

Abstract
Altered metabolism is a common feature of new and recurring malignancy. In this issue of Cancer Cell, Reina-Campos and colleagues report upregulation of the serine, glycine, one-carbon (SGOC) metabolic network is required for neuroendocrine prostate cancer, a castration-resistant aggressive form of the disease, and presents a targetable vulnerability.
Acquired and intrinsic resistance to targeted therapies including androgen deprivation in prostate cancer is often lethal. Thus, understanding resistance mechanisms or non-oncogenic vulnerabilities such as dysregulated cellular metabolism is important for the development of new therapeutic strategies. Indeed, targeting metabolism in cancer is a promising approach (Luengo et al., 2017). However, establishing a therapeutic window requires the identification of specific pathways and contexts where it may be effective. In this issue of Cancer Cell, Reina-Campos and colleagues define a mechanism whereby loss of protein kinase C (PKC) λ/ι and subsequent upregulation of the serine, glycine, one-carbon (SGOC) metabolic network in early-stage prostate cancer leads to the development of neuroendocrine prostate cancer (NEPC), an aggressive, particularly lethal form that lacks effective therapeutic options (Figure 1). Of significance, targeting the SGOC metabolic network is effective in reducing NEPC formation in a murine prostate cancer tumor model, providing proof-of-concept that SGOC metabolism is a viable target in aggressive prostate cancer (Reina-Campos et al., 2019).
Figure 1. Schematic of how SGOC metabolism creates a vulnerability in NEPC.
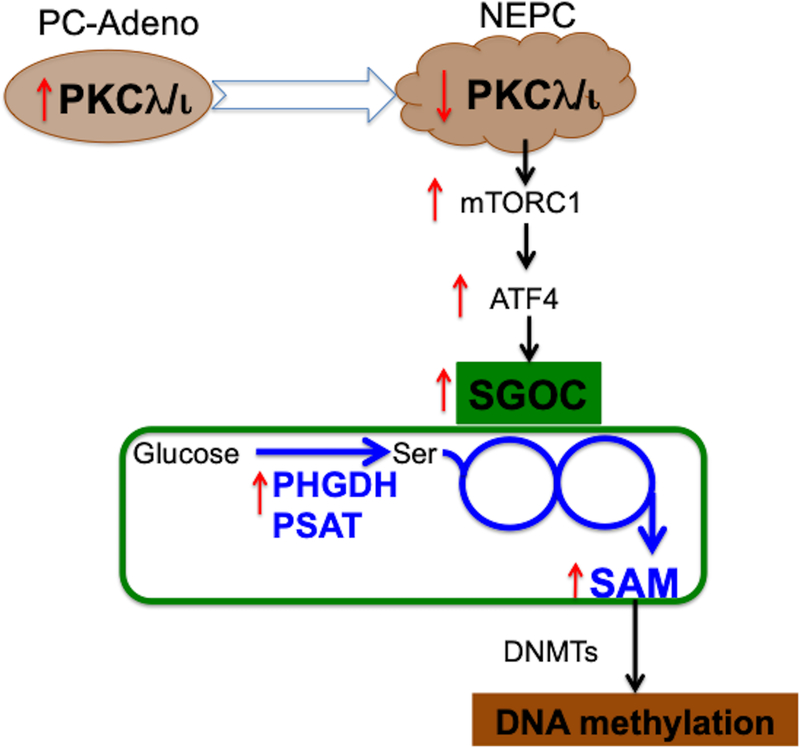
Downregulation of protein kinase C (PKC)λ/ι in neuroendocrine prostate cancer (NEPC) leads to rewiring of the serine, glycine, one-carbon (SGOC) metabolic network through an mTORC1/ATF4-dependent pathway. The upregulation of de novo serine biosynthesis results in increased intracellular S-Adenosylmethionine (SAM) levels that alters DNA methylation status and supports the development of NEPC. PC-Adeno (Prostate adenocarcinoma); DNMTs (DNA methyltransferases); PHGDH (phosphoglycerate dehydrogenase); PSAT1 (phosphoserine aminotransferase 1); Ser (serine).
Using bioinformatics, the authors examined the expression of the PKCλ/ι-coding gene, PRKCI, in human prostate tumors. Intriguingly, PRKCI gene expression was lower in patients with NEPC, an aggressive form of late-stage prostate cancer, compared to earlier-stage prostate adenocarcinomas. This led the authors to propose that prostate cancer progression and NEPC development can occur in response to PKCλ/ι deficiency. Consistent with this hypothesis, knockout of Prkci in a mouse prostate cancer model and PRKCI ablation in human prostate cancer cell lines recapitulated histological, gene expression, and disease outcome profiles found in NEPC patient tumors. These results provided compelling evidence that that loss of PKCλ/ι in prostate cancer leads to a more aggressive form of the disease.
The authors then investigated how PKCλ/ι loss may lead to NEPC formation. Using transcriptomic, proteomic, and biochemical approaches, they identified a sequence of events whereby PKCλ/ι deficiency leads to ATF4-mediated transcriptional reprogramming with mTORC1 activation as an intermediate. In addition to upregulating genes required for NEPC differentiation, ATF4 also promoted the expression of numerous genes in the SGOC network including PHGDH, PSAT1, and MTHFD2, suggesting this metabolic pathway may play a causal role in NEPC development. Importantly, stable isotope tracing and mass spectrometry confirmed an increase of flux through the SGOC pathway when PKCλ/ι was deleted.
Having established that PKCλ/ι loss leads to NEPC formation via an mTORC1/ATF4/SGOC axis, the authors aimed to understand how upregulation of SGOC metabolism could possibly drive NEPC development. Because the SGOC network can sometimes be coupled to the methionine cycle, which is important for generating the methyl donor S-Adenosylmethionine (SAM) used by histone and DNA methyltransferase enzymes, the authors hypothesized that changes in chromatin biology directly contributed to NEPC formation. Indeed, genome-wide increases in DNA methylation levels were observed in PKCλ/ι-deficient cells compared to control, which were correlated with NEPC transcriptional changes and could be rescued by knockdown of the enzyme PHGDH which is the first committed step in serine synthesis. Of particular interest, the differentially methylated regions in PKCλ/ι-null cells significantly overlap with CpG areas found hypermethylated in NEPC patients and highly aggressive prostate cancers. Finally, the authors demonstrate targeting DNA methyltransferase activity or the SGOC pathway was sufficient to reduce NEPC formation.
In all, the study by Reina-Campos et al. is a compelling example of how alterations in cellular metabolism are relevant to cancer pathology. It is well established that cancer cells have different nutritional requirements due to enhanced metabolic demands and that alterations of metabolic networks can lead to growth and fitness advantages. As demonstrated by this study, rewiring of metabolism by cancer cells can be used to circumvent targeted therapies.
Reina-Campos and colleagues describe a mechanism whereby nutritional status drives disease by regulating chromatin biology. In this manuscript, the authors reported the cellular availability of the methionine-derived metabolite SAM as one of the major causative factors of aggressive prostate cancer development. The levels of SAM, a methyl-donor and substrate for methyltransferase enzymes, were elevated in response to upregulation of the SGOC metabolic network and promoted hypermethylation of DNA, which ultimately led to transcriptional programming that favored NEPC formation. These findings extend the cancer contexts in which this mechanism has been attributed to the pathogenesis of cancer. Previous reports have identified loss of LKB1 in a type of pancreatic cancer (Kottakis et al., 2016) and the evolution of chemoresistance in breast cancer (Deblois, 2018) leading to a similar mechanism.
These studies also raise the intriguing possibility that dietary interventions could have therapeutic consequences. For example, histone methylation is dynamically regulated by SAM levels, which can be controlled by the manipulation of dietary methionine intake (Mentch et al., 2015). Moreover, inhibition of histone methylation has been shown to reduce prostate cancer development and increase therapeutic response (Ku et al., 2017). Thus, it would be interesting to explore how dietary methionine availability could influence the formation and progression of this cancer. Furthermore, it is tempting to speculate that change to dietary amino acid intake such as serine, glycine or methionine could influence prostate cancer outcome - possibly through these proposed mechanisms. Indeed, dietary depletion of serine and glycine produces therapeutic responses in aggressive pre-clinical cancer models (Maddocks et al., 2017).
Finally, this work opens additional research directions for the role of the SGOC metabolic network in the progression of prostate and other cancers. Besides contributing to methylation reactions, the SGOC network is important for redox balance and the biosynthesis of nucleotides, proteins, and lipids (Mehrmohamadi et al., 2014), all of which are key components required for cell viability and proliferation and may be co-opted by aggressive cancers. Moreover, the SGOC enzyme PHGDH has been shown to be coupled to the anabolic fluxes of the TCA cycle and pentose phosphate pathway in certain contexts (Reid et al., 2018). Whether this occurs in response to PHGDH upregulation during NEPC development is intriguing and may lead to further therapeutic considerations. Furthermore, the closely related PKCλ/ι family member, PKCζ, was previously shown to negatively regulate the SGOC enzyme PHGDH and exhibit control over glutamine metabolism (Ma et al., 2013). Accordingly, it would be important to explore other metabolic changes that may occur during progression of cancer or in response to drug resistance. Finally, while gene expression analyses which account for overall network expression can sometimes be sufficient to characterize the activity of metabolic pathways (Mehrmohamadi et al., 2014), other approaches such as metabolomics and metabolic flux analysis can quantitatively measure metabolic phenotypes, and warrant future consideration in contexts where metabolic vulnerabilities may be identifiable.
ACKNOWLEDGEMENTS
Support from the Canadian Institutes of Health Research (146818 to X.G.), the National Institutes of Health (R01CA193256 to J.W.L.), and the American Cancer Society (131615-PF-17–210-01-TBE to M.A.R.) are gratefully acknowledged.
REFERENCES
- Deblois G, Tonekaboni SAM, Kao Y, Tai F, Liu X, Ettayebi I, Grillo G, Guilhamon P, Ba-alawi W, Fedor A, et al. (2018). Metabolic adaptations underlie epigenetic vulnerabilities in chemoresistant breast cancer. bioRxiv (Preprint). [Google Scholar]
- Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T , et al. (2016). LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J , et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Gui DY, and Vander Heiden MG (2017). Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. (2013). Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell 152, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- Mehrmohamadi M, Liu X, Shestov AA, and Locasale JW (2014). Characterization of the usage of the serine metabolic network in human cancer. Cell Rep 9, 1507–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE , et al. (2015). Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab 22, 861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Allen AE, Liu S, Liberti MV, Liu P, Liu X, Dai Z, Gao X, Wang Q, Liu Y, et al. (2018). Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun 9, 5442. [DOI] [PMC free article] [PubMed] [Google Scholar]