

Supplemental Digital Content is available in the text.
Keywords: acetylsalicylic acid, aspirin, endotoxemia, endotoxin tolerance, immunoparalysis, sepsis
Abstract
Objective:
To investigate immunostimulatory effects of acetylsalicylic acid during experimental human endotoxemia and in sepsis patients.
Design:
Double-blind, randomized, placebo-controlled study in healthy volunteers and ex vivo stimulation experiments using monocytes of septic patients.
Setting:
Intensive care research unit of an university hospital.
Subjects:
Thirty healthy male volunteers and four sepsis patients.
Interventions:
Healthy volunteers were challenged IV with endotoxin twice, at a 1-week interval, with each challenge consisting of a bolus of 1 ng/kg followed by continuous administration of 1 ng/kg/hr during 3 hours. Volunteers were randomized to acetylsalicylic acid prophylaxis (80 mg acetylsalicylic acid daily for a 14-d period, starting 7 d before the first endotoxin challenge), acetylsalicylic acid treatment (80 mg acetylsalicylic acid daily for the 7-d period in-between both endotoxin challenges), or the control group (receiving placebo). Furthermore, monocytes of sepsis patients were incubated with acetylsalicylic acid preexposed platelets and were subsequently stimulated with endotoxin.
Measurements and Main Results:
Acetylsalicylic acid prophylaxis enhanced plasma tumor necrosis factor-α concentrations upon the first endotoxin challenge by 50% compared with the control group (p = 0.02) but did not modulate cytokine responses during the second endotoxin challenge. In contrast, acetylsalicylic acid treatment resulted in enhanced plasma levels of tumor necrosis factor-α (+53%; p = 0.02), interleukin-6 (+91%; p = 0.03), and interleukin-8 (+42%; p = 0.02) upon the second challenge, whereas plasma levels of the key antiinflammatory cytokine interleukin-10 were attenuated (–40%; p = 0.003). This proinflammatory phenotype in the acetylsalicylic acid treatment group was accompanied by a decrease in urinary prostaglandin E metabolite levels (–27% ± 7%; p = 0.01). Ex vivo exposure of platelets to acetylsalicylic acid increased production of tumor necrosis factor-α (+66%) and decreased production of interleukin-10 (–23%) by monocytes of sepsis patients.
Conclusions:
Treatment, but not prophylaxis, with low-dose acetylsalicylic acid, partially reverses endotoxin tolerance in humans in vivo by shifting response toward a proinflammatory phenotype. This acetylsalicylic acid–induced proinflammatory shift was also observed in septic monocytes, signifying that patients suffering from sepsis-induced immunoparalysis might benefit from initiating acetylsalicylic acid treatment.
Sepsis is defined as a life-threatening organ dysfunction caused by a dysregulated host response to infection (1). Despite decades of research, sepsis incidence is increasing (2), the associated healthcare costs are enormous, and sepsis represents the leading cause of in-hospital mortality (3). The pathophysiology of sepsis is highly complex, as the host response between patients is variable and can comprise both hyperinflammatory and immunosuppressive phenotypes (4). The latter phenotype, often referred to as sepsis-induced immunoparalysis, is increasingly recognized as the overriding immune dysfunction in septic patients (5, 6). Sepsis-induced immunoparalysis is characterized by impaired innate and adaptive immune responses, including exhaustion and apoptosis of lymphocytes, diminished capacity of monocytes and macrophages to produce cytokines, and decreased HLA-DR expression on the cell surface of monocytes (5, 7, 8). This suppressed immune function may contribute to adverse outcome, as the host is unable to control the primary infection, and is increasingly susceptible toward secondary infections (6, 9–13). As such, these patients might benefit from immune stimulatory therapy to restore host defence.
In several large observational studies, prehospital use of low-dose acetylsalicylic acid (ASA or aspirin) was associated with reduced mortality in patients admitted to the ICU with sepsis (14–21). The most recent meta-analysis performed in 17,065 patients showed a 7% reduction (range, 2–12%) in mortality in patients taking aspirin prior to sepsis onset (22). A large randomized controlled trial (AspiriN To Inhibit SEPSIS [ANTISEPSIS]) is currently being conducted to evaluate the effect of prehospital ASA use on outcome in the sepsis population (23).
Although it is often proposed that antiinflammatory properties of ASA may account for the observed beneficial effects, ASA has been shown to potentiate, not attenuate, leukocytic cytokine production. For instance, previous data of our group show that a short course of oral aspirin in volunteers increases ex vivo cytokine production capacity (24). Furthermore, a 7-day treatment with low-dose ASA in healthy volunteers resulted in a more pronounced increase in plasma levels of proinflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-8 during experimental human endotoxemia (25). Although subjectively counterintuitive, these findings are to be expected considering the inhibitory impact of aspirin on prostaglandins, a known negative regulator of (proinflammatory) cytokine production (26, 27). This indicates that low-dose ASA exerts proinflammatory effects during systemic inflammation. As a consequence, ASA may alleviate sepsis-induced immunoparalysis, which could contribute to the improved survival found in the abovementioned observational studies. However, the effects of ASA on the development of immunoparalysis have hitherto not been investigated.
In the present study, we investigated whether a course of low-dose ASA can prevent or reverse immunoparalysis in humans in vivo. Similar to previous work of our group, we used repeated experimental human endotoxemia as a model for sepsis-induced immunoparalysis (28). We investigated whether ASA can prevent immunoparalysis by treating subjects 7 days before the first challenge until the second endotoxin challenge. Also, ASA’s potential to reverse immunoparalysis was investigated by treating subjects after the first endotoxin challenge. Furthermore, we investigated whether this ASA-induced pathway is still amenable in monocytes of septic patients.
MATERIALS AND METHODS
Subjects
After approval of the local ethics committee (CMO Arnhem-Nijmegen; reference no. NL57410.091.16 and 2016–2550), 30 healthy male volunteers between 18–35 years old were recruited. All subjects gave written informed consent and medical history, physical examination, laboratory tests, and a 12-leads electrocardiogram did not reveal any abnormalities. Smoking, medication use (in particular the use of cyclooxygenase [COX] inhibitors), previous participation in experimental human endotoxemia, or signs of acute illness within 3 weeks prior to the start of the study were exclusion criteria. All study procedures were performed in accordance with the declaration of Helsinki, including the latest revisions. The study was registered at ClinicalTrials.gov (NCT02922673). For ex vivo stimulation experiments, we obtained monocytes from four patients with sepsis according to the Sepsis-3 criteria (1) and platelets from three healthy volunteers. These experiments were approved by the local ethics committee (CMO Arnhem-Nijmegen; reference number 2016–2923 and 2010–104).
Endotoxemia Study Design
We performed a randomized, double-blind, placebo-controlled study, of which the design is depicted in Figure 1. All subjects were challenged IV with endotoxin twice, on study days 7 and 14. The second challenge was used to quantify the extent of endotoxin tolerance. Study medication was prescribed in two 7-day courses prior to both endotoxin challenges and was encapsulated to maintain the double-blinded design. Subjects were randomized to one of three groups (n = 10 per group). The ASA prophylaxis group received 80 mg ASA once daily starting 1 week prior to the first endotoxin challenge and continuing until the second endotoxin challenge. The ASA treatment group received placebo once daily in the week prior to the first endotoxin challenge and 80 mg ASA once daily in the week prior to the second endotoxin challenge. The control group received placebo in the weeks prior to both endotoxin challenges. In both the ASA prophylaxis and ASA treatment groups, the first ASA dose administered was a loading dose of 160 mg, consistent with our previous work (25) and clinical use of ASA. Therapy compliance was verified by diaries, pill counts, and urinary 11-dehydro-thromboxane (TX) B2 concentrations.
Figure 1.
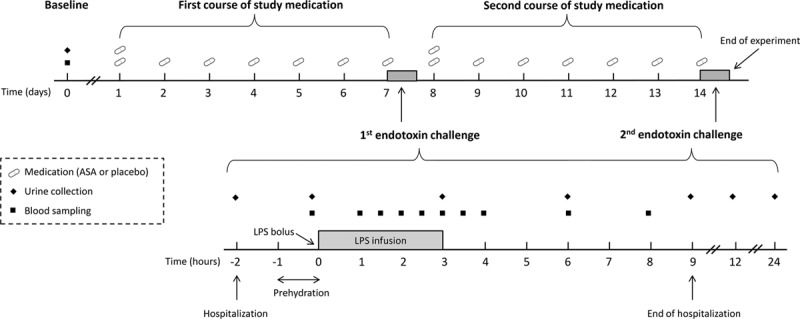
Endotoxemia study design. Procedures on the day of the first and second endotoxin challenge are similar. ASA = acetylsalicylic acid, LPS = lipopolysaccharide.
All additional study procedures, including ex vivo experiments with monocytes of sepsis patients, and analysis methods are provided in the Supplemental Digital Content.
RESULTS
Subject Characteristics
Baseline demographic characteristics of the study population are listed in Supplementary Table 1 (Supplemental Digital Content 1, http://links.lww.com/CCM/E307) and reveal no significant differences between the three study groups. Apart from the well-known endotoxin-induced symptoms, no adverse events occurred during the study. There were no baseline differences in urinary 11-dehydro-TXB2 levels between groups (Fig. S1, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). Therapy compliance was 100%, as verified by diaries and pill counts and confirmed by urinary 11-dehydro-TXB2 concentrations, which were greatly reduced in all ASA-treated subjects (before ASA: 184 ± 28, after ASA: 41 ± 8 pg/mL/creat; p < 0.0001) (Fig. S1, Supplemental Digital Content 1, http://links.lww.com/CCM/E307).
Plasma Cytokines
The first endotoxin challenge resulted in a profound inflammatory response, illustrated by an increase in plasma levels of all cytokines (TNF-α, IL-6, IL-8, IL-10, and IL-1 receptor antagonist [RA] are depicted in Fig. 2, and monocyte chemoattractant protein [MCP]-1, macrophage inflammatory protein [MIP]-1α, and MIP-1β in Fig. S2, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). Individual data of the area under the time-cytokine concentration curves are depicted in Figure S3 (Supplemental Digital Content 1, http://links.lww.com/CCM/E307). Plasma levels of IL-1β, IL-4, and IL-13 were below the detection limit of the assay in the great majority of samples obtained on both endotoxemia days (data not shown). Prophylactic use of ASA enhanced plasma concentrations of TNF-α by 50% compared with the control group upon the first endotoxin challenge (p = 0.02) (Fig. 2A; and Fig. S3A, Supplemental Digital Content 1, http://links.lww.com/CCM/E307) but did not significantly affect the other cytokines measured. Following the second endotoxin challenge, the development of endotoxin tolerance was illustrated by a severely blunted plasma cytokine response upon the second endotoxin challenge in the control group (decrease in area under the time concentration curve of TNF-α: 58% ± 11%, p = 0.003) (Fig. 2A); IL-6: 73% ± 11%; p = 0.004 (Fig. 2B); IL-8: 65% ± 10%, p = 0.003 (Fig. 2C); IL-10: 56% ± 11%; p = 0.0034 (Fig. 2D); IL-1RA: 54% ± 14%; p = 0.003 (Fig. 2E); MCP-1: 38% ± 11%; p = 0.007 (Fig. S2A, Supplemental Digital Content 1, http://links.lww.com/CCM/E307); MIP-1α: 48% ± 5%; p = 0.001 (Fig. S2B, Supplemental Digital Content 1, http://links.lww.com/CCM/E307); MIP-1β: 55% ± 11%; p = 0.01 (Fig. S2C, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). Prophylactic treatment with ASA did not significantly affect the development of endotoxin tolerance (Fig. 2; and Figs. S2 and S3, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). In the ASA treatment group, endotoxin tolerance was less pronounced, illustrated by significantly higher plasma levels of TNF-α (+53%, p = 0.02) (Fig. 2A; and Fig. S3A, Supplemental Digital Content 1, http://links.lww.com/CCM/E307), IL-6 (+91%; p = 0.03) (Fig. 2B; and Fig. S3B, Supplemental Digital Content 1, http://links.lww.com/CCM/E307), and IL-8 (+42%; p = 0.02) (Fig. 2C; and Fig. S3C, Supplemental Digital Content 1, http://links.lww.com/CCM/E307) upon the second endotoxin challenge compared with the control group. The shift toward a more proinflammatory phenotype in the treatment group was further exemplified by lower plasma levels of the key antiinflammatory cytokine IL-10 compared with the control group (–40%; p = 0.003) (Fig. 2D; and Fig. S3D, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). No between-group differences were found upon the second endotoxin challenge for IL-1RA, MCP-1, MIP-1α, and MIP-1β (Fig. 2E; and Figs. S2 and S3E–H, Supplemental Digital Content 1, http://links.lww.com/CCM/E307).
Figure 2.
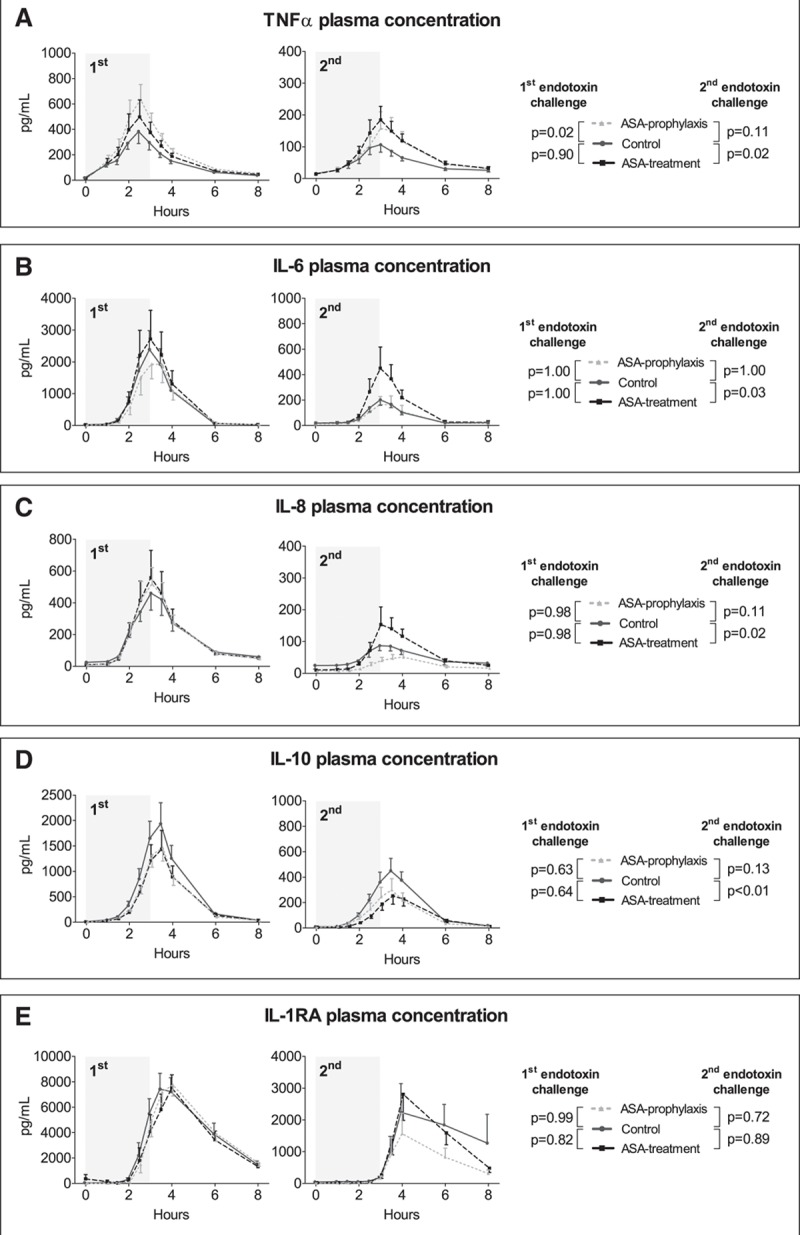
Plasma levels of tumor necrosis factor (TNF)-α (A), interleukin (IL)-6 (B), IL-8 (C), IL-10 (D), and IL-1 receptor antagonist (RA) (E), upon the first and second endotoxin challenge. Cytokine concentrations over time are depicted as mean and sem. The gray area indicates the 3-hr endotoxin administration period. p values represent the interaction term of repeated measures two-way analysis of variance. ASA = acetylsalicylic acid.
HLA-DR Expression on Monocytes
ASA prophylaxis did not affect mHLA-DR expression in the absence of systemic inflammation (Fig. 3A). The first endotoxin challenge expectedly resulted in decreased mHLA-DR expression in subjects not (yet) exposed to ASA (the control and treatment groups, p < 0.01 and p < 0.001), whereas no significant decrease was observed in the ASA prophylaxis group. The second endotoxin challenge resulted in significantly attenuated mHLA-DR expression levels in all groups (p < 0.001) (Fig. 3A).
Figure 3.
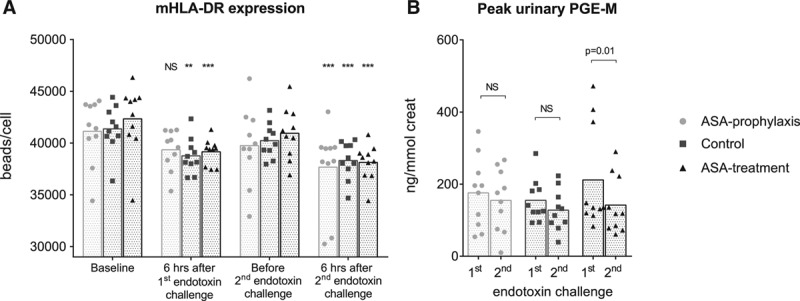
A, Monocytic human leukocyte antigen-DR (mHLA-DR) expression before (baseline) and 6 hr after start of the first and second endotoxin challenge. B, Peak urinary prostaglandin E metabolite (PGE-M) concentrations upon the first and second endotoxin challenge. Data are depicted as bar (mean) and scatter plots. **p < 0.01 and ***p < 0.001 compared with baseline in A (calculated using repeated measures one-way analysis of variance with Dunnett’s post hoc tests). Data (B) were analyzed using paired Student’s t tests. ASA = acetylsalicylic acid, NS = non significant.
PGE-M
We did not observe differences in baseline urinary prostaglandin E metabolite (PGE-M) levels between the three groups (data not shown). Endotoxemia resulted in increased urinary PGE-M concentrations in all groups upon both endotoxin challenges, with peak levels observed in urine collected during the first 3 hours after the start of endotoxin administration. Peak PGE-M levels did not differ upon the first and second challenge in the control and ASA prophylaxis groups (Fig. 3B). In the ASA treatment group, peak PGE-M concentrations were significantly attenuated upon the second challenge compared with the first (–27% ± 7%; p = 0.01) (Fig. 3B).
Oxidative Stress
Plasma levels of malondialdehyde, a product of lipid peroxidation due to oxidative stress, increased upon both endotoxemia days. Malondialdehyde concentrations did not differ between the first and second endotoxin challenges within any of the groups. Furthermore, no between-group differences were observed (Fig. 4).
Figure 4.
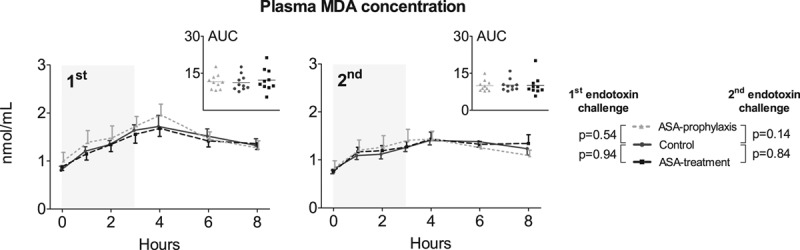
Oxidative stress expressed by plasma levels of malondialdehyde (MDA) upon the first and second endotoxin challenge. MDA concentrations over time are depicted as mean and sem. The inserts depict the individual area under the time concentration curves (AUC) data expressed as nmol × hr/mL. The gray area indicates the 3-hr endotoxin administration period. p values represent the interaction term of repeated measures two-way analysis of variance. ASA = acetylsalicylic acid.
Hematologic and Clinical Variables
Transient changes in hematologic variables were observed during both challenges; however, changes were less outspoken upon the second challenge. ASA prophylaxis or treatment did not affect endotoxin-induced changes in cell counts or differentiation (Fig. S4, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). The first endotoxin challenge caused a profound increase in body temperature, symptoms, and heart rate and a decrease in mean arterial pressure (MAP) (Fig. S5, Supplemental Digital Content 1, http://links.lww.com/CCM/E307). The presence of endotoxin tolerance was characterized by less pronounced effects on clinical variables upon the second challenge in the control group (peak body temperature of 39.3°C ± 0.3°C vs 38.1°C ± 0.2°C, peak symptom score of 8 ± 1 vs 3 ± 1, peak heart rate of 102 ± 3 vs 91 ± 2 beats/min, and nadir MAP 69 ± 2 vs 75 ± 3 mm Hg, for the first and second endotoxin challenge, respectively). There were no differences in vital variables between the ASA prophylaxis or ASA treatment and the control group upon both endotoxin challenges.
Ex Vivo Monocyte Stimulation Experiments
We previously showed that ASA potentiates monocytic TNF-α production in healthy volunteers in a platelet-dependent manner (25). We set out to investigate whether these effects also apply to monocytes of sepsis patients. Patient characteristics (n = 4) are listed in Supplementary Table 2 (Supplemental Digital Content 1, http://links.lww.com/CCM/E307). Three patients exhibited mHLA-DR levels less than 12,000 antibodies/cell, consistent with immune suppression (29). Compared to coincubation with naive platelets, coincubation with ASA preexposed platelets resulted in enhanced production of TNF-α (+66%, 1,365 pg/mL [333–2,744 pg/mL] vs 2,272 pg/mL [453–3,800 pg/mL]; p = 0.02) by lipopolysaccharide (LPS)-stimulated monocytes, whereas production of IL-10 was decreased (–23%, 363 pg/mL [100–400 pg/mL] vs 281 pg/mL [70–347 pg/mL]; p < 0.001) (Fig. 5). Coincubation of monocytes with ASA alone (without platelets) did not affect LPS-stimulated cytokine production (data not shown).
Figure 5.
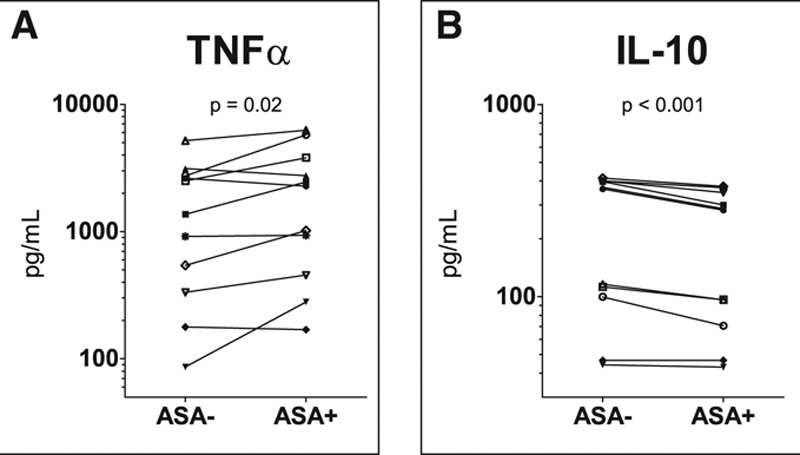
Production of tumor necrosis factor (TNF)-α (A) and interleukin (IL)-10 (B) by monocytes obtained from septic patients after ex vivo 24 hr stimulation with lipopolysaccharide. Differences in cytokine production of monocytes coincubated with ASA preexposed platelets (ASA+) and naive platelets (ASA–) were analyzed using paired Student’s t tests on log-transformed data. ASA = acetylsalicylic acid.
DISCUSSION
In the current study, we show that continuous administration of endotoxin to healthy volunteers results in the development of profound endotoxin tolerance, exemplified by a severely blunted cytokine response, fewer symptoms, and less changes in vital variables upon the second endotoxin challenge 1 week later. Prophylaxis with a clinically relevant dose of ASA (80 mg) resulted in an augmented proinflammatory response upon the first challenge but did not prevent or otherwise modulate the development of endotoxin tolerance observed during the second challenge. Treatment with a 7-day course of low-dose ASA in between both endotoxin challenges resulted in partial reversal of endotoxin tolerance, exemplified by a more proinflammatory phenotype upon the second endotoxin challenge compared with the control group. These data confirm previous reports that ASA augments the innate immune response (24, 25, 30, 31) and demonstrate that ASA treatment partially reverses endotoxin tolerance in humans in vivo. Furthermore, our ex vivo results reveal that this pathway is still amenable in sepsis patients, as the ASA-induced proinflammatory shift was also observed in monocytes obtained from these patients.
ASA treatment resulted in a distinct shift toward a more proinflammatory phenotype upon the second endotoxin challenge, exemplified by enhanced plasma levels of proinflammatory cytokines TNF-α, IL-6, and IL-8 compared with the control group, whereas the antiinflammatory IL-10 response was further decreased. Levels of another antiinflammatory cytokine, IL-1RA, did not reveal a pattern similar to IL-10 but were also not enhanced like the proinflammatory mediators. Interestingly, an imbalance in the IL-6/IL-10 ratio was shown to correlate with an increased risk toward secondary infections in sepsis patients, and this effect was already observed when levels of IL-6 and IL-10 deviated greater than 30–50% from the mean values obtained in sepsis patients (32). In our study, the ASA-induced changes in cytokine levels/production in healthy volunteers in vivo (TNF-α: +53%, IL-6: +91%, IL-8: +42%, and IL-10: –40%) and in septic patients ex vivo (TNF-α: +66% and IL-10: –23%) were in the same order of magnitude or greater. Therefore, it appears plausible that the observed ASA-induced shifts are of clinical relevance.
Although the beneficial effects of ASA are most frequently attributed to its antiinflammatory properties, proinflammatory characteristics of ASA were demonstrated decades ago in vitro (24, 30, 31), but also more recently in vivo by our group (25). These ASA-mediated effects appear to be dose specific, as in vitro work has revealed that low ASA dosages enhance cytokine responses, whereas very high dosages inhibit cytokine production (33). Low-dose ASA potently acetylates the constitutively active enzyme COX-1, thereby irreversibly inhibiting the conversion of arachidonic acids into prostaglandins (34). In addition, we have previously shown that platelets are critically involved in ASA’s proinflammatory effects, as coincubation of monocytes of healthy volunteers with ASA preexposed platelets augmented monocytic LPS-stimulated TNF-α production, whereas incubation of monocytes with ASA alone did not exert any effects (25). In the current study, we show that this ASA-induced pathway is still amenable in monocytes of sepsis patients and confirm that this effect is platelet dependent, as incubation with solely ASA did not exert any effects on monocytic cytokine production. Others have shown that platelets exert antiinflammatory effects during sepsis via the COX-1- prostaglandin E (PGE2)-pathway (35), and it is well established that PGE2 inhibits the release of TNF-α and IL-6 (27, 36), whereas it stimulates production of IL-10 (26). In accordance, we previously demonstrated that addition of PGE2 attenuated the proinflammatory effects of ASA in a dose-dependent manner (25). The present work reveals that the endotoxin-induced increase in urinary PGE-M concentrations (as a surrogate measure of in vivo systemic PGE2 synthesis) was significantly attenuated upon the second endotoxin challenge in the ASA-treatment group. Taken together, we hypothesize that the ASA-induced COX-1 inhibition results in decreased levels of PGE2 and thereby abrogates the suppressive effects on LPS-induced proinflammatory cytokine production, ultimately resulting in increased production of these mediators (27).
Beneficial effects of antiplatelet therapy, and in particular of ASA, in the context of sepsis have been extensively described (18, 20, 37). In a recent meta-analysis using propensity matching, the use of low-dose ASA prior to sepsis onset was associated with a 7% reduction in mortality (22). Similar results were found in a Bayesian network meta-analysis, where prehospital use of ASA was associated with lower hospital mortality in septic patients (38). In accordance with previous work of our group (25), the present study demonstrates that ASA prophylaxis enhances TNF-α response upon the first endotoxin challenge. However, we now show that ASA prophylaxis does not affect the development of endotoxin tolerance. This shift toward a more proinflammatory profile upon the initial challenge could nevertheless represent an explanation for the abovementioned beneficial effects observed in sepsis patients on prehospital low-dose ASA treatment, as a more potent host response may enhance clearance of pathogens (39). In addition, ASA did not result in increased oxidative stress, this might indicate that the ASA-induced shift toward a more proinflammatory phenotype does not come at the expense of increased collateral damage.
Unfortunately, most epidemiologic studies that have evaluated the effects of prehospital (low-dose) ASA on sepsis outcome merely assessed clinical variables. To the best of our knowledge, only one study assessed circulating cytokine levels, and no effects of prehospital ASA use on host-response variables were observed (40). Clearly, assessment of ASA’s putative immunomodulatory effects from these retrospective patient data is difficult. Interpatient variability is extensive, and it could be argued that, due to an enhanced initial immune response, prehospital ASA use renders patients less vulnerable to develop a severe sepsis that requires hospitalization (41). Therefore, it would be interesting to investigate the effects of prehospital ASA use on sepsis susceptibility. Interestingly, a large randomized clinical trial (ANTISEPSIS) is currently underway that will address this question (23).
The effects of ASA in this study were mainly exerted at cytokine production capacity level, while other immune variables were less strongly affected. In line with previous data of our group (28), experimental endotoxemia resulted in decreased mHLA-DR expression. This effect was similar across all groups and during both endotoxin challenges, although it did not reach statistical significance in the ASA prophylaxis group upon the first endotoxin challenge, which might be another reflection of the enhanced proinflammatory profile observed in this group. Despite the fact that the effects of ASA on HLA-DR expression were relatively small, they could still be of clinical relevance, as an increase of 4.8% in HLA-DR expression was shown to be associated with improved survival in patients suffering from severe sepsis (42).
The ASA-induced shift toward a more proinflammatory phenotype was not reflected in the assessed clinical variables. This is likely explained by the fact that cytokine levels do not directly correlate with clinical variables in the experimental human endotoxemia model, as we have shown before (43). Rather, there appears to be a threshold effect, in which clinical variables only show changes when cytokine responses are profoundly altered (43).
This study has limitations which should be acknowledged. First, only male subjects and patients were studied. Pertaining to the endotoxemia results, females were previously shown to mount a more pronounced proinflammatory immune response upon endotoxin challenge (44), meaning that including both sexes would result in more interindividual variation and impair statistical power of the study (44). Furthermore, fluctuating hormone levels during the menstrual cycle are also known to influence immune variables (45, 46), which would further increase variation. This would necessitate a larger sample size, which is undesirable for the very labor- and cost-intensive endotoxemia studies. Despite the fact that there are no indications that the immunologic response to ASA is different between males and females, the use of only males limits the generalizability of our work. Second, only young subjects were studied, whereas ASA therapy is used mostly in the aged, and sepsis patients are usually older as well. Again, there is no reason to assume that the immunologic response to ASA is different in older subjects, and our ex vivo experiments using monocytes of (also older) sepsis patients corroborate this. Third, it should be noted that endotoxin tolerance only partially resembles sepsis-induced immunoparalysis. For instance, in sepsis patients also profound adaptive immune system derangements, such as lymphocyte apoptosis and dysfunction occur (7, 47). Administration of an endotoxin mainly results in activation of the innate immune system; there is no antigen presentation and no classic adaptive response, nor profound lymphocyte dysfunction and/or apoptosis as observed in sepsis. Nevertheless, human endotoxemia is the only human model available that captures several important hallmarks of both sepsis and sepsis-induced immunoparalysis, and the only human model to study these conditions. Furthermore, although our model mimics infection with Gram-negative bacteria, transcriptomic analyses have revealed that most sepsis response pathways are common and independent of pathogen or source of infection (48). Both the considerable overlap in signaling pathways downstream of pathogen-specific pattern recognition receptors and inflammation-induced translocation of endotoxin from the gut to the circulation may explain these common responses (49). This suggests that our findings may also be relevant for infections with other pathogens. Fourth, it has been reported that the efficacy of low-dose ASA varies between patients according to their lean body mass (50). Whereas this is not relevant for our study results (body mass index was equally distributed among groups), it might be relevant for patients. Furthermore, a certain part of the population appears to be resistant to the antiplatelet aggregation effects of low-dose ASA (51); however, it remains to be determined whether this also applies to the proinflammatory effects of ASA.
CONCLUSIONS
Treatment, but not prophylaxis, with low-dose ASA partially reverses endotoxin tolerance in humans in vivo by causing a shift toward a more proinflammatory phenotype. This effect likely involves inhibition of PGE2. This ASA-induced shift toward a proinflammatory phenotype is also observed in monocytes of sepsis patients, which signifies that patients suffering from sepsis-induced immunoparalysis may benefit from initiation of ASA treatment in the hospital. Although prophylaxis with ASA did not prevent the development of endotoxin tolerance, the ASA-associated survival benefits in observational studies might be related to an augmented proinflammatory response in the early phase of sepsis, resulting in improved bacterial clearance of their primary infection. As such, the current study provides a potential explanation of the benefits of ASA use in sepsis patients. If the upcoming ANTISEPSIS trial (23) confirms the association between ASA use and survival benefits once again, a prospective trial with ASA in sepsis patients is highly warranted.
ACKNOWLEDGMENTS
We would like to thank Yvonne Kaspers, Christopher Geven, Quirine Habes, and Roger van Groenendael for their help during the endotoxin challenge days.
Supplementary Material
Footnotes
*See also p. 603.
Drs. Leijte, Netea, Kox, and Pickkers designed the study. Drs. Leijte, Kiers, Jansen, and Boerrigter included the subjects, performed the experiments, and processed the samples. Drs. Leijte and van der Heijden performed the ex vivo experiments. Drs. van der Heijden and Gerretsen performed the laboratory analyses. Dr. Leijte performed the statistical analyses and drafted the article. Drs. Kiers, van der Heijden, Netea, Kox, and Pickkers critically revised the article and supervised the research. All authors read and approved the final article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/ccmjournal).
Dr. Netea was funded by a Spinoza grant of the Netherlands Organization for Scientific Research, and a Competitiveness Operational Program Grant of the Romanian Ministry of European Funds. The remaining authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016; 315:801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleischmann C, Scherag A, Adhikari NK, et al. ; International Forum of Acute Care Trialists: Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193:259–272 [DOI] [PubMed] [Google Scholar]
- 3.Liu V, Escobar GJ, Greene JD, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 2014; 312:90–92 [DOI] [PubMed] [Google Scholar]
- 4.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 2013; 369:840–851 [DOI] [PubMed] [Google Scholar]
- 5.van der Poll T, van de Veerdonk FL, Scicluna BP, et al. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 2017; 17:407–420 [DOI] [PubMed] [Google Scholar]
- 6.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat Rev Immunol 2013; 13:862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011; 306:2594–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamers L, Kox M, Pickkers P. Sepsis-induced immunoparalysis: Mechanisms, markers, and treatment options. Minerva Anestesiol 2015; 81:426–439 [PubMed] [Google Scholar]
- 9.Osuchowski MF, Welch K, Siddiqui J, et al. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol 2006; 177:1967–1974 [DOI] [PubMed] [Google Scholar]
- 10.Monneret G, Venet F, Pachot A, et al. Monitoring immune dysfunctions in the septic patient: A new skin for the old ceremony. Mol Med 2008; 14:64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamayo E, Fernández A, Almansa R, et al. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw 2011; 22:82–87 [DOI] [PubMed] [Google Scholar]
- 12.Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, et al. Sepsis: Multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev 2013; 93:1247–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Vught LA, Klein Klouwenberg PM, Spitoni C, et al. ; MARS Consortium: Incidence, risk factors, and attributable mortality of secondary infections in the intensive care unit after admission for sepsis. JAMA 2016; 315:1469–1479 [DOI] [PubMed] [Google Scholar]
- 14.Sossdorf M, Otto GP, Boettel J, et al. Benefit of low-dose aspirin and non-steroidal anti-inflammatory drugs in septic patients. Crit Care 2013; 17:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lösche W, Boettel J, Kabisch B, et al. Do aspirin and other antiplatelet drugs reduce the mortality in critically ill patients? Thrombosis 2012; 2012:720254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winning J, Neumann J, Kohl M, et al. Antiplatelet drugs and outcome in mixed admissions to an intensive care unit. Crit Care Med 2010; 38:32–37 [DOI] [PubMed] [Google Scholar]
- 17.Winning J, Reichel J, Eisenhut Y, et al. Anti-platelet drugs and outcome in severe infection: Clinical impact and underlying mechanisms. Platelets 2009; 20:50–57 [DOI] [PubMed] [Google Scholar]
- 18.Falcone M, Russo A, Farcomeni A, et al. Septic shock from community-onset pneumonia: Is there a role for aspirin plus macrolides combination? Intensive Care Med 2016; 42:301–302 [DOI] [PubMed] [Google Scholar]
- 19.Eisen DP, Reid D, McBryde ES. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med 2012; 40:1761–1767 [DOI] [PubMed] [Google Scholar]
- 20.Tsai MJ, Ou SM, Shih CJ, et al. Association of prior antiplatelet agents with mortality in sepsis patients: A nationwide population-based cohort study. Intensive Care Med 2015; 41:806–813 [DOI] [PubMed] [Google Scholar]
- 21.Otto GP, Sossdorf M, Boettel J, et al. Effects of low-dose acetylsalicylic acid and atherosclerotic vascular diseases on the outcome in patients with severe sepsis or septic shock. Platelets 2013; 24:480–485 [DOI] [PubMed] [Google Scholar]
- 22.Trauer J, Muhi S, McBryde ES, et al. Quantifying the effects of prior acetyl-salicylic acid on sepsis-related deaths: An individual patient data meta-analysis using propensity matching. Crit Care Med 2017; 45:1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisen DP, Moore EM, Leder K, et al. AspiriN To Inhibit SEPSIS (ANTISEPSIS) randomised controlled trial protocol. BMJ Open 2017; 7:e013636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Netea MG, Puren AJ, Dinarello CA. A short course of oral aspirin increases IL-18-induced interferon-gamma production in whole blood cultures. Eur Cytokine Netw 2000; 11:379–382 [PubMed] [Google Scholar]
- 25.Kiers D, van der Heijden WA, van Ede L, et al. A randomised trial on the effect of anti-platelet therapy on the systemic inflammatory response in human endotoxaemia. Thromb Haemost 2017; 117:1798–1807 [DOI] [PubMed] [Google Scholar]
- 26.Strassmann G, Patil-Koota V, Finkelman F, et al. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J Exp Med 1994; 180:2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zasłona Z, Pålsson-McDermott EM, Menon D, et al. The induction of pro-IL-1β by lipopolysaccharide requires endogenous prostaglandin E2 production. J Immunol 2017; 198:3558–3564 [DOI] [PubMed] [Google Scholar]
- 28.Leentjens J, Kox M, Koch RM, et al. Reversal of immunoparalysis in humans in vivo: A double-blind, placebo-controlled, randomized pilot study. Am J Respir Crit Care Med 2012; 186:838–845 [DOI] [PubMed] [Google Scholar]
- 29.Döcke WD, Höflich C, Davis KA, et al. Monitoring temporary immunodepression by flow cytometric measurement of monocytic HLA-DR expression: A multicenter standardized study. Clin Chem 2005; 51:2341–2347 [DOI] [PubMed] [Google Scholar]
- 30.Endres S, Whitaker RE, Ghorbani R, et al. Oral aspirin and ibuprofen increase cytokine-induced synthesis of IL-1 beta and of tumour necrosis factor-alpha ex vivo. Immunology 1996; 87:264–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osterud B, Olsen JO, Wilsgård L. Increased lipopolysaccharide-induced tissue factor activity and tumour necrosis factor production in monocytes after intake of aspirin: Possible role of prostaglandin E2. Blood Coagul Fibrinolysis 1992; 3:309–313 [DOI] [PubMed] [Google Scholar]
- 32.Frencken JF, van Vught LA, Peelen LM, et al. ; MARS Consortium: An unbalanced inflammatory cytokine response is not associated with mortality following sepsis: A prospective cohort study. Crit Care Med 2017; 45:e493–e499 [DOI] [PubMed] [Google Scholar]
- 33.Härtel C, von Puttkamer J, Gallner F, et al. Dose-dependent immunomodulatory effects of acetylsalicylic acid and indomethacin in human whole blood: Potential role of cyclooxygenase-2 inhibition. Scand J Immunol 2004; 60:412–420 [DOI] [PubMed] [Google Scholar]
- 34.Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res 2003; 110:255–258 [DOI] [PubMed] [Google Scholar]
- 35.Xiang B, Zhang G, Guo L, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun 2013; 4:2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinz B, Brune K, Pahl A. Prostaglandin E(2) upregulates cyclooxygenase-2 expression in lipopolysaccharide-stimulated RAW 264.7 macrophages. Biochem Biophys Res Commun 2000; 272:744–748 [DOI] [PubMed] [Google Scholar]
- 37.Toner P, McAuley DF, Shyamsundar M. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit Care 2015; 19:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du F, Jiang P, He S, et al. Antiplatelet therapy for critically ill patients: A pairwise and Bayesian network meta-analysis. Shock 2018; 49:616–624 [DOI] [PubMed] [Google Scholar]
- 39.Kleinnijenhuis J, Quintin J, Preijers F, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A 2012; 109:17537–17542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiewel MA, de Stoppelaar SF, van Vught LA, et al. ; MARS Consortium: Chronic antiplatelet therapy is not associated with alterations in the presentation, outcome, or host response biomarkers during sepsis: A propensity-matched analysis. Intensive Care Med 2016; 42:352–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisen DP. Manifold beneficial effects of acetyl salicylic acid and nonsteroidal anti-inflammatory drugs on sepsis. Intensive Care Med 2012; 38:1249–1257 [DOI] [PubMed] [Google Scholar]
- 42.Wu JF, Ma J, Chen J, et al. Changes of monocyte human leukocyte antigen-DR expression as a reliable predictor of mortality in severe sepsis. Crit Care 2011; 15:R220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiers D, Koch RM, Hamers L, et al. Characterization of a model of systemic inflammation in humans in vivo elicited by continuous infusion of endotoxin. Sci Rep 2017; 7:40149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Eijk LT, Dorresteijn MJ, Smits P, et al. Gender differences in the innate immune response and vascular reactivity following the administration of endotoxin to human volunteers. Crit Care Med 2007; 35:1464–1469 [DOI] [PubMed] [Google Scholar]
- 45.O’Brien SM, Fitzgerald P, Scully P, et al. Impact of gender and menstrual cycle phase on plasma cytokine concentrations. Neuroimmunomodulation 2007; 14:84–90 [DOI] [PubMed] [Google Scholar]
- 46.Gornicsar K, Mózes T, Grósz A, et al. TNFα variation during the menstrual cycle and thereafter: A new explanation for gender-based disparities in ICU admission rates, trauma outcomes, and general mortality. Shock 2017; 47:416–421 [DOI] [PubMed] [Google Scholar]
- 47.Chang K, Svabek C, Vazquez-Guillamet C, et al. Targeting the programmed cell death 1: Programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care 2014; 18:R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burnham KL, Davenport EE, Radhakrishnan J, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med 2017; 196:328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marshall JC, Foster D, Vincent JL, et al. ; MEDIC study: Diagnostic and prognostic implications of endotoxemia in critical illness: Results of the MEDIC study. J Infect Dis 2004; 190:527–534 [DOI] [PubMed] [Google Scholar]
- 50.Rothwell PM, Cook NR, Gaziano JM, et al. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: Analysis of individual patient data from randomised trials. Lancet 2018; 392:387–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westphal ES, Rainka M, Amsler M, et al. Prospective determination of aspirin sensitivity in patients resistant to low dose aspirin: A proof of concept study. J Clin Pharmacol 2018; 58:1157–1163 [DOI] [PubMed] [Google Scholar]