

Abstract
A combination of liquid chromatography, ion mobility spectrometry, mass spectrometry, and database searching techniques were used to characterize the proteomes of four biological replicates of adult Drosophila melanogaster heads at seven time points across their lifespans. Based on the detection of tryptic peptides, the identities of 1281 proteins were determined. An estimate of the abundance of each protein, based on the three most intense peptide ions, shows that the quantified species vary in concentration over a factor of ~103. Compared to initial studies in the field of Drosophila proteomics, our current results show an eight-fold higher temporal protein coverage with increased quantitative accuracy. Across the lifespan, we observe a range of trends in the abundance of different proteins, including: an increase in abundance of proteins involved in oxidative phosphorylation, and the tricarboxylic acid cycle; a decrease in proteasomal proteins, as well as ribosomal proteins; and, many types of proteins, which remain relatively unchanged. For younger flies, proteomes are relatively similar within their age group. For older flies, proteome similarity decreases within their age group. These combined results illustrate a correlation between increasing age and decreasing proteostasis.
Keywords: Liquid chromatography, Ion mobility spectrometry, Tandem mass spectrometry, Proteomics, Aging, Proteostasis, Label free quantification
Graphical Abstract
1. Introduction
The fruit fly, Drosophila melanogaster (D. melanogaster), is a well-studied model organism because of its short lifespan, high-reproductive rate, and the availability of an extensive variety of genotypes [1,2]. Nearly 75% of genes associated with human neurological diseases are found in D. melanogaster [3]. Transgenic animals expressing human proteins can be used in models of neurodegenerative diseases [4]. Expression of the human amyloid precursor protein (APP) in D. melanogaster leads to animals that display neurodegeneration and a shortened lifespan; [5] such models are relevant to understanding Alzheimer’s Disease. In the case of many neurodegenerative diseases, pathological progression is associated with the accumulation of aberrant proteins, as seen with APP cleavage products that oligomerize to form amyloid fibrils [5,6] or with the protein huntingtin in Huntingtin’s disease that leads to inclusion bodies [7]. The accumulation of specific proteins, as well as the loss of functional proteins, is a hallmark of aging and is associated with numerous diseases [8,9].
In order to understand the overall effect of disease, it is necessary to characterize the protein expression in wild-type organisms. In 2007, Sowell et al. [10] reported an in depth proteome study using multiple techniques including liquid chromatography (LC), ion mobility spectrometry (IMS), mass spectrometry (MS), and database searching. This study determined which proteins were present by identifying peptides that were produced from tryptic digestion of proteins extracted from the heads of 25 animals. The results were a first attempt to comprehensively map what proteins were present and how the abundances of species changed with aging. However, the methodologies involved were first generation prototypes, therefore providing only a limited view of age-induced changes. Overall, this and other work [11–14] has suggested that protein abundance varies throughout an organism’s life.
Since Sowell’s early investigation, many technological advances have been made. In the work presented below, we reexamine the wild-type aging studies, using a more robust commercially available LC-IMS–MS instrument to characterize the proteomes of four biological replicates of adult animals at seven time points across their lifespan. Using the new methods, we have identified and quantified 1281 proteins extracted from the heads of three animals per sample. This study appears to be sensitive to concentrations which vary by over a factor of ~103. Trends in abundances of different proteins across the organism’s lifespan are reported. For example, proteins involved in oxidative phosphorylation and the tricarboxylic acid cycle appear to increase in abundance with age, while proteasomal proteins, as well as ribosomal proteins decrease. Finally, many types of proteins, e.g., heat shock proteins, remain relatively unchanged. In general, we find stronger protein correlations in younger flies compared with older animals. It therefore appears that proteostasis is more difficult to maintain with increasing age. Here we present the results of our current studies, compare these results to prior work in the field, and discuss possible interpretations for the observed changes.
2. Methods
2.1. Sample preparation, protein extraction and digestion
Populations of D. melanogaster female adult virgins were grown at 24 °C in 12 h light and dark cycles. Flies were transferred to fresh media every four days. Flies were collected at five day intervals over 30 days.
Flies at individual time points were dissected to isolate the heads [10]. Populations of fly heads were then homogenized in 8 M urea (Sigma Aldrich, St. Louis, MO) with 100 mM ammonium bicarbonate (Sigma Aldrich, St. Louis, MO, pH 7.8) using a glass mortar and pestle. SDS-Page gel with Coomassie blue staining was initially used to estimate a protein yield of approximately 10 μg per fly head. Denatured proteins were then reduced using 5 mM tris(2-carboxyethyl)phosphine (TCEP, Sigma Aldrich, St. Louis, MO) for 1 h at 56 °C. Reduced proteins were then alkylated using 5 mM iodoacetamide (Sigma Aldrich, St. Louis, MO) for 1 h at 23° C in the dark. Fly head homogenate was buffer exchanged with 100 mM ammonium bicarbonate to a final concentration of 1 M urea using a 30 kDa molecular weight cut off filter (Millipore, Billerica, MA). Proteins were digested with trypsin (Promega, Madison, WI) for 12 h at 37° C, using a 5% w/w enzyme to protein ratio. Peptides were desalted using C18 Zip-Tips (Agilent Technologies, Santa Clara, CA) and lyophilized to dryness.
2.2. LC-IMS–MS/MS analysis
Peptides where reconstituted in LC buffer A (0.1% formic acid, water, Fisher Scientific, Hanover, NH) with an internal standard spike of digested equine alcohol dehydrogenase (500 fmol/uL, Sigma Aldrich, St. Louis, MO). Peptides were separated on a nanoAcquity ultra high pressure liquid chromatography system (UPLC) (Waters, Milford, MA) by first loading onto a trap column (Symmetry C18, 180 μm × 20 mm, Waters, Milford, MA) at a flow rate of 10 μL/min at 97% buffer A for 5 min. Peptides were then separated on a UPLC column (1.8 μm HSS T3, 75 μm × 250 mm, Waters, Milford, MA). A linear gradient of 3%–34% buffer B (0.1% formic acid, acetonitrile, Fisher Scientific, Hanover, NH) was applied over 180 min. Peptides eluting from the column were electrosprayed in positive mode into a Synapt G2S HDMS (Waters, Milford, MA) instrument using a universal nanospray source. Data were acquired in an ion mobility supplemented MSe manner (HDMSe). Drift specific collision energies were optimized using a digest of mouse synaptic proteins and is described further in the results section.
MS datasets were processed and database searched using ProteinLynx Global Server 2.5.3 (PLGS, Waters, Milford, MA). The intensity thresholds for low energy and high energy were optimized individually using PLGS threshold inspector. Databank searches were completed using the D. melanogaster proteome (downloaded from Uniprot on 10/2016) with the default false discovery rate (FDR) of 4% [15]. Using this FDR setting, our data was searched against a randomized concatenated database and returned an empirical protein FDR equal to 1.7%. For the purposes of label free quantification, exact mass retention time tables were aligned and analyzed using IsoQuant [16]. This allowed for relative quantification based upon the three most intense peptides ions per protein (Top3 quantification). The complete quantitative data for all time points and replicates is provided in Supplementary Table S1 in the online version at DOI: 10.1016/j.ijms.2018.01.003. Listed in this table are the information regarding protein name, gene name and accession number as well as its quantitation results in that sample. Using the internal standard, equine alcohol dehydrogenase, allowed for determination of absolute protein levels.
Further analysis was completed using Microsoft Office Excel. The David Bioinformatics [17] server was used to determine gene ontology terms. When we selected a group of proteins belonging to a given KEGG pathway, we did not require that individual members were exclusive to a single pathway/process. Heat shock proteins were used as annotated in Uniprot. Cluster 3.0 [18] was used to group proteins with similar temporal patterns.
3. Results
3.1. Systematic optimization of drift time-specific collision energy
Mass spectrometry (MS) based quantitative proteomics has emerged as the central technology for the characterization of peptides generated from proteins originating in complex mixtures/cells/tissues. In addition to mass spectrometry, ion mobility spectrometry (IMS) is another gas phase separation which discriminates based on an ion’s shape and charge. Because these techniques are largely orthogonal to each other, combining IMS with MS yields a higher peak capacity, thereby allowing for an increase in the number of identified peptides [13,19,20].
Developments in data independent acquisition modes (DIA), such as SWATH [21] or MSe [22,23], have also resulted in increased peptide sequencing capabilities. During each one second cycle of an MSe acquisition, sequential low and high collision energy scan events are collected across the full mass range. This sequential acquisition mode results in peaklists of precursor ions and their corresponding product ions. High energy scans occurring after an ion mobility separation generate product ions that can be drift time aligned with their precursors. This provides an additional increase in peak capacity [24,25]. Data acquired using IMS-MS in a MSe acquisition mode has been termed HDMSe [22].
In our experimental workflow, precursor peptides elute from the IMS cell into the transfer cell at specific drift times prior to mass analysis by a time-of-flight mass spectrometer (TOF, Fig. 1A). During each acquisition cycle, as mentioned above, the transfer cell alternates between low and high collision energies (Fig. 1A). When in the low-energy state, precursor ions pass through the transfer cell without fragmenting. When in the high-energy state, precursor ions are fragmented to generate product ions. The optimal collision energy to fragment a peptide increases with that compound’s mass (Fig. 1B). Because there is a relationship between mass and drift time, there is also a correlation between drift time and optimal collision energy (Fig. 1C). Previous work has shown that drift time specific collision energy ramps can increase peptide IDs substantially, although only a subset of possible collision energy ramps were investigated [16].
Fig. 1.

Systematic determination of the optimal drift bin specific collision energy ramp. (A) A series of HDMSe runs were acquired with a fixed high energy collision value for fragmentation in the transfer cell. Ions were detected using a time-of-flight (TOF) mass spectrometer. For each subsequent run, the collision energy was increased. When a peptide was identified at multiple collision energies, the collision energy producing the highest scoring match was retained. (B) Peptides were binned by the (M + H) of the precursor, and within each bin, the average best collision energy for those peptides was determined. Error bars represent one standard deviation. (C) For precursor ions, there is a linear dependence observed between drift time and mass-to-charge. (D–E) The distribution of collision energies used to generate product ions at a particular drift bin window is shown. Black bars represent the average collision energy for that drift bin window. (F) The average collision energies for peptides eluting from all drift bin windows are plotted as black circles. The intensities of the red circles are proportional to the number of individual peptide identifications. Error bars represent one standard deviation. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We sought to systematically evaluate the entire drift time –collision energy landscape with the goal of determining the most efficient collision energy setting for each drift time. This was carried out with a standard proteome digest. During this optimization, a series of runs were acquired where the collision energy in the transfer cell was held constant for a given run during the elevated energy portion of the scan cycle. A series of individual runs were acquired at fixed high energy collision voltages, with varied between 20 and 55 V, in 2 V increments. All acquisitions were then searched against a database. The entire set of peptide IDs along with their collision energies and drift times were extracted. A series of drift bin ranges were constructed, and the distribution of collision energies leading to positive peptide identifications were determined (Fig. 1C–D, black bars, average value black line). For all peptides identified within a given drift bin, the average collision energy was calculated (Fig. 1F). While we expect an additional dependence of optimal collision energy on charge state, there is currently no way to select specific charge states within the drift time distribution using this instrument. Using this data, a drift time specific look up table was determined and used in further analyses.
3.2. Top3 protein quantification
We employed a label free protein quantification approach to characterize the D. melanogaster proteome, by averaging the three most abundant peptides signals (Top3 peptides) from a given protein (Fig. 2A) to infer a protein’s abundance (Fig. 2B). Previous research has demonstrated a linear relationship between the Top3 value for a given protein and the amount of protein present [22,26]. An internal standard of known concentration was used to convert Top3 values to absolute amounts of each protein per condition (Fig. 2C).
Fig. 2.

Label-free protein quantification. (A) An example of peptides from odorant binding protein (Q7K084). For each peptide, the precursor intensity as a function of day was plotted. The four peptides identified were SPANEWAFR (open circle), TFDYPDDDITR (triangle), GFKVENLVAQLGQGKEDK (cross diamond), and VENLVAQLGQGKEDK (center dot diamonds). Peptides show good correlation with each other across the time points. (B) The top 3 peptide intensities were then averaged together to determine the protein intensity at each day. (C) Using an internal standard, equine alcohol dehydrogenase, an absolute abundance can also be determined. (D–I) Lifespan median absolute abundance data were plotted for all proteins in rank abundance order along the X axis. Black squares represent specific groups of proteins associated with biochemical pathways; including proteasome (D), ribosome (E), heat shock (F), TCA cycle (G), oxidative phosphorylation (H), and glycolysis (I).
Using PLGS in combination with Isoquant allowed for both identification and quantification of the temporal D. melanogaster proteome. In our collected dataset, 11,085 peptides were identified belonging to 1281 different proteins (1269 proteins were identified with three or more peptides). On average, nine peptides were reported per protein, and the average protein sequence coverage was 36%. Across adult D. melanogaster lifespan, the fold change per protein was determined to have a median of 2.1 and an average of 4.1. Overall biological percent relative standard deviation showed a median of 23%, lower relative standard deviations were observed on days 0, 5 and 20. Approximately 80% of total proteins were identified in each individual analysis by PLGS. For those peptides not identified in every run, the analysis software, Isoquant, is nevertheless able to quantify them. When PLGS identifies a high-confidence peptide in any run, Isoquant uses that ion’s retention time, drift time and accurate mass to quantify the species in other runs where it may not have been identified by PLGS [16].
For each protein, we calculated its median abundance across the fly’s lifespan, and these values varied over three orders of magnitude (Fig. 2D). For comparison, the dynamic range of tissue-specific proteomes is reported to be three to four orders of magnitude [27,28]. We were particularly interested in the abundance of proteins associated with specific biochemical pathways (i.e. ribosome, TCA cycle, and oxidative phosphorylation; Fig. 2E, G, H, respectively).
3.3. Comparison with our previous temporal proteome map
Sowell et al. [10] measured the temporal proteome of D. melanogaster at 9 time points over its lifespan, from day 0 to day 60. This work combined several forms of analysis, including liquid chromatography-tandem mass spectrometry (LC–MS), strong cation exchange fractionation followed by LC–MS, and also ion mobility spectrometry mass spectrometry (IMS-MS), allowing for evidence of 5902 unique peptides and 1699 proteins. The identified proteins were quantified using spectral counting [28].
In some instances, protein temporal abundance patterns were similar between these two studies. One example, arginine kinase isoform E shows a similar age-dependent increase in abundance between the two datasets (Fig. 3A and B). To gain a broader understanding of how these datasets compared, we ranked co-identified proteins in the current study using their summed intensity (Top3) and in Sowell’s data using summed spectral counts. The proteins’ rank positions in both experiments were then be plotted as scatterplots (Fig. 3C, D). The top 50 most abundant proteins in Sowell’s dataset at day zero showed a roughly linear relationship when compared to their abundances the present work (Fig. 3C). However, this correlation is lost when the full dataset is compared (Fig. 3D). We repeated this analysis at each individual day (data not shown), and in all cases, the relative abundances of the full datasets showed poor correlation.
Fig. 3.

Quantitative mass spectrometric comparison to previous work by Sowell et al. [10]. Arginine kinase isoform E from Sowell et al. (A) and current dataset (B) were plotted using spectral matches and Top3 intensity measurements, respectively, over the lifespan of the fly. Four replicate analysis are averaged in the current dataset (B) with error bars representing SEM. The 50 most abundant (C) and all mutually quantified proteins (D) were rank ordered in each dataset and plotted as scattergrams. Heat maps showing the day and protein normalized data are shown for Sowell et al. (E) and current dataset, demonstrating increased coverage of protein abundance values in the current dataset. Each day in the current dataset contains biological replicates which are denoted by tick marks (F). Color represents the relative abundance from low (green) to high (red). Empty data points remain white. Ribosome proteins were extracted from the heat map analysis and pairwise Pearson cross correlations were determined. These are plotted as histograms for the number of protein pairs correlated at a specific correlation value for Sowell et al. (G) and the current dataset (H). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Sample handling practices, instrumentation and methods of data analysis have improved greatly since the original acquisition of the D. melanogaster temporal proteome. In the original work by Sowell et al. ~60% of the time, an individual protein was not quantified on a given day. For the current dataset, only ~7% of this data is missing. The difference in data coverage can be visualized using a heat map representation (Fig. 3E and F) and likely results from methodological advances mentioned previously. We then sought to determine which method allowed for a more accurate quantitative representation of the proteins. We chose to focus on the proteins involved in the ribosome for two reasons: many unique proteins are required (therefore many protein measurements exist); and these proteins physically interact in a protein complex. Our rationale is that physically interacting proteins should have very similar temporal protein abundance patterns. The dataset with better protein quantification should display a higher correlation between ribosomal associated proteins. In the work completed by Sowell et al., 55 ribosomal proteins were identified, with data at roughly 50% of all time points. In the current dataset, we identified 64 ribosomal proteins, with data at roughly ~98% of all time points and replicates. If we normalize the data within a protein and within a day and then correlate all pairwise protein temporal patterns, we would expect interacting proteins would have higher Pearson correlation coefficients. We completed this for the proteins involved in the ribosome from both Sowell et al. and our current data (Fig. 3G and H, respectively). In the original work by Sowell et al. the distribution of ribosomal protein correlations showed a median value of negative 0.01. This indicates no systematic correlation between the observed ribosomal temporal patterns. In contrast, our current data has a median correlation of 0.71. This suggests that our current data more accurately quantifies the temporal proteome.
3.4. The effect of aging on energy consumption: TCA cycle, oxidative phosphorylation, and glycolysis
Eukaryotic energy production, via oxidative phosphorylation or the TCA cycle, occurs primarily in the mitochondria. The highly reactive environment within this organelle results from the generation of metabolites, such as NAD+, acetyl-CoA, and ATP. Oxidative modification of mitochondrial proteins has been shown to increase during aging [29,30], which could result from increases in the above mentioned reactive species. We observed an increased abundance relative to day zero for 53/58 oxidative phosphorylation proteins (out of a total of 144 proteins in this pathway, Fig. 4A, black line). Of these proteins, Cytochrome b c1 subunit 9 showed the largest increase in abundance with time (Fig. 5A, circles). This accessory protein assists in correctly orienting a heme group in complex 3 of the pathway [31]. Overexpression of this protein has also been able to rescue complexes lacking Cytochrome b c1 subunit 6 [32]. We also observed Cytochrome b c1 subunit 7, from this same complex, which shows a similar protein abundance pattern (Fig. 5A, squares). Finally, one other protein, stunted isoform B (Fig. 5A, triangles), also showed increased temporal abundance. Mutation of this protein can increase longevity [33]. This protein is expressed and binds the G-protein coupled receptor in the synapse called methuselah, which has been shown to alter lifespan [34]. Correlated abundance patterns for these proteins is not unexpected as they are all part of a contiguous metabolic pathway.
Fig. 4.

Normalized temporal abundances for proteins of various biological function. Proteins annotated as oxidative phosphorylation (A), proteasome (B), TCA cycle (C), ribosome (D), glycolysis (E), and heat shock proteins (F) using the bioinformatics gene annotation program David are shown (HSPs annotations were extracted from Uniprot). For all classes, the median protein abundance is plotted as the thick black line with error bars denoting SEM. Within each line plot, all individual proteins are shown as thin grey lines.
Fig. 5.

Average protein abundance measurements. Cytochrome b c1 subunit 7 and 9 are shown as squares and circles, respectively along with protein stunted isoform B in triangles. (A) Proteasome core subunits alpha 7 and beta 1 are shown as open circles and open triangles, respectively. (B) An accessory proteasome protein Ter94 is shown as squares. (B) Aconitase hydratase, pyruvate carboxylase and ATP citrate synthase are given as squares, circles, and triangles, respectively. (C) Ribosome protein subunit 17 and 24 are shown as circles and squares, respectively. (D) Aldose 1 epimerase, phosphotransferase, and triosephosphate isomerase are squares, circles and triangle, respectively. (E) Heat shock protein 27, 60 kDa heat shock protein mitochondrial homolog, and the HSP 70 kDa protein are shown as squares, circles, and triangles, respectively. (F) Error bars represent SEM.
Another pathway in mitochondria is the tricarboxylic acid (TCA) cycle, which generates NADH equivalents used in the oxidative phosphorylation pathway. Of the 43 proteins in the TCA cycle, we observed 31. Of these, 24 showed increased abundance with time. The median TCA protein abundance and standard deviation are shown in Fig. 4C (black line). Pyruvate carboxylase (Fig. 5C, circles), which converts pyruvate to oxaloacetate, showed great overlap with the median value (R = 0.85). Citrate synthase and aconitase both show similar abundance patterns, which were distinct from that of the median TCA protein (Fig. 5C, triangles and squares respectively). These enzymes catalyze the sequential conversion of oxaloacetate to citrate and then to cis-aconitate.
The glycolysis pathway is also used for energy generation and storage. Of the 54 proteins in the glycolysis pathway, we observed 28. Of these, 24 showed an increased abundance with time. The median abundance of this biochemical pathway is shown (Fig. 4E, black line). The first enzyme in this pathway, which converts sugar to pyruvate, is phosphotransferase, whose temporal pattern matches this pathway’s median protein abundance (Fig. 5E, circles). However, the protein showing greatest overlap with the median abundance is triosephosphate isomerase (Fig. 5E, triangle, R = 0.98). This protein interconverts dihydroxyacetone phosphate and glyceraldehyde-3-phosphate (GAP). Subsequent conversion of GAP to a reactive methylglyoxal compound can lead to erroneous modifications of proteins and DNA. Deficiencies in triosephosphate isomerase have been linked to neurological pathologies [35]. One protein that shows a decreased abundance with time is aldose 1 epimerase (Fig. 5E, squares). This protein is involved in the conversion of alpha-glucose to beta-glucose, either of which can enter early in the glycolysis pathway.
3.5. Effect of aging on proteostasis: the ribosome and the proteasome
Cellular proteostasis is the combined processes of protein production, maintenance and degradation. Proper proteostasis is critical for the optimal function of complex biological pathways [36]. Proteostatic dysregulation has been observed with aging as well as numerous diseases. One example is the accumulation of plaques, observed in Alzheimer’s Disease, as a result of aberrant protein degradation [37]. Given the complexity of cellular proteostasis, it is important to develop a better understanding of how this process is regulated in an age dependent manner for otherwise healthy individuals. This will provide a framework for interpreting which changes are critical during disease progression.
Overall, we observed 330 proteins which showed a 50% or greater decrease in abundance at some time point relative to day zero. Gene ontology analysis revealed that these proteins were enriched for a range of biological functions, including pathways for the ribosome and proteasome, as well as heat shock proteins. Given the central role of proteasomal processing in proteostasis, we examined these components in more detail. In total, we quantified the abundance of 28 of the 50 annotated proteasome components. Of those components, all 28 decreased in abundance (Fig. 4B). Both core (13/13) and regulatory (15/15) subunits were down regulated over time. Fig. 5B shows the core alpha subunit 4 and beta subunit 6 as well as the regulatory ER ATPase Ter94 protein. Ter94, which has previously been linked to apoptosis [38], showed one of the largest decreases in abundance of proteasomal proteins.
Of the 94 D. melanogaster proteins annotated in the ribosomal pathway, we quantified 48. All of the observed ribosomal proteins showed a decrease in abundance of at least 50% as the organism aged (Fig. 4D), with a median decrease of 53%. The overall trend of ribosomal abundance is shown in Fig. 4D, where the median ribosomal protein level is shown as a black line. For additional reference, ribosomal proteins S17 and S24 are shown (Fig. 5D), as circles and squares, respectively. The S24 ribosomal protein, which has previously been linked to ribosomal biogenesis via ribosomal RNA pre-processing [39], showed the most drastic decreases amongst ribosomal proteins.
Because proper folding is critical for protein function, organisms have evolved a series of mechanisms to maintain this aspect of protein structure. These mechanisms involve pathways to aide in protein folding, refolding and (in the case of misfolded proteins) unfolding. Chaperones are one class of proteins which promote protein folding during expression and coordinate refolding events during the protein’s lifespan. Heat shock proteins (HSP), a sub-class of chaperones, mediate a host of functions including preventing aggregation, facilitating protein transport to organelles, refolding misfolded proteins and binding partially unfolded proteins [40]. We analyzed 14 of the 37 annotated heat shock proteins (in Uniprot), and seven showed a temporal decrease in abundance. This group of proteins, while sharing a common set of functions, are not for the most part physically associated. As a consequence, we would not expect their temporal abundance patterns to be as highly correlated as those of the proteasome or ribosome. Overall, the median heat shock protein abundance showed an overall decrease at all time points relative to day zero, shown as a black line in Fig. 4F. A proteasome accessory protein, small heat shock protein 27 decreased 93%, showing the greatest change in protein abundance of this group (Fig. 5F, squares). This protein has been shown to accelerate the rate of degradation by the proteasome via enhancement of the NF-κB pathway [41]. In contrast to other HSPs, HSP60c and the HSP 70 kDa protein showed small increases in abundance. HSP60c is incorporated into the mitochondrial matrix, where it recognizes misfolded or damaged proteins (i.e. by oxidation) for refolding or removal [42]. It is particularly interesting that this HSP, one of the few incorporated into mitochondria, shows a similar temporal pattern to that of mitochondrial proteins, rather than correlating with other HSPs. The HSP 70 kDa protein has been linked to oxidative stress [43] and response to neuronal protein aggregation [44].
3.6. Age dependent loss of proteostasis
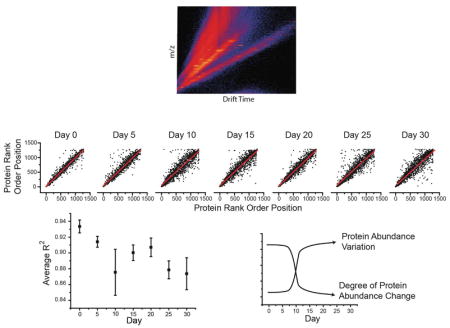
We hypothesized our data could demonstrate that the ability to maintain proteostasis decreases as an organism becomes older [8]. Multiple biological replicates at each time point allowed us to ask if protein variation between individuals increased with age. For a given protein and time point, we measured the variation in protein abundance between biological replicates and plotted this as a function of time, an example of which is shown in Fig. 6A for ribosomal protein subunit 31. We observed a notable increase in the variation of this protein particularly at day 30. To investigate protein level changes more globally, we calculated the relative standard deviation (%RSD) for each protein across the biological replicates at a given day. For each protein, we determined the day at which it was most variable and plotted this data as a histogram (Fig. 6B). We observe a clear day dependent increase in protein variability. Day ten and day 30 showed the highest degree of variability with ~300 proteins being most variable at each of these days.
Fig. 6.

Loss of correlated protein abundance with time. (A) A scattergram of the average (black line) and individual protein-level abundance measurements (black points) for 60S ribosomal protein L31 (RpL31) across four biological replicates. RpL31 shows an increased spread of abundance with increasing time. (B) To systematically assess this, we calculated the intraday relative standard deviation for all proteins at all days. For each protein, we then determined the day at which it displayed the largest %RSD. This data was then plotted as a histogram where for each day the total number of proteins having highest variation at that day was shown. The rank order of a protein within one replicate can be compared to its order in the other replicates at the same day. An example of this for each day is shown as a scattergram (C). To get a broader picture of the degree to which individual proteins vary with age, we calculated the R2 values for the graphs shown in C along with the other possible pairs of replicates not shown. These average R2 values at a given day are shown as a scatter plot, with error bars representing SEM (D). These results support a hypothesis where protein abundance variation increases during aging (E).
It has been proposed that as an organism ages, the proteome changes in response to the unique environment experienced by that individual [45]. Overall, we observe that proteins become more variable with time, particularly when compared to day zero post-eclosion. Changes in protein expression can have variable effects on protein pathways depending on whether other proteins in that pathway also show a similar change in abundance. We sought to determine if our data could provide evidence to address this issue. We acquired four biological replicates at each day throughout the time course, allowing for us to determine the similarity between replicate datasets. One way to quantify their similarity is to rank order the observed proteins by their abundance. We can then plot these data as a scatter plot (Fig. 6C) and calculate the R2 value. We show a single comparison at each day of our analysis. Interestingly, the scatter increases up to day 10 and then remains relatively constant during the remaining days. If we complete this analysis for the remaining biological replicates and days we generate six R2 values at each day. We then plot the averaged R2 value (Fig. 6D), which shows a decrease as the fly ages.
4. Discussion
We successfully developed a method that reproducibility detects and quantifies 1281 proteins from D. melanogaster. Our temporal proteome experiments included seven time points between day zero and 30 post-eclosion, with a total of four biological replicates. These data allowed us to characterize co-regulated protein abundance patterns within specific biological pathways. We observed a decrease in both ribosomal and proteasomal protein abundances, which we believe results in increased stress on an organism’s ability to maintain proteostasis. We provide evidence that impairments in maintaining the relationship between protein synthesis and catabolism should be considered a hallmark of aging.
4.1. Comparison to previous studies
Since the original analysis by Sowell et al. [10] many technological advances have increased our ability to profile an organism’s proteome. The initial success of that study was based on multiple analytical techniques, including fractionation using strong cation exchange. Furthermore, spectral counting allowed for approximate quantification of the identified species. Our current technique, based upon Top3 quantification appears to be much more accurate for these types of samples. Furthermore, instrument advances have also allowed for increased sensitivity and decreased analysis time. While the original work by Sowell et al. helped set the foundation for proteome analysis across lifespan, the presented data advances and showcases the power of this technique to interrogate important biological aging phenotypes.
4.2. Technological advancement
Our results demonstrate the utility of uHDMSe for large scale D. melanogaster protein quantification. Furthermore, we describe a method to systematically define the optimal collision energy across an IMS separation. Our optimization is based off of the relationship between collision energy and m/z combined with that between m/z and drift time. The resulting collision energy profile maximizes the amount of informative fragmentation, thereby increasing the number of peptides identified in a single acquisition. This approach to optimize collision energy can be applied across instrument platforms. We expect these settings to vary for different instruments and laboratories (due to factors such as gas pressures, voltage stability, etc.), however once optimized should be independent of sample origin.
In part due to the increased technological capabilities, our data shows a ~103 dynamic range, which agrees well with that reported for other organisms and tissues [46]. Biochemical pathways function in concert, such as subunits of the ribosome during protein translation. It is therefore not surprising that proteins belonging to a pathway show similar abundances at a particular age. This observation could potentially be used to further define functional protein pathways using proteomic data [47]. Future work should be done to determine if age dependent changes in abundance spread, provide insight into the overall fitness or efficacy of particular pathways.
4.3. Analysis of the temporal proteome pattern
In general, one would expect protein species involved in specific biological pathways to show similar temporal protein abundance patterns. Our quantitative data were accurate enough to demonstrate such patterns. Across a range of biological processes, we show that components of a specific pathway correlate strongly with others in that pathway. Of course, most biochemical pathways have multiple points of regulation. Therefore, it seems likely that subsets of an individual biochemical pathway may be differentially regulated. We observe this phenomenon particularly in the case of glycolytic and heat shock proteins, where not all components showed the same time dependent changes.
We observe that pathways involved in both energy generation and metabolism, mainly localized to the mitochondria, are highly correlated and increase with age, consistent with previous transcriptomic analysis in mouse brain [48]. The oxidizing environment within the mitochondria could lead to cumulative protein or mitochondrial DNA damage. This damage could lead to mutations in amino acid sequence, and further cause protein misfolding, or decreased enzymatic function. The overall increase in protein abundance may be a mechanism to compensate for the decreased activity over time in the oxidizing environment. One other explanation is that energy requirements in adult flies increase with age, and eventually reach a steady state. To address potential impairments in mitochondrial function [49] or the increased energy demands [50] one could directly characterize the activity level of isolated mitochondria as a function of age [51].
It has become increasingly appreciated that protein aggregation, occurring via many mechanisms, plays a key role in aging and disease. The most fundamental of these mechanisms include protein unfolding [52], and deficiencies in protein degradation pathways [53,54]. Our data shows that the abundance of proteins involved in the ribosome as well as those involved in the proteasome decrease with age. Proteostasis can be thought of as a balance between protein production via the ribosome, and protein degradation via the proteasome. Our data points toward two possible mechanisms connecting the changes we observed in the ribosome and the proteasome. In the first, age dependent decreases in ribosomal protein abundance could be sensed by mechanisms regulating proteasome levels, with the decreased level of protein synthesis causing a decreased need for protein degradation. Alternatively, deficits in proteasome level or function would lead to increased protein accumulation, activating a similar negative homeostatic feedback for protein synthesis via the ribosome. An isotopic pulse-chase analysis could delineate the extent to which ribosomal and proteasomal function is impaired with age.
4.4. Misregulation increases with aging
Our data demonstrates that as an organism ages, its proteome become less regulated, and therefore proteome dysregulation could be a phenotype associated with normal, disease-free aging. One explanation for this dysregulation is stochastic epigenetic changes which accumulate with time [8,45,55]. This decreased correlation becomes apparent at a fairly early age, approximately day 10–15 (Fig. 6). This time range corresponds to D. melanogaster’s peak fecundity [56]. It is interesting to speculate that when an organism can no longer reproduce, there is little evolutionary pressure to maintain specific protein temporal patterns. Additional misregulation, as may occur during specific disease states, would exacerbate the proteome changes noted above. Our data represents an initial attempt to probe this phenomenon, and further investigations of the interplay of disease and aging will be particularly revealing.
In general, protein abundances show the largest changes between day 0 and day 15, with changes after day 15 being of smaller magnitude (Figs. 4 and 5). This likely reflects the fact that younger organisms in general show a greater degree of change with time compared to their older counterparts. In addition, we demonstrate that older organisms show significantly more variations in their proteome than younger organisms (Fig. 6). While older organisms may be changing more slowly, the effects of individual deviations in protein expression have had more time to accumulate, and thus become greater in magnitude.
5. Conclusion
A combination of liquid chromatography, ion mobility spectrometry, mass spectrometry and database searching techniques were used to map the proteome isolated from the heads of D. melanogaster. A key aspect of our analysis was the systematic optimization of drift specific collision energies promoting successful precursor fragmentation. We determined the abundance patterns for 1281 proteins, which vary over a factor of ~103 throughout the adult organism’s lifespan. Examination of the data revealed individual biochemical pathways displayed unique temporal patterns. We focused on those changes involved in the ribosome, proteasome, heat shock proteins, oxidative phosphorylation, TCA cycle, and glycolysis. These data also show protein abundances from the younger fly populations were more similar to each other than corresponding abundances from the older fly populations. In other words, younger flies were very similar to each other, but older flies were more unique. This suggests that proteostasis becomes less regulated with age in the population. Proteome dysregulation has previously been linked with disease and aging [8,57]. Finally, we hope this study will establish an updated frame work to probe the intimate connection between disease and aging.
Supplementary Material
Acknowledgments
We thank Melissa Phelps and Robert (Tank) Eisman for outstanding scientific support and insightful discussion and acknowledge the Bloomington Drosophila Stock Center for providing the D. melanogaster stocks used in these experiments. This research was funded by the National Institutes of Health grant 5R01GM117207.
References
- 1.Chintapalli VR, Wang J, Dow JAT. Nat Genet. 2007;39(6):715. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 2.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. Genome Res. 2001;11(6):1114. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandey UB, Nichols CD. Pharmacol Rev. 2011;63(2):411. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fossgreen A, Brückner B, Czech C, Masters CL, Beyreuther K, Paro R. PNAS. 1998;95(23):13703. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. Mol Cell Neurosci. 2004;26(3):365. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Neuron. 2003;39(3):409. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 7.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Cell. 1997;90(3):549. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 8.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. Cell. 2013;153(6):1194. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner RS, Suzuki N, Chyung ASC, Younkin SG, Lee VMY. J Biol Chem. 1996;271(15):8966. doi: 10.1074/jbc.271.15.8966. [DOI] [PubMed] [Google Scholar]
- 10.Sowell RA, Hersberger KE, Kaufman TC, Clemmer DE. J Proteome Res. 2007;6(9):3637. doi: 10.1021/pr070224h. [DOI] [PubMed] [Google Scholar]
- 11.Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, Hartl FU. Cell. 2015;161(4):919. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson RAS, Kellie JF, Kaufman TC, Clemmer DE. Mech Ageing Dev. 2010;131(9):584. doi: 10.1016/j.mad.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taraszka JA, Gao X, Valentine SJ, Sowell RA, Koeniger SL, Miller DF, Kaufman TC, Clemmer DE. J Proteome Res. 2005;4(4):1223. doi: 10.1021/pr050038g. [DOI] [PubMed] [Google Scholar]
- 14.Pérez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, Ward W, Richardson A, Chaudhuri A. PNAS. 2009;106(9):3059. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shliaha PV, Bond NJ, Gatto L. J Proteome. 2013;12(6):2323. doi: 10.1021/pr300775k. [DOI] [PubMed] [Google Scholar]
- 16.Distler U, Kuharev J, Navarro P, Levin Y, Schild H, Tenzer S. Nat Publ Group. 2014;11(2):167. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 17.Da Wei Huang, Sherman BT, Lempicki RA. Nat Protoc. 2009;4(1):44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.de Hoon MJL, Imoto S, Nolan J, Miyano S. Bioinformatics. 2004;20(9):1453. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 19.Hilderbrand A. J Am Soc Mass Spectrom. 2003:1. doi: 10.1016/j.jasms.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Lee YJ, Hoaglund-Hyzera CS, Srebalus Barnes CA, Hilderbrand AE, Valentine SJ, Clemmer DE. J Chromatogr B. 2002;782(1–2):343. doi: 10.1016/s1570-0232(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 21.Schubert OT, Gillet LC, Collins Ben C, Navarro P, Rosenberger G, Wolski WE, Lam H, Amodei D, Mallick P, MacLean B, Aebersold R. Nat Protoc. 2015;10(3):426. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 22.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, Geromanos S. Anal Chem. 2005;77(7):2187. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 23.Silva JC, Gorenstein MV, Li GZ, Vissers JPC, Geromanos SJ. Mol Cell Proteomics. 2006;5(1):144. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Hoaglund-Hyzer CS, Li J, Clemmer DE. Anal Chem. 2000;72(13):2737. doi: 10.1021/ac0000170. [DOI] [PubMed] [Google Scholar]
- 25.Merenbloom SI, Koeniger SL, Valentine SJ, Plasencia MD, Clemmer DE. Anal Chem. 2006;78(8):2802. doi: 10.1021/ac052208e. [DOI] [PubMed] [Google Scholar]
- 26.Baker ES, Burnum Johnson KE, Ibrahim YM, Orton DJ, Monroe ME, Kelly RT, Moore RJ, Zhang X, Théberge R, Costello CE, Smith RD. Proteomics. 2015;15(16):2766. doi: 10.1002/pmic.201500048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CAK, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, Feilitzen, von K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F, Uhlen M. Mol Cell Proteomics. 2014;13(2):397. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borg J, Campos A, Diema C, Omenaca N, de Oliveira E, Guinovart J, Vilaseca M. Clin Proteomics. 2011;8(1):6. doi: 10.1186/1559-0275-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong SY, Ng LT, Ng LF, Inoue T, Tolwinski NS. PLoS One. 2016;11(12):e0168752. doi: 10.1371/journal.pone.0168752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kauppila TES, Kauppila JHK, Larsson NG. Cell Metab. 2016;25(1):1. [Google Scholar]
- 31.Phillips JD, Graham LA, Trumpower BL. J Biol Chem. 1993;268(16):11727. [PubMed] [Google Scholar]
- 32.Schmitt ME, Trumpower BL. J Biol Chem. 1991;266(23):14958. [PubMed] [Google Scholar]
- 33.Cvejic S, Zhu Z, Felice SJ, Berman Y, Huang XY. Nat Cell Biol. 2004;6(6):540. doi: 10.1038/ncb1133. [DOI] [PubMed] [Google Scholar]
- 34.Song W, Ranjan R, Dawson-Scully K, Bronk P, Marin L, Seroude L, Lin YJ, Nie Z, Atwood HL, Benzer S, Zinsmaier KE. Neuron. 2002;36(1):105. doi: 10.1016/s0896-6273(02)00932-7. [DOI] [PubMed] [Google Scholar]
- 35.Gnerer JP, Kreber RA, Ganetzky B. PNAS. 2006;103(41):14987. doi: 10.1073/pnas.0606887103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartl FU, Bracher A, Hayer-Hartl M. Nature. 2011;475(7356):324. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 37.Greeve I, Kretzschmar D, Tschäpe JA, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. J Neurosci. 2004;24(16):3899. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, Kakizuka A. Cell Death Differ. 2002;9(3):264. doi: 10.1038/sj.cdd.4400955. [DOI] [PubMed] [Google Scholar]
- 39.Choesmel V, Fribourg S, Aguissa-Touré AH, Pinaud N, Legrand P, Gazda HT, Gleizes PE. Hum Mol Genet. 2008;17(9):1253. doi: 10.1093/hmg/ddn015. [DOI] [PubMed] [Google Scholar]
- 40.Sarto C, Binz PA, Mocarelli P. Electrophoresis. 2000;21(6):1218. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1218::AID-ELPS1218>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 41.Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantôme A, Plenchette S, Khochbin S, Solary E, Garrido C. Mol Cell Biol. 2003;23(16):5790. doi: 10.1128/MCB.23.16.5790-5802.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruschi SA, Lindsay JG, Crabb JW. PNAS. 1998;95(23):13413. doi: 10.1073/pnas.95.23.13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calabrese V, Scapagnini G, Ravagna A, Fariello RG, Giuffrida Stella AM, Abraham NG. J Neurosci Res. 2002;68(1):65. doi: 10.1002/jnr.10177. [DOI] [PubMed] [Google Scholar]
- 44.Evans CG, Wisén S, Gestwicki JEJ. Biol Chem. 2006;281(44):33182. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 45.Jaenisch R, Bird A. Nat Genet. 2003;33(3s):245. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 46.Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1(11):845. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 47.Dhondt I, Petyuk VA, Bauer S, Brewer HM, Smith RD, Depuydt G, Braeckman BP. Mol Cell Proteomics. 2017;16(9):1621. doi: 10.1074/mcp.RA117.000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang CH, Tsien JZ, Schultz PG, Hu Y. PNAS. 2001;98(4):1930. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Navarro A, Boveris A. Am J Physiol Cell Physiol. 2007;292(2):C670. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein S, Ballantyne SR, Robson AL, Moerman EJ. J Cell Physiol. 1982;112(3):419. doi: 10.1002/jcp.1041120316. [DOI] [PubMed] [Google Scholar]
- 51.Chandel NS, Budinger G, Choe SH, Schumacker PT. J Biol Chem. 1997;272(30):18808. doi: 10.1074/jbc.272.30.18808. [DOI] [PubMed] [Google Scholar]
- 52.Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, Beyreuther K, Masters CL, Barrow CJ, Collins SJ, Cappai R. J Neurochem. 1999;73(4):1557. doi: 10.1046/j.1471-4159.1999.0731557.x. [DOI] [PubMed] [Google Scholar]
- 53.Dawson TM, Dawson VL. Science. 2003;302(5646):819. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 54.Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. PNAS. 2000;97(6):2898. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fraga MF, Esteller M. Trends Genet. 2007;23(8):413. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Lints FA, Lints CV. Exp Gerontol. 1971;6(6):417. doi: 10.1016/0531-5565(71)90021-0. [DOI] [PubMed] [Google Scholar]
- 57.Ben-Zvi A, Miller EA, Morimoto RI. PNAS. 2009;106(35):14914. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.