

Abstract
Our goal is to identify the genetic underpinnings of bicuspid aortic valve and aortopathy in Turner syndrome. We performed whole exome sequencing on 188 Turner syndrome study subjects from the GenTAC registry. A gene-based burden test, SKAT-O, was used to evaluate the data using bicuspid aortic valve (BAV) and aortic dimension z-scores as covariates. This revealed that TIMP3 was associated with BAV and increased aortic dimensions at exome-wide significance. It had been previously shown that genes on chromosome Xp contribute to aortopathy when hemizygous. Our analysis of Xp genes revealed that hemizygosity for TIMP1, a functionally redundant paralogue of TIMP3, increased the odds of having BAV aortopathy compared to individuals with more than one TIMP1 copy. The combinatorial effect of a single copy of TIMP1 and TIMP3 risk alleles synergistically increased the risk for BAV aortopathy to nearly 13-fold. TIMP1 and TIMP3 are tissue inhibitors of matrix metalloproteinases (TIMPs) which are involved in development of the aortic valve and protection from thoracic aneurysms. We propose that the combination of TIMP1 haploinsufficiency and deleterious variants in TIMP3 significantly increases the risk of BAV aortopathy in Turner syndrome, and suggest that TIMP1 hemizygosity may play a role in euploid male aortic disease.
Keywords: Aortic disease, Bicuspid aortic valve, Congenital cardiovascular disease, Polygenic trait
The goal of this project is to identify the genetic basis of bicuspid aortic valve (BAV) and associated aortopathy in Turner syndrome. Turner syndrome is an aneuploid condition resulting from the complete or partial loss of the second sex chromosome. The resulting deficiency of sex chromosome genes predisposes individuals with Turner syndrome to diseases with a sex bias in the general population, including BAV. BAV is the most common cardiovascular birth defect in humans, affecting about 2 to 3 percent of euploid males and around 0.05 percent of euploid females, whereas it affects 25–30% of individuals born with Turner syndrome, which is a 50–100 fold increase over the euploid population. The high incidence in Turner syndrome and the substantial sex bias towards males in the euploid population suggests that there is protection conferred by having a second X chromosome.
Our study was designed to determine the genetic basis of BAV in Turner syndrome with the presumption that there are contributions from autosomal gene variants in addition to an X chromosome component. Importantly, about 95 percent of individuals with Turner syndrome who have an aortic dissection also have BAV. So there is a clear association between BAV and later onset aortopathy in Turner syndrome. In addition to identifying genes associated with BAV, we also want to find the genetic intersection between BAV and aortopathy.
We use the disease threshold model shown in figure 1 to provide a framework for thinking about how BAV in Turner syndrome may occur. In the general euploid population an unknown number of genetic variants and/or other risk factors must be present in order to breach the theoretical threshold of disease. On the other hand, individuals with Turner syndrome are genetically predisposed to having BAV aortopathy as the result of having a complete or partial loss of the second sex chromosome. The sex chromosome gene deficiency is not alone sufficient to push individuals with Turner syndrome above the disease threshold, but we hypothesized that it will take few additional risk factors for them to manifest the disease. In fact, one additional risk factor may suffice.
Figure 1. Disease threshold model to illustrate polygenic disease and the role of effect size of risk variants.

The horizontal dotted line represents a theoretical threshold of disease. Factors, such as genetic variants are represented by boxes forming vertical columns. Columns that cross the dotted line breach the disease threshold indicating that a sufficient burden of risk factors exists to cause the disease, in this case BAV. In Turner syndrome the loss of the second sex chromosome is a risk factor of very large effect size. However, it is not sufficient alone to breach the disease threshold for BAV as not everyone with Turner syndrome has BAV. However, it is likely that fewer risk factors are required to cause BAV compared to the euploid population, which has an inherently low risk.
We tested this hypothesis using a whole exome sequencing study of individuals from the GenTAC registryTurner syndrome cohort, with and without BAV. (Eagle & GenTAC Investigators, 2009; Kroner et al., 2011) We used BAV status and aortic Z-scores as the phenotypic variables for the analysis(Corbitt, Maslen, Prakash, Morris, & Silberbach, 2018; Quezada, Lapidus, Shaughnessy, Chen, & Silberbach, 2015), with aortic enlargement with a Z-score of greater than 2 as a proxy for aortic aneurysm formation. The whole exome sequencing on 188 Turner syndrome samples was done by the NHLBI Resequencing and Genotyping Service at the University of Washington.
We used a sequence kernel associated test, or SKAT-O to analyze the data.(Wu et al., 2011) In SKAT-O, which is a gene burden by phenotype association test, genetic variants are weighted based on allele frequency as a proxy for potential deleteriousness and clustered into genes for an association analysis. The power to detect a signal is increased because the multiple testing burden is based on the number of genes tested instead of the traditional single variant by phenotype testing where every variant is an individual test. Figure 2 shows the Manhattan plots resulting from the SKAT-O analyses. Figure 2A is the result using BAV as the variable.(Corbitt, Morris, et al., 2018) A single gene, TIMP3, reached exome-wide significance. Adding in aortic root dimensions or ascending aortic dimensions as continuous covariates, we found exome wide significance with BAV and aortic root Z-scores, where the TIMP3 signal actually rose in significance, indicating that it is associated with both BAV and aortic dilation (Figure 2B). The associated Q-Q plots are shown in figure 3.
Figure 2. SKAT-O Analysis Shows that TIMP3 Variants are Associated with BAV/TAD.
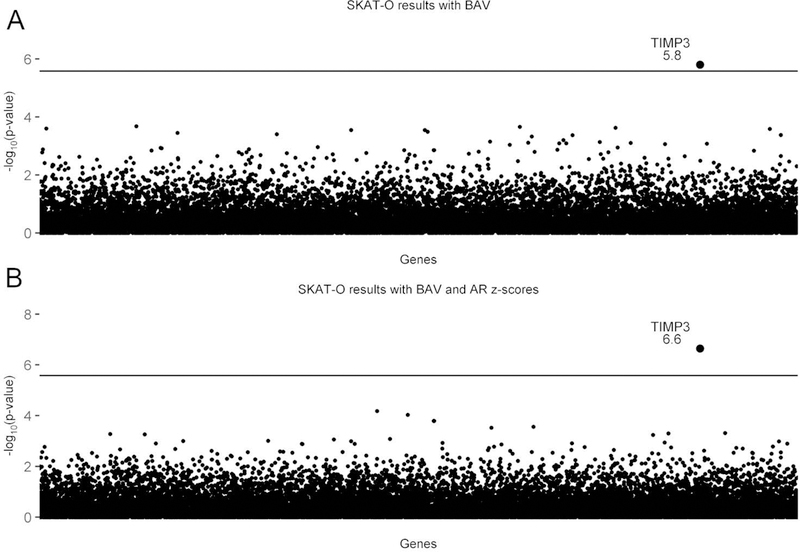
Manhattan plots showing the exome-wide significant finding that TIMP3 variants are associated with BAV, and with AR enlargement as an indicator of TAD. The horizontal line is the threshold for exome- wide significance (based on testing 19,392 genes, the exome-wide significance p-value=2.578×10–6). TIMP3 is the only gene that exceeds exome-wide significance. It is notable that no other genes approach the significance line. A) Shows the association with BAV as the sole predictor (p=1.58×10–6). B) Shows the association results for BAV and aortic root (AR) z-scores as covariates (p=2.27×10–7). The significance level for TIMP3 increases nearly 10-fold when AR z-scores were added. (From Corbitt H et al. (2018) TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genetics, 2018 Oct 3;14(10):e1007692. doi: 10.1371/journal.pgen.1007692.)
Figure 3. Quantile-Quantile (Q-Q) Plots for the SKAT-O Analyses.
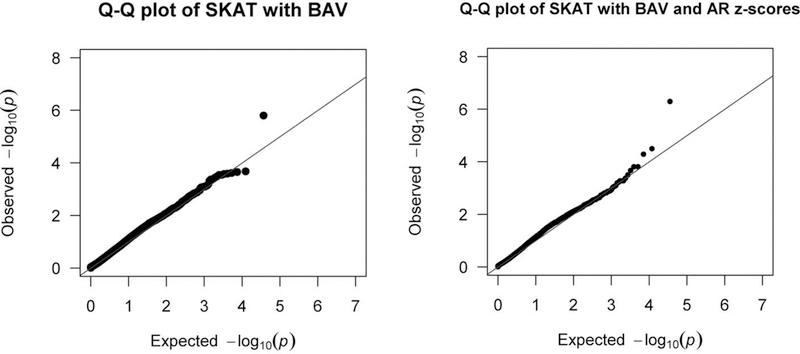
Q-Q plots for SKAT-O analysis of BAV and BAV with AR Z-scores, which shows no significant deviation from the normal distribution.
We analyzed the variants identified in TIMP3 to determine what was driving the association. There were a total of four variants identified in all of the exomes, so a small amount of variation is tolerated in this gene. We found that one variant in particular, rs11547635, which is a synonymous single nucleotide variant (SNP) in the codon for amino acid position at p.Ser87, was the significant driver of association here. It was present at a higher than expected frequency in cases compared to controls, and it was also predicted to be highly deleterious. We used CADD scores as a measure of the degree of deleteriousness, with a score of >15 indicating that a variant is in the top 5% of most deleterious variants in the genome(Kircher et al., 2014). All of the variants and their allele frequencies are shown in Table 1.
Table 1. Summary of the TIMP3 Variants Associated with BAV/TAD6.
(From Corbitt H et al. (2018) TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genetics, 2018 Oct 3;14(10):e1007692. doi: 10.1371/journal.pgen.1007692.)
| rsID | Protein Change | CADD Score | Expected Allele Frequency (Alleles/Total) | Observed Frequency in Cases (Cases/Total) | Observed Allele Frequency in Cases (Alleles/Total) | Observed Frequency in Controls (Controls/Total) | Observed Allele Frequency in Controls (Alleles/Total) |
|---|---|---|---|---|---|---|---|
| rs9862 | p.His83= | 2.597 | 49.1% (32790/66734) | 64.8% (57/88) | 43.8% (77/176) | 71.0% (71/100) | 50.5% (101/200) |
| rs11547635 | p.Ser87= | 16.67 | 7.1% (4735/66738) | 23.8% (21/88) | 11.9% (21/176) | 6.0% (6/100) | 3.5% (6/200) |
| rs149161075 | p.Phe172= | 5.801 | 0.3% (203/66736) | 2.3% (2/88) | 1.1% (2/176) | 0% (0/100) | 0% (0/200) |
| rs369072080 | p.Gly173= | 0.002 | 0% (0/66738) | 1.1% (1/99) | 0.6% (1/176) | 0% (0/100) | 0% (0/200) |
All of the TIMP3 variants identified through whole exome sequencing of subjects in our Turner syndrome cohort are listed. The dbSNP rs identifier is listed, along with the consequence of the change, ExAC expected allele frequencies (European non-Finnish), CADD score, the number of subjects that had a BAV (case) or a normal valve (control), and the allele frequency of each variant, reported as percentages.
Two other variants also contributed to the association. They were both very rare, observed only in cases. We also observed that the p.Ser87 variant is always accompanied by a synonymous SNP at the codon for amino acid position 83, p.His83, which appears to have functional significance.
We wished to replicate the TIMP3 association, which we were able to do thanks to Dr. Claus Gravholt, who contributed samples from his Danish Turner syndrome registry. We did a SKAT-O analysis after sequencing the exons of TIMP3 and found that the association replicated with the p.Ser87 position still acting as the driver, as shown in Table 2, which was statistically significant even though it was a small replication cohort. And we also saw the co- segregation with p.His83, and one of the very rare variants, also only in cases.
Table 2. Replication Cohort Sequencing Results6.
(From Corbitt H et al. (2018) TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genetics, 2018 Oct 3;14(10):e1007692. doi: 10.1371/journal.pgen.1007692.)
| Chromosome | Position | ID | REF | ALT | MAF cases | MAF controls |
|---|---|---|---|---|---|---|
| 22 | 33253280 | rs9862 | T | C | 50.00% | 50.00% |
| 22 | 33253292 | rs11547635 | C | T | 14.29% | 6.40% |
| 22 | 33255244 | rs149161075 | C | T | 3.57% | 0.00% |
| Gene | P value | N variants in Test | ||||
| TIMP3 | 0.03734* | 3 | ||||
Variants identified in Danish cohort by Sanger sequencing of TIMP3 exons.
ID; rs identifier from dbSNP
MAF; minor allele frequency
P value; calculated by SKAT-O gene-based association test
reaches the significance threshold of <0.05
N; number
Based on this association we are interested in understanding the functional consequences of these TIMP3 variants. We know from the cancer literature that the linked p.His83 and p.Ser87 SNPs are associated with reduced levels of TIMP3 in plasma.(Bashash et al., 2013; Jackson, Defamie, Waterhouse, & Khokha, 2017; Su et al., 2015) Our preliminary data from western blot analysis of recombinant TIMP3 from constructs harboring the p.His83 and p.Ser87 SNPs, together and separately appears to support this association (Figure 4). Expression constructs were made that carried each of the variants alone, and together. Western blot analysis of protein secreted from HEK293 cells transfected with the various TIMP3 constructs shows an overall reduction in secreted TIMP3 when pHis83= and/or p.Ser87= are present, particularly of the glycosylated form. Consequently, we propose that a reduction in extracellular TIMP3 is the biologically relevant mechanism of action for these SNPs, which is important in maintaining structural integrity of the aorta, as TIMP3 is a member of the tissue inhibitor of matrix metalloproteinase family. Matrix metalloproteinases (MMPs) are enzymes that are known to break down extracellular matrix. MMP activity is controlled by TIMPs through direct binding, which is critical to maintaining tissue homeostasis.
Figure 4. TIMP3 variants decrease expression.
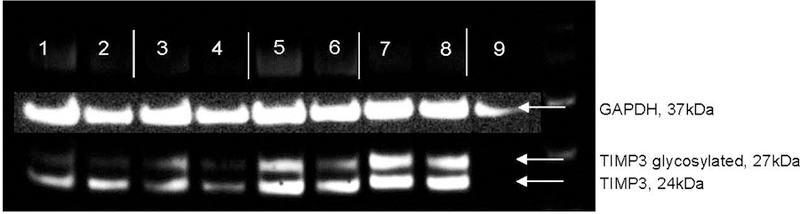
Western blot of secreted protein from TIMP3 constructs, in duplicates. Lanes 1–2 His83/Ser87 double variant; Lanes 3–4, His83; Lanes 5–6 Ser87; Lanes 7–8 wild type; Lane 9, empty vector. GAPDH loading control is superimposed. Note that the His83 and Ser87 variants significantly reduce secreted TIMP3 expression, particularly the glycosylated form.
TIMP3 is known to control MMPs 2, 3, 7, 9, 13, and 14. Significantly, MMPs 2 and 9 are the most highly expressed MMPs in thoracic aortic aneurysms, and an imbalance between TIMPs and MMPs is thought to be the underlying pathogenic mechanism in thoracic aortic aneurysms and progression to dissection.(Abaci, Kocas, Kilickesmez, Uner, & Kucukoglu, 2013; Allaire, Forough, Clowes, Starcher, & Clowes, 1998; Ikonomidis et al., 2004; Koullias, Ravichandran, Korkolis, Rimm, & Elefteriades, 2004; Li et al., 2010; Rabkin, 2014; Zhang, Shen, & LeMaire, 2009; Zhang et al., 2014) This occurs when increased MMP activity in the tunica media of the aorta disrupts the usually highly ordered elastin-based extracellular matrix. As the matrix is degraded, macrophages are allowed to infiltrate, and aneurysms form.
Going back to our disease threshold model (Figure 1), we’re proposing that the p.His83 and p.Ser87 variants in TIMP3 exist as common variants in the general population and are presumably benign unless triggered by another genetic event where they can become pathogenic. In particular, these variants are associated with progression of certain cancers. In Turner syndrome we propose that there is a pathogenic interaction between these TIMP3 variants and hemizygosity of a gene or genes on the X chromosome that is the underlying etiology for BAV aortopathy in that population. To address this, we did a bioinformatic analysis to identify candidate genes. We know from prior publications that the region of the X chromosome associated with BAV and aortic aneurysms is actually the short arm, Xp.(Bondy et al., 2013; Prakash et al., 2016) We established inclusion criteria based on the premise that reduced dosage of a gene or genes on Xp increase risk of BAV aortopathy. There were three criteria to consider. One, the gene must be to unique to the X chromosome. There can’t be a homolog elsewhere in the genome based on our premise that it’s a reduction in dosage that is involved, specifically on the Y chromosome. The gene also must escape X inactivation, so the gene on the second X chromosome has the capacity to be expressed. Finally, any candidate gene needs to be expressed in the developing aortic valve to be involved with bicuspid aortic valve and in the mature aorta to be associated with later onset aortopathy.
In our analysis, out of the 247 Xp genes, only 3 met those criteria. And they are UBA1, RBM3, and TIMP1. UBA1 is associated in mouse models and in humans with spinal and muscular atrophy, and there is no cardiovascular phenotype. Likewise, RBM3 is associated with neurodegenerative disease both in mouse models and in humans. But once again, there is no cardiovascular phenotype, even though both are expressed at low levels in the aorta. On the other hand, reduced expression of TIMP1 is associated in humans in thoracic aortic aneurysms and in mouse models where the TIMP1-deficient mouse is susceptible to developing aortic aneurysms. TIMP1 is expressed in the aorta(Akahane, Akahane, Shah, & Thorgeirsson, 2004), but also in the developing aortic valve(Dreger, Taylor, Allen, & Yacoub, 2002), and its expression level is tenfold higher than UBA1 or RBM3. Consequently, we propose that TIMP1 is the most likely Xp gene to be involved in BAV aortopathy. Importantly, TIMP1 and TIMP3 are functionally redundant in the aorta, and both control MMPs 2 and 9, which are known to be upregulated in thoracic aortic aneurysms, so we consider this association to be functionally very important.
Next we asked the question, is TIMP1 copy number associated with aortic disease and, most importantly, bicuspid aortic valve? We were able do this because Turner syndrome is not always monosomy X, but in some cases there is just a partial loss of the second sex chromosome, and our GenTAC cohort reflected this diversity. About 50 percent have a complete monosomy X. Around 30 percent have a partial deletion or rearrangement of a second X chromosome. Twenty percent are mosaic for a second sex chromosome, and a few have some residual Y chromosome material. In addition, since there is no Y chromosome TIMP1 homologue males only have a single copy of TIMP1.
We used our whole exome data to construct molecular karyotypes for our entire cohort so that we could determine the copy number of TIMP1, including accounting for mosaicism where some cells may have one copy of TIMP1 and other may have two copies. For example, if 50 percent of cells are 45,XO, and 50 percent are 46,XX, then the average copy number would be 1.5, which would be expected to result in a higher average plasma protein level as it is a secreted protein. When we did the association study, we showed that TIMP1 copy number is associated with bicuspid aortic valve, as shown in figure 5. Having only one copy of TIMP1 confers a greater than fourfold increased risk for having a bicuspid aortic valve compared to more than one copy of TIMP1.
Figure 5. TIMP1 copy number is associated with the risk of BAV and BAV with TAD.
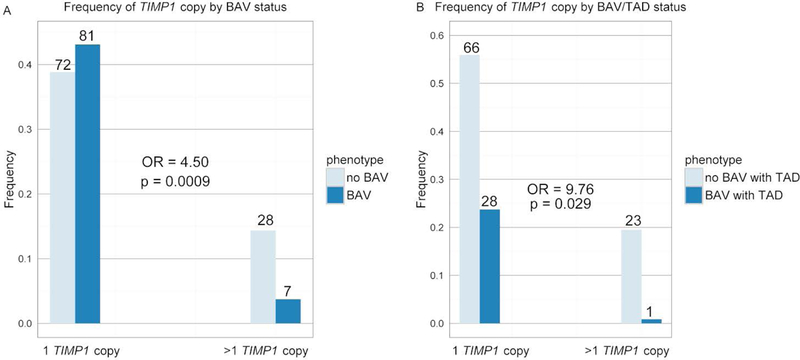
Bar graph showing the frequency of BAV with or without TAD when only one copy of TIMP1 is present compared to the frequency when there is more than one copy of TIMP1. A) The dark blue bars represent individuals with a diagnosis of BAV and the light blue bars represent individuals without a BAV. B) The dark blue bars represent individuals with a diagnosis of BAV with TAD and the light blue bars represent individuals without a BAV with TAD. For both a logistic regression model was run, with TIMP1 copy number as the categorical predictor and the phenotype as the response variable. Odds ratio and p-value were calculated and showed that having a single copy of TIMP1 significantly increased the odds of having a BAV and a BAV with TAD. (From Corbitt H et al. (2018) TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genetics, 2018 Oct 3;14(10):e1007692. doi: 10.1371/journal.pgen.1007692.)
As expected, TIMP1 expression is reflective of copy number as the second copy is known to polymorphically escape X inactivation.(Anderson & Brown, 1999) Figure 6 shows TIMP1 protein levels in plasma from individuals in our Turner syndrome cohort. The expression level associated with a single copy of TIMP1 is consistently low. However, when there is more than one copy, the expression level is generally increased but is also highly variable. This is based on the fact that TIMP1 polymorphically escapes X inactivation in humans, and expression levels are significantly different.
Figure 6. TIMP1 protein levels in plasma.
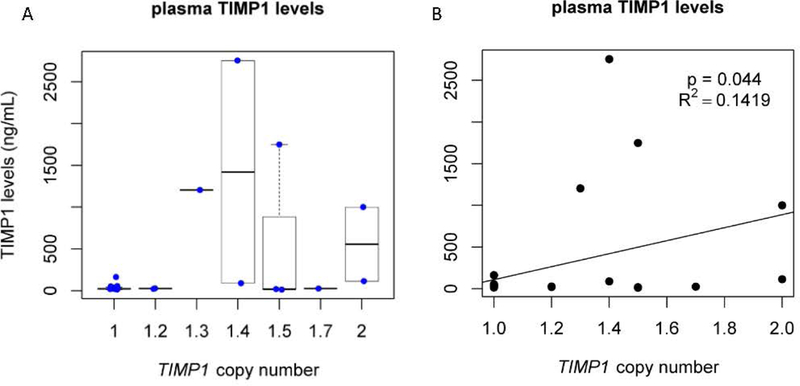
Expression of TIMP1 levels in plasma as measured by ELISA plotted by the number of TIMP1 gene copy number. Note that TIMP1 copy number is significantly correlated with TIMP1 protein levels in plasma. The samples with only one copy of TIMP1 are tightly clustered with little variability, whereas the samples with more than 1 copy are highly variable, as has been previously described.
We then determined if there was a combinatorial effect between TIMP1 copy number and the TIMP3 risk alleles. As shown in Table 3, there is a synergistic effect of having both the TIMP3 risk alleles and only one copy of TIMP1, with a nearly 13-fold increase in risk for BAV and aortic root dilation compared to having more than one copy of TIMP1 and no TIMP3 risk allele. Figure 7 shows the influence of TIMP1 copy number on aortic dimensions. The mean aortic root Z-score is significantly greater in individuals with only one copy of TIMP1, compared to those with greater than one TIMP1 copy number (Figure 7A). Likewise, the mean ascending aorta Z-score is significantly larger with only one copy of TIMP1 (Figure 7B).
Table 3. Combinatorial Effects of TIMP1 Copy Number and TIMP3 SNP rs11547635 on the Outcome of BAV or BAV with TAD6.
(From Corbitt H et al. (2018) TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genetics, 2018 Oct 3;14(10):e1007692. doi: 10.1371/journal.pgen.1007692.)
| Groups, outcome of BAV with TAD | N | N affected | % affected | OR | 95% CI | P value |
|---|---|---|---|---|---|---|
| no rs11547635 & >1 TIMP1 copy | 22 | 2 | 9.09% | 1.00** | - | - |
| no rs11547635 & only 1 TIMP1 copy | 78 | 20 | 25.64% | 3.45 | 0.89 – 22.80 | 0.115 |
| yes rs11547635 & >1 TIMP1 copy* | 2 | 0 | 0.00% | - | - | - |
| yes rs11547635 & only 1 TIMP1 copy | 16 | 9 | 56.25% | 12.86 | 2.57 – 99.39 | 0.004 |
Logistic regression model for the combinatorial effect of have both rs11547635 and one copy of TIMP1 on the outcome of having a BAV with TAD.
BAV and TAD are defined as having a BAV and at least one z-score ≥ 1.9 (rounded to the 10th decimal place); those without an AR or AAO measurement were excluded.
For this group, the sample size was too small, so an accurate odds ratio could not be calculated.
N; total number of subjects.
N affected; number of subjects with BAV or BAV and TAD.
Reference group
Figure 7. TIMP1 copy number vrs aortic z-scores.

Panel A shows a box plot representing study subject aortic root z-scores plotted against TIMP1 copy number. Panel B shows the box plot for ascending aorta z-scores plotted against TIMP1 copy number. Note that for both aortic root and ascending aorta z-scores there is a significant difference in aortic diameters comparing individuals with only one copy of TIMP1 to those with greater than one copy of TIMP1.
And finally, one more piece of data that we find very compelling, and this was done by Drs. Michael Silberbach and Hal Dietz looking at a Turner syndrome aorta that was donated to Dr. Silberbach from a young woman with Turner syndrome who died from a type B aortic dissection. The pathology is shown in Figure 8. Panels A and C show an aortic section from a normal sex- and age-related control. Panels B and D are from the aorta of the Turner syndrome individual. The aorta section from the Turner syndrome patient was from non- aneurysmal tissue. The aortas from panels A and B are stained with an antibody against connective tissue growth factor (CTGF). The staining shows that CTGF expression is greatly increased in the Turner syndrome aorta compared to the normal control. Panels C and D are stained with trichrome, which shows a significant amount of fibrosis compared to the normal control, likely as a result of the increase in CTGF expression.
Figure 8. TS aorta CTGF and extracellular matrix staining.
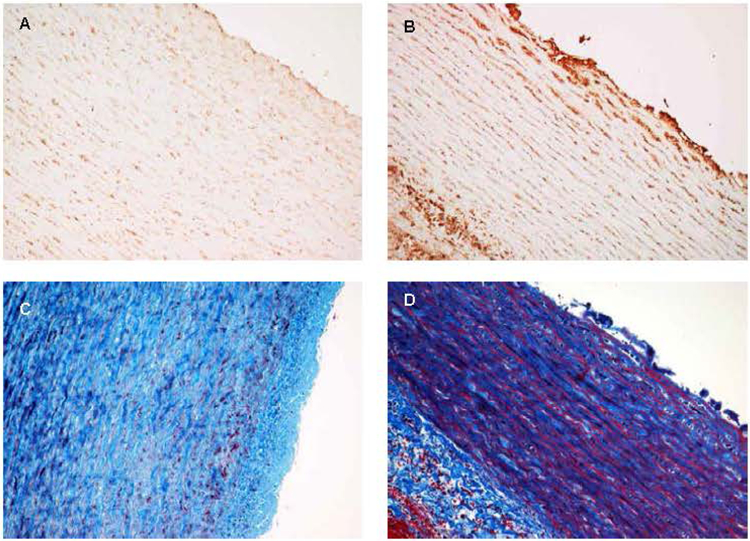
Analysis of the same aortic tissue from a young woman with TS who died from a dissection of the ascending aorta to visualize extracellular matrix components. The top two panels (A, B) are stained with an antibody to connective tissue growth factor (CTGF). Panel A is from an age and gender matched normal control aorta. Note the lack of staining for CTGF. Panel B is from a non-aneurysm section of aorta from the TS patient. Note the significantly increased level of CTGF indicating an activation of connective tissue production. Panels C and D are stained with trichrome, staining collagens blue. Note the substantial increase in staining in the TS aorta (D) compared to the control (C), showing increased collagen deposition in the TS aorta (fibrosis), which is thought to be compensatory for aortic wall instability.
Figure 9 shows the same aortas immunohistochemically stained to detect SMAD4 as a proxy for TGF-beta signaling. Panels A and C are from the control aorta, and panels B and D are the Turner syndrome aorta. Turner syndrome aorta shows an increased level of SMAD4 expression compared to the normal control, indicating that there is an increase in TGF-β signaling. This is a familiar scenario in genetically-triggered thoracic aortic aneurysms. An increase in TGF-β signaling was originally noted in aneurysmal tissue from the aortas on individuals with Marfan syndrome.(Neptune et al., 2003) Although the specific significance of this process is still under investigation, it gives a framework for developing a pathogenic model for the development of thoracic aortic aneurysms in Turner syndrome, as shown in Figure 10. Our data indicates that in individuals with Turner syndrome there can be an inherent imbalance in TIMPs and MMPs due to hemizygosity of TIMP1, which is further exacerbated in the presence of TIMP3 risk alleles. As a result, there is a loss of inhibition of MMPs 2 and 9 which proteolytically degrades the extracellular matrix (ECM) of the aortic wall. Degradation of the ECM releases active TGF-β, which is held in its latent inactive form in the ECM under conditions of tissue homeostasis. As TGF-β activity increases there is a process by which fibrosis and inflammation occurs, including an increase in MMP activity, eventually resulting in an aortic aneurysm.
Figure 9. TS aorta SMAD staining.
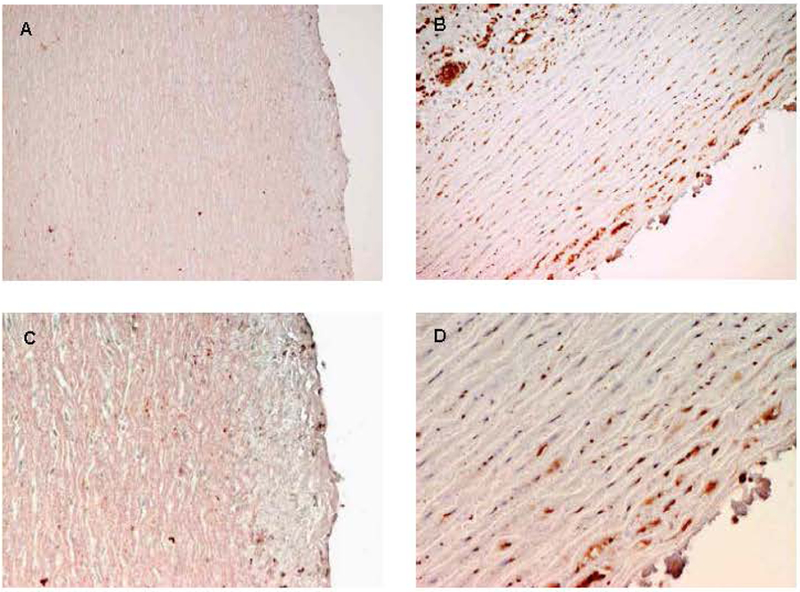
Immunohistochemical analysis the same aortas shown in figure 6 to assess levels of TGF-β signaling. The two left panels (A, C) are from control aortas. The two right panels are sections from the TS aorta. All are stained with p-SMAD antibody. SMADs are downstream products of TGF-β signaling. Note the increase in staining (reddish-brown) in the TS aorta (Panels B and D) compared to a normal control (Panels A and C), indicating an increase in p-SMAD production as a result of increased TGF-β signaling. Excessive TGF-β signaling is seen in aortas from individuals with Marfan syndrome and is thought to contribute to aortic wall instability. This is the first demonstration of excessive TGF-β signaling in TS.
Figure 10. Theoretical model of aortic disease pathogenesis in TS.
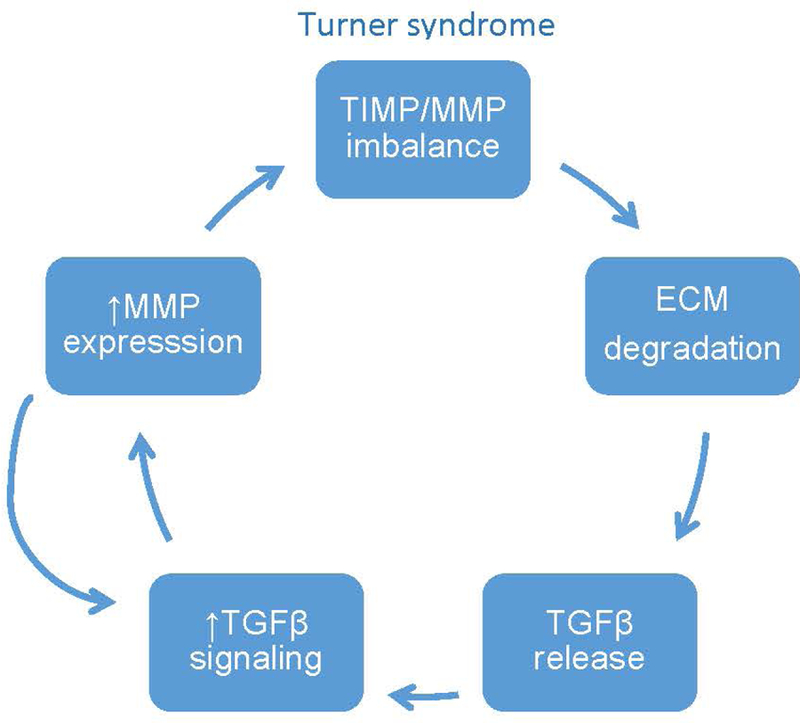
This model depicts the proposed pathogenic events underlying aortopathy in Turner syndrome. We propose that there is an inherent imbalance in the TIMP/MMP ratio that leads to the degradation of the ECM, including fibrillin-1 microfibrils. As in Marfan syndrome this releases active TGFβ resulting in an increase in TGFβ signaling. One of the consequences of TGFβ signaling is an increase in MMP expression, which feeds into the TIMP/MMP imbalance. There is also a feedback loop through which increased MMP activity proteolytically cleaves latent TGFβ producing the active form.
We can now speculate that the sex bias that predisposes euploid males to bicuspid valve aortopathy may, in part, be due to a relative impairment of TIMP1/MMP signaling pathways triggered by TIMP1 hemizygosity in males.
In conclusion, we’ve shown based on whole exome sequencing that deleterious variants in TIMP3 are associated with BAV and aortic enlargement and that TIMP1 is likely the Xp chromosome sensitizing factor for BAV and aortopathy in Turner syndrome. We propose that during valve morphogenesis inherent reductions in TIMP1 copy number and SNP-associated TIMP3 expression increase risk for BAV. In the mature aorta, TIMP1 deficiency due to hemizygosity coupled with reduced TIMP3 expression sensitizes the aortic wall, leading to aortopathy.
Acknowledgments
We’d like to thank the many Turner syndrome study subjects and their families for their generous donation of time and body to our studies and to the Turner Syndrome Society of the U.S. for facilitating our interactions with their members over the years. Thanks to Drs. Shaine Morris, Claus Gravholt, Kristian Mortensen, Rebecca Tippner-Hedges, and the GenTAC Investigators for their contributions to this work. This work was supported by technical assistance from Jessica Kushner, Brian Booty, Carrie Farrar, Sarah Egan, and Dr. Harry C. Dietz. This work was supported by a grant from OHSU, funds from the Knight Cardiovascular Institute (to CLM), the Friends of Doernbecher (to HC), and the Ravelle Research Fund of the Turner Syndrome Society of the U.S. (to MS). The whole exome sequencing was provided through the NHLBI Resequencing and Genotyping Service at the University of Washington (D. Nickerson, director), NHLBI Grant number: 268201100037C-0–0-1.
Footnotes
Conflict of Interest
The authors do not have any conflicts of interest.
References
- Abaci O, Kocas C, Kilickesmez KO, Uner S, & Kucukoglu S (2013). Matrix metalloproteinase-2 and −9 levels in patients with dilated ascending aorta and bicuspid aortic valve. Echocardiography, 30(2), 121–126. doi: 10.1111/echo.12004 [DOI] [PubMed] [Google Scholar]
- Akahane T, Akahane M, Shah A, & Thorgeirsson UP (2004). TIMP-1 stimulates proliferation of human aortic smooth muscle cells and Ras effector pathways. Biochemical and Biophysical Research Communications, 324(1), 440–445. doi: 10.1016/j.bbrc.2004.09.063 [DOI] [PubMed] [Google Scholar]
- Allaire E, Forough R, Clowes M, Starcher B, & Clowes AW (1998). Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. Journal of Clinical Investigation, 102(7), 1413–1420. doi: 10.1172/JCI2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CL, & Brown CJ (1999). Polymorphic X-chromosome inactivation of the human TIMP1 gene. Am J Hum Genet, 65(3), 699–708. doi: 10.1086/302556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashash M, Shah A, Hislop G, Treml M, Bretherick K, Janoo-Gilani R, … Brooks-Wilson A (2013). Genetic polymorphisms at TIMP3 are associated with survival of adenocarcinoma of the gastroesophageal junction. PLoS One, 8(3), e59157. doi: 10.1371/journal.pone.0059157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy C, Bakalov VK, Cheng C, Olivieri L, Rosing DR, & Arai AE (2013). Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J Med Genet, 50(10), 662–665. doi: 10.1136/jmedgenet-2013-101720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbitt H, Maslen C, Prakash S, Morris SA, & Silberbach M (2018). Allometric considerations when assessing aortic aneurysms in Turner syndrome: Implications for activity recommendations and medical decision-making. Am J Med Genet A, 176(2), 277–282. doi: 10.1002/ajmg.a.38584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbitt H, Morris SA, Gravholt CH, Mortensen KH, Tippner-Hedges R, Silberbach M, GenTAC Research Investigators. (2018). TIMP3 and TIMP1 are risk genes for bicuspid aortic valve and aortopathy in Turner syndrome. PLoS Genet, 14(10), e1007692. doi: 10.1371/journal.pgen.1007692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreger SA, Taylor PM, Allen SP, & Yacoub MH (2002). Profile and localization of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in human heart valves. J Heart Valve Dis, 11(6), 875–880; discussion 880. [PubMed] [Google Scholar]
- Eagle KA, & GenTAC. (2009). Rationale and design of the National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC) [DOI] [PMC free article] [PubMed]
- American Heart Journal, 157(2), 319–326. doi: 10.1016/j.ahj.2008.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidis JS, Gibson WC, Butler JE, McClister DM, Sweterlitsch SE, Thompson RP, … Spinale FG (2004). Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation, 110(11 Suppl 1), II268–273. doi: 10.1161/01.CIR.0000138384.68947.20 [DOI] [PubMed] [Google Scholar]
- Jackson HW, Defamie V, Waterhouse P, & Khokha R (2017). TIMPs: versatile extracellular regulators in cancer. Nat Rev Cancer, 17(1), 38–53. doi: 10.1038/nrc.2016.115 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet, 46(3), 310–315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koullias GJ, Ravichandran P, Korkolis DP, Rimm DL, & Elefteriades JA (2004). Increased tissue microarray matrix metalloproteinase expression favors proteolysis in thoracic aortic aneurysms and dissections. Annals of Thoracic Surgery, 78(6), 2106–2110; 10.1016/j.athoracsur.2004.05.088 [DOI] [PubMed] [Google Scholar]
- Kroner BL, Tolunay HE, Basson CT, Pyeritz RE, Holmes KW, Maslen CL, … Eagle KA (2011). The National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC): results from phase I and scientific opportunities in phase II. American Heart Journal, 162(4), 627–632 10.1016/j.ahj.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Shao AZ, Jiang HT, Dong GH, Xu B, Yi J, & Jing H (2010). The prominent expression of plasma matrix metalloproteinase-8 in acute thoracic aortic dissection. J Surg Res, 163(2), e99–104. doi: 10.1016/j.jss.2010.05.030 [DOI] [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, … Dietz HC (2003). Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet, 33(3), 407–411. doi: 10.1038/ng1116 [DOI] [PubMed] [Google Scholar]
- Prakash SK, Bondy CA, Maslen CL, Silberbach M, Lin AE, Perrone L, … Milewicz DM (2016). Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am J Med Genet A, 170(12), 3157–3164. doi: 10.1002/ajmg.a.37953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada E, Lapidus J, Shaughnessy R, Chen Z, & Silberbach M (2015). Aortic dimensions in Turner syndrome. Am J Med Genet A, 167A(11), 2527–2532. doi: 10.1002/ajmg.a.37208 [DOI] [PubMed] [Google Scholar]
- Rabkin SW (2014). Differential expression of MMP-2, MMP-9 and TIMP proteins in thoracic aortic aneurysm - comparison with and without bicuspid aortic valve: a meta-analysis. Vasa, 43(6), 433–442. doi: 10.1024/0301-1526/a000390 [DOI] [PubMed] [Google Scholar]
- Su CW, Huang YW, Chen MK, Su SC, Yang SF, & Lin CW (2015). Polymorphisms and Plasma Levels of Tissue Inhibitor of Metalloproteinase-3: Impact on Genetic Susceptibility and Clinical Outcome of Oral Cancer. Medicine, 94(46), e2092. doi: 10.1097/MD.0000000000002092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, & Lin X (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet, 89(1), 82–93. doi: 10.1016/j.ajhg.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shen YH, & LeMaire SA (2009). Thoracic aortic dissection: are matrix metalloproteinases involved? Vascular, 17(3), 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu D, Choi JC, Minard CG, Hou X, Coselli JS, … LeMaire SA (2014). Matrix metalloproteinase levels in chronic thoracic aortic dissection. J Surg Res, 189(2), 348–358. doi: 10.1016/j.jss.2014.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]