

Abstract
Trifluoromethoxy (OCF3) group exhibits unique properties, which render it a highly desirable structural motif in life and materials sciences. The numbers of newly synthesized trifluoromethyl ethers are booming as new synthetic methods are constantly being developed to access these valuable compounds. This Review summarizes recent catalytic approaches towards preparation of trifluoromethyl ethers. Alkene, allylic, benzylic, and aryl trifluoromethoxylation reactions are discussed herein.
Keywords: Trifluoromethoxylation, Transition metal catalysis, Photoredox catalysis, Fluorine, Radical
Incorporation of fluorinated functional groups to fine-tune physicochemical properties of drug candidates is an important strategy in drug discovery and development process. Among the fluorine-containing substituents, the trifluoromethoxy (OCF3) group has recently become an increasingly common structural motif of pharmaceutical agents [1–3] because its introduction into organic molecules exerts profound effects on their stability, lipophilicity, and membrane permeability [4]. Notably, the OCF3containing aromatic compounds, due to hyperconjugative interaction and the steric bulk of the CF3 group, usually adopt a conformation with the O–CF3 bond in the plane orthogonal to the arene ring (Scheme 1a) [5], which enriches molecular three-dimensional complexity and provides additional binding affinity to active sites of a target [4]. Owing to its unique properties, trifluoromethyl ethers have found numerous applications as pharmaceuticals [6–9], agrochemicals [10–14], and functional materials [15,16]. Selected examples of medicinally and agrochemically important OCF3-bearing compounds include Riluzole® (anticonvulsant), Odomzo® (treatment of basal skin carcinoma), Triflumuron® (insecticide), and Thifluzamide® (fungicide) (Scheme 1b).
Scheme 1.

(a) Structural properties of OCF3-bearing arenes. (b) Selected examples of OCF3-containing pharmaceuticals and agrochemicals. (c) Summary of recent developments in the field of catalytic trifluoromethoxylation.
Despite their favorable characteristics, easy, direct access to OCF3-containing compounds remained elusive for many decades. On the contrary, their preparation usually required laborious functional group interconversions, substrate pre-functionalization, toxic reagents, and harsh reactions conditions [17–19]. In addition, many of the conventional approaches also exhibited limited substrate scope and functional group tolerance. Development of trifluoromethoxylation reactions have been hindered by the low reactivity and intrinsic instability of trifluoromethoxide anion, −OCF3, which readily decomposes to fluoride anion, F−, and fluorophosgene [17,20–22]. The scarcity of reports for transferring the OCF3 group from metal centers into organic compounds arise from the propensity of transition-metal-OCF3 complexes towards β-fluoride elimination [23]. Over the last few years, however, several innovative and promising approaches for the synthesis of these prominent compounds have been reported. The purpose of this Review is to provide an overview of the recent developments of catalytic approaches towards preparation of trifluoromethyl ethers. Alkene, allylic, benzylic, and aryl trifluromethoxylation reactions are discussed herein (Scheme 1c).
Direct introduction of the OCF3 group into olefins and (hetero) aromatic systems has long been recognized as a formidable challenge in organic synthesis, and it was not until very recently that such transformations were successfully achieved. The first catalytic trifluoromethoxylation reaction of unactivated alkenes under mild conditions was developed by Liu and co-workers in 2015. They reported a palladium-catalyzed regioselective synthesis of 3-trifluoromethoxylated piperidines from N-tosylate alkenylamines (Scheme 2) [24]. This intramolecular aminotrifluoromethoxylation process was enabled by the use of Pd(MeCN)2Cl2 as a catalyst, AgOCF3 as a trifluoromethoxide source, and SelectFluor- as an oxidant. The authors observed that the reaction worked best at low temperature (20 C), at which the extent of the competing fluoroformylation reaction of sulfonamide was significantly decreased. The aminotrifluoromethoxylation procedure was applicable to the synthesis of a variety of OCF3-substituted piperidines, which were obtained in 37–82% yields. Gem-disubstituted N-tosylate alkenylamines as well as substrates with a substituent on allylic position or on the carbon adjacent to nitrogen were all compatible with the reaction conditions. The aminotrifluoromethoxylation also enabled the synthesis of various spiro- and bicyclic piperidine derivatives.
Scheme 2.

Palladium-catalyzed intramolecular aminotrifluoromethoxylation of alkenes. [24]a Ratio of trans and cis isomers. bRatio of 3,5-cis and 3,5-trans isomers.
Based on a deuterium-labelling experiment, in which 3.1 afforded 3.4 as a single isomer, the authors postulated that the reaction proceeds via a reversible trans-aminopalladation, which generates a secondary alkyl-PdII intermediate 3.2 (Scheme 3a). Oxidation of 3.2 by SelectFluor- and transmetallation with AgOCF3 leads to the formation of a high-valent alkyl-PdIV(OCF3) complex 3.3, which undergoes C(sp3)–OCF3 reductive elimination to yield 3.4. The evidence that supports facile reductive elimination from PdIV–OCF3 complex to form sp3 C–OCF3 bond came from the studies of reactivity of palladium complex 3.5 (Scheme 3b). Upon treatment with SelectFluor- at 0˚C, 3.5 underwent full conversion to PdIVF complex 3.6. After addition of AgOCF3, the NMR signals of 3.6 disappeared and a quantitative formation of a new species was observed by 19F and 1 H NMR. The new signals were attributed to the PdIV(OCF3) complex 3.7. When a solution of 3.7 in MeCN was warmed to room temperature, C(sp3)–OCF3 reductive elimination took place, and Pd(II) complex 3.9 was observed (43% yield). In addition to 3.9, the reaction also afforded a small amount of 3.8, which was the product of β-fluoride elimination/C(sp3)–F reductive elimination sequence. The structure of 3.9 was further confirmed by (i) isolation and characterization of its derivatives, complexes 3.10 and 3.11, (ii) formation of a hydrogenation product 3.12 upon treatment of 3.9 with HCO2Na. In summary, Liu’s work is the first example of a catalytic trifluoromethoxylation reaction, in which the successful transfer of the trifluoromethoxy group from the metal into the organic substrate is enabled by the use of a highvalent palladium center. In this instance, reductive elimination to form C–OCF3 bond is favored over β-fluoride elimination due to the lack of vacant coordination sites at Pd(IV)
Scheme 3.
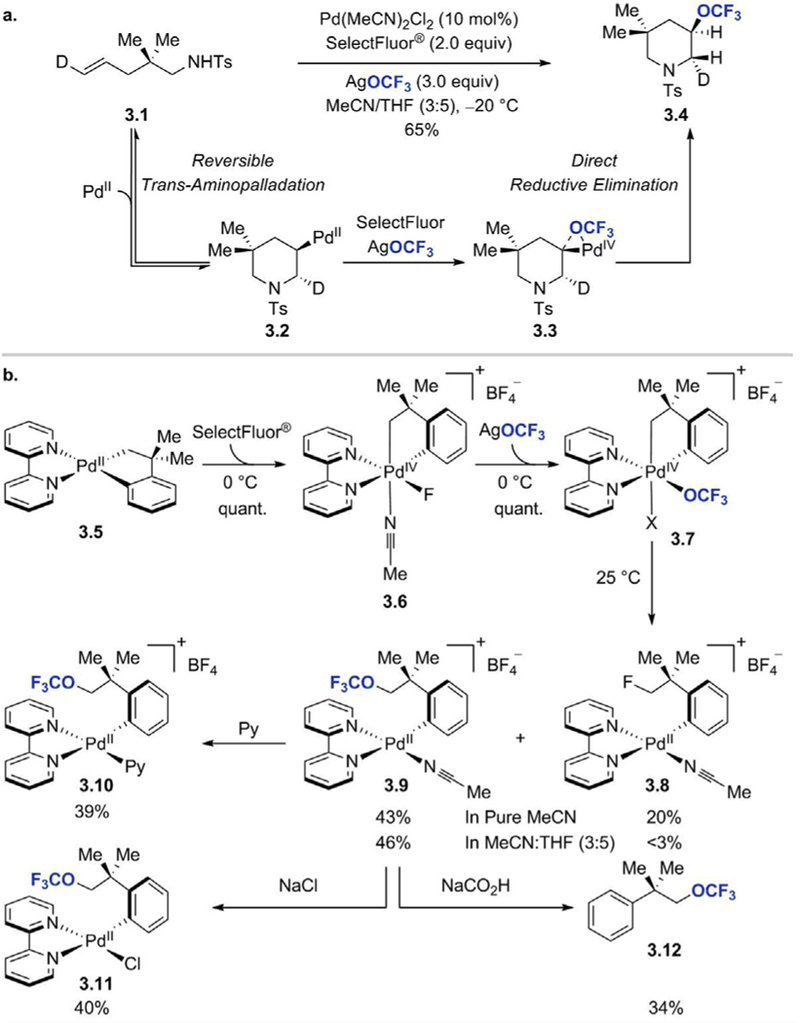
(a) Deuterium-labelling study/proposed mechanism for palladiumcatalyzed aminotrifluoromethoxylation of alkenes. (b) Trifluoromethoxylation reaction of palladium complex 3.5 as evidence supporting the proposed C–OCF3 reductive elimination from Pd(IV) [24]. Adapted with permission from [24]. Copyright 2015 American Chemical Society
Following the development of the first catalytic oxidative trifluoromethoxylation reaction of alkenes, Liu’s group devised a new method to convert allyl (hetero)arenes into allylic trifluoromethyl ethers (Scheme 4) [25]. The oxidative trifluoromethoxylation of allylic C–H bonds was achieved using PdCl2(PhCN)2 as a catalyst, 1,4-benzoquinone (BQ) as an oxidant, and 2,4,6-trimethylbenzoic acid as an additive. CsOCF3 and AgBF4 salts were employed to generate AgOCF3 in situ, whereas extraneous AgF was added to impede the decomposition of AgOCF3 to silver fluoride and fluorophosgene. Under these conditions, a variety of allyl (hetero)arenes decorated with either electron-donating or electronwithdrawing substituents on the arene ring afforded allylic trifluoromethyl ethers in moderate to good yields. The reaction proceeded at room temperature and tolerated a range of common functionalities including halides, ketone, ester, nitrile, ether, alkyl, and the CF3 group. In addition to allyl benzene derivatives, allyl heteroarenes such as indole, coumarin, pyridine, and benzothiophene were also suitable substrates for the C–H trifluoromethoxylation reaction. Substrates with more complex molecular architectures, for instance, steroid scaffold with a free hydroxyl group, or an estrone derivative, also afforded the trifluoromethoxylated products in good yields. The allylic C–H trifluoromethoxylation reaction was found to be limited to allyl (hetero)arenes; terminal alkenes and cyclic aliphatic alkenes which lacked the acidic C–H benzylic proton were not converted to their trifluoromethoxylated derivatives under the reaction conditions.
Scheme 4.
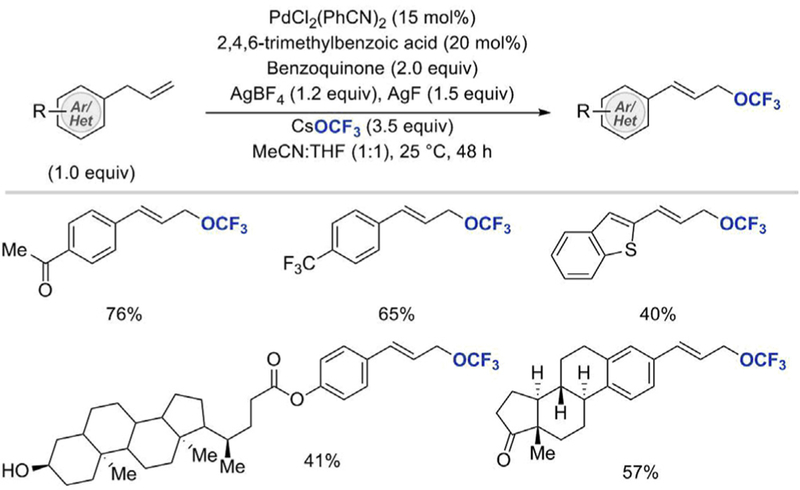
Selected examples of the allylic C–H trifluoromethoxylation [25]
The proposed mechanism for the allylic trifluoromethoxylation is depicted in Scheme 5. The reaction begins with the coordination of the substrate to Pd(II) catalyst and is followed by a turn-over limiting allylic C(sp3)–H bond activation. The rate of formation of the π-allyl palladium complex 5.2 is accelerated by the addition of the catalytic amount of 2,4,6-trimethylbenzoic acid, which is involved in the deprotonation/metalation step. A nucleophilic attack by trifluoromethoxide on the π-allyl Pd(II) intermediate 5.3 affords the desired trifluoromethoxylation product. The released Pd(0) catalyst is reoxidized to Pd(II) by BQ, which, apart from being an oxidant, also acts as a π-acceptor ligand activating the π-allyl intermediate towards OCF3 attack. AgOCF3, the active trifluoromethoxide reagent, is formed in situ from a poorly soluble and stable CsOCF3. The slow release of AgOCF3 ensures its low concentration throughout the reaction and slows down its decomposition. Since the trifluoromethoxylation reaction of 5.5 proceeded with stereoinversion to afford trans-5.6 as the major product (trans/cis = 4:1), Liu and co-workers postulated that the C–OCF3 bond-forming step occurs through an outer-sphere nucleophilic substitution pathway by AgOCF3, rather than C–O reductive elimination from a Pd(II) center.
Scheme 5.
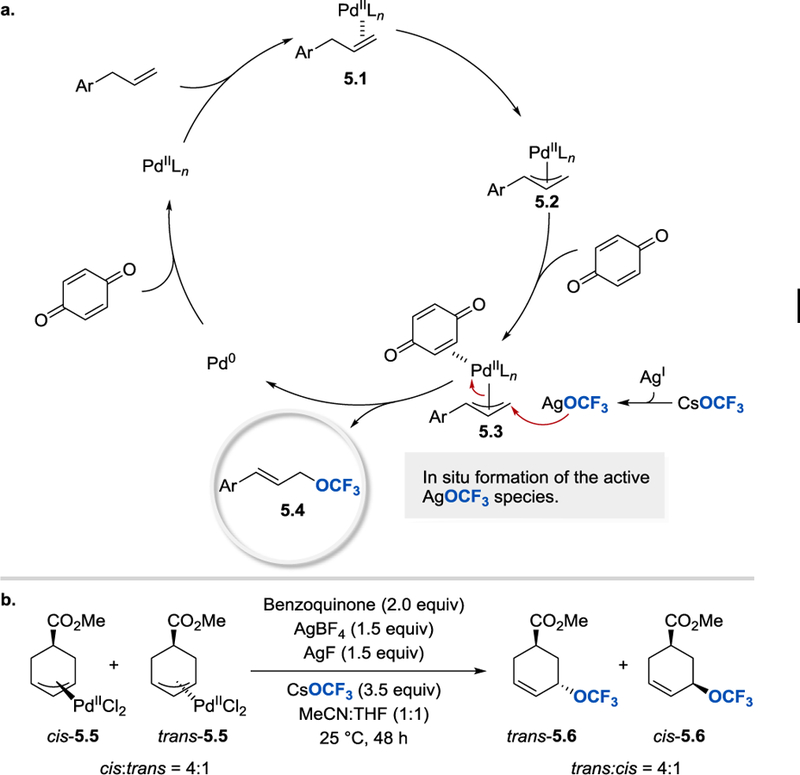
(a) Proposed mechanism of the palladium-catalyzed allylic C(sp3)–H bond trifluoromethoxylation. (b) Stereochemistry of the trifluoromethoxylation reaction [25].
As a continuation of their work aimed at development of catalytic trifluoromethoxylation of olefins, Liu and co-workers disclosed a novel Pd-catalyzed intermolecular ditrifluoromethoxylation reaction of unactivated alkenes in 2018 (Scheme 6) [26]. The vicinal difunctionalization was performed at 20 C using Pd(iPrCN)2Cl2 as a catalyst, SelectfluorCN-OTf as a strong oxidant, and AgOCF3 as a trifluoromethoxide source. The reaction was applicable to a wide range of aliphatic terminal alkenes substituted with imide, ester, nitro, halide, ether, nitrile, phosphate ester, hydroxyl, ketal, and aldehyde moieties and afforded ditrifluoromethoxylated products in moderate to good yields. More complex substrates, for instance, those containing heterocycles, a bicyclic amine, or a sugar scaffold in their structure were well tolerated under the reaction conditions
Scheme 6.
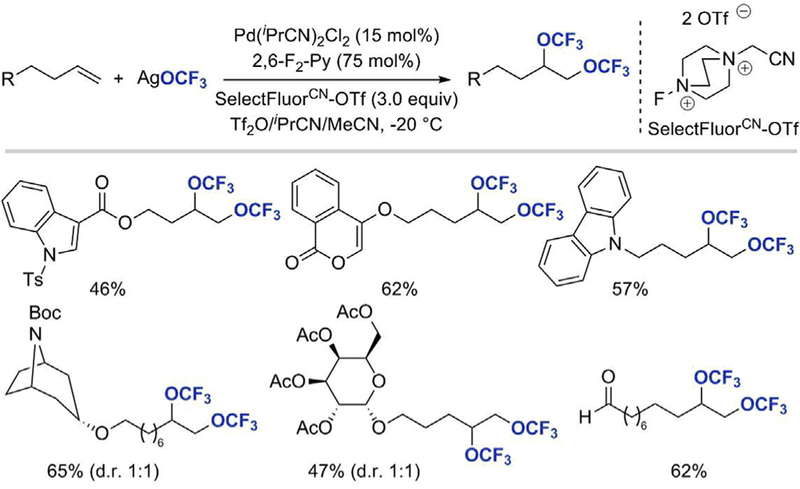
Palladium-catalyzed intermolecular ditrifluoromethoxylation of unactivated alkenes [26].
Based on preliminary mechanistic studies, Liu et al. proposed that the reaction involves a high-valent palladium species and proceeds via cis-trifluoromethoxypalladation of olefin (Scheme 7). The addition of Pd(IV)–OCF3 7.2 onto the double bond leads to the formation of the first C–OCF3 bond, whereas the second C–OCF3 bond is furnished, in analogy to the aminotrifluoromethoxylation process (vide supra), through C–O reductive elimination from alkylPd(IV)OCF3 intermediate 7.4.
Scheme 7.
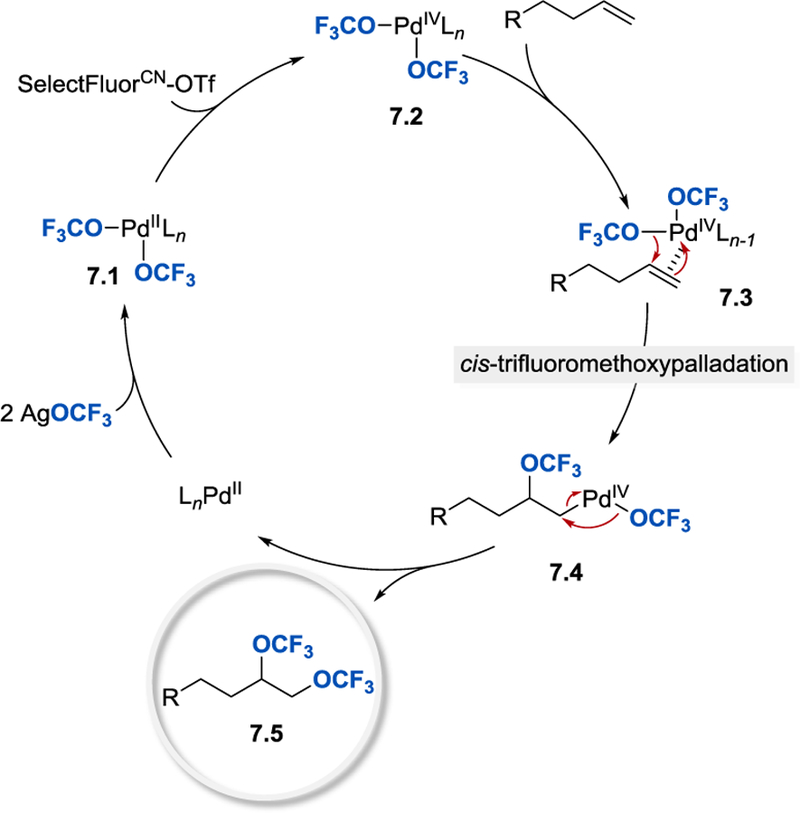
Proposed mechanism for palladium-catalyzed intermolecular ditrifluoromethoxylation of unactivated alkenes [26].
In 2017, Tang and co-workers reported the first catalytic asymmetric intermolecular bromotrifluoromethoxylation reaction of alkenes (Scheme 8) [27]. Their groundbreaking work was enabled by the development of a novel reagent, trifluoromethyl arylsulfonate (TFMS), which is easy to prepare and exhibits high thermal stability. While its structural analog, trifluoromethyl triflate (TFMT) is a very reactive, volatile liquid (bp = 19˚C), TFMS can be readily handled at room temperature and was reported to be inert towards some nucleophiles such as phenyl lithium [28]. In addition, the formation of the trifluoromethoxide anion from TFMS takes place under very mild conditions; it can be generated in situ upon treatment of TFMS with fluoride salts in the presence of a catalytic amount of silver fluoride. Tang was able to circumvent the issues usually associated with the use of the OCF3 anion (its low nucleophilicity and low stability) by an in situ formation of a very reactive electrophile. Although precise mechanistic details of the transformation are not yet fully understood, the reaction is likely to proceed via a nucleophilic attack by trifluoromethoxide on a highly electrophilic species generated by the interaction of an alkene with the brominating reagent. This activation of the π-bond with 1,3- dibromo-5,5-dimethylhydantoin (DBDMH) has extended the scope of trifluoromethoxylation to a wide range of substrates and enabled achievement of 39–95% yields of bromotrifluoromethoxylated products. In addition to styrenes, the reaction is applicable to non-activated olefins, including mono-, di-, tri-, and even tetra-substituted alkenes. Whereas trifluoromethoxylation via SN2 with TFMT is limited to primary and activated secondary alkyl halides [21], Tang’s approach enables the synthesis of trifluoromethoxylated molecules containing secondary and even tertiary carbon centers bearing the OCF3 group. Moreover, the bromine atom allows further structural elaborations on the difunctionalized product. The mild reaction conditions tolerated diverse functional groups including ester, amide, nitrile, nitro, and halides. The highest enantioselectivity was obtained with styrene derivatives, in particular with those bearing electron-withdrawing substituents. Monosubstituted alkenes afforded products with lower regioisomeric ratios (r.r) than tri-substituted alkenes. While bromotrifluoromethoxylated products were obtained from cyclic olefins in high yields and with excellent diastereoselectivity for the trans-product, no enantioselectivities were observed for these substrates. Remarkably, complex molecules such as estrone, cinchonine, and taxol derivatives have also undergone the bromotrifluoromethoxylation reaction in good yield, yet with low diastereoselectivities. The achievement of good enantioselectivities of bromotrifluoromethoxylation reaction was made possible thanks to the use of the dimeric cinchona alkaloid (DHQD)2PHAL as a chiral ligand. In order to account for the observed enantioselectivity of the reaction, the authors proposed that the alkene substrate is located in a chiral pocket and binds to the quinoline parts of the catalyst via π,π-stacking. The catalyst also interacts with the brominating reagent and the in situ formed silver trifluoromethoxide. One of the tertiary amines of the chiral ligand reacts with DBDMH to form an asymmetric brominating reagent, which activates the alkenes towards -OCF3 attack. Coordination of the silver ion by the Lewis basic nitrogen atom of the quinoline likely directs the approach of the trifluoromethoxide anion.
Scheme 8.
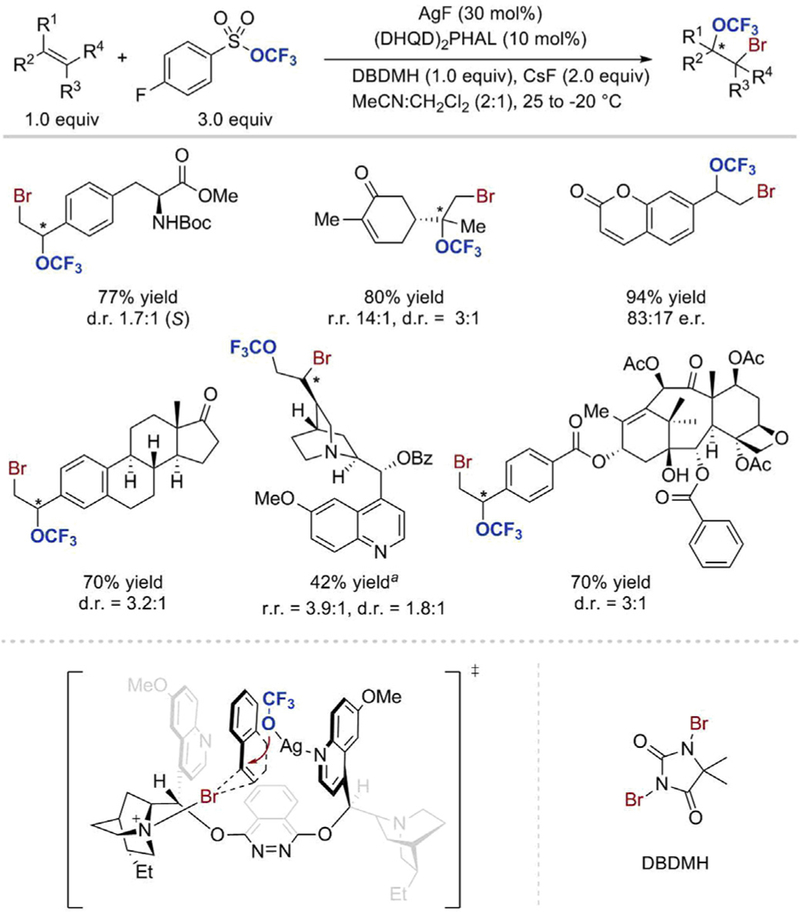
Asymmetric silver-catalyzed intermolecular bromotrifluoromethoxylation of alkenes. a 2.0 equiv of DBDMH were used [27]. Adapted with permission from [27]. Copyright 2017 Nature Publishing Group.
A structurally analogous trifluoromethoxylating reagent (trifluoromethyl 4-methylbenzenesulfonate) was later used by Tang and coworkers to achieve azidotrifluoromethoxylation of styrenes under synergistic visible-light mediated photoredox and silver catalysis (Scheme 9) [29]. In this transformation, Zhdankin reagent, which is known to generate ·N3 radical with photoredox catalysts, was employed as an azide source. Among the photoredox catalysts tested, Ru(bpy)3(PF6)2 gave the highest yield of the azidotrifluoromethoxylated product. As in the bromotri fluoromethoxylation reaction, reactive AgOCF3 species was formed from aryl trifluoromethyl sulfonate and silver(I) fluoride salt. Tang’s azidotrifluoromethoxylation protocol was applicable to a wide panel of styrenes, as well as thianaphthene and quinolone analogs. In general, higher yields were obtained with styrenes bearing electron-donating substituents as compared to those with electron-withdrawing groups. Unfortunately, this method could not be readily applied to aliphatic olefins, which afforded the desired products in low yields. Nonetheless, the azidotrifluoromethoxylated compounds formed in this transformation were shown to be versatile synthetic intermediates, which can be functionalized further, for example, via azide group reduction, or copper-catalyzed click reaction
Scheme 9.
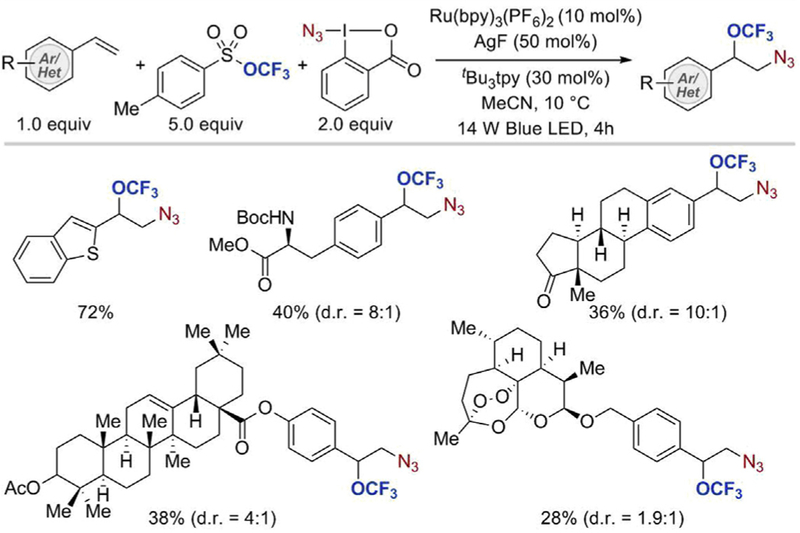
Ag/Ru-catalyzed azidotrifluoromethoxylation of styrenes [29].
In order to gain insights into the reaction mechanism, the authors performed Stern-Volmer luminescence quenching experiments and found that the Zhdankin reagent was the only quencher of excited ruthenium photoredox catalyst. Intermediacy of the putative ·N3 radical was corroborated by a radical trap experiment with TEMPO. In the presence of an excess of TEMPO, no azidotrifluoromethoxylated product was formed, but TEMPO-N3 adduct was isolated instead. In addition, the authors performed the reaction with (E) and (Z)-styrene derivatives and observed the same diastereoselectivities and comparable yields with both isomers, which suggested that a benzylic radical species was a reaction intermediate. Based on these experiments, azidotrifluoromethoxylation of styrenes was proposed to proceed as depicted in Scheme 10. Upon absorption of a photon of visible light, is excited to provide a long-lived triplet excited state, . Reduction of the Zhdankin reagent (10.1) by via a SET generates a strongly oxidizing and delivers the azide radical, ·N3. Addition of ·N3 affords benzylic radical 10.2, which is subsequently oxidized to benzylic carbocation 10.3 by . Benzylic carbocation 10.3 reacts with silver(I) trifluoromethoxide, generated in situ from trifluoromethyl 4-methylbenzenesulfonate and AgF to afford the desired product 10.4.
Scheme 10.
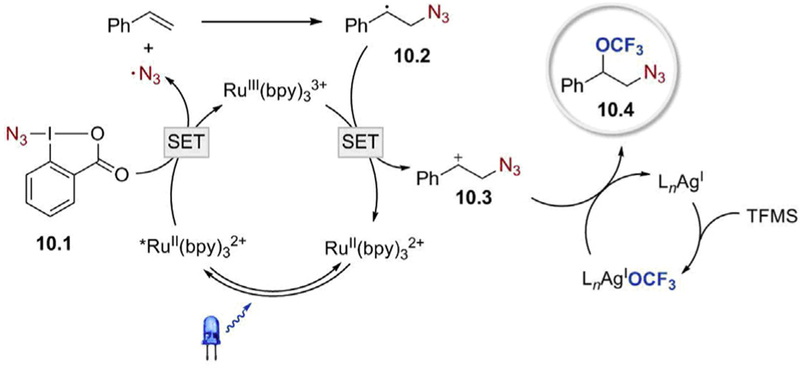
Proposed mechanism for the Ag/Ru-catalyzed azidotrifluoromethoxylation of styrenes [29]. Adapted with permission from [29]. Copyright 2018 Royal Society of Chemistry.
In addition to enabling bromo- and azidotrifluoromethoxylation of olefins, Tang’s TFMS reagent was recently employed in benzylic C–H trifluoromethoxylation reaction (Scheme 11) [30]. The desired reactivity was achieved upon treatment of methylated arene derivatives with TFMS, AgOTf, 1,10-phenanthroline, K2S2O8, and CsF in DMC at 50˚C. The reaction conditions were varied based on the electronic properties of the substituents on the aromatic ring. For instance, trifluoromethoxylation of substrates containing electronwithdrawing groups was performed using AgF2 as both the oxidant and silver salt, while in other cases the transformation worked better when F-TEDA-OTf was used as the oxidant. Functional groups such as halides, ether, ester, ketone, nitrile, nitro, sulfonamide were tolerated under the reaction conditions, but no desired products were obtained for substrates containing free hydroxyl, amine, or acid substituents. Arenes containing both electrondonating and electron-withdrawing groups provided the corresponding trifluoromethoxylated products in moderate to good yields (28–81% yield); the reaction was also applicable to heterocyclic structures as well as more complex molecular scaffolds including a lithocholic acid derivative and a trisubstituted pyrazole.
Scheme 11.
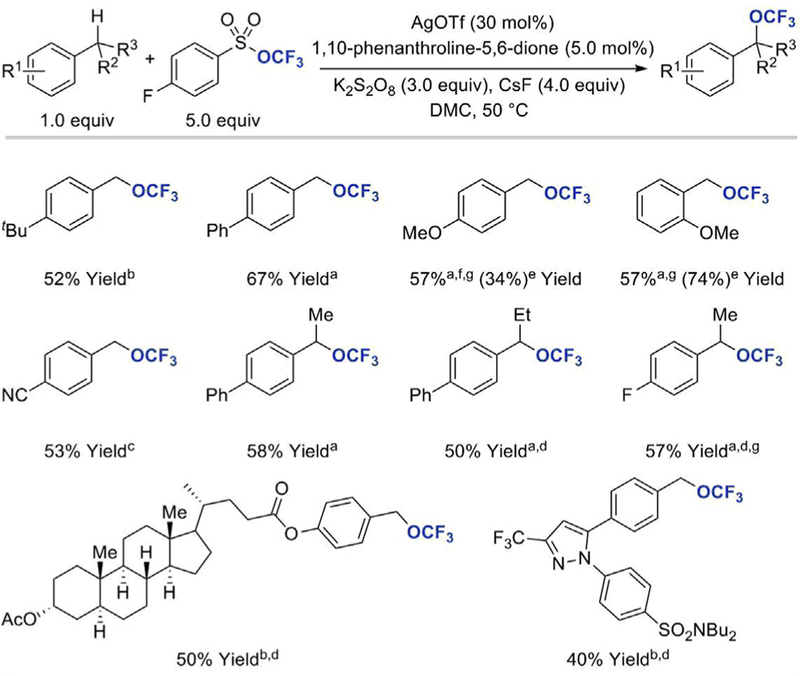
Silver-promoted oxidative benzylic C–H trifluoromethoxylation [30]a Standard conditions. b Reaction conditions: benzylic substrates (1.0 equiv), TFMS (3.0 equiv), F-TEDA-OTf (3.0 equiv), AgOTf (30 mol%), 1,10-phenanthroline-5,6-dione (5 mol %), 4,7-diphenyl-1,10-phenanthroline (5 mol%), CsF (4.0 equiv), 25˚C, DMC, N2. c Reaction conditions: benzylic substrates (1.0 equiv), TFMS (4.0 equiv), AgF2 (6.0 equiv), 4,4׳ -dimethoxy-2,2׳ - bipyridine (10 mol%), di(2-pyridinyl)-methanone (10 mol %), 25˚C, MeCN, N2. d0.5 equiv of AgOTf was used. e1.0 equiv of AgOTf was used. f The reaction was conducted at 40˚C. gYields were determined by integration of the 19F NMR spectrum using trifluoromethylthiobenzene or benzotrifluoride as internal standards. F-TEDA-OTf = 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(trifluoromethanesulfonate).
The proposed mechanism for the benzylic C–H trifluoromethoxylation is illustrated in Scheme 12. Ag(I)F, generated in situ from AgOTf and CsF, is oxidized by peroxodisulfate to Ag(II) F2, which then reacts with TFMS to form FAg(II)OCF3. Oxidation of the benzylic C–H bond by FAg(II)OCF3 generates benzylic radical, whose subsequent reaction with FAg(II)OCF3 affords the desired product (Path B). Based on DFT calculations, formation of benzylic cation upon oxidation of benzylic radical by FAg(II)OCF3 (Path A) is energetically favored, which suggests the reaction proceeds via trapping of benzylic cation by the OCF3 anion. The benzylic C–H trifluoromethoxylation is the third example of a reaction reported by Tang, in which achievement of moderate to good yields of compounds containing OCF3-group was made possible due to the intermediacy of a very reactive electrophilic species reacting with trifluoromethoxide along the reaction pathway.
Scheme 12.
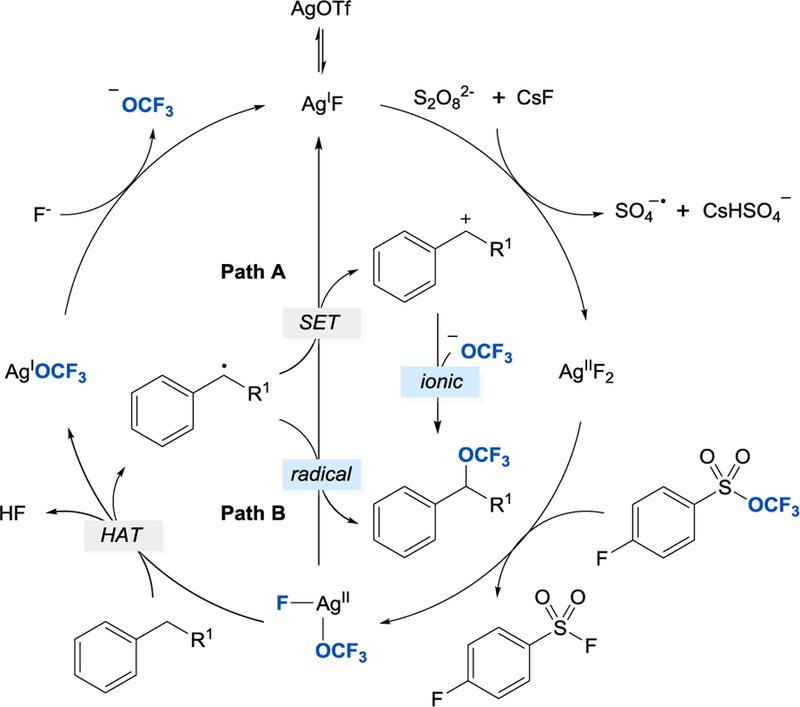
Proposed mechanism for the silver-promoted oxidative benzylic C–H trifluoromethoxylation [30]. Adpated with permission from [30]. Copyright 2018 Wiley-VCH.
In 2018, Ngai et al. reported the first catalytic intermolecular C(sp2)–H trifluoromethoxylation of (hetero)arenes under photocatalytic conditions [31]. In contrast to the nucleophilic trifluoromethoxylation strategies, which employ reagents capable of releasing the -OCF3 anion, Ngai’s approach relies on underexplored and previously difficult to access the •OCF3 radical intermediate and involves its addition onto aromatic π-system in the central C–OCF3 bond-forming step (see Scheme 13) [32]. The key to success of this distinctly different transformation was development of an easy-tohandle, bench-stable, photoactive trifluoromethoxylating reagent I, capable of liberating the •OCF3 radical under unprecedentedly mild reaction conditions. The benzimidazole derivative I was prepared in three steps from simple aromatic building blocks and Togni reagent I. Upon exposure of a mixture of reagent I, (hetero)arene, and Ru(bpy)3(PF6)2 (0.03 mol%) in MeCN to a 10 W LED light (λem = 402 nm) for 16 h, desired (hetero)aryl trifluoromethyl ethers were formed in moderate to good yields along with 3–5% of the Narylated side products. Due to over-trifluoromethoxylation of substrates, (hetero)arenes were used in excess (10 equiv) to prevent generation of complex mixtures of products. Ngai’s trifluoromethoxylation protocol was applicable to simple arenes containing common function groups including halides, carboxylic acids, ketones, esters, ethers, nitriles, carbonates, and phosphine oxides. Substrates with benzylic moieties as well as heteroarenes such as pyridine, pyrimidine, and thiophene were all well tolerated under the reaction conditions. More structurally elaborated compounds, including derivatives of fructose, and trans-androsterone were also successfully trifluoromethoxylated using 1 equivalent of substrates. Because of the nature of radical reactivity, in most cases, (hetero)aryl trifluoromethyl ethers were obtained as a mixture of isomers. While being a hurdle from a large-scale synthesis standpoint, this feature can be highly beneficial from the perspective of drug development since easy access to a range of regioisomers enables more rapid SAR studies of trifluoromethoxylated analogs
Scheme 13.
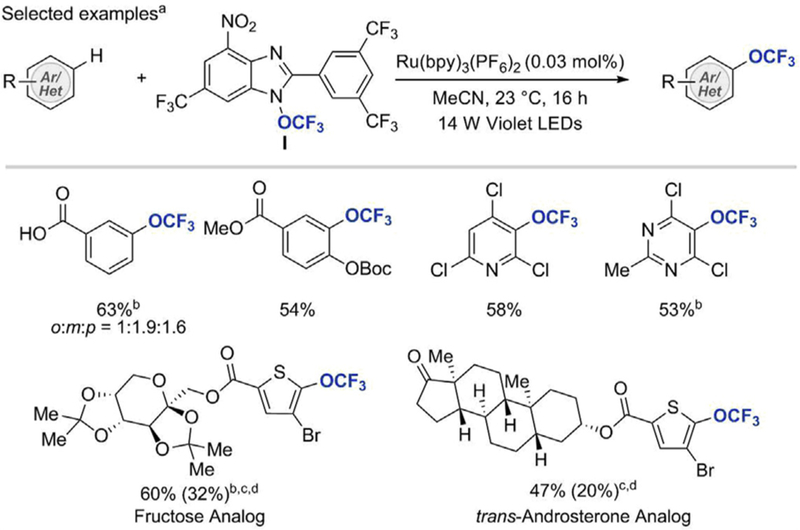
Intermolecular C–H trifluoromethoxylation of arenes with Reagent I. aReaction was performed using 10 equivalents of the substrate and 1 equivalent of the Reagent I. bReaction was performed at 40˚C. c1 equivalent of the substrate was used. dYield of isolated product based on the recovered starting material, yield in parenthesis is the yield of isolated product [31].
In order to shed light upon the reaction mechanism, the authors attempted the trifluoromethoxylation process using a bandpass filter (λ = 488 ± 2 nm). The filter was employed to block all the light required for the photoexcitation of reagent I, but to allow the passage of blue light needed for generation of the excited Ru species, . Under this conditions, 93% of reagent I was recovered and<5% of trifluoromethoxylation product was formed, which indicated that formation of the•OCF3 radical via the reduction of reagent I by excited Ru(bpy)3(PF6)2 is unlikely. In addition, DFT calculations showed that even if the anion radical of I (I-•) was formed, it would preferentially undergo mesolytic cleavage of the N-O bond to generate •NR1 R2 , rather than the•OCF3 radical. Thus, Ngai and Liu proposed an alternative reaction pathway, which begins with the photoexcitation of the OCF3-reagent I with near-UV light (Scheme 14). This direct excitation is followed by a homolytic cleavage of the NeOCF3 bond to afford the•OCF3 radical and an Ncentered benzimidazole radical. The •OCF3 then adds to an arene to afford the cyclohexadienyl radical 14.5, while •NR1 R2 is reduced by to benzimidazole anion 14.6. This reduction of the N centered radical by the excited photoredox catalyst suppresses competing radical amination reaction and thus allows high selectivity for the trifluoromethoxylation product. The subsequent single electron transfer (SET) from 14.5 to Ru(III) affords cyclohexadienyl cation 14.7, deprotonation of which restores the aromaticity and yields the desired aryl trifluoromethyl ether (14.9). It is noteworthy that other redox-active catalysts such as Cu(I) or Cu(II) salts could be used as well, although the selectivity between the desired product and the N-arylation product decreases.
Scheme 14.
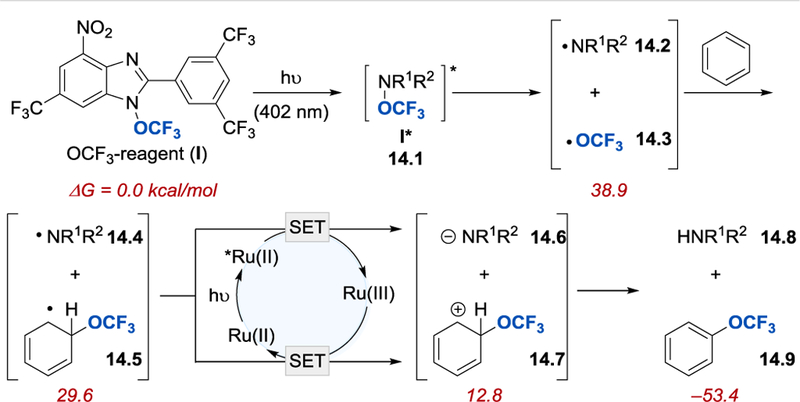
Proposed mechanism of trifluoromethoxylation of arenes with Reagent I [31]. All energies are Gibbs free energies (kcal/mol) with respect to the ground state reagent I and benzene calculated at the M06–2X/6–311++G(d,p)/SMD(MeCN)//M06– 2X/6–31 +G(d,p) level of theory. Adapted with permission from [31]. Copyright 2018 Wiley-VCH.
Following the development of photoactive reagent I, which required violet light (λ = 402 nm) for the formation of the•OCF3 radical along with benzimidazole radical, Ngai et al. concentrated their efforts on finding a reagent capable of forming the desired •OCF3 radical in a catalytic and selective manner. Later in 2018, they reported a redox-active cationic reagent II capable of accepting an electron from photoredox catalysts to liberate the •OCF3 radical exclusively (see Scheme 15). This reagent is also applicable to the catalytic (hetero)aryl C–H trifluoromethoxylation [33]. Upon irradiation with blue light, a broad array of (hetero)arenes, including a number of biorelevant compounds, for example Metronidazole®, Chlorpropramide®, Baclofen® derivatives, were successfully converted into their trifluoromethoxylated analogs. Although the reagent II requires three-step synthesis, the reaction tolerates air and moisture environment and does not produce N-arylated side products.
Scheme 15.
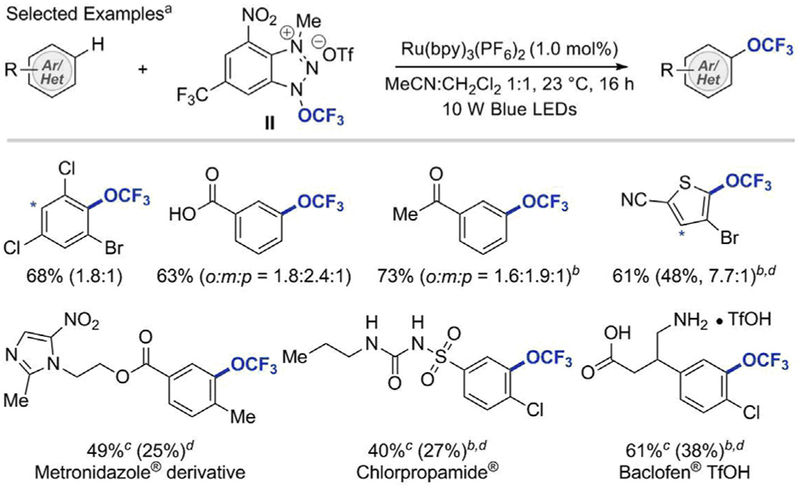
Photoredox-catalyzed intermolecular C–H trifluoromethoxylation of arenes with Reagent II33 aReactions were performed using 1 equivalent of reagent II and 10 equivalents of (hetero)arenes. Yields and regioselectivity were determined by 19F NMR using PhCF3 as an internal standard. bMeCN was used as the solvent. cReactions were performed using 1 equivalent of substrates and 2 equivalents of reagent II. The isolated yield based on the recovered starting material. dYield in parenthesis is of isolated yield.
It was postulated that aryl C–H trifluoromethoxylation with II proceeds via a mechanistic pathway that is different from the hypothesis outlined for the synthesis of (hetero)aryltrifluoromethyl ethers with the photoactive reagent I. The methylated cationic N–OCF3 reagent II (Ep =+ 0.140 V, vs SCE in CH3CN) is a better electron acceptor than I, and thus its single electron reduction by is energetically favorable (see Scheme 16). The SET from to II affords and a neutral radical 16.1 that fragments to liberate the •OCF3 radical exclusively. The N–OCF3 bond cleavage to form the undesired N-centered benzotriazole cation radical is energetically disfavored, which successfully prevents generation of N-arylated side products. The exothermic (∆G = 9.2 kcal/mol) addition of the •OCF3 radical to an arene gives cyclohexadienyl radical 16.3, which is subsequently oxidized by to afford cyclohexadienyl cation 16.4, deprotonation of which produces the desired trifluoromethoxylation product.
Scheme 16.
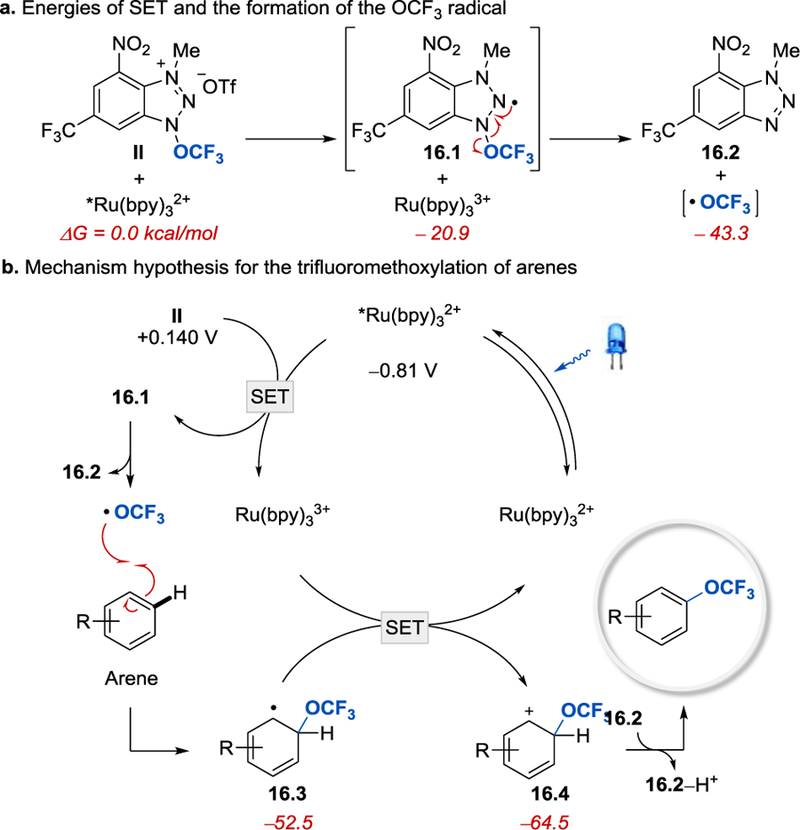
(a) Energies of SET and the formation of the•OCF3 radical from reagent II. All energies are Gibbs free energies (kcal/mol) with respect to the ground state reagent II and benzene calculated at the M06–2X/6–311 + +G(d,p)/SMD(MeCN)//M06–2X/6– 31 +G(d,p) level of theory. (b) Proposed mechanism for the photoredox-catalyzed C–H trifluoromethoxylation of arenes with Reagent II [33]. Reprinted with permission from [33]. Copyright 2018 Wiley-VCH.
Concurrently, Togni and co-workers disclosed their elegant work on radical trifluoromethoxylation of (hetero)arenes (see Scheme 17) [34]. Their approach uses a pyridinium-based reagent (III), first developed by Umemoto and Hu [35], for the generation of the•OCF3 radical under photoredox conditions. III was reported to be thermally stable, could be manipulated outside of a glovebox, and stored at ambient conditions for up to a year without a significant degree of decomposition. More importantly, III was readily synthesized in 63% yield on a multi-gram scale by trifluoromethylation of 4-cyanopyridine N-oxide with Togni reagent I. Aryl C(sp2)–H trifluoromethoxylation was performed upon irradiation of a mixture of III (1 equiv), arene (5 equiv), and a catalytic amount of Ru(bpy)3(PF6)2 (5.0 mol%) with 350 W blue LED light for 1 h. A wide array of simple aryl trifluoromethyl ethers bearing common functional groups (halides, ketones, esters, amides, acids, nitriles, sulfonamides, imides, boronic esters, and phosphonates), as well as more challenging substituents (benzylic moieties and aldehydes) was prepared using Togni’s trifluoromethoxylation protocol. The reaction was also applicable to more complex structures including Femara® (a breast cancer drug), Metalaxyl (an acylalanine fungicide), Phenytoin (an anti-seizure medication), and Procymidone (a fruit fungicide). Although, Togni et al. pointed out that their trifluoromethoxylation reaction using reagent III suffers from background N-aryl pyridination (~15%), trifluoromethoxylation of 4-cyanopyridine that is the byproduct of reagent III, and sensitivity of III to adventitious water, the easy accessibility of reagent III is advantageous.
Scheme 17.
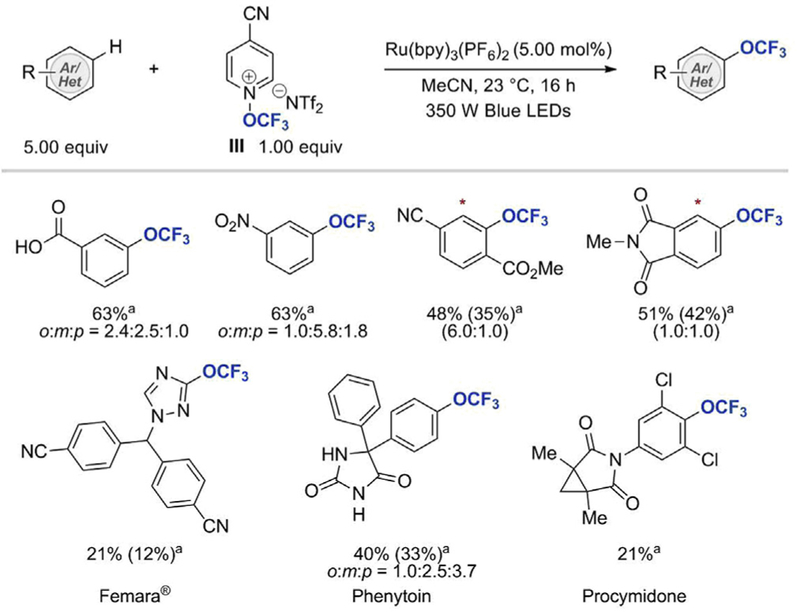
Visible light photoredox-catalyzed intermolecular C–H trifluoromethoxylation of arenes with Reagent III. aYields determined by 19F NMR, yield in parenthesis is the yield of isolated product [34].
Togni et al. proposed that the trifluoromethoxylation of arenes under photoredox conditions proceeds as depicted in Scheme 18. The process begins with the absorption of a photon of blue light by , which generates the long-lived triplet-excited state of (t1/2 =1.1 ms) [36]. Oxidative quenching of by the pyridinium cation III affords and 4- cyanopyridium radical 18.1. According to DFT calculations, fragmentation of 18.1 to generate the desired trifluoromethoxy radical and neutral pyridine 18.2 is more thermodynamically favorable than the formation of a trifluoromethoxide and a pyridinium radical cation. Following the addition of the •OCF3 radical to the arene, the cyclohexadienyl radical 18.3 is then oxidized by , and the resulting cation 18.4 is deprotonated by 4- cyanopyridine 18.2 to afford the final trifluoromethoxylated product 18.5.
Scheme 18.
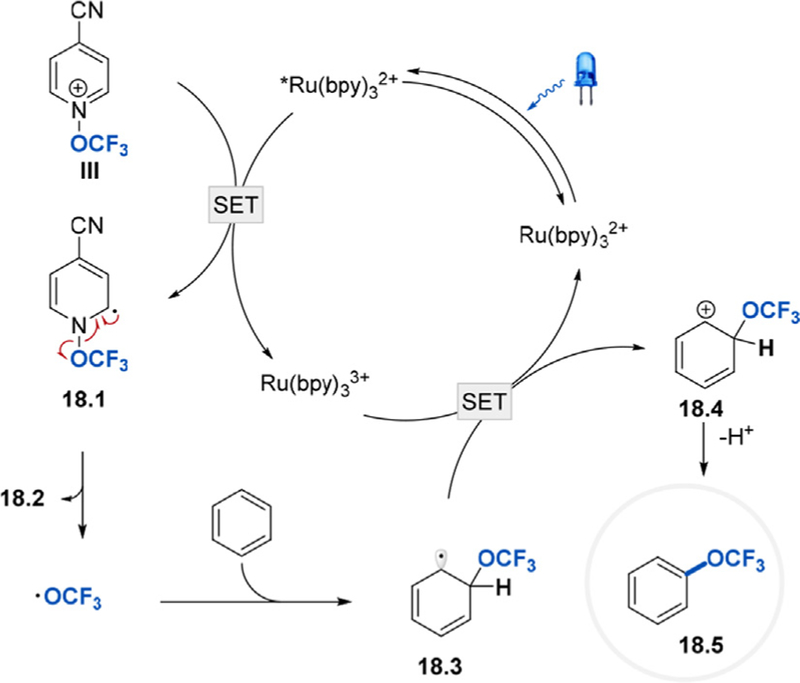
Proposed catalytic cycle for trifluoromethoxylation of arenes with Reagent III under visible light photoredox conditions [34].
Overall, the radical approach towards (hetero)aryl trifluoromethyl ethers independently developed by Ngai and Togni enables direct C–H trifluoromethoxylation of simple (hetero)aromatic substrates, which is highly advantages in the context of latestage functionalization of complex molecules. By obviating the need for prefunctionalization of starting materials and/or laborious functional group interconversions, the radical trifluoromethoxylation provides an extremely short, convenient route to a wide array of OCF3-derivatives. Despite its lack of regioselectivity, one might envision that an easy access to (hetero)aryl trifluoromethyl ethers via • OCF3 radical addition will greatly expedite drug development and screening process and thus will be highly impactful in the field of medicinal chemistry.
In summary, recent years have witness a tremendous progress in the area of catalytic synthesis of trifluoromethyl ethers. The very fast advancements in preparative approaches towards OCF3- bearing compounds were enabled by development of new, easy-tohandle, and bench-stable OCF3 reagents as well as a judicious choice of metal catalysts and ligands. Trifluoromethoxylation of alkenes, allylic and benzylic trifluoromethoxylation, as well as direct C–H trifluoromethoxylation of (hetero)arenes were performed for the first time in moderate to good yields under unprecedentedly mild conditions. Even though the newly developed transformations will probably not be employed for large scale manufacture of trifluoromethyl ethers due to either insufficient yields, high catalyst loadings, or the requirement for expensive reagents, they will undeniably be useful for discovery purposes. Based on the increasing interest in OCF3-substituted compounds, one might expect further fascinating breakthroughs in the field. The newly prepared trifluoromethyl ethers are, in turn, likely to find a broad range of applications in different fields and contribute to a rapid progress in life sciences to materials chemistry research
Acknowledgments
We thank NIGMS (R35GM119652) in support of this work
References
- [1].Purser S, Moore PR, Swallow S, Gouverneur V, Chem. Soc. Rev 37 (2008) 320–330. [DOI] [PubMed] [Google Scholar]
- [2].Ojima I, Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing Ltd, 2009. [Google Scholar]
- [3].Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed 52 (2013) 8214–8264. [DOI] [PubMed] [Google Scholar]
- [4].Muller K, Faeh C, Diederich F, Science 317 (2007) 1881–1886. [DOI] [PubMed] [Google Scholar]
- [5].Federsel D, Herrmann A, Christen D, Sander S, Willner H, Oberhammer H, J. Mol. Struct 567 (2001) 127–136. [Google Scholar]
- [6].Delorenzi FG, Pulm. Pharmacol 7 (1994) 129–135.8081073 [Google Scholar]
- [7].Landelle G, Panossian A, Leroux FR, Curr. Top. Med. Chem 14 (2014) 941–951. [DOI] [PubMed] [Google Scholar]
- [8].Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR, Nature 405 (2000) 962–966. [DOI] [PubMed] [Google Scholar]
- [9].Valsasina B, Beria I, Alli C, Alzani R, Avanzi N, Ballinari D, Cappella P, Caruso M, Casolaro A, Ciavolella A, Cucchi U, De Ponti A, Felder E, Fiorentini F, Galvani A, Gianellini LM, Giorgini ML, Isacchi A, Lansen J, Pesenti E, Rizzi S, Rocchetti M, Sola F, Moll J, Mol. Cancer Therapeut 11 (2012) 1006–1016. [DOI] [PubMed] [Google Scholar]
- [10].Amir OG, Peveling R, J. Appl. Entomol 128 (2004) 242–249. [Google Scholar]
- [11].Chen Y, Zhang AF, Wang WX, Zhang Y, Gao TC, Ann. Appl. Biol 161 (2012) 247–254. [Google Scholar]
- [12].Howard J, Wall R, Bull. Entomol. Res 85 (1995) 71–77. [Google Scholar]
- [13].Sagheer M, Yasir M, Hasan M, Ashfaq M, Pakistan J Agric. Sci 49 (2012) 173–178. [Google Scholar]
- [14].Santel HJ, Bowden BA, Sorensen VM, Mueller KH, in: Brighton Conference: Weeds, vols 1–3, 1999, pp. 23–28, 1999. [Google Scholar]
- [15].Kirsch P, Bremer M, Angew. Chem. Int. Ed 39 (2000) 4217–4235. [DOI] [PubMed] [Google Scholar]
- [16].Mamada M, Shima H, Yoneda Y, Shimano T, Yamada N, Kakita K, Machida T, Tanaka Y, Aotsuka S, Kumaki D, Tokito S, Chem. Mater 27 (2015) 141–147. [Google Scholar]
- [17].Leroux FR, Manteau B, Vors JP, Pazenok S, Beilstein J Org. Chem 4 (2008) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee KN, Lee JW, Ngai MY, Synlett 27 (2016) 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tlili A, Toulgoat F, Billard T, Angew. Chem. Int. Ed 55 (2016) 11726–11735. [DOI] [PubMed] [Google Scholar]
- [20].Kolomeitsev AA, Vorobyev M, Gillandt H, Tetrahedron Lett 49 (2008) 449–454. [Google Scholar]
- [21].Marrec O, Billard T, Vors JP, Pazenok S, Langlois BR, J. Fluorine Chem 131 (2010) 200–207. [Google Scholar]
- [22].Marrec O, Billard T, Vors JP, Pazenok S, Langlois BR, Adv. Synth. Catal 352 (2010) 2831–2837. [Google Scholar]
- [23].Zhang CP, Vicic DA, Organometallics 31 (2012) 7812–7815. [Google Scholar]
- [24].Chen C, Chen P, Liu G, J. Am. Chem. Soc 137 (2015) 15648–15651. [DOI] [PubMed] [Google Scholar]
- [25].Qi X, Chen P, Liu G, Angew. Chem. Int. Ed 56 (2017) 9517–9521. [DOI] [PubMed] [Google Scholar]
- [26].Chen C, Luo Y, Fu L, Chen P, Lan Y, Liu G, J. Am. Chem. Soc 140 (2018) 1207–1210. [DOI] [PubMed] [Google Scholar]
- [27].Guo S, Cong F, Guo R, Wang L, Tang P, Nat. Chem. 9 (2017) 546–551. [DOI] [PubMed] [Google Scholar]
- [28].Koller R, Huchet Q, Battaglia P, Welch JM, Togni A, Chem. Commun (2009) 5993–5995. [DOI] [PubMed] [Google Scholar]
- [29].Cong F, Wei Y, Tang P, Chem. Commun 54 (2018) 4473–4476. [DOI] [PubMed] [Google Scholar]
- [30].Yang H, Wang F, Jiang X, Zhou Y, Xu X, Tang P, Angew. Chem. Int. Ed (2018), 10.1002/anie.201807144. [DOI] [PubMed] [Google Scholar]
- [31].Zheng W, Morales-Rivera CA, Lee JW, Liu P, Ngai MY, Angew. Chem. Int. Ed 57 (2018) 9645–9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sahoo B, Hopkinson MN, Angew. Chem. Int. Ed 57 (2018) 7942–7944. [DOI] [PubMed] [Google Scholar]
- [33].Zheng W, Lee JW, Morales-Rivera CA, Liu P, Ngai MY, Angew. Chem. Int. Ed (2018), 10.1002/anie.201808495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jelier BJ, Tripet PF, Pietrasiak E, Franzoni I, Jeschke G, Togni A, Angew. Chem. Int. Ed (2018), 10.1002/anie.201806296. [DOI] [PubMed] [Google Scholar]
- [35].Umemoto T, Zhou M, Hu J, Faming Zhuanli Shenqing CN, 2015, 105017143. [Google Scholar]
- [36].Juris A, Balzani V, Belser P, von Zelewsky A, Helv. Chim. Acta 64 (1981) 2175–2182. [Google Scholar]