

Abstract
Influenza is a viral respiratory tract infection responsible for up to 5 million cases of severe infection and nearly 600,000 deaths worldwide each year. While treatments for influenza exist, diagnostics for the virus at the point of care are limited in their sensitivity and ability to differentiate between subtypes. We have developed an integrated two-dimensional paper network (2DPN) for the detection of the influenza virus by the surface glycoprotein, hemagglutinin. The hemagglutinin assay was developed using proteins computationally designed to bind with high affinity to the highly-conserved sialic acid binding site. The integrated 2DPN uses a novel geometry that allows automated introduction of an enzymatic amplification reagent directly to the detection zone. This assay was integrated into a prototype device and demonstrated successful detection of clinically relevant virus concentrations spiked into 70 μL of virus-free pediatric nasal swab samples. Using this novel geometry, we found improved assay performance on the device (compared to a manually-operated dipstick method), with a sensitivity of 4.45 × 102 TCID50/mL on device.
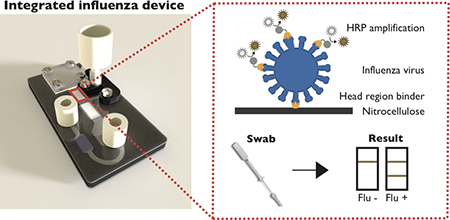
Graphical Abstract
1. Introduction.
Influenza is a viral respiratory tract infection responsible for between 3–5 million cases of severe illness and 291,000–646,000 deaths worldwide each year (1,2). While influenza primarily impacts young children, pregnant women, the elderly, and people with chronic medical conditions, it can also lead to severe respiratory disease in otherwise-healthy adults. There are three strains of the influenza virus that circulate in the human population, influenza A, B, and C. Influenza A, the strain commonly responsible for both seasonal and pandemic flu, can be further characterized based on two surface proteins that are constantly undergoing antigenic drift: hemagglutinin and neuraminidase (3,4). Antiviral medications are available for the treatment of most influenza strains, and have been shown to reduce the severity of symptoms, shorten the duration of illness, and decrease the risk of complications (5). However, these medications are generally effective when prescribed within the first 48 hours of symptoms. Therefore, timely and accurate diagnosis of the influenza virus is key for successful disease management.
Hemagglutinin is the rapidly mutating glycoprotein responsible for binding to sialic acids on human epithelial cells, as well as the subsequent fusion of the viral and endosomal membranes(6). This surface protein is of particular interest as a diagnostic target due to its role in virulence and immune response, as it is constantly evolving to escape the host immune system. While hemagglutinin protein classification provides both diagnostic and epidemiological information, available methods for screening and detection of the hemagglutinin protein are limited at the point of care. We have previously published the development of an assay that targets the hemagglutinin protein using computationally-designed affinity proteins (7,8). These designed protein “binders” target specific epitopes on the hemagglutinin surface protein with higher affinity than traditionally-used antibodies (9,10). With higher thermostability and lower production costs than antibodies, computationally-designed affinity proteins would be optimal for integration into diagnostic devices.
Today, clinical diagnosis of influenza is most commonly carried out by reverse transcriptase polymerase chain reaction (RT-PCR) or protein-based rapid influenza diagnostic tests (11). While diagnostic methods like RT-PCR are highly effective, they are challenging to implement at the point of care due to their need for trained laboratory personnel, external machinery, and refrigeration of reagents (12). Rapid influenza diagnostic tests, a form of paper-based lateral flow assay (LFA), have helped bridge the gap between the laboratory and the patient. Commercially-available tests for the detection of influenza virus rely on the detection of the RNA-associated nucleoprotein (13). Not only have these tests proved to be limited in their clinical sensitivity (at least in the US), but they also are unable to differentiate between influenza strains (14,15). Strain differentiation is generally beneficial for a few reasons: to enable epidemiological tracking, to direct antiviral therapy and to influence clinical outcomes.
LFAs rely on nitrocellulose membranes as the primary substrate, generally referred to as “paper” in this manuscript, with integrated dried reagents to allow visual detection of an analyte in an easy-to-use format (16). Instead of requiring syringe pumps or multiple pipetting steps to move fluids through a device, LFAs use capillary action to enable flow of assay reagents through the porous substrate (17). LFAs provide rapid results for the detection of many pathogens without the need for well-equipped and well-staffed clinical laboratories (18). The implementation of LFAs at the point of care has been highly successful due to their low cost and ease of use.
Despite the commercial success of LFAs, they generally lack the ability to perform additional chemistry and multistep assays that are often required for more complex laboratory assays (19). The conventional enzyme-linked immunosorbent assay (ELISA), for example, requires manual delivery of each reagent to enable protein detection. While ELISAs are sensitive protein assays, the need for refrigeration, electricity, and trained personnel complicate their implementation at the point of care (20). The traditional LFA involves one assay step, where the sample rehydrates the conjugate pad and travels downstream to the test line, where a capture antibody is immobilized. This works well for sandwich assays that use colorimetric detection, such as by gold nanoparticles, but proves challenging when attempting to integrate assays that use signal amplification chemistries. There has been an emphasis on the development of strategies that allow more complex fluidic manipulation within the context of paper-based diagnostic systems.
Various approaches to introduce more complex fluidic manipulation using paper microfluidics have been demonstrated. The Whitesides group spearheaded the use of paper for diagnostics through the layering of paper and tape to create three-dimensional paper microfluidic devices that deliver different reagents at each individual layer (21). The Filipe group described the use of dry pullulan films for the sequential delivery of reagents in a lateral flow or flow through format (22). Alternatively, the Ying group has used the stacked flow immunoassay to significantly reduce non-specific signal due to substances present in the patient sample (23). Our lab has developed a platform to address these limitations using a two-dimensional paper network (2DPN). The 2DPN allows the sequential delivery of reagents across a test line in a fully automated manner (24). The 2DPN platform has been demonstrated for the integration of assays using enzymatic amplification for more sensitive detection of an analyte, including gold enhancement and horseradish peroxidase (HRP) for signal amplification of an immunoassay (25–27).
However, 2DPN geometries can be limited in their ability to control the rate and direction of flow for delivery of multiple fluidic steps. Our previously-demonstrated 2DPN architecture for enzymatic amplification required that the last assay step flow through a long section of paper (26,27). This has led to reduced performance in integrated devices; specifically, long assay run times and decreases in sensitivity due to reagent loss to the porous material, and increased background outside of the capture lines due to nonspecific adsorption of enzyme-labelled conjugates upstream of the capture line.
Recent advances in the understanding of fluid flow in paper microfluidics have shown that understanding the effects of partial saturation are critical to predicting fluid flow in LFTs, and particularly in complex multicomponent 2DPNs (28). Controlled partial saturation effects enable the assay developer to sequence fluid delivery using specific material choices. This understanding allows for the design of devices that can control flow rates of subsequent assay steps independently and provide better spatial control of reagent transport. This understanding of membrane wetting and fluid flow has enabled the implementation of more complex fluidic handling into integrated paper microfluidics. Other applications using partial saturation include the automation of DNA extraction from a urine sample and concentration from large sample volumes and an automated dilution series for downstream immunoassay detection (28).
Here we describe the development of a prototype device that integrates computationally designed hemagglutinin head region binders for the detection of intact influenza virus, Figure 1. This assay uses enzymatic amplification with HRP to visualize hemagglutinin binding at the test line. The original binder-based hemagglutinin assay relied on gold nanoparticles for signal development; however, poor sensitivity of the assay necessitated the need for enzymatic signal amplification (29). The modified 2DPN platform presented here harnesses partial saturation to create two distinct, independently controllable fluidic paths, enabling more precise control over assay timing, reagent concentrations, and fluid flow rates. This modified platform also enables reagent delivery while minimizing interaction between the enzyme and its colorimetric substrate before reaching the test line. This novel device architecture is demonstrated for the sensitive detection of influenza hemagglutinin from nasal swab samples.
Figure 1.

Schematic of the device used for automated hemagglutinin detection. (A) The two-dimensional paper network (2DPN) used in this device. This network contains a nitrocellulose test strip, glass fiber paths for the delivery of sample, conjugate, wash and amplification buffers, and a cellulose wicking pad. (B) The fully assembled device. (C) The expanded view of the device, containing the two-dimensional paper network (2DPN) and automatic release mechanism for the amplification tube. The automatic release mechanism consists of a pinch valve that is held closed until the water-soluble paper wets and releases the “arm”. Once the arm is released, the amplification buffer is delivered to the paper network.
2. Materials and Methods
2.1. Recombinant hemagglutinin binder preparation.
Two computationally-designed proteins were used for this work, both iterations of the influenza hemagglutinin head-region binders previously described (TriHSB.1.C and TriHSB.2) (8). The hemagglutinin binders were cloned, expressed in Escherichia coli, and purified using metal affinity chromatography using Ni-NTA resin that recognizes a C-terminal His tag (10). Thiolated versions of these binders were made by mutating a select amino acid to cysteine to introduce a single thiol moiety for site-specific immobilization to a thiol-reactive agent, as previously published [7]. This allowed for the direct conjugation of HRP to the head region binder, with a range of 0 to 3 HRP molecules conjugated to each binder using a commercial conjugation kit (ThermoFisher, Waltham, MA, USA). All additions and mutations were made to the recombinant hemagglutinin binders through the Rosetta software suite (www.rosettacommons.org), which allowed minimal impact on the HA-binding functionality of these proteins.
2.3. Dipstick assay for hemagglutinin detection
A dipstick assay was used for high throughput assay development and optimization. Test strips for the dipstick assay were made by cutting nitrocellulose membranes (GE FF80 HP, GE Healthcare, Piscataway, NJ, USA) into 3 mm wide by 24 mm tall strips using a CO2 laser cutter (VLS3.60, Universal Laser Systems, Scottsdale, AZ, USA). Sets of four test legs were cut together and connected by a 6mm section at the top of the device, enabling rapid batch assembly and processing. Protein solutions were filtered through a centrifugal filter with a 0.2 μm nylon membrane (VWR, Radnor, PA, USA) at 8000g for 5 minutes before spotting. Head region binder was prepared at 0.62 mg/mL and anti-HRP antibody (ThermoFisher) was prepared at 0.5 mg/mL. A piezoelectric printer (sciFLEXARRAYER S3, Scienion AG, Berlin, Germany) was used to deposit capture proteins onto the nitrocellulose membranes. Each line was made by the deposition of 12 spots of the reagent, each of which were separated by 250 μm, at 30 drops per spot and 450–500 pL per drop. Each test line received approximately 300 nL of total protein solution. The spotted membranes were stored overnight in the desiccator and were tested within 7 days. Influenza A virus of the following strains was obtained from the International Reagent Resource (IRR), Solomon Islands 2006 H1N1 (FR331), Switzerland 2013 H3N2 (FR1416), Victoria 2011 H3N2 (1027). Influenza B virus for the Yamagata (FR1373, FR1370) and Victoria (FR20, FR663) strains were also obtained from the IRR.
The prepared nitrocellulose membranes described above were assembled into holding cards with cellulose wicking pads (CFSP223, Merck Millipore, Billerica, MA, USA) to maintain consistent contact across the entire width of the device, Figure S2. These comb-shaped devices were prepared the same day as the assay was run. Each assay was performed using a 96-well plate, which had been preloaded with the reagents required for each step. Assay reagents were delivered sequentially by moving each test leg from one well to the next when all 20 μL of each step was visually depleted from the well. Each step for the head region binder assay consisted of the following reagents delivered in order (1) Sample containing influenza virus or negative control (2) Wash (3) Head region binder directly conjugated to HRP diluted to 100 nM (4) Wash (5) Amplification buffer with 0.0125% H2O2 from sodium percarbonate and 144 mg/mL of 3,3’-diaminobenzidine (DAB) as the substrate for HRP. Phosphate-buffered saline (PBS) + 0.1% v/v Tween −20 (Thermo Fisher Scientific, Waltham, MA) (PBST) was used as the wash buffer in all cases. All non-wash reagents were diluted in a running buffer of PBST +1% w/v bovine serum albumin (BSA) (Sigma-Aldrich, Saint Louis, MO). The total assay before the optimization of wash steps took 58±5 minutes.
2.4. Two-dimensional paper network construction and testing
Prototype device “cards” were comprised of layered plastic with a single-side adhesive (T-5501–10/1, Melinex, Fralock, Valencia, CA, USA) used as backing and spacer layers. Glass fiber membrane (8964, Ahlstrom-Munksjö, Stockholm, Sweden) was used for the glass fiber and conjugate pad regions of the network shown in Figure 1. 5mm nitrocellulose strips were used for the assay region, and cellulose (CFSP223, Merck Millipore, Billerica, MA, USA) for the wicking pad. Nitrocellulose membranes were prepared similarly to the dipstick assay described above. Because the nitrocellulose strips were 2mm wider on the integrated device, 20 spots of reagent were deposited across the width of the strip.
Glass fiber conjugate pads were prepared using a previously published method (30). Pads were pre-blocked by soaking in 10mM PBS with 1% BSA and 0.01% Tween 20 for 30 minutes. Once blocked, pads were stored in a desiccator at 25°C to dry overnight. A master mix was made containing 300nM TriHSB.2-HRP, 0.1% BSA, 4% trehalose 0.005M FeSO4 and 0.01M EDTA. 20μL of the master mix was added to each pre-blocked conjugate pad (5mm by 12mm), and flash frozen in liquid nitrogen. The frozen pads were then placed onto a lyophilizer at room temperature 24 hours before being stored with desiccant until use.
Devices were run by the manual application of the wet assay components onto the assembled device. Volumes of 70μL, 100μL, and 80μL were applied for the sample, rehydration/wash buffer, and amplification buffer, respectively. For the assays described here, colorimetric signal was generated by the precipitation of oxidized DAB. Device runs were considered complete after all glass fiber on device no longer visibly contained liquid.
2.5. qRT-PCR
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to determine the concentration of the different influenza virus stocks used in this paper. UltraSense One-Step Quantitative RT-PCR assay mix (Life Technologies, Carlsbad, CA) was used with primer and probe sequences published previously (31,32). 20μL reactions were run on CFX96 Touch (BioRad) using the following protocol: 50°C for 15 min, 95°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 55 s. Concentrations were determined using a standard curve developed from influenza A RNA of known copy number.
2.6. Clinical nasal swab specimens
Clinical nasal swab specimens were acquired from Seattle Children’s Hospital between January and March 2016 from pediatric patients with suspected influenza infection after signed parental informed consent. Each Nylon flocked mid-nasal swab was collected in the hospital or emergency department and placed into 750μL of PBS, 0.05% Tween-20 and 0.01% sodium azide. The diluted nasal swab sample was used for RT-PCR and immunoassay detection of influenza. In order to determine the minimum dilution of nasal swab required for this device, diluted nasal swab samples were concentrating using a protein concentrator with molecular weight cut-off of 5kDa (VivaSpin). Seattle Children’s Hospital Institutional Review Board approved the sample collection and analysis of specimens. Written consent was obtained from a parent or legal guardian, as approved by the Seattle Children’s Institutional Review Board, with paper copies given to both a parent and maintained under secure storage by the research team.
2.7. Fully integrated system for hemagglutinin detection
The base of the automated device (Figure 1) was built from 1/4 inch black acrylic (8505K754, McMaster-Carr, Santa Fe Springs, CA, USA) cut using a CO2 laser into a 3.8cm by 7.4cm rectangle. An automatic, saturation-dependent fluid transfer element (the “valve”) was designed to deliver the amplification buffer after the first three assay steps were complete. To do so, water-soluble paper (Water Soluble Paper, Edmund Scientific, Tonawanda, NY, USA) was placed above the cellulose wicking pad between the wicking pad and the clear acrylic cover. This paper holds closed a pinched ~10 mm length section of silicone tubing (STHT-C-093–2; Saint-Gobain, Valley Forge, PA, USA) that prevents the flow of amplification buffer out of the cut off syringe and onto the secondary membrane. As the water-soluble paper dissolves, the pinch is released, thereby allowing fluid to flow into the glass fiber pad below. The amplification buffer then flows through the overlap junction and into the nitrocellulose test strip, Figure 2.
Figure 2.

Amplification buffer delivery using the novel paper network geometry involving two dissimilar porous materials and pinch valve. The membrane overlap between the first and second fluidic paths enables delivery of each set of fluids orthogonally. The device uses water-soluble (WS) paper surrounding a lever arm that holds a pinch valve close (A). The first three steps; sample, conjugate, and wash steps, are delivered across the primary fluidic path (B). As the wicking pad wets, the water-soluble paper that is placed in contact with it wets until the force of the lever arm overcomes the strength of the paper. This leads to the release of the lever arm and opening the pinch valve (C). Now open, the amplification buffer is delivered via the secondary fluidic path and onto the nitrocellulose test membrane (D).
The device assembly is split into two main components – preparation of the 2DPN and integration of the 2DPN into fluidic delivery system. Assembly of the 2DPN is described in Section 2.4. The 2DPN is integrated into the full device, which incorporates the pinch valve, water-soluble paper, and lever arm to enable automated delivery of the amplification buffer. The 2DPN is placed onto a thick acrylic platform and screwed into the platform. The pinch valve stopper is screwed through the 2DPN and the acrylic backing and a nut is used on the backside to ensure that the stopper does not rotate during use. The lever arm is attached to the acrylic platform via a peg at the fulcrum. A piece of water-soluble paper is wrapped around the end of the lever arm and placed overtop the cellulose wicking pad. The pinch valve is then inserted between the lever arm and the pinch valve stopper. The lever arm is held tight while a clear acrylic piece is screwed into the platform such that it clamps down onto the spy paper and the wicking pad. Finally, small ports are added to the sample and buffer inlet ports and secured with adhesive.
2.8. Signal quantification and analysis
For the assays described here, colorimetric signal is generated by the precipitation of DAB. All membranes are scanned at 48-bit HDR color, 600 dpi, gamma=1 using a desktop scanner (Perfection V700 Photo Scanner, Epson, Long Beach, CA) while the membranes were still wet. The blue channel of the RGB scanned image was used for quantification. All reported signal intensities were quantified using a previously-published custom script in MATLAB (MathWorks, Natick, MA, USA) (33). This script computes the average pixel intensity in a region of interest manually drawn around the test line. This value is then normalized based on computed average pixel intensity of a region directly downstream from the test line, resulting in a series of values between 0 and 1 which represent the minimum and the maximum respectively.
All significance testing utilized Welch’s t-test, which is also known as a Student’s t-test with unequal variances, and a significance threshold of α=0.05. The limits of detection (LODs) were calculating using the signal intensity quantified as described above, looking specifically at the 5% chance of false-positive signal and the 5% chance of false-negative signal. This was then mapped to the associated HA concentration using a four-parameter logistic curve, with the data presented alongside a 95% confidence interval for each estimate. All statistical comparisons were performed using the open-source statistical package R (64 bit, version 3.3.2) (34).
3. Results and discussion
3.1. Two-dimensional paper network
The novel two-dimensional paper network described in this work was designed to automate the steps for enzymatic amplification in a controlled manner. Enzymatic amplification has been used to improve sensitivity in lateral flow assays, however the additional steps required for the assay increase the overall complexity of the end device. Previous devices to automate enzymatic amplification have relied on delivery of each subsequent reagent over the region that had delivered the previous reagents. While these devices have been demonstrated in the literature, the large fluid travel distances can lead to reagent loss to adsorption to the paper along the device upstream of the test line (27). In addition, the fluid resistance increases with the length of the membrane through which that flow must pass. By forcing the last reagent to flow through the entire downstream paper network, the time to assay completion increases, while limiting the ability to individually tune assay steps. We thereby aimed to deliver amplification buffer across the test line orthogonally after the sample, conjugate, and wash steps were complete.
Orthogonal delivery was accomplished using an overlapping section of glass fiber membrane, which has larger pores and lower suction pressure than the nitrocellulose assay membrane. This allows exploitation of partial saturation to create two distinct, independently controllable fluidic paths, Figure 2. This fluid phenomenon has been previously demonstrated for use in processing large fluid samples, like urine (35). The device is activated by the addition of the sample, wash buffer, and amplification buffer to their respective ports by the user. The solution addition steps, specifically wash and amplification buffers, can be simply automated in a commercial product (36). The first three solutions are delivered sequentially in the traditional manner: the sample, rehydrated conjugate solution, and wash buffer respectively. When a sufficient volume of fluid has moved into the wicking pad, water-soluble paper in contact with the wicking pad dissolves and releases the arm pinching the tubing preventing amplification buffer flow. This releases the amplification buffer into the glass fiber, which subsequently flows through the detection strip. Images of this process using food coloring are shown in Figure 3.
Figure 3.

Images of the operational assay device showing the automated delivery of each reagent across the test region. Food coloring was used to represent each reagent; red for sample, clear for rehydration/wash buffer, and blue for amplification buffer. (t=0) device before the beginning of the assay (t=1) sample applied to the sample pad (t=2) conjugate rehydration by wash buffer (t=3) conjugate and wash buffer delivered across the test lines (t=4) amplification buffer applied after the water-soluble membrane releases the pinch valve, occurring at about the 29.4±3.6 minute mark and (t=5) the test line develops as the amplification buffer is applied to the tests line, which begins at the 35 minute mark, on average.
This device uses a paper-based fluid delivery system to automatically deliver the amplification buffer to the test line at the right time, after the sample, conjugate, and wash buffer steps are all complete. Once the cellulose wicking pad wets a defined amount, water soluble paper in contact with the wicking pad as shown in Figure 2 dissolves enough to lose tensile integrity, ripping due to the force applied by the pinch valve arm holding the silicone tubing closed. The pinch valve arm can then rotate, enabling the amplification buffer to pass through the silicone tubing and be delivered to the test line through 2nd glass fiber fluidic path. The location, width, and length of the water-soluble paper can be adjusted to tune the release of the amplification buffer. Optimization of these parameters are shown in Table S1. For the final dimensions of the prototype device, release of the amplification buffer occurred at 29.4± 3.6 minutes.
3.2. Assay optimization in dipstick
This assay had been previously demonstrated for influenza A virus capture using a gold nanoparticle detection system (7). Here, we replaced the gold nanoparticles with HRP covalently attached to the hemagglutinin binder, enabling enzymatic amplification of signal. For this assay, the capture and detection binders are designed to target the same epitope on the head region of hemagglutinin. Therefore, sequential delivery of the sample and detection binders is required to prevent the detection binder from blocking the capture event. The head-region binder assay with enzymatic amplification was demonstrated for successful capture and detection of the influenza virus using the flu assay shown in Figure 4. In the presence of detergents capable of lysing the virion, the signal from this assay is lost completely; this supports the hypothesis that intact influenza virus is captured on the test line, Figure S3.
Figure 4.

Illustration of the influenza hemagglutinin (HA) sandwich assay using computationally designed binders for the capture and detection of the virus. This assay uses horseradish peroxidase (HRP) and 3,3’ diaminobenzidine (DAB), which form a brown precipitate at the test line in the presence of H202. The black bar represents the nitrocellulose surface.
A panel of influenza A and B strains was tested to evaluate assay specificity. Figure S4 shows the normalized signal intensity and a representative test line for each virus strain. This assay was found to selectively detect the Solomon Islands 2006 strain of H1N1 and the Switzerland 2013 strain of H3N2. This selective detection is due to structures in the sialic acid binding pocket that are specific to these two strains. While influenza B also has the hemagglutinin protein on its surface, there was no cross reactivity seen for this assay due to differences in protein structure. It is important to note that while the Solomon Islands strain does not currently circulate in the general population, the Switzerland H3 strain continues to circulate in the general population and therefore is the most clinically relevant of the tested Influenza A strains of the virus (37). This specificity indicates that mutations in the vicinity of the sialic acid binding pocket will influence the ability of the binder to effectively bind to different strains of the virus. Conversely, due to the specificity of binding in the head region, there is potential for subtyping with the appropriate binder designs.
The quantification of influenza virus in a sample or stock solution has traditionally been characterized using either infectious units, protein amount or activity, or RNA content. In this work we report influenza virus infectivity using two different units: tissue culture infectious dose 50% endpoint (TCID50/mL) or chicken embryo infectious dose 50% endpoint (CEID50/mL). However, the magnitude of these infectivity units is dependent on the cell line the viruses were grown in and the cell line used to determine infectious titer. Clinical influenza virus loads in nasopharyngeal washes have been reported to peak at 103 to 107 TCID50/mL (38). By RNA copy number, the virus load in nasopharyngeal washes have been found to range between 105 and 107 copies/mL for H1N1 and 106 to 108 copies/mL for H3N2 (39). The correlation of different units used to characterize the influenza virus will vary depending on the virus strain and the cell line used for growing the virus.
To determine the analytical sensitivity of the assay for the strains detected by the head region binder, dilution series of the A/H1N1/Solomon Islands 2006 and A/H3N2/Switzerland 2013 influenza viruses were run. The assays for H1N1 and H3N2 were found to yield statistical LODs of 1.35 × 107 CEID50/mL and 1.11 × 107 CEID50/mL respectively, with 95% confidence intervals of [7.26 × 106,1.90 × 107] and [6.85 × 106, 1.50 × 107], Figure S5. These results are consistent with the gold nanoparticle based assay that was published previously for detection of H1N1, with a sensitivity improvement that can be attributed to enzymatic amplification (7). The assay for H1N1 was tested with a new stock of virus grown in Madin-Darby Kidney cells (MDCK) and was found to have a statistical LOD of 1.15 × 103 TCID50/mL, with a 95% confidence interval of [7.56 × 102, 2.18 × 103]. The variation in LOD between virus stocks suggested the need for further characterization of the virus samples. RNA copy number was determined for each virus stock using qRT-PCR, and the LODs by copy number were found to be statistically similar, Figure S6. With the LOD of 1.15 × 103 TCID50/mL, this assay is capable of detecting the lowest clinical range during peak infection.
Colorimetric detection of influenza virus by the hemagglutinin protein has been previously demonstrated by other groups using either sialic acid modified gold nanoparticles or polymerized liposomes containing sialic acid; each with a limit of detection around 4500 particles per μL (40,41). In both cases, sialic acid is utilized to bind HA by the highly conserved sialic acid binding pocket of the influenza hemagglutinin. While each interaction has low affinity, the overall interaction is driven by avidity effects. This is due to the presence of a high number of both sialic acid residues and hemagglutinin in close proximity to one another. While the head region binder only binds to one trimeric hemagglutinin protein at a time, it takes advantage of the trimeric form of HA to benefit from avidity effects (8). The computationally-designed binder used in this work targets the highly conserved sialic acid binding pocket while additionally interacting with portions of the surrounding head region, leading to more energetically favourable interactions that are ideally strain specific. The calculated limits of detection for this assay are lower than previously published work for the detection of hemagglutinin, which can be explained by coupling the improved affinity to the protein with enzymatic amplification techniques, Figure S3.
3.3. Assay integration into the prototype device
Each assay step required for influenza hemagglutinin detection was then integrated into the device shown in Figure 1. In the dipstick format, five steps were delivered sequentially to enable capture and enzymatic amplification to take place. Optimization of the required number of wash steps found that the first wash was not necessary for assay performance, Figure 5a. Therefore, the only wash step required was built into the device to occur after delivery of the HRP-binder conjugate. This was accomplished by using a larger buffer volume than needed for the rehydration of all HRP-binder conjugate in the lyophilized pad. The HRP-binder conjugate was stored on device using a lyophilized pad containing additives to improve long-term stability and storage. The optimization of the concentration of components in the lyophilized conjugate pad is shown in Figure S7 and S8. Timing of the pinch valve was tuned to enable delivery of the reagents for the final amplification step after all previous steps had been completed.
Figure 5.

Assay optimization in the dipstick format to prepare for device integration. Optimization of wash steps (A) demonstrated that the second wash alone was sufficient for reducing background. Dilution of nasal swab samples (B) showed signal restoration after a 10-fold dilution of influenza negative nasal swabs spiked with influenza virus. 1X represents a swab diluted into 70 μL of PBS, the volume required to cover the entire swab. As the patient sample is diluted, signal begins to be restored at the 1/10X dilution, which translates to 700 μL of PBS. For comparison, nasal swabs are typically diluted in 3mL of a solution such as universal transfer medium (UTM) (42). Data points are mean ± standard deviation, n=4.
Using a new stock of influenza virus, the limit of detection for the assay on device and in the dipstick format was determined. The limit of detection for the device was calculated to be 4.45 × 102 TCID50/mL (95% CI: [2.29 × 102, 6.26 × 102]) on device, as compared 1.15 × 103 TCID50/mL (95% CI: [7.56 × 102, 2.18 × 103]) for the dipstick format using the same virus stock, Figure 6. While the total assay time required for the dipstick averaged 54 ± 3 minutes, the device was able to deliver larger reagent volumes in 41 ± 4 minutes. One major difference between the dipstick and the device assay is the volume of sample. The volume used on device, 70 μL, was the amount required to cover the nasal swab; alternatively, the standard 20 μL was used for sample delivery in the dipstick format. Directly comparing the same volume between the two assay formats, we found comparable results in significantly less time, requiring 94 minutes to run the dipstick format with 70 μL of sample. When corrected for volume, the limit of detection for device was statistically similar as compared to the dipstick format, with a steeper slope of the concentration curve, Figure S9. Not only does this prototype device enable processing of larger volumes of nasal swab sample with comparable limit of detection, the steeper slope of the concentration curve translates to improved analytical sensitivity, as defined by the International Union of Pure and Applied Chemistry (IUPAC) (43).
Figure 6.

Limit of detection analysis for the device (blue) and dipstick (orange) assays (A) Representative scans of device test strips. (B) Normalized pixel intensity for the assay run on device and in the dipstick format. The limits of detection were found to be 4.45 × 102 TCID50/mL (95% CI: [2.29 × 102,6.26 × 102]) and 1.15 × 103 TCID50/mL (95% CI: [7.56 × 102, 2.18 × 103]) in the on device and dipstick formats respectively. The solid lines indicate the calculated LOD, while the dashed lines indicate the 95% confidence interval. Data points are mean ± standard error, n=6.
3.4. Detection of influenza virus from clinical samples
Nasal swabs are most commonly used for the detection of influenza virus from a patient presenting with influenza-like illness (44). Because true sensitivity of a diagnostic requires that known concentrations of target analyte are tested in the context of a clinical sample, it is crucial to validate the head region binder assay in a complex sample matrix. Nasal swab samples contain a wide variety of proteins and other components that might interfere with the hemagglutinin assay, including albumin, antibodies and mucins (45). Initial testing with nasal swab samples was completed with influenza negative patient samples received from Seattle Children’s hospital during the 2015–2016 flu season. This testing demonstrated two primary modes of failure for the assay; poor flow of the viscous sample within the porous network and non-target binding on the test line. To address both modes of failure, we explored dilution with a sample buffer containing surfactant (Tween 20) and 1% BSA to reduce the effect of the nasal swab sample by improving flow and reducing non-target binding respectively.
Using a serial dilution of patient sample, we found that high concentrations of the influenza-negative patient sample lead to assay failure. This was primarily due to a combination of problems with solute and solvent transport to the test line. Due to the viscosity of the nasal swab sample at high concentrations, the analyte and conjugate both were becoming trapped where they first met the nitrocellulose membrane. However, a 10-fold dilution was sufficient to regain assay performance, Figure 6b. This is the equivalent of placing the nasal swab into a tube with 700 μL of buffer. This is significantly less dilution than the current standard, which involves dilution in approximately 3 mL of universal transport medium (UTM) or other buffer (42). A ten-fold dilution of the nasal swab is significantly less than the dilution currently required for commercially available tests for the detection of the influenza nucleoprotein (14). However, the dilution of sample does lead to a reduction in the concentration of analyte delivered to the test line. Future work will involve the investigation of preparation techniques to treat nasal swabs samples that limit overall sensitivity losses.
Negative nasal swab samples spiked with the 2006 Solomon Islands strain of influenza A virus were run on the automated prototype device using the 10-fold dilution factor determined in Figure 5b. The automated delivery of these reagents led to successful detection of influenza virus spiked into a human nasal sample, Figure 7. This prototype device fully automated the delivery of a multi-step enzymatic assay, demonstrating sensitive detection of influenza virus within an average of 45 minutes. We have herein demonstrated the successful integration of an automated paper microfluidic device to detect influenza virus in a nasal swab sample.
Figure 7.

Spiked patient samples tested on device. The device was tested with influenza virus spiked into nasal swabs samples that tested negative for influenza, at 6.32 × 103 TCID50/mL for 70 μL of sample. Representative scans of the devices run (A) and normalized pixel intensity of these scans (B) are shown. Negative patient samples were run with n=3, and spiked patient sample was run with n=4.
Specific detection of hemagglutinin is useful for epidemiological tracking, to inform clinical treatment, and as a companion diagnostic. However, there is currently no commercially available test to detect this viral glycoprotein. As the hemagglutinin protein undergoes a high degree of antigenic drift due to pressure to evade the host immune system, targeting this protein enables characterization of these viral mutations in a way other protein assay, specifically the nucleoprotein, are otherwise unable. Not only does this viral glycoprotein provide information about the virus, detection of the hemagglutinin protein has the potential to simplify the design of a point of care device. By targeting the readily available viral surface protein, we limit the need to integrate complex sample preparation steps, such as solubilization and purification, into the microfluidic system.
Conclusions
In this paper, we describe the development of a novel device architecture to enhance the sensitivity of a chemically-amplified sandwich assay. Here, we use that device to integrate a binder-based influenza assay. This architecture relies on recent advances in the understanding of flow in porous materials to introduce fluid from a secondary fluidic path using a material with different pore sizes and capillary pressure. We demonstrate the development of a 2DPN that integrates enzymatic amplification in a manner that improves assay performance over the gold standard, dipstick method. The full assay was integrated into the novel 2DPN geometry to demonstrate the detection of intact influenza virus from a spiked patient sample. We believe this to be the first demonstration of an integrated prototype device for the detection of hemagglutinin from intact influenza virus.
Lateral flow assays are often restricted in their ability to integrate more complex chemistries within their simple one-dimensional architecture. While the traditional 2DPN addresses some of these concerns, there are limitations related to control over timing and interaction of reagents before reaching the test region. The device architecture demonstrated here reduces the potential for unwanted interaction between the three reactive species, HRP, H2O2 and DAB, upstream of the test region. In addition, the device takes into account the different timing constraints for each step, allowing sufficient time for analyte and conjugate binding while then delivering the amplification buffer orthogonally with more control over reagent concentrations and flow rates. By taking into consideration the needs for each step of this complex assay, the integrated device outperforms the conventional dipstick assay without requiring user input or timing. This work demonstrates the possibility for fully integrated, sensitive enzymatic assays that can be used for detection at the point of care.
The proof of concept, microfluidic device shown in this manuscript proved successful for the detection of influenza virus from spiked nasal swab samples. Future work with this device will focus on adapting the design for longer-term storage stability studies and testing with suspected influenza positive nasal swab samples. In addition, new iterations of this paper-based microfluidic device can be adapted to other multistep analytical assays that are currently limited in their use at the point of care. By enabling the integration of these more complex assays into automated, paper-based device, we believe it will open the door to more sensitive and specific diagnostics that can be easily implemented at the point of care.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Jesse Bloom from the Fred Hutchinson Cancer Research Center for helping supply influenza virus and are grateful for helpful discussions with colleagues in the Yager and Baker groups. Specifically, we would like to thank Drs. Joshua Bishop and Lisa Lafleur for their conversations surrounding device design. This material is based upon work supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI096184, and by the University of Washington Department of Bioengineering. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest
The authors have no conflict of interest to declare.
Notes and references
- 1.CDC. Seasonal flu death estimate increases worldwide | CDC Online Newsroom | CDC [Internet]. 2017. [cited 2018 Feb 20]. Available from: https://www.cdc.gov/media/releases/2017/p1213-flu-death-estimate.html
- 2.WHO | Influenza (Seasonal). WHO [Internet]. World Health Organization; 2018. [cited 2018 Feb 20]; Available from: http://www.who.int/mediacentre/factsheets/fs211/en/ [Google Scholar]
- 3.Fields BN, Knipe DM, David M, Howley PM. Fields virology [Internet]. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2007. [cited 2017 Sep 27]. 86 p. Available from: https://books.google.ca/books/about/Fields_Virology.html?id=5O0somr0w18C&redir_esc=y [Google Scholar]
- 4.Eisfeld AJ, Neumann G, Kawaoka Y. Influenza A virus isolation, culture and identification. Nat Protoc [Internet]. NATURE PUBLISHING GROUP, MACMILLAN BUILDING, 4 CRINAN ST, LONDON N1 9XW, ENGLAND; 2014. November [cited 2016 Mar 3];9(11):2663–81. Available from: http://apps.webofknowledge.com/full_record.do?product=WOS&search_mode=CitingArticles&qid=3&SID=4FPs72c1BACL4UC1ZuU&page=2&doc=12&cacheurlFromRightClick=no [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonner AB, Monroe KW, Talley LI, Klasner AE, Kimberlin DW. Impact of the rapid diagnosis of influenza on physician decision-making and patient management in the pediatric emergency department: results of a randomized, prospective, controlled trial. Pediatrics [Internet]. 2003. August [cited 2017 Sep 27];112(2):363–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12897288 [DOI] [PubMed] [Google Scholar]
- 6.Skehel John J, Wiley Don. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu Rev Biochem. 2000;69. [DOI] [PubMed] [Google Scholar]
- 7.Anderson CE, Holstein CA, Strauch M, Bennett S, Chevalier A, Nelson J, et al. Rapid Diagnostic Assay for Intact Influenza Virus Using a High Affinity Hemagglutinin Binding Protein. Anal Chem [Internet]. American Chemical Society; 2017. June 20 [cited 2017 Oct 30];89(12):6608–15. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.analchem.7b00769 [DOI] [PubMed] [Google Scholar]
- 8.Strauch EM, Bernard SM, La D, Bohn AJ, Lee PS, Anderson CE, et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat Biotechnol [Internet]. 2017. [cited 2017 Sep 27];35(7):667–71. Available from: http://research.fhcrc.org/content/dam/stripe/bloom/labfiles/publications/Strauch2017.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleishman SJ, Whitehead T a, Ekiert DC, Dreyfus C, Corn JE, Strauch E-M, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science [Internet]. 2011. May 13 [cited 2014 Sep 29];332(6031):816–21. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3164876&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol [Internet]. 2012. May 27 [cited 2017 Sep 27];30(6):543–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22634563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hui DS, Lee N, Chan PKS. Clinical management of pandemic 2009 influenza A(H1N1) infection. Chest [Internet]. Elsevier; 2010. April 1 [cited 2018 Feb 11];137(4):916–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20022969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez AW, Phillips ST, Whitesides GM, Carrilho E. Diagnostics for the Developing World: Microfluidic Paper-Based Analytical Devices. Anal Chem [Internet]. 2010. January [cited 2016 Jul 4];82(1):3–10. Available from: http://pubs.acs.org/doi/abs/10.1021/ac9013989 [DOI] [PubMed] [Google Scholar]
- 13.CDC. Rapid Diagnostic Testing for Influenza: Information for Clinical Laboratory Directors | Seasonal Influenza (Flu) | CDC [Internet]. 2018. [cited 2018 Feb 20]. Available from: https://www.cdc.gov/flu/professionals/diagnosis/rapidlab.htm
- 14.CDC. Evaluation of 11 Commercially Available Rapid Influenza Diagnostic Tests - United States. Morb Mortal Wkly Rep. 2012;61:873–6. [PubMed] [Google Scholar]
- 15.van Doorn HR, van Kinh N, Tuan HM, Tuan TA, Minh NNQ, Bryant JE, et al. Clinical validation of a point-of-care multiplexed in vitro immunoassay using monoclonal antibodies (the MSD influenza test) in four hospitals in Vietnam. J Clin Microbiol [Internet]. American Society for Microbiology; 2012. May 1 [cited 2018 Feb 20];50(5):1621–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22357497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutz BR, Trinh P, Ball C, Fu E, Yager P. Two-dimensional paper networks: programmable fluidic disconnects for multi-step processes in shaped paper. Lab Chip [Internet]. The Royal Society of Chemistry; 2011. November 23 [cited 2017 Oct 27];11(24):4274 Available from: http://xlink.rsc.org/?DOI=c1lc20758j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Wang S, Wang L, Li F, Pingguan-Murphy B, Lu TJ, et al. Advances in paper-based point-of-care diagnostics. Biosens Bioelectron [Internet]. Elsevier; 2014. April 15 [cited 2018 Feb 20];54:585–97. Available from: https://www.sciencedirect.com/science/article/pii/S095656631300777X [DOI] [PubMed] [Google Scholar]
- 18.Zhao W, Ali MM, Aguirre SD, Brook MA, Li Y. Paper-Based Bioassays Using Gold Nanoparticle Colorimetric Probes. Anal Chem [Internet]. American Chemical Society; 2008. November 15 [cited 2018 Feb 20];80(22):8431–7. Available from: http://pubs.acs.org/doi/abs/10.1021/ac801008q [DOI] [PubMed] [Google Scholar]
- 19.Fridley GE, Holstein C a., Oza SB, Yager P. The evolution of nitrocellulose as a material for bioassays. MRS Bull [Internet]. 2013. April 12 [cited 2014 Sep 25];38(04):326–30. Available from: http://www.journals.cambridge.org/abstract_S0883769413000602 [Google Scholar]
- 20.Nayak S, Blumenfeld NR, Laksanasopin T, Sia SK. Point-of-Care Diagnostics: Recent Developments in a Connected Age. Anal Chem [Internet]. American Chemical Society; 2017. January 3 [cited 2017 Sep 6];89(1):102–23. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.analchem.6b04630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez AW, Phillips ST, Nie Z, Cheng C-M, Carrilho E, Wiley BJ, et al. Programmable diagnostic devices made from paper and tape. Lab Chip [Internet]. 2010. October 7 [cited 2018 Feb 20];10(19):2499 Available from: http://xlink.rsc.org/?DOI=c0lc00021c [DOI] [PubMed] [Google Scholar]
- 22.Jahanshahi-Anbuhi S, Kannan B, Pennings K, Monsur Ali M, Leung V, Giang K, et al. Automating multi-step paper-based assays using integrated layering of reagents. Lab Chip [Internet]. The Royal Society of Chemistry; 2017. February 28 [cited 2017 Oct 24];17(5):943–50. Available from: http://xlink.rsc.org/?DOI=C6LC01485B [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Bai J, Ying JY. A stacking flow immunoassay for the detection of dengue-specific immunoglobulins in salivary fluid. Lab Chip [Internet]. 2015. March 21 [cited 2018 Feb 20];15(6):1465–71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25608951 [DOI] [PubMed] [Google Scholar]
- 24.Fu E, Liang T, Houghtaling J, Ramachandran S, Ramsey SA, Lutz B, et al. Enhanced Sensitivity of Lateral Flow Tests Using a Two-Dimensional Paper Network Format. Anal Chem [Internet]. American Chemical Society; 2011. October 15 [cited 2017 Nov 20];83(20):7941–6. Available from: http://pubs.acs.org/doi/abs/10.1021/ac201950g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu E, Kauffman P, Lutz B, Yager P. Chemical signal amplification in two-dimensional paper networks. Sens Actuators B Chem [Internet]. NIH Public Access; 2010. August 6 [cited 2017 Sep 27];149(1):325–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20706615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant BD, Smith CA, Karvonen K, Richards-Kortum R. Highly Sensitive Two-Dimensional Paper Network Incorporating Biotin–Streptavidin for the Detection of Malaria. Anal Chem [Internet]. American Chemical Society; 2016. March [cited 2017 Oct 23];88(5):2553–7. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.analchem.5b03999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang S, Abe K, Bennett S, Liang T, Ladd PD, Yokobe L, et al. Disposable Autonomous Device for Swab-to-Result Diagnosis of Influenza. Anal Chem [Internet]. 2017. June 6 [cited 2017 Sep 20];89(11):5776–83. Available from: http://pubs.acs.org/doi/abs/10.1021/acs.analchem.6b04801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buser JR. Heat, Fluid, and Sample Control in Point-of-Care Diagnostics [Internet]. 2016. [cited 2017 Nov 7]. Available from: https://digital.lib.washington.edu/researchworks/handle/1773/38064
- 29.Anderson CE, Shah KG, Yager P. Sensitive Protein Detection and Quantification in Paper-Based Microfluidics for the Point of Care. Methods Enzymol [Internet]. Academic Press; 2017. January 1 [cited 2017 Sep 21];589:383–411. Available from: http://www.sciencedirect.com/science/article/pii/S0076687917300447 [DOI] [PubMed] [Google Scholar]
- 30.Ramachandran S, Fu E, Lutz B, Yager P. Long-term dry storage of an enzyme-based reagent system for ELISA in point-of-care devices. Analyst [Internet]. The Royal Society of Chemistry; 2014. March 21 [cited 2016 Feb 20];139(6):1456–62. Available from: http://pubs.rsc.org/en/content/articlehtml/2014/an/c3an02296j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuypers J, Wright N, Ferrenberg J, Huang M-L, Cent A, Corey L, et al. Comparison of Real-Time PCR Assays with Fluorescent-Antibody Assays for Diagnosis of Respiratory Virus Infections in Children. J Clin Microbiol [Internet]. 2006. July 1 [cited 2018 Jun 6];44(7):2382–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16825353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casado B, Pannell LK, Iadarola P, Baraniuk JN. Identification of human nasal mucous proteins using proteomics. Proteomics [Internet]. Wiley-Blackwell; 2005. July [cited 2018 Jun 6];5(11):2949–59. Available from: http://doi.wiley.com/10.1002/pmic.200401172 [DOI] [PubMed] [Google Scholar]
- 33.Holstein CA, Griffin M, Hong J, Sampson PD. Statistical Method for Determining and Comparing Limits of Detection of Bioassays. Anal Chem [Internet]. American Chemical Society; 2015. October 6 [cited 2018 Feb 20];87(19):9795–801. Available from: http://pubs.acs.org/doi/10.1021/acs.analchem.5b02082 [DOI] [PubMed] [Google Scholar]
- 34.R Development Core Team. R: A language and environment for statistical computing. R Founation for Statistical Computing, Vienna, Austria: ISBN 3–900051-07–0, URL R-project.org. [Internet]. Foundation for Stastical Computing. 2010. [cited 2017 Nov 12]. Available from: https://www.r-project.org/ [Google Scholar]
- 35.Byrnes SA, Buser JR, Kline EC, Bishop JD, Wheeler MD, Yager P. An integrated sample preparation system for large volume processing at the point of care. In: MicroTAS Dublin, Ireland; 2016. [Google Scholar]
- 36.Lafleur LK, Bishop JD, Heiniger EK, Gallagher RP, Wheeler MD, Kauffman P, et al. A rapid, instrument-free, sample-to-result nucleic acid amplification test. Lab Chip [Internet]. 2016. [cited 2017 Sep 20];16(19):3777–87. Available from: http://xlink.rsc.org/?DOI=C6LC00677A [DOI] [PubMed] [Google Scholar]
- 37.CDC. Weekly U.S. Influenza Surveillance Report | Seasonal Influenza (Flu) | CDC [Internet]. 2017. [cited 2017 Dec 6]. Available from: https://www.cdc.gov/flu/weekly/index.htm
- 38.Fields BN, Knipe DM (David M, Howley PM. Fields virology. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013. 82 p. [Google Scholar]
- 39.Wang J, Tai W, Angione SL, John AR, Opal SM, Artenstein AW, et al. Subtyping clinical specimens of influenza A virus by use of a simple method to amplify RNA targets. J Clin Microbiol [Internet]. American Society for Microbiology; 2013. October 1 [cited 2018 Feb 20];51(10):3324–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23903546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C, Gaston MA, Weiss AA, Zhang P. Colorimetric viral detection based on sialic acid stabilized goldnanoparticles. Biosens Bioelectron. 2013;42:236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charych DH, Nagy JO, Spevak W, Bednarski MD. Direct colorimetric detection of a receptor-ligand interaction by a polymerized bilayer assembly. Science [Internet]. 1993. July 30 [cited 2017 Dec 5];261(5121):585–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8342021 [DOI] [PubMed] [Google Scholar]
- 42.Hassan F, Nguyen A, Formanek A, Bell JJ, Selvarangan R. Comparison of the BD Veritor System for Flu A+B with the Alere BinaxNOW influenza A&B card for detection of influenza A and B viruses in respiratory specimens from pediatric patients. J Clin Microbiol [Internet]. American Society for Microbiology; 2014. March 1 [cited 2017 Nov 20];52(3):906–10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24391198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borysiak MD, Thompson MJ, Posner JD. Translating diagnostic assays from the laboratory to the clinic: analytical and clinical metrics for device development and evaluation. Lab Chip [Internet]. 2016. April 21 [cited 2017 Nov 12];16(8):1293–313. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27043204 [DOI] [PubMed] [Google Scholar]
- 44.Robinson JL, Lee BE, Kothapalli S, Craig WR, Fox JD. Use of Throat Swab or Saliva Specimens for Detection of Respiratory Viruses in Children. Clin Infect Dis [Internet]. Oxford University Press; 2008. April 1 [cited 2018 Feb 20];46(7):e61–4. Available from: https://academic.oup.com/cid/article-lookup/doi/10.1086/529386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomazic PV, Birner-Gruenberger R, Leitner A, Obrist B, Spoerk S, Lang-Loidolt D. Nasal mucus proteomic changes reflect altered immune responses and epithelial permeability in patients with allergic rhinitis. J Allergy Clin Immunol [Internet]. 2014. March [cited 2018 Feb 20];133(3):741–50. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0091674913014899 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.