP, Parker JJ & Waziri A The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. J. Neurooncol 23, 405–412 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Roesch S, Rapp C, Dettling S & Herold-Mende C When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci 19, E436 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okolie O et al. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol 18, 1622–1633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pencheva N et al. Identification of a druggable pathway controlling glioblastoma invasiveness. Cell Rep 20, 48–60 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Aspelund A et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med 212, 991–999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boussiotis VA & Charest A Immunotherapies for malignant glioma. Oncogene 15, 1121–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thuringer D et al. Transfer of functional microRNAs between glioblastoma and microvascular endothelial cells through gap junctions. Oncotarget 7, 73925–73934 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong X, Sin WC, Harris AL & Naus CC Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 6, 15566–15577 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balça-Silva J et al. The expression of connexins and SOX2 reflects the plasticity of glioma stem-like cells. Transl Oncol 10, 555–569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tkach M & Théry C Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Maas SL, Breakefield XO & Weaver AM Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol 27, 172–188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minciacchi VR et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 6, 11327–11341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rilla K et al. Hyaluronan production enhances shedding of plasma membrane-derived microvesicles. Exp. Cell Res 319, 2006–2018 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Lai CP et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 8, 483–494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fonseca P, Vardaki I, Occhionero A & Panaretakis T Metabolic and signaling functions of cancer cell-derived extracellular vesicles. Int. Rev. Cell. Mol. Biol 326, 175–199 (2016). [DOI] [PubMed] [Google Scholar]
- 19.D’Asti E, Chennakrishnaiah S, Lee TH & Rak J Extracellular vesicles in brain tumor progression. Cell. Mol. Neurobiol 36, 383–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redzic J, Balaj L, van der Vos K & Breakefield XO Extracellular RNA mediates and marks cancer progression. Semin. Cancer Biol 28, 14–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Veruki ML, Bukoreshtliev NV, Hartveit E & Gerdes HH Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl Acad. Sci. USA 107, 17194–17199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vignais ML, Caicedo A, Brondello JM & Jorgensen C Cell connections by tunneling nanotubes: effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int 2017, 6917941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osswald M et al. Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Weil S et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol 19, 1316–1326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Vos KE et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol 18, 58–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowman RL et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17, 2445–2459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 77, 2266–2278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ajami B, Bennett JL, Krieger C, Tetzlaff W & Rossi FMV Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci 10, 1538–1543 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Ginhoux F et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hickman SE et al. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller A, Brandenburg S, Turkowski K, Müller S & Vajkoczy P Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 137, 278–288 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Hambardzumyan D, Gutmann DH & Kettenmann H The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci 19, 20–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W & Graeber MB The molecular profile of microglia under the influence of glioma. Neuro Oncol 14, 958–978 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou W et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol 17, 170–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alieva M et al. Preventing inflammation inhibits biopsy-mediated changes in tumor cell behavior. Sci. Rep 7, 7529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang AL et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res 76, 5671–5682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P & de Gruijl TD Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim. Biophys. Acta 1846, 560–575 (2014). [DOI] [PubMed] [Google Scholar]
- 38.de Vrij J et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 137, 1630–1642 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Ransohoff RM A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci 19, 987–991 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Mantovani A, Sozzani S, Locati M, Allavena P & Sica A Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23, 549–555 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Xue J et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabrusiewicz K et al. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 6, e23902 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szulzewsky F et al. Glioma-associated microglia/ macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 10, e0116644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim CC, Nakamura MC & Hsieh CL Brain trauma elicits non-canonical macrophage activation states. J. Neuroinflamm 13, 117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kessenbrock K, Plaks V & Werb Z Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du R et al. Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GB. Neuro Oncol 10, 254–264 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu F et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/ macrophages Toll-like receptor 2 signaling. Neuro Oncol 17, 200–210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandenburg S et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol 131, 365–378 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Nijaguna MB et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J. Biol. Chem 290, 23401–23415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res 74, 7285–7297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fossati G et al. Neutrophil infiltration into human gliomas. Acta Neuropathol 98, 349–354 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Põlajeva J et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE 6, e25222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kolaczkowska E & Kubes P Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Sionov RV, Fridlender ZG & Granot Z The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron 8, 125–158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Põlajeva J et al. Glioma-derived macrophage migration inhibitory factor (MIF) promotes mast cell recruitment in a STAT5-dependent manner. Mol. Oncol 8, 50–58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roy A et al. Glioma-derived plasminogen activator inhibitor-1 (PAI-1) regulates the recruitment of LRP1 positive mast cells. Oncotarget 6, 23647–23661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Attarha S, Roy A, Westermark B & Tchougounova E Mast cells modulate proliferation, migration and stemness of glioma cells through downregulation of GSK3β expression and inhibition of STAT3 activation. Cell. Signal 37, 81–92 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Compston A & Coles A Multiple sclerosis. Lancet 372, 1502–1517 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Dalmau J & Rosenfeld MR Paraneoplastic syndromes of the CNS. Lancet Neurol 7, 327–340 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berger JR & Koralnik IJ Progressive multifocal leukoencephalopathy and natalizumab—unforeseen consequences. N. Engl. J. Med 353, 414–416 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Calzascia T et al. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity 22, 175–184 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Galea I, Bechmann I & Perry VH What is immune privilege (not)? Trends Immunol 28, 12–18 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lohr J et al. Effector T cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin. Cancer Res 17, 4296–4308 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Kim YH et al. Tumour-infiltrating T cell subpopulations in glioblastomas. Br. J. Neurosurg 26, 21–27 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Kmiecik J et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol 264, 71–83 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Han S et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 110, 2560–2568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Donson AM et al. Increased immune gene expression and immune cell infiltration in high-grade astrocytoma distinguish long-term from short-term survivors. J. Immunol 189, 1920–2197 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cserr HF & Knopf PM Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: a new view. Immunol. Today 13, 507–512 (1992). [DOI] [PubMed] [Google Scholar]
- 70.Reboldi A et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol 10, 514–523 (2009). [DOI] [PubMed] [Google Scholar]
- 71.Schwyzer M & Fontana A Partial purification and biochemical characterization of a T cell suppressor factor produced by human glioblastoma cells. J. Immunol 134, 1003–1009 (1985). [PubMed] [Google Scholar]
- 72.Masson F et al. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J. Immunol 179, 845–853 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Thomas DA & Massagué J TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Weller M et al. CD95-dependent T cell killing by glioma cells expressing CD95 ligand: more on tumor immune escape, the CD95 counterattack, and the immune privilege of the brain. Cell Physiol. Biochem 7, 282–288 (1997). [Google Scholar]
- 75.Berghoff AS et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 17, 1064–1075 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wainwright DA et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res 18, 6110–6121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bao S et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Valdor R et al. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 8, 68614–68626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Speiser DE, Ho PC & Verdeil G Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol 16, 599–6110 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinez GJ et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bauer CA et al. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest 124, 2425–2450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Park BV et al. TGFβ1-mediated SMAD3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov 6, 1366–1381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voron T et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med 212, 139–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spranger S et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl Med 5, 200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parsa AT et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med 13, 84–88 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Reardon DA et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res 4, 124–135 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Snyder A, Wolchok JD & Chan TA Genetic basis for clinical response to CTLA-4 blockade. N. Engl. J. Med 372, 783 (2015). [DOI] [PubMed] [Google Scholar]
- 89.Daud AI et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Invest 126, 3447–3452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Omuro A et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase 1 cohorts of CheckMate 143. Neuro. Oncol 20, 674–686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reardon DA et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab versus bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro. Oncol 19 (Suppl. 3), iii21 (2017). [Google Scholar]
- 92.Long GV et al. A randomized phase II study of nivolumab or nivolumab combined with ipilimumab in patients (pts) with melanoma brain metastases (mets): the Anti-PD1 Brain Collaboration (ABC) (abstract 9508). J. Clin. Oncol 35 (Suppl. 15), 9508 (2017). [Google Scholar]
- 93.Sharma A & Shiras A Cancer stem cell-vascular endothelial cell interactions in glioblastoma. Biochem. Biophys. Res. Commun 473, 688–692 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Treps L, Perret R, Edmond S, Ricard D & Gavard J Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 6, 1359479 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rupp T et al. Tenascin-C orchestrates glioblastoma angiogenesis by modulation of pro- and anti-angiogenic signaling. Cell Rep 17, 2607–2619 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Soda Y et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl Acad. Sci. USA 108, 4274–4280 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guelfi S, Duffau H, Bauchet L, Rothhut B & Hugnot JP Vascular transdifferentiation in the CNS: a focus on neural and glioblastoma stem-like cells. Stem Cells Int 2016, 2759403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mei X, Chen YS, Chen FR, Xi SY & Chen ZP Glioblastoma stem cell differentiation into endothelial cells evidenced through live-cell imaging. Neuro Oncol 19, 1109–1118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu B et al. Epigenetic activation of WNT5A drives glioblastoma stem cell differentiation and invasive growth. Cell 167, 1281–1295.e1218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peterson TE et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl Acad. Sci. USA 113, 4470–4475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Watkins S et al. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun 5, 4196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wen L et al. VEGF-mediated tight junctions pathological fenestration enhances doxorubicin-loaded glycolipid-like nanoparticles traversing BBB for glioblastoma-targeting therapy. Drug Deliv 24, 1843–1855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Treps L et al. Extracellular vesicle-transported semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 35, 2615–2623 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Xu B et al. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Rep 27, 882–897 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miller JJ & Wen PY Emerging targeted therapies for glioma. Expert Opin. Emerg. Drugs 21, 441–452 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Buckingham SC et al. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med 17, 1269–1274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sattler R et al. Increased expression of glutamate transporter GLT-1 in peritumoral tissue associated with prolonged survival and decreases in tumor growth in a rat model of experimental malignant glioma. J. Neurosurg 119, 878–886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vazana U et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. J. Neurosci 36, 7727–7739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wei Z et al. Full-coverage landscape of extracellular RNAs, coding and non-coding, released by human glioma stem cells. Nat. Commun 10.1038/s41467-017-01196-x (2017). [DOI] [PMC free article] [PubMed]
- 110.Kim H et al. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc. Natl Acad. Sci. USA 107, 2183–2188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Teplyuk NM et al. MicroRNA-10b inhibition reduces E2F1-mediated transcription and miR-15/16 activity in glioblastoma. Oncotarget 6, 3770–3783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Herrup K & Yang Y Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci 8, 368–378 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Absalon S, Kochanek DM, Raghavan V & Krichevsky AM MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci 33, 14645–14659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Takano T et al. Glutamate release promotes growth of malignant gliomas. Nat. Med 7, 1010–1015 (2001). [DOI] [PubMed] [Google Scholar]
- 115.Ishiuchi S et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci 27, 7987–8001 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.El-Habr EA et al. A driver role for GABA metabolism in controlling stem and proliferative cell state through GHB production in glioma. Acta Neuropathol 133, 645–660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Venkatesh HS et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Venkatesh HS et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li F, Liu X, Sampson JH, Bigner DD & Li CY Rapid reprogramming of primary human astrocytes into potent tumor-initiating cells with defined genetic factors. Cancer Res 76, 5143–5150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jahani-Asl A et al. Control of glioblastoma tumorigenesis by feed-forward cytokine signaling. Nat. Neurosci 19, 798–806 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Biasoli D et al. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 3, e123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lemée JM, Clavreul A & Menei P Intratumoral heterogeneity in glioblastoma: don’t forget the peritumoral brain zone. Neuro Oncol 17, 1322–1332 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mangiola A et al. Gene expression profile of glioblastoma peritumoral tissue: an ex vivo study. PLoS ONE 8, e57145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leiss L et al. Tumour-associated glial host cells display a stem-like phenotype with a distinct gene expression profile and promote growth of GB xenografts. BMC Cancer 17, 108 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Iwadate Y, Fukuda K, Matsutani T & Saeki N Intrinsic protective mechanisms of the neuron-glia network against glioma invasion. J. Clin. Neurosci 26, 19–25 (2016). [DOI] [PubMed] [Google Scholar]
- 126.El Fatimy R, Subramanian S, Uhlmann EJ & Krichevsky AM Genome editing reveals glioblastoma addiction to microRNA-10b. Mol. Ther 25, 368–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yuan JX, Bafakih FF, Mandell JW, Horton BJ & Munson JM Quantitative analysis of the cellular microenvironment of glioblastoma to develop predictive statistical models of overall survival. J. Neuropathol. Exp. Neurol 75, 1110–1123 (2016). [DOI] [PubMed] [Google Scholar]
- 128.Rath BH, Fair JM, Jamal M, Camphausen K & Tofilon PJ Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS ONE 8, e54752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roos A, Ding Z, Loftus JC & Tran NL Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Front. Oncol 7, 120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Asslaber M et al. Native oligodendrocytes in astrocytomas might inhibit tumor proliferation by WIF1 expression. J. Neuropathol. Exp. Neurol 76, 16–26 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Peferoen L, Kipp M, van der Valk P, van Noort JM & Amor S Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141, 302–313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Trajkovic K et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Dinkins MB, Dasgupta S, Wang G, Zhu G & Bieberich E Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 1792–1800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Asai H et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci 18, 1584–1593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Phuyal S, Hessvik NP, Skotland T, Sandvig K & Llorente A Regulation of exosome release by glycosphingolipids and flotillins. FEBS J 281, 2214–2227 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Atai NA et al. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J. Neurooncol 115, 343–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jansen F et al. Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler Thromb. Vasc. Biol 32, 1925–1935 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Chen Q et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ulrich TA, de Juan Pardo EM & Kumar S The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res 69, 4167–4174 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nuti E et al. Bifunctional inhibitors as a new tool to reduce cancer cell invasion by impairing MMP-9 homodimerization. ACS Med. Chem. Lett 8, 293–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Barker HE, Paget JT, Khan AA & Harrington KJ The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pyonteck SM et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Quail DF et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Butowski N et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol 18, 557–564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brennan CW et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Prins RM et al. Gene expression profile correlates with T cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res 17, 1603–1615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Reardon DA et al. Randomized phase 3 study evaluating the efficacy and safety of nivolumab versus bevacizumab in patients with recurrent glioblastoma: checkmate 143. Neuro. Oncol 19 (suppl. 3), iii21 (2017). [Google Scholar]
- 149.Sampson JH et al. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498. J. Clin. Oncol 34 (Suppl. 15), TPS2079 (2016). [Google Scholar]
- 150.Weller M et al. A randomized phase 2, single-blind study of temozolomide (TMZ) and radiotherapy (RT) combined with nivolumab or placebo (PBO) in newly diagnosed adult patients (pts) with tumor O6-methylguanine DNA methyltransferase (MGMT)-methylated glioblastoma (GBM)—CheckMate-548. Ann. Oncol 27 (Suppl. 6), 356TiP (2016). [Google Scholar]
- 151.Bouffet E et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol 34, 2206–2211 (2016). [DOI] [PubMed] [Google Scholar]
- 152.Saha D, Martuza RL & Rabkin SD Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell 32, 253–267.e255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jiang H et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res 77, 3894–3907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Khasraw M, Ameratunga MS, Grant R, Wheeler H & Pavlakis N Antiangiogenic therapy for high-grade glioma. Cochrane Database Syst. Rev 9, CD008218 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Park JS et al. Normalization of tumor vessels by Tie2 activation and Ang2 inhibition enhances drug delivery and produces a favorable tumor microenvironment. Cancer Cell 30, 953–967 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Infante J, Burris HA & Lewis N A multicenter phase Ib study of the safety, pharmacokinetics, biological activity and clinical efficacy of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17. Breast Cancer Res. Treat 106, S269 (2007). [Google Scholar]
- 157.Friedman S et al. Clinical benefit of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17, in combination with trastuzumab in metastatic HER2 positive breast cancer patients. Cancer Res 69, 5056 (2014). [Google Scholar]
- 158.Kim SS, Pirollo KF & Chang EH Isolation and culturing of glioma cancer stem cells. Curr. Protoc. Cell Biol 67, 10.21–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hubert CG et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res 76, 2465–2477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hira VVV et al. Periarteriolar glioblastoma stem cell niches express bone marrow hematopoietic stem cell niche proteins. J. Histochem. Cytochem 66, 155–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Calabrese C et al. A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82 (2007). [DOI] [PubMed] [Google Scholar]
- 162.Xu Z, Kader M, Sen R & Placantonakis DG Orthotopic patient-derived glioblastoma xenografts in mice. Methods Mol. Biol 1741, 183–190 (2018). [DOI] [PubMed] [Google Scholar]
- 163.William D et al. Optimized creation of glioblastoma patient derived xenografts for use in preclinical studies. J. Transl Med 15, 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Oh T et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl Med 12, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hambardzumyan D, Parada LF, Holland EC & Charest A Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia 59, 1155–1168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ben-David U et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet 49, 1567–1575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baysan M et al. Micro-environment causes reversible changes in DNA methylation and mRNA expression profiles in patient-derived glioma stem cells. PLoS ONE 9, e94045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bigner SH, Mark J & Bigner DD Chromosomal progression of malignant human gliomas from biopsy to establishment as permanent lines in vitro. Cancer Genet. Cytogenet 24, 163–176 (1987). [DOI] [PubMed] [Google Scholar]
- 169.Beutler AS, Banck MS, Wedekind D & Hedrich HJ Tumor gene therapy made easy: allogeneic major histocompatibility complex in the C6 rat gliomamodel. Hum. Gene Ther 10, 95–101 (1999). [DOI] [PubMed] [Google Scholar]
- 170.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang Q et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Louis DN et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 173.Flavahan WA et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Parsons DW et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Frattini V et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet 45, 1141–1149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]