

Supplemental Digital Content is available in the text.
Keywords: atrial fibrillation, atrial remodeling, histone deacetylase inhibitor, proteomics, valproic acid
Abstract
Background:
A structural, electrical and metabolic atrial remodeling is central in the development of atrial fibrillation (AF) contributing to its initiation and perpetuation. In the heart, HDACs (histone deacetylases) control remodeling associated processes like hypertrophy, fibrosis, and energy metabolism. Here, we analyzed, whether the HDAC class I/IIa inhibitor valproic acid (VPA) is able to attenuate atrial remodeling in CREM-IbΔC-X (cAMP responsive element modulator isoform IbΔC-X) transgenic mice, a mouse model of extensive atrial remodeling with age-dependent progression from spontaneous atrial ectopy to paroxysmal and finally long-lasting AF.
Methods:
VPA was administered for 7 or 25 weeks to transgenic and control mice. Atria were analyzed macroscopically and using widefield and electron microscopy. Action potentials were recorded from atrial cardiomyocytes using patch-clamp technique. ECG recordings documented the onset of AF. A proteome analysis with consecutive pathway mapping identified VPA-mediated proteomic changes and related pathways.
Results:
VPA attenuated many components of atrial remodeling that are present in transgenic mice, animal AF models, and human AF. VPA significantly (P<0.05) reduced atrial dilatation, cardiomyocyte enlargement, atrial fibrosis, and the disorganization of myocyte’s ultrastructure. It significantly reduced the occurrence of atrial thrombi, reversed action potential alterations, and finally delayed the onset of AF by 4 to 8 weeks. Increased histone H4-acetylation in atria from VPA-treated transgenic mice verified effective in vivo HDAC inhibition. Cardiomyocyte-specific genetic inactivation of HDAC2 in transgenic mice attenuated the ultrastructural disorganization of myocytes comparable to VPA. Finally, VPA restrained dysregulation of proteins in transgenic mice that are involved in a multitude of AF relevant pathways like oxidative phosphorylation or RhoA (Ras homolog gene family, member A) signaling and disease functions like cardiac fibrosis and apoptosis of muscle cells.
Conclusions:
Our results suggest that VPA, clinically available, well-tolerated, and prescribed to many patients for years, has the therapeutic potential to delay the development of atrial remodeling and the onset of AF in patients at risk.
WHAT IS KNOWN?
Atrial remodeling contributes to initiation and perpetuation of atrial fibrillation. CREM-IbΔC-X (cAMP responsive element modulator isoform IbΔC-X) transgenic mice progressively develop atrial remodeling preceding spontaneous onset of atrial fibrillation.
HDACs (histone deacetylases) control remodeling associated processes like hypertrophy, fibrosis, and energy metabolism, and HDAC inhibition was inter alia linked to attenuation of atrial fibrosis and reduced inducibility of atrial arrhythmic episodes in preclinical models.
WHAT THE STUDY ADDS?
Valproic acid, a known anticonvulsant and HDAC class I/IIa inhibitor, attenuated the progressive atrial remodeling in CREM-IbΔC-X transgenic mice (atrial dilatation, cardiomyocyte elongation, ultrastructural disorganization, fibrosis, action potential alterations, and thrombus formation) finally delaying the spontaneous onset of atrial fibrillation.
Valproic acid specifically counter-regulated remodeling associated proteomic changes in CREM-IbΔC-X transgenic mice while it had little effects on wild-type mice.
Valproic acid, clinically available and well-tolerated, may have the therapeutic potential to delay the development of atrial remodeling and the onset of atrial fibrillation in patients at risk.
Atrial fibrillation (AF) is the most common arrhythmia in the elderly and increases the rate of death (2-fold), stroke (5-fold), heart failure, and hospitalization.1 Because of population aging, the number of patients with AF has been estimated to more than double in the next decades which will have major public health implications.2 AF progresses from rare paroxysmal episodes, often unrecognized by patients, to long-lasting persistent and finally permanent stages. Simultaneously, the atrium undergoes a complex remodeling process comprising structural and functional alterations of myocytes, extracellular matrix, and vasculature forming vicious circles culminating in the perpetuation of the disease.3 The mechanisms underlying this extensive remodeling are not fully understood, and the current treatment of AF is limited to rate control or prevention of stroke with anticoagulation but misses finally to prevent the perpetuation of the disease.1,3,4
Decreased expression of targets of the CREB/CREM/ATF (cAMP response element-binding protein/cAMP responsive element modulator/activating transcription factor) transcription factor family was linked to AF susceptibility in humans,5 and the CREM repressor isoform CREM-IbΔC-X is upregulated in human AF6. Mice with heart-specific expression of CREM-IbΔC-X represent a unique model of spontaneous onset AF preceded by the development of an extensive atrial remodeling including atrial dilatation, atrial myocyte hypertrophy, and fibrosis.7–10 In this study, we investigated whether atrial remodeling in CREM-IbΔC-X transgenic mice can be attenuated by valproic acid (VPA), which is an inhibitor of HDACs (histone deacetylases). Histone deacetylation by HDACs leads to chromatin condensation and gene silencing whereas histone acetylation by HATs (histone acetyltransferases) leads to chromatin relaxation and facilitates gene transcription. During transcriptional activation, CREB recruits coactivators (p300/CBP [CREB binding protein]) which contain HAT domains.11 Binding of CREM repressors to the common DNA binding sequence cAMP response element prevents this recruitment which might shift the balance to histone deacetylation by HDACs. Moreover, the isoform CREMα has even been shown to recruit HDAC1 to the cAMP response element, facilitating deacetylation of histones.12
In the heart, HDACs control processes like hypertrophy, fibrosis, apoptosis, and energy metabolism,13 which are objects of AF related remodeling.3 HDAC inhibitors targeting different HDAC classes have been shown to act as antifibrotic and antihypertrophic therapeutics in the heart in preclinical models14–16 and have clinically already been tested for anticancer treatment. Recent studies even demonstrated the effectiveness of HDAC inhibitors in reducing atrial fibrosis and atrial arrhythmia inducibility in mice and electrical remodeling in tachypaced dogs suggesting HDAC inhibition as a promising therapeutic approach for AF.17,18 Thus, we hypothesized that HDAC inhibition might beneficially affect the AF phenotype in transgenic mice.
Here, we show that the known anticonvulsant VPA, which is a clinically available and well-tolerated HDAC class I/IIa inhibitor, delays the development of atrial remodeling and the onset of AF in transgenic mice. Our results suggest a promising role for HDAC inhibition in the treatment of AF and propose VPA as a therapeutic option to be tested in humans.
Methods
Details on Methods are available in the Data Supplement.
Data and methodical details not available within the article, and its Data Supplement files are available from the corresponding author on reasonable request.
Animals and Experimental Design
Mice with cardiomyocyte-specific expression of CREM-IbΔC-X have been described before.7–10 Adult male transgenic (TG) and wild-type (WT) mice were randomly assigned to vehicle ([VEH] WTVEH, TGVEH) or VPA-treatment groups (WTVPA, TGVPA). VPA administration started at an age of 5 weeks for short-term (7 weeks) or long-term (25 weeks) treatment (Figure 1A). Mice with cardiomyocyte-specific knockout of HDAC2 (HDAC2KO) are specified in Methods in the Data Supplement. All experiments on animals conformed to the Directive 2010/63/EU of the European Parliament and were approved by the local animal welfare authorities (LANUV [Landesamt für Natur, Umwelt und Verbraucherschutz]; North Rhine-Westphalia, Germany; permit AZ 84-02.04.2011.A155; 84-02.04.2015.A418).
Figure 1.
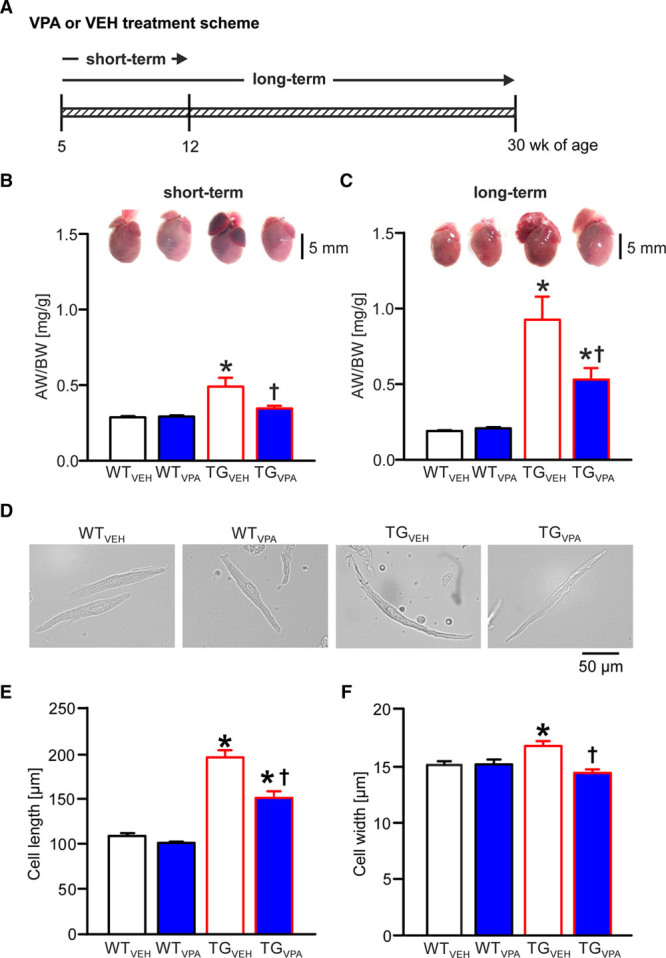
Valproic acid (VPA) treatment attenuates atrial dilatation and cardiomyocyte hypertrophy in CREM-IbΔC-X transgenic (TG) mice. A, Short-term (7 wk) and long-term (25 wk) treatment scheme for mice receiving VPA or vehicle (VEH). B and C, Representative images of mouse hearts and statistical analysis of AW/BW ratio (atrial weight/body weight) illustrate age-dependent atrial dilatation in TGVEH animals and its attenuation by VPA after both short-term and long-term treatment (n=22–34 animals/group). D, Representative images of isolated atrial cardiomyocytes from all 4 treatment groups (short-term). E, Mean cell length and (F) cell width. n=5–7 isolations/group; average of 50 cells/isolation; *P<0.05 vs wild-type (WT); †P<0.05 vs VEH. CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
Preparation of Drugs for Mouse Treatment
VPA dosing was chosen to achieve serum levels reported for HDAC inhibition.19,20 VPA sodium salt (0.71% wt/vol) dissolved in drinking water (vehicle) was available ad libitum.21
Electrocardiography Recordings
ECGs were recorded in mice during long-term treatment every 2 weeks starting from week 10 of age as described.22 AF was defined by absence of P-waves in combination with an irregular ventricular rate.
Serum Analysis
Blood was collected directly from the heart with a syringe immediately after mice have been euthanized. Serum VPA concentration was determined at the Center for Laboratory Medicine, University Hospital of Münster.
Atrial Tissue Preparation
Mouse atria were dissected from whole heart preparations in ice-cold saline solution (0.9 %), atrial thrombi were removed, and then atria were frozen in liquid nitrogen and weighed after freezing. Atrial tissue was used for Western blot, proteome analysis, and chromatin immunoprecipitation.
Western Blots
Immunoblotting of atrial lysates (40 µg protein per sample) was performed as described.23 Anti-Histone H4 (acetyl K8) antibody (ab15823; Abcam) and anti-Histone H4 (dilution: 1:1.000, ab10158; Abcam) were used as primary antibody, ECL HRP-conjugated anti-rabbit or mouse IgG antibody (GE Healthcare) were used as secondary antibodies (dilution: 1:5.000).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed as described23 modified for atrial tissue. Genomic DNA from transgenic or WT mouse atria was precipitated by affinity-purified rabbit polyclonal HA tag antibody (ab9110; Abcam). Enrichment of genomic DNA fragments (anti-HA transgenic versus anti-HA WT) was tested by quantitative real-time polymerase chain reaction.
Isolation and Sizing of Atrial Cardiomyocytes
After short-term treatment, atrial cardiomyocytes were isolated according to Bögeholz et al24 with minor modifications. Only spindle-shaped, clearly striated myocytes were used for subsequent analyses. Length and width of 50 to 60 randomly seeded atrial cardiomyocytes were determined and averaged per preparation using a wild-field microscope (Nikon Eclipse Ti-E, Nikon Instruments) equipped with a motorized stage and a CCD camera (stitching of 5×5 images, ×20 magnification, NIS elements; Nikon Instruments).
Patch-Clamp Experiments
Atrial cardiomyocytes were slowly adapted to 1 mmol/L Ca2+ with perfusion of Tyrode solution. Action potentials (APs), triggered at 1 Hz, were recorded using the perforated patch current clamp technique as described.22 Three to 5 consecutive APs were averaged for AP parameter analysis. Additionally, APs and Na+ currents (voltage-clamp mode) were recorded under basal conditions and after acute application of 1 mmol/L VPA (500 ms test pulse duration; −80 to +70 mV, Δ10 mV, −80 mV holding potential).
Acute VPA Treatment of Atrial Cardiomyocytes and Heart Preparations
Atrial cardiomyocytes were treated with vehicle (Tyrode solution) or VPA (1 mmol/L, VPA sodium salt in Tyrode solution) at 37°C for 20 minutes. Explanted hearts were treated accordingly by retrograde Langendorff-perfusion.
Analysis of Fibrosis and Ultrastructure
Masson trichrome staining was performed on 5 µm sections of paraffin-embedded hearts for estimation of interstitial fibrosis which was quantified with Image-Pro Plus software (Media Cybernetics Incorporation, Rockville, MD).
Ultra-thin sections of atria were stained with uranyl acetate and lead citrate and analyzed with a Philips EM 208S transmission electron microscope. In 12 to 18 electron microscope images per animal (100 µm2 in size), the relative areas (%) with sarcomere structure, mitochondria, and collagen were estimated using ImageJ software.
Proteome Analysis of Atrial Tissue
Atrial tissue preparation (n=5–8/group), protein digestion, and liquid chromatography–tandem mass spectrometry were performed as described.10,25 Ingenuity Software (Qiagen, Hilden, Germany) and the Kyoto Encyclopedia of Genes and Genomes database were used to assign altered proteins in atrial tissues to biological pathways.
Statistical Analysis
If not indicated otherwise, data are presented as mean±SEM. Normally distributed data were analyzed with unpaired 2-tailed Student t test or ANOVA, nonnormally distributed data with Mann-Whitney U test or Kruskal-Wallis ANOVA. The Log-Rank test was used for Kaplan-Meier analysis of AF development. Fisher exact test was used for analysis of frequency distribution. Analyses were performed using SigmaPlot (Systat Software, Erkrath, Germany) or Origin Pro (OriginLab, Northampton). A P value <0.05 was considered statistically significant.
Results
VPA Reduced Atrial Enlargement in CREM-IbΔC-X Transgenic Mice
Our VPA-treatment scheme (Figure 1A) resulted in mean VPA serum levels in mice that were close to therapeutic levels in human (0.3–1 mmol/L/43–144 µg/mL)19 (n=10–30 animals/group; Figure I in the Data Supplement). The atrial weight/body weight ratio in TGVEH was increased by 69% and 396% versus WTVEH mice, at 12 and 30 weeks of age, respectively (P<0.05; Figure 1B and 1C), reflecting atrial enlargement in transgenic mice. At the same time, ventricular weights did not differ between groups (P >0.05; data not shown). VPA short-term treatment (Figure 1B) almost normalized the increased atrial weight/body weight ratio (P<0.05), whereas long-term treatment (Figure 1C) still reduced the atrial weight/body weight ratio in TGVPA mice by 42% (P<0.05; n=22–34 animals/group).
Atrial enlargement in transgenic mice is associated with atrial cardiomyocyte elongation.9 Atrial cardiomyocyte length and width (Figure 1D through 1F) were increased in TGVEH mice at an age of 12 weeks (P<0.05). Short-term-VPA treatment led to a reduced elongation of transgenic atrial cardiomyocytes (P<0.05; Figure 1E) and completely prevented the increase in cell width (P<0.05; Figure 1F; n=5–7 isolations/group).
VPA Treatment Attenuated Ultrastructural Disarrangement of Atria in Transgenic Mice
Electron microscopy analyses revealed noticeable ultrastructural remodeling in transgenic mice at the age of 7 and more prominent at the age of 16 weeks (Figure 2). In line with Seidl et al,10 we observed a loss of sarcomere structure (P<0.05), reduced organization and amount of mitochondria (P<0.05), and increased occurrence of collagen fibers (P<0.05), glycogen, and lipofuscin granules (P<0.05) culminating in a completely disarranged cell and tissue structure. VPA treatment was able to delay the development of this ultrastructural remodeling in transgenic mice. The tissue structure was considerably more organized in TGVPA versus TGVEH after both treatment periods and the percentage distribution of sarcomeres and collagen fibers (P<0.05 versus TGVEH) was closer to WT (WTVPA examples: Figure II in the Data Supplement; n=3–4 animals/group).
Figure 2.
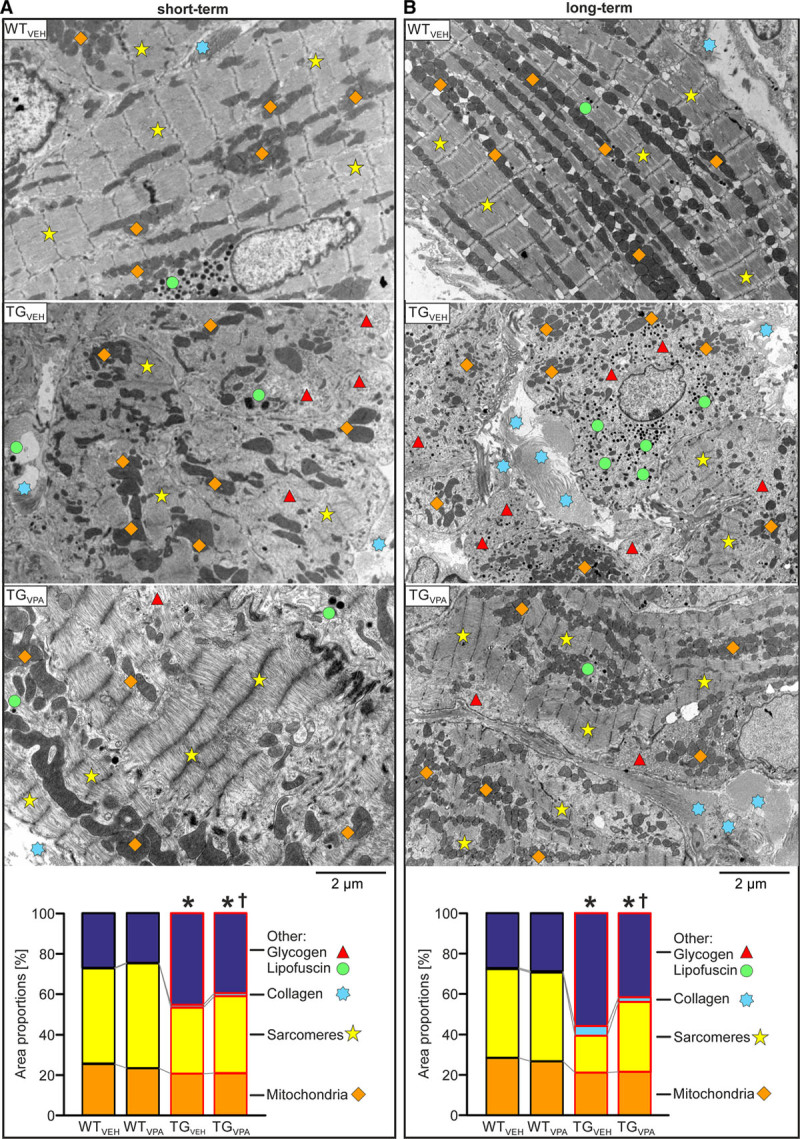
Valproic acid (VPA) treatment attenuates ultrastructural remodeling in CREM-IbΔC-X transgenic (TG) mice atria. Representative electron microscopic images of atrial tissue and quantitative analysis (area proportions in %) of sarcomeres, mitochondria, collagen fibers, lipofuscin granules, and glycogen in the indicated groups during (A) short-term (7th wk) and (B) long-term (16th wk) treatment. With age, myolysis dominated in TGVEH atrial cardiomyocytes and accumulation of collagen fibers between cells was enhanced. VPA attenuated this ultrastructural disarrangement after short- and long-term treatment in transgenic atria (TGVPA; n=3–4 animals/group; average of 16–18 pictures/animal; *P<0.05 vs wild-type (WT; all areas); †P<0.05 vs vehicle (VEH; other, collagen, sarcomeres), mitochondria: not significant). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
VPA Reduced the Amount of Fibrosis and the Occurrence of Thrombi in Transgenic Atria
Atrial fibrosis is a hallmark of arrhythmogenic structural remodeling. TGVEH mice displayed distinct atrial fibrosis at the age of 16 weeks (TGVEH: 11% versus WTVEH: 2%; P<0.05). VPA treatment markedly reduced the amount of atrial fibrosis in TGVPA to 6% (P<0.05; Figure 3A through 3C; n=8–9 animals/group).
Figure 3.
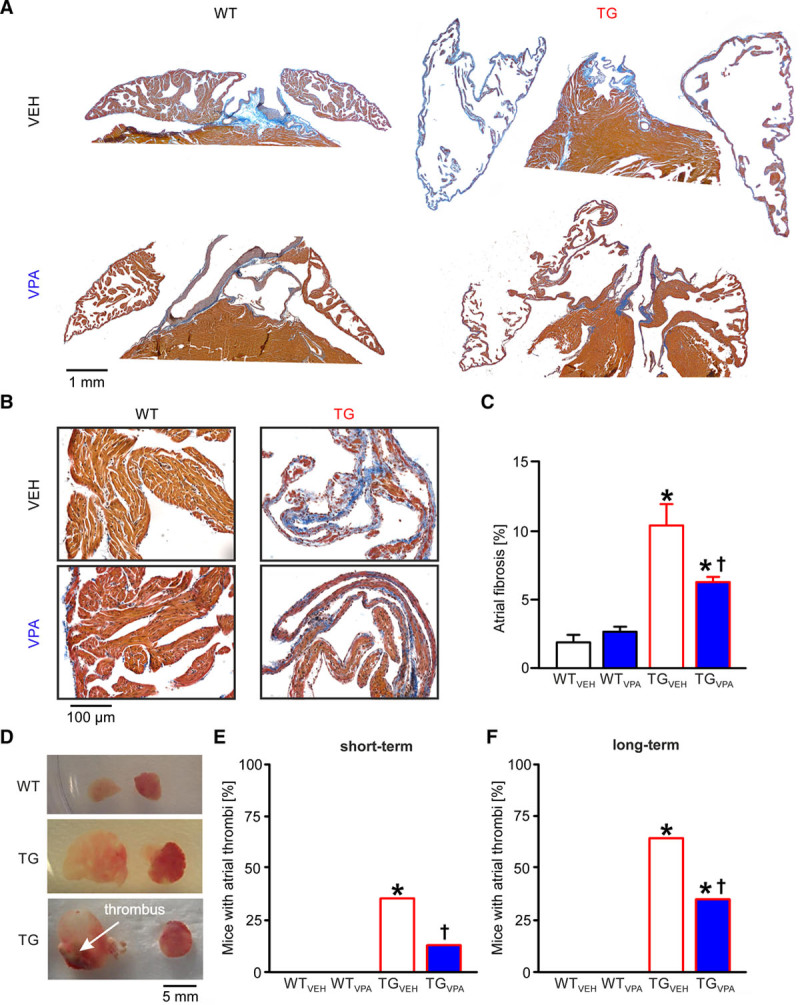
Valproic acid (VPA) treatment attenuates fibrosis and thrombus occurrence in CREM-IbΔC-X transgenic (TG) mice atria. A, Exemplary images of Masson trichrome–stained atrial sections for all groups during long-term treatment (16th wk), collagen (fibrosis) is stained blue. B, Exemplary image sections for all groups at higher magnification. The wall of transgenic atria was extremely thin containing just a few trabeculae in contrast to the thick atrial wall of wild-type (WT) mice. C, Mean atrial fibrosis in % (n=8–9 animals/group; *P<0.05 vs WT; †P<0.05 vs vehicle [VEH]). D, Representative images of WT and transgenic atria, the latter with and without thrombus. E and F, Portion of mice with atrial thrombi after short- and long-term treatment (n=30–34 animals/group; *P<0.05 vs WT; †P<0.05 vs VEH). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
As in human, AF in transgenic mice is associated with the occurrence of atrial thrombi.26 The proportion of thrombus-positive TGVEH mice increased with age from 35% (12 weeks) to 65% (30 weeks; P<0.05; Figure 3D through 3F). VPA clearly reduced the occurrence of thrombi in TGVPA atria (short-term: 13%; long-term: 35%; P<0.05; n=30–34 animals/group).
VPA Prevented AF-Associated Electrical Remodeling in Transgenic Mice
Electrical remodeling leading to changes in AP duration (APD) is thought to facilitate or stabilize AF. The APD until 50, 70, and 90% of repolarization (APD50, APD70, and APD90) measured in atrial cardiomyocytes from TGVEH mice was increased by 54% to 100% versus WTVEH (P<0.05; Figure 4A through 4F). VPA short-term treatment completely prevented AP prolongation in TGVPA, whereas APs measured in atrial cardiomyocytes from WTVPA mice were not different versus WTVEH (P >0.05; n=21–35 cardiomyocytes/5–10 isolations). In line, no arrhythmogenic alterations were observed in ECG parameters even after VPA long-term treatment in WT mice underlining VPA’s cardiac safety (n=8 animals per group; Figure III in the Data Supplement).
Figure 4.
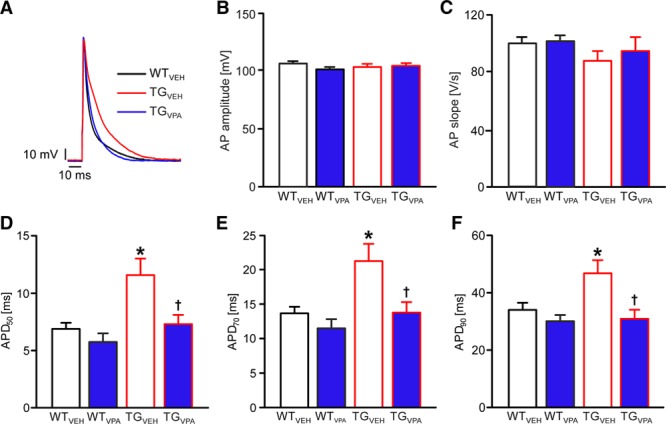
Valproic acid (VPA) prevents action potential (AP) prolongation in CREM-IbΔC-X transgenic (TG) mice. A, Representative APs recorded from isolated atrial cardiomyocytes of indicated groups. B, Mean AP amplitude (C) slope and (D–F) action potential duration (APD) until 50, 70, and 90% of repolarization (APD50, APD70, and APD90) in atrial cardiomyocytes after short-term treatment. VPA treatment prevented the APD increase in transgenic myocytes (n=21–35 cardiomyocytes/5–10 isolations; *P<0.05 vs wild-type (WT); †P<0.05 vs vehicle [VEH]). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
VPA Delayed the Time Until Spontaneous Onset of AF in Transgenic Mice
Because VPA attenuated both the structural and electrical remodeling, we questioned whether these effects would affect the onset of AF. Periodic ECG recordings confirmed the onset of AF before week 10 of age in TGVEH mice. However, VPA treatment led to a delay in the time until onset of AF in TGVPA mice (P<0.05; Figure 5A and 5B) indicated by a right-shift of the Kaplan-Meier curve, depicting the time-dependent increase of AF in transgenic mice (n=19–20 [WT]; 32–36 [transgenic] animals per group).
Figure 5.
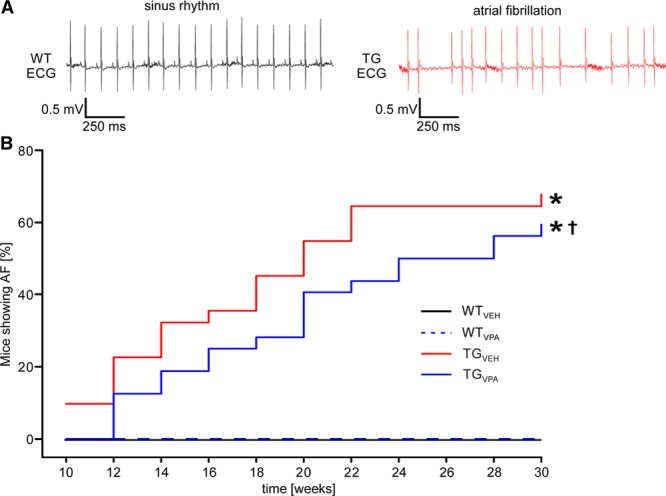
Valproic acid (VPA) delays the onset of atrial fibrillation (AF) in CREM-IbΔC-X transgenic (TG) mice. A Representative ECG traces of a wild-type (WT) mouse in sinus rhythm (left) and a transgenic mouse showing AF (right). B, AF increased with age in TGVEH as demonstrated by Kaplan-Meier plots showing the survival of mice with AF. VPA treatment delayed the onset of spontaneous AF in TGVPA mice. No AF was observed in WT animals (n=19–20 [WT]; 32–36 [transgenic] animals/group). *P<0.05 vs WT; †P<0.05 vs vehicle (VEH). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
VPA Increased Histone H4-Acetylation in Transgenic Atria After Short- and Long-Term Treatment
To confirm in vivo HDAC inhibition in mouse hearts by the VPA-treatment regime used in this study, we analyzed H4-acetylation in atrial homogenates of all groups at the end of both VPA-treatment periods. In atrial homogenates of TGVPA mice H4-acetylation was increased by 30% after VPA short-term treatment and by 59% after VPA long-term treatment versus WTVEH (P<0.05; n=5–10/group; Figure 6A and 6B). H4-acetylation was not different between the WT groups (P >0.05). Thus, prolonged VPA treatment increased H4-acetylation exclusively in atria of transgenic mice.
Figure 6.
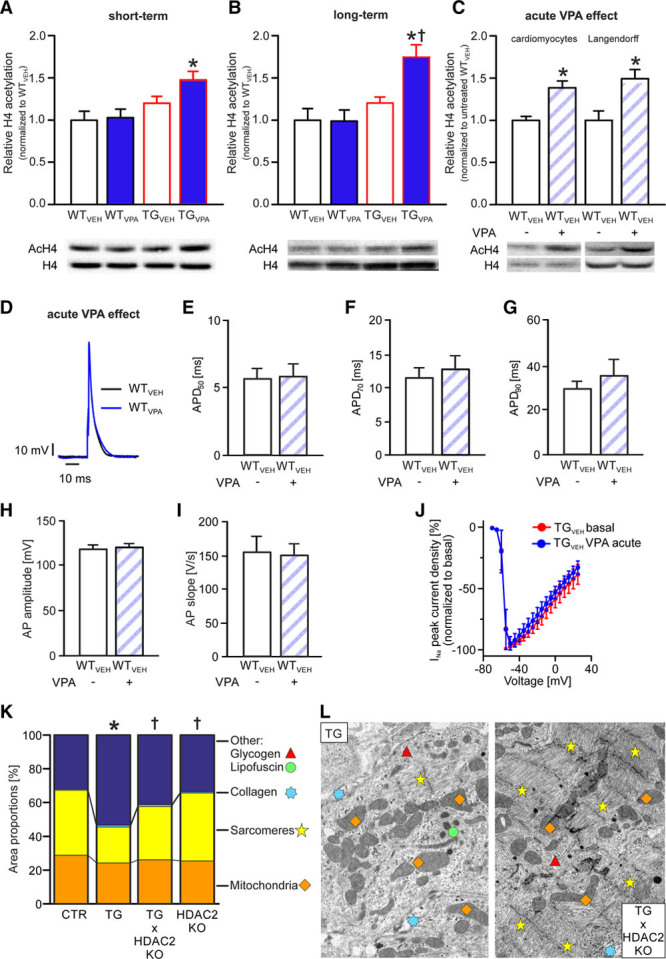
Valproic acid (VPA) operates as an HDAC (histone deacetylase) inhibitor. A–C, Representative Western blot images and quantitative analysis of acetylated histone H4 (AcH4) and H4. In vivo VPA treatment increased H4-acetylation in CREM-IbΔC-X (cAMP responsive element modulator isoform IbΔC-X) transgenic (TG) mice atrial homogenates after (A) short- and (B) long-term treatment (n=5–10/group; *P<0.05 vs wild-type [WT]; †P<0.05 vs vehicle [VEH]). C, Increased H4-acetylation was also visible when VPA was applied acutely to atrial cardiomyocytes and perfused atria from WT mice (n=4–5 isolations or hearts/group; *P<0.05 vs untreated). D, Representative action potentials (APs) recorded from atrial cardiomyocytes (WTVEH; age: 12 wk) before and during acute administration of VPA. E–G, Acutely administered VPA did not alter AP duration (APD)50, APD70, and APD90, the mean AP amplitude H, slope I (n=6 cardiomyocytes/3 isolations) or J the amplitude of INa in atrial TGVEH cardiomyocytes (n=6 cardiomyocytes/5 isolations). K, Genetic inactivation of HDAC2 (HDAC2KO) in TG×HDAC2KO mice lead to a similar attenuation of the ultrastructural disarrangement than VPA treatment (see Figure 2; TG×HDAC2KO, transgenic: n=5 animals/group; HDAC2KO, control [CTR]: n=3 animals/group; average of 12 images/animal; Sarcomeres, Other: *P<0.05 vs CTR and †<0.05 vs transgenic; mitochondria, collagen: not significant). L, Representative images.
Acute VPA Application Did Not Affect AP Kinetics or Na+ Current
We recorded APs in WT atrial cardiomyocytes before and during acute application of VPA in a dose reported to affect ionic currents in neurons (1 mmol/L).27 Whereas we did observe an increased histone H4-acetylation in WT isolated atrial cardiomyocytes and whole heart preparations acutely treated with 1 mmol/L VPA (P<0.05; n=4–5 isolations or hearts/group; Figure 6C), we did not observe any acute changes in AP parameters (amplitude, slope, durations; P >0.05; n=6 cardiomyocytes/3 isolations; Figure 6D through 6I). Moreover, Na+ currents (INa) directly measured in atrial cardiomyocytes from TGVEH mice were not affected at this dose (P >0.05; n=6 cardiomyocytes/5 isolations; Figure 6J). Thus, noteworthy acute effects of VPA on AP relevant cardiac ion channels up to a concentration of 1 mmol/L are unlikely.
Genetic Inactivation of HDAC2 Attenuated Ultrastructural Disarrangement of Atria in Transgenic Mice Comparably With VPA
Electron microscopy analyses of atria from transgenic mice crossbred with heart-specific HDAC2KO mice (age 12–14 weeks) revealed an attenuation of the ultrastructural remodeling that mimicked the effect of VPA in transgenic mice. Whereas the area with sarcomeres was reduced on average by 44% and the amount of areas with lipofuscin and glycogen granules increased by 66% in transgenic versus WT mice (P<0.05), TG×HDAC2KO atria were significantly less disarranged versus Transgenic and not statistically different versus controls (P >0.05 versus WT or HDAC2KO; n=3 animals/group; Figure 6K and 6L, Figure IV in the Data Supplement). This underlines the role of HDAC2 inhibition for the attenuation of the ultrastructural remodeling in transgenic mice.
VPA Treatment Reversed Protein Dysregulation in Transgenic Atria
With a proteome analysis, we identified 1374 proteins differing in amount among groups after VPA short-term treatment (n=8 atria/group). Changes in protein levels observed in TGVEH versus WTVEH inversely correlated with protein changes induced by VPA in transgenic mice (Figure 7A) but did not correlate with changes induced by VPA in WT (Figure 7B), indicating a disease-specific counter-regulatory effect of VPA in transgenic mice. A detailed intersection analysis is given in Figure V in the Data Supplement. We focused further analysis on strongly regulated proteins in transgenic mice (TGVEH versus WTVEH ratio: <0.5 or >2; P<0.05) that were at the same time significantly altered by VPA (P<0.05), and it turned out that these 295 proteins were consistently counter-regulated by VPA in transgenic mice (Figure 7C; Table I in the Data Supplement).
Figure 7.
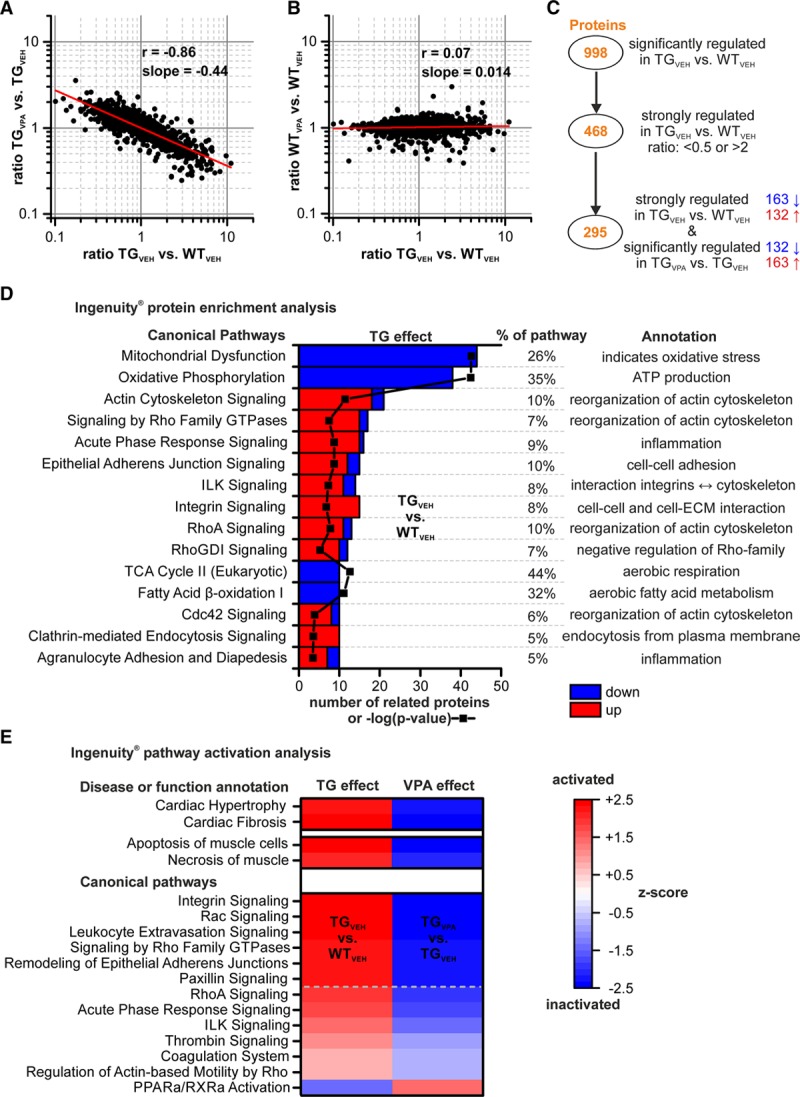
Analysis of proteomic changes induced by CREM-IbΔC-X transgenic (TG) expression and the valproic acid (VPA) treatment. Correlations of protein ratios of 1374 identified proteins between (A) TGVPA/TGVEH and TGVEH/WTVEH, (B) WTVPA/WTVEH and TGVEH/WTVEH after short-term treatment (12 wk of age; red line: linear regression curve) demonstrate a disease-specific counter-regulatory effect of VPA on altered proteins in transgenic mice (Pearson r: −0.86). C, Two hundred and ninety-five proteins, strongly regulated in transgenic mice (TGVEH vs WTVEH, ratio <0.5 or >2) and significantly affected by VPA, were consistently counter-regulated by VPA. D, Enriched top 15 pathways according to Ingenuity Pathway Analysis on these 295 proteins, including number of related proteins (red=upregulated, blue=downregulated vs WTVEH), enrichment P value (black line/symbols; as –log; shared x axis) and proportion of regulated proteins within the indicated pathways. E, Activation state of selected pathways and disease functions in TGVEH atria and in response to VPA (red=activation, blue=inactivation; Z scores >2 and <−2 are considered significant [above dashed line]). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X; ECM, extracellular matrix; ILK, integrin linked kinase; PPAR, peroxisome proliferator-activated receptor; RhoA, Ras homolog gene family, member A; RXRa, retinoid X receptor alpha; TCA, tricarboxylic acid cycle; VEH, vehicle; and WT, wild-type.
We matched these 295 proteins with a recently published independent dataset derived from transgenic mice before the onset of AF at an age of 7 weeks.10 Ninety-eight percent of those proteins identified in both datasets (102/104 matching proteins; indicated by * in all datasets) were already altered at 7 weeks of age (Figure IV in the Data Supplement).
An Ingenuity Pathway Analysis revealed that the 295 proteins are part of and enriched (P<0.05) in pathways related to actin cytoskeleton organization, cell-cell and cell-matrix interaction, metabolism, inflammation, and dysfunction of mitochondria (Figure 7D; Table II in the Data Supplement). It further revealed that relevant disease functions (eg, cardiac hypertrophy, cardiac fibrosis, necrosis of muscle) and pathways (eg, integrin, RhoA, and Rac (Ras-related C3 botulinum toxin substrate) signaling) were activated in TGVEH atria and inhibited by VPA treatment (Figure 7E). Illustrative Kyoto Encyclopedia of Genes and Genomes pathway maps for the pathways oxidative phosphorylation, focal adhesion, hypertrophic cardiomyopathy, and coagulation cascade are shown in Figures VII through X in the Data Supplement. Selected proteins identified by Kyoto Encyclopedia of Genes and Genomes pathway mapping and directly related to AF’s pathology are summarized in Table.
Table.
Selected Proteins Directly Related to the Pathology of AF
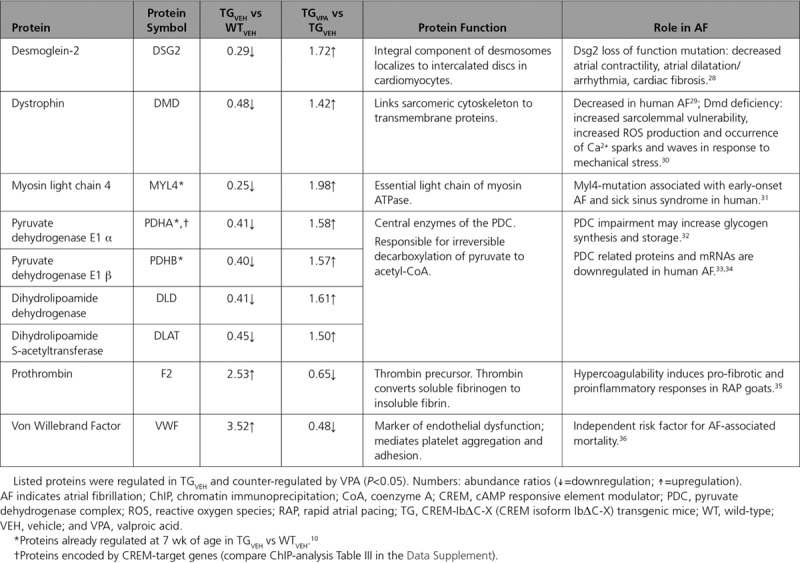
Sixty of the 295 most strongly affected proteins in TGVEH versus WTVEH were, in turn, more than halved or doubled by VPA (TGVPA versus TGVEH ratio: <0.5 and >2; P<0.05). A heatmap of these 60 proteins (Figure 8) illustrates that VPA, for example, increased reduced levels of sarcomeric proteins (MYL7 [myosin light chain 7], TNNI3 [cardiac troponin I, cardiac 3]) and proteins involved in energy metabolism (UQCR10 [ubiquinol-cytochrome c reductase, complex III subunit X], ATP5L [ATP synthase subunit g]), whereas it decreased elevated levels of fibrosis associated proteins (COL1A2 [collagen, type I, alpha 2], ELN [elastin]) and of proteins involved in the coagulation system (VWF [Von Willebrand factor], FGG [fibrinogen gamma chain]) in transgenic atria. We identified 9 genes (Atp5l, Ces1d, Myl7, Ndufa12, Ndufa8, Ndufs7, Pdha1, Tnni3, Uqcr10), whose corresponding proteins were downregulated in TGVEH and reversed by VPA, as direct CREM-IbΔC-X target genes by chromatin immunoprecipitation (Table III in the Data Supplement).
Figure 8.
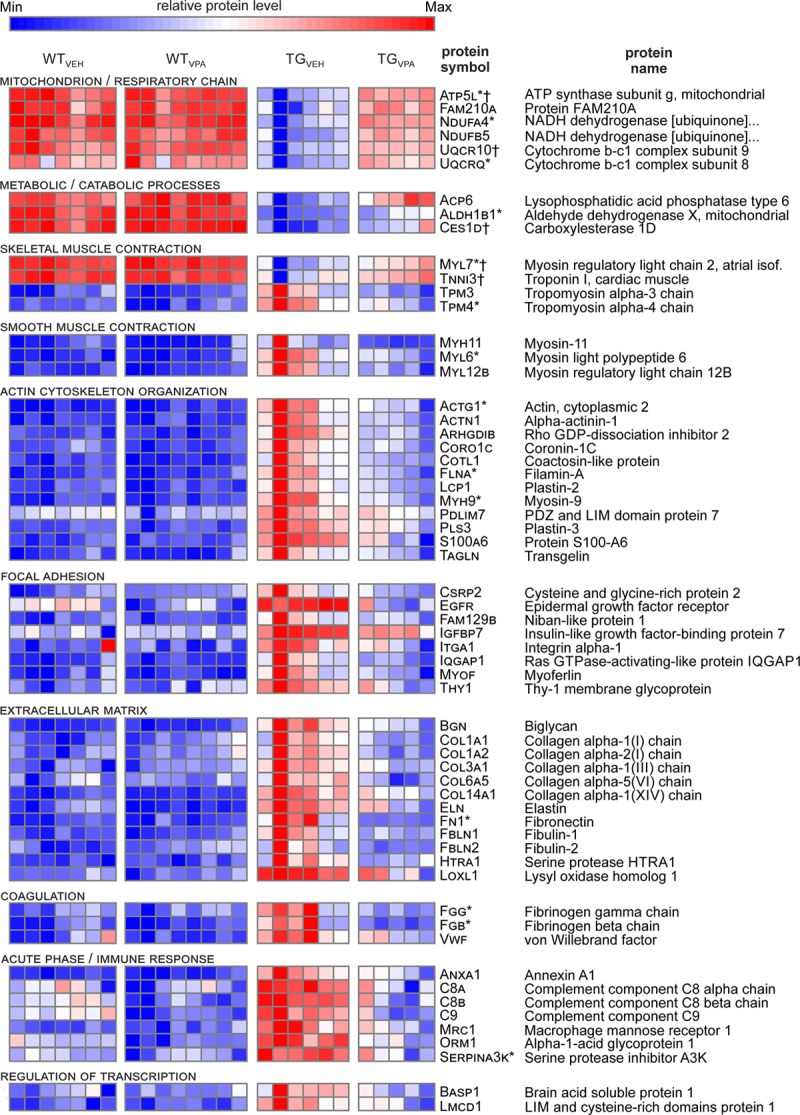
Valproic acid (VPA) counteracts protein deregulation in CREM-IbΔC-X transgenic (TG) mice atria. 60 of the 295 selected proteins (compare Figure 7C) were strongest counter-regulated by VPA (n=5–8/treatment group). Each row corresponds to the protein stated on the right. Colors: relative protein levels (blue=minimum; red=maximum). Proteins are grouped according to their assigned pathways. *Proteins already regulated at 7 wk in TGVEH vs WTVEH.10 †Proteins encoded by CREM-target genes (compare chromatin immunoprecipitation analysis Table III in the Data Supplement). CREM-IbΔC-X indicates CREM (cAMP responsive element modulator) isoform IbΔC-X.
Discussion
Atrial remodeling promotes the initiation and especially perpetuation of AF3. CREM-IbΔC-X mice show an extensive atrial remodeling finally leading to the spontaneous development of AF without additional pharmacological or electrical provocation protocols.6,9,10 In this model, the mild HDAC class I/IIa inhibitor VPA attenuates atrial remodeling and delays the onset of AF by counter-regulating the imbalance of proteins and associated pathways that are well linked to the pathophysiology of AF.
VPA Attenuates the Remodeling of AF Related Substrates in Transgenic Mice
VPA attenuated atrial remodeling by affecting AF related substrates that have been described already in previous studies on transgenic mice. These include atrial enlargement (Figure 1B and 1C),7,9 atrial cardiomyocyte hypertrophy (Figure 1D through 1F),9 atrial fibrosis (Figure 3A through 3C),9 occurrence of thrombi (Figure 3D through 3F),26 and AP prolongation (Figure 4A through 4F).9,10 The ultrastructural remodeling in transgenic (Figure 2) comprising myolysis, loss and disarrangement of mitochondria, glycogen deposition, and increased formation of collagen fibers was recently described in 7-and 12-week-old transgenic mice10 and showed further progression with age in the current study.
All these alterations have been identified as substrates responsible for initiation or perpetuation of AF in human and other animal AF models in previous studies. Atrial dilatation and fibrosis have been reported in animal models with AF induced by rapid atrial pacing.37,38 Left atrial dilatation was associated with incidence of AF in an asymptomatic patient population.39 Increased atrial fibrosis was linked to lower effectiveness of AF catheter ablation.40 Loss of sarcomeres, glycogen accumulation, and disorganization of mitochondria were described in atrial myocardium from patients with AF and in the rapid atrial pacing goat model of sustained AF.38,41 AP prolongation as observed in transgenic mice was associated with an increased risk for atrial torsades-de-pointes-like arrhythmias in patients with long QT–syndrome.42 In human AF, the AP is reportedly shortened leading to a decreased atrial refractory period, potentially facilitating reentry. In transgenic mice, AF is present along with prolongation of the AP, which rather serves as a trigger for AF in this model.
In conclusion, all these substrates will likely contribute to the progressive development of AF observed in transgenic mice (Figure 5), and thus, their consistent attenuation by VPA well explains the delay in onset of AF in TGVPA mice.
VPA Mediated a Disease-Specific, Counter-Regulatory Effect on Altered Protein Levels and Pathways Linked to Remodeling in AF
Changes on protein levels induced by transgenic expression of CREM-IbΔC-X inversely correlated with VPA-mediated changes in transgenic mice (Figure 7A) but not in WT (Figure 7B), indicating a disease-specific, counter-regulatory effect of VPA in transgenic mice. Consequently, 295 of 468 strongly regulated proteins in the transgenic group (TGVEH versus WTVEH ratio: <0.5 or >2) were significantly counter-regulated by VPA (Figure 7C; Table I in the Data Supplement). We focused the proteome analysis (Ingenuity Pathway Analysis) on these 295 proteins because the related pathways and functions should be involved in both, the remodeling in Transgenic mice and its attenuation by VPA.
The analysis revealed that these 295 proteins are enriched in pathways related to cytoskeleton organization, cell-cell and cell-matrix interaction, metabolism, inflammation, and dysfunction of mitochondria (Figure 7D). The disorganization of mitochondria in transgenic mice (Figure 2), for example, is reflected by the downregulation of 44 and 38 mitochondrial proteins—enriched in the pathways Mitochondrial dysfunction and Oxidative phosphorylation, respectively (Figure 7D, Table II and Figure VII in the Data Supplement).
The Ingenuity Pathway Analysis further indicated activation of disease functions in transgenic mice like cardiac hypertrophy and fibrosis, apoptosis and necrosis of muscle cells, and activation of pathways like integrin, Rac, RhoA, and paxillin signaling (Figure 7E). These pathways are involved in the interaction between myocytes and the extracellular matrix and organization of the cytoskeleton. RhoA and Rac are involved in the promotion of stress fibers (F-actin), which play a role in coupling between cardiomyocytes and myofibroblasts and contribute to conduction disturbances.43 In line, mice with transgenic expression of RhoA show pronounced atrial enlargement and occasionally AF.44
By Kyoto Encyclopedia of Genes and Genomes pathway mapping, we identified several proteins that were altered in transgenic mice and counter-regulated by VPA, which have been directly linked to remodeling and the pathophysiology of AF in previous studies (Table). The dysfunction or deficiency of desmoglein-2, dystrophin, and myosin light chain 4, for example, which are downregulated in transgenic atria, goes along with atrial remodeling and AF in human and animal models.28–31 Hypercoagulability was shown to promote AF in rapid atrial pacing goats and thrombin to cause pro-fibrotic and proinflammatory responses in atrial fibroblasts.35 Thus, the observed upregulation of prothrombin and the Von Willebrand factor in transgenic mice (Table), as well as the increased occurrence of thrombi (Figure 3D through 3F) in transgenic atria can be directly linked to the structural remodeling and onset of AF in transgenic mice.
Downregulation of proteins of the pyruvate dehydrogenase complex (PDHA1 [pyruvate dehydrogenase E1 α], PDHB [pyruvate dehydrogenase E1 β], DLD [dihydrolipoamide dehydrogenase], DLAT [dihydrolipoamide S-acetyltransferase]; Table) and the fatty acid β-oxidation (Figure 7D) along with inhibition of the PPAR (peroxisome proliferator-activated receptor) pathway (Figure 7E) and glycogen accumulation (Figure 2) was present in transgenic mice in the current study. Similar metabolic alterations were observed in 7-week-old transgenic mice recently.10 They indicate a metabolic remodeling, as has been described in AF45, with activation of the fetal gene program leading to a metabolic switch from fatty acid to carbohydrate oxidation and accumulation of glycogen because of an impairment of the pyruvate dehydrogenase complex and the PPAR pathway, a pathway which plays an important role in maintaining fatty acid metabolism.10,46 In general, there is high redundancy in identified pathways between the current dataset derived at 12 weeks of age (AF already detectable) and our previous study performed at 7 weeks of age (before onset of AF).10 Because 98% of all matching proteins were already altered in young mice (Figure VI in the Data Supplement), the alterations of the identified pathways in this study start before the onset of AF and thus should be especially responsible for its initiation.
It should be emphasized that the multitude of different functions and pathways altered in transgenic mice (Figure 7D and 7E) well reflects the actual complexity of remodeling in AF that hardly can be attributed to a single pathway.3,45,47 However, the most important finding is that VPA mediates a wide-ranging effect on these pathways and functions rather than targeting a single pathway. This clearly indicates the interaction with a general regulative mechanism as VPA’s mode of action in this model.
VPA Restored Expression of Repressed CREM-Target Genes
The extensive remodeling in transgenic mice is in accordance with other mouse models with heart-specific repression of cAMP-dependent transcription.48–50 Moreover, decreased expression of targets of the CREB/CREM/ATF transcription factor family was linked to AF susceptibility in humans,5 and the repressor isoform CREM-IbΔC-X itself is upregulated in human AF6. Thus, we analyzed whether VPA is able to derepress repressed CREM-target genes. Among the top 60 regulated proteins that were both >2-fold regulated in transgenic mice, as well as >2-fold counter-regulated by VPA (Figure 8), 9 were downregulated in transgenic mice, and 5 of them (Atp5l, Uqcr10, Myl7, Tnni3, Ces1d [carboxylesterase 1]) identified to be encoded by CREM-target genes (Table III in the Data Supplement). This points to a direct role of CREM-IbΔC-X in reducing the level of muscle-related (Myl7, Tnni3) and mitochondrial (Atp5l, Uqcr10) proteins and thus for the loss and disorganization of mitochondria and sarcomeres observed in AF-associated remodeling.
VPA Operated as an HDAC Inhibitor in Transgenic Mice
VPA has been used for the treatment of epilepsy and mood disorders for >40 years, and its effects on neuronal ionic currents and brain GABA (γ-aminobutyric acid) levels have been proposed as a mechanism for its anti-convulsive activity. Although the number of cellular targets that have been proven to be directly affected by VPA is small,51 it has been repeatedly demonstrated that VPA acts as an epigenetic modulator by direct inhibition of HDACs.19,20,52 In this capacity, VPA is, for example, currently tested in clinical trials for anticancer therapy along with other HDAC inhibitors.53
In the heart, studies suggest diverging roles for different HDAC classes. Reportedly, class IIa HDACs (HDAC4, 5, 7, and 9) promote antihypertrophic effects, whereas class I HDACs (HDAC1, 2, 3, and 8) promote cardiac hypertrophy or hyperplasia.13 Consequently, inhibition of class I HDACs is thought to mediate beneficial effects on cardiac remodeling, whereas inhibition of class IIa HDACs may promote hypertrophy. In line, HDAC2 protein level was upregulated in TGVEH versus WTVEH (ratio: 1.65). Cardiomyocyte-specific inactivation of just this single HDAC isoform in TG×HDAC2KO mice already significantly attenuated the ultrastructural remodeling in transgenic mice (Figure 6K and 6L) similar to the VPA treatment (Figure 2). This is strong support for both, an important role for HDAC2 in the observed remodeling in transgenic mice and HDAC inhibition as the predominant mechanism of VPA in this context.
HDAC inhibitors act as epigenetic modifiers by increasing acetylation of histones and nonhistones, and beneficial effects in the heart seem to be mediated by derepression of protective genes and repression of inductors of remodeling and inflammation.16 Consequently, HDAC inhibitors modulate a plethora of pathways and functions. In good accordance, VPA affected a huge number of proteins and associated pathways in transgenic mice, inter alia by derepressing identified CREM-IbΔC-X target genes.
VPA serum levels in mice in this study reached reported therapeutic levels in humans (0.3–1 mmol/L/43–144 µg/mL19,54) and were close to reported IC50 (half maximal inhibitory concentration) values for VPA-mediated HDAC inhibition.19,20,55 Because for VPA, a fatty acid, a remarkably high uptake in the heart has been demonstrated in primates,56 tissue levels may even exceed serum levels. VPA is a semiselective HDAC inhibitor that inhibits HDAC class I>IIa (IC50 [half maximal inhibitory concentration] class I: <1 mmol/L; class IIa: >1 mmol/L).20 Interestingly, Zhang et al57 recently demonstrated converse roles for class I and IIa HDACs in tachypaced HL-1 cardiomyocytes (HL-1 atrial muscle cell line). HDAC3 (class I) overexpression induced cardiomyocyte dysfunction and HDAC5 and 7 (class IIa) overexpression exerted protection against tachypacing-induced myocyte dysfunction. This suggests that the HDAC inhibitory profile of VPA might be especially beneficial in the context of AF by inhibiting preferentially class I HDACs while leaving the potential protective effect of class IIa HDACs largely intact. Zhang et al18,58 also suggested an important role for HDAC6 (class IIb) as a key modulator in AF progression responsible for the derailment of cardiomyocyte proteostasis, and they consequently proposed HDAC6 inhibition as a promising target to treat AF. However, because VPA does not inhibit class IIb HDACs at therapeutic levels20 HDAC6 inhibition is not part of VPAs profile.
Effective in vivo HDAC inhibition in our study was confirmed by demonstrating increased histone H4-acetylation in atrial tissue of transgenic mice after VPA treatment (Figure 6A and 6B).
VPA mediated a disease-specific effect on the proteome and thus remodeling in this study (Figure 7A and 7B). Such a disease-specific effect on cardiac remodeling has also been shown for another HDAC inhibitor, the pan-HDAC inhibitor Trichostatin A (TSA), in the context of hypertrophy. Along preventing pathological cardiac remodeling, TSA reversed promoter specific histone acetylation in mice induced by transverse aortic constriction. Changes in promoter acetylation negatively correlated between TSA and transverse aortic constriction, whereas there was no significant correlation in sham-operated animals.59
VPA and TSA attenuated ventricular hypertrophy in mice with transgenic expression of Hop (homeodomain protein), a homeodomain protein that represses SRF (serum response factor)-dependent transcription by recruiting the class I isoform HDAC2.60 In that model, TSA and the class I HDAC inhibitor CI-994 reduced atrial fibrosis and inducibility of atrial arrhythmic episodes provoked by programmed electrical stimulation.21,61 HDAC inhibitors were shown to block development of fibrosis by blocking cardiac fibroblast cell cycle progression.62 Moreover, VPA was recently shown to block pericyte-myofibroblast trans-differentiation and thereby fibrosis development in rats, which could be attributed to the inhibition of HDAC4.63
Furthermore, VPA effectively induced tPA (tissue-type plasminogen activator) along with other HDAC inhibitors64 and reduced thrombus formation in a mouse model of vascular injury,65 which clearly links the reduced occurrence of thrombi in TGVPA to HDAC inhibition by VPA.
VPA and TSA have been shown to activate AMPK (AMP-activated protein kinase).66,67 Activated AMPK reduces fatty acid synthesis, stimulates fatty acid oxidation, and inhibits cascades involved in protein synthesis, hypertrophic growth, proliferation, and possibly fibrosis.68 It was suggested that AMPK activation might be beneficial in AF by optimizing the balance between energy production and consumption.45,68 AMPK increases the fatty acid transporter CD36 (platelet glycoprotein 4).69 CD36 is considerably downregulated in transgenic mice and significantly counter-regulated by VPA (Table I in the Data Supplement; ratio TGVEH/WTVEH: 0.4 and TGVPA/TGVEH: 1.48). This might point to a long-term AMPK activation by VPA-mediated HDAC inhibition in TGVPA hearts—beneficially affecting hypertrophy, fibrosis, and metabolism in transgenic mice in our study.
An acute effect of VPA on ion channels as reported in neurons27 was not visible in atrial cardiomyocytes (Figure 6D through 6J) up to 1 mmol/L, which is clearly above the mean serum level observed in transgenic mice. Though we cannot formally exclude the contribution of further mechanisms of VPA to the attenuation of atrial remodeling in transgenic mice, the evidence for HDAC inhibition, as the predominant mechanism of VPA in this study, is obvious. The effects on hypertrophy, fibrosis, thrombus occurrence, and metabolism that can be attributed to HDAC inhibition by VPA form a beneficial combination aiming at many proposed therapeutic targets in atrial remodeling and AF.
VPA Showed No Detrimental Effects in WT
Although VPA attenuated atrial remodeling in transgenic mice, structural or electrophysiological changes in WTVPA mice could not be observed. This is in line with an unaltered H4-acetylation in atria from WTVPA mice, which might point to a possible long-term compensatory mechanism because H4-acetylation was increased acutely in WT atria by VPA. Recently, Lugenbiel et al70 reported AP prolongation in HL-1 cardiomyocytes after 12-hour treatment with the pan-HDAC inhibitor TSA. Such an effect seems not to be present after prolonged treatment with VPA in mice, which might depend on VPAs HDAC inhibitory profile or efficacy. Long-term VPA treatment had no detrimental effects on ECG parameters in WT (Figure III in the Data Supplement). In good accordance, VPA is recommended as antiepileptic drug for patients with epilepsy and cardiovascular comorbidities71 underlining its relative cardiac safety.
Limitations of the Study
We started the treatment of transgenic mice at a time point where first atrial ectopies are visible in transgenic mice and AF starts to develop9 to test whether VPA is able to affect the development of atrial remodeling along the progression of AF. Future studies will have to address whether treatment with VPA or more specific HDAC inhibitors beneficially affect atrial remodeling and development of AF detected at more progressed stages of the disease. We investigated only male mice in our study, which showed a more pronounced phenotype. Thus, the impact of VPA on female mice cannot be addressed. VPA did not stop progression of atrial remodeling in our study, and readers may question the clinical relevance of the delay in onset of AF observed in this study on mice. In this context, one has to keep in mind that a delay in the onset of AF of a few weeks in face of the rapid development of the disease in transgenic mice, and an average expected life expectancy in mice of 2 years is a lot. We did not perform hemodynamic measurements in this study because previous investigations in transgenic mice revealed an increased left ventricular contractility (dp/dtmax) and accelerated speed of relaxation in hemodynamic measurements,7 unchanged left ventricular function and wall dimensions in echocardiographic investigations,6 and no hypertrophy of ventricular cardiomyocytes.22 Consequently, the atrial phenotype in transgenic mice cannot be explained by an impaired ventricular function and it is rather unlikely that the attenuation of atrial remodeling in transgenic mice is caused by an improvement of ventricular function by VPA.
With respect to the model and our initial hypothesis, we expected to observe reduced histone H4-acetylation in TGVEH mice because CREM-IbΔC-X as a repressor isoform is not able to recruit p300/HATs. However, CREM-IbΔC-X overexpression starts early in development which may induce compensatory mechanisms. Nevertheless, more important in the context of this study is the clearly visible effect of VPA on histone acetylation as an evidence for effective HDAC inhibition.
Conclusions
In summary, we report that the clinical available and well-tolerated HDAC class I/IIa inhibitor VPA is able to retard the development of atrial remodeling in CREM-IbΔC-X transgenic mice, a unique model of atrial remodeling with spontaneous onset AF. VPA reverses the dysregulation of proteins in transgenic mice that are involved in many AF relevant pathways. This, in turn, results in beneficial effects on a multitude of AF promoting and maintaining substrates, which finally delays the time until the onset of AF. Currently, no available drug in the treatment of human AF has proven to achieve this.
Acknowledgments
We thank Stefanie Triebel, Sophie Eisenlöffel, and Cordula Westermann for technical assistance, as well as Vishnu Dhople for assistance in mass spectrometry.
Sources of Funding
This study was funded by European Union Frame project 7 (EUTRAF to Dr Müller) and Deutsche Forschungsgemeinschaft (DFG Mu1376/11–3 to Dr Müller).
Disclosures
None.
Supplementary Material
Footnotes
Drs Scholz, Schulte, and Hamer are joint first authors.
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCEP.118.007071.
References
- 1.January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC, Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1–e76. doi: 10.1016/j.jacc.2014.03.022. doi: 10.1016/j.jacc.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 2.Krijthe BP, Kunst A, Benjamin EJ, Lip GY, Franco OH, Hofman A, Witteman JC, Stricker BH, Heeringa J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur Heart J. 2013;34:2746–2751. doi: 10.1093/eurheartj/eht280. doi: 10.1093/eurheartj/eht280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 4.Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012;11:275–291. doi: 10.1038/nrd3682. doi: 10.1038/nrd3682. [DOI] [PubMed] [Google Scholar]
- 5.Deshmukh A, Barnard J, Sun H, Newton D, Castel L, Pettersson G, Johnston D, Roselli E, Gillinov AM, McCurry K, Moravec C, Smith JD, Van Wagoner DR, Chung MK. Left atrial transcriptional changes associated with atrial fibrillation susceptibility and persistence. Circ Arrhythm Electrophysiol. 2015;8:32–41. doi: 10.1161/CIRCEP.114.001632. doi: 10.1161/CIRCEP.114.001632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Müller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XHT. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–1285. doi: 10.1161/CIRCULATIONAHA.113.006611. doi: 10.1161/CIRCULATIONAHA.113.006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Müller FU, Lewin G, Baba HA, Bokník P, Fabritz L, Kirchhefer U, Kirchhof P, Loser K, Matus M, Neumann J, Riemann B, Schmitz W. Heart-directed expression of a human cardiac isoform of cAMP-response element modulator in transgenic mice. J Biol Chem. 2005;280:6906–6914. doi: 10.1074/jbc.M407864200. doi: 10.1074/jbc.M407864200. [DOI] [PubMed] [Google Scholar]
- 8.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Müller FU, Schmitz W, Schotten U, Anderson ME, Valderrábano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T, Schulte K, Hanstein J, Schulte JS, Vogel M, Mougenot N, Laakmann S, Fortmueller L, Eckstein J, Verheule S, Kaese S, Staab A, Grote-Wessels S, Schotten U, Moubarak G, Wehrens XH, Schmitz W, Hatem S, Müller FU. Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol. 2013;166:366–374. doi: 10.1016/j.ijcard.2011.10.057. doi: 10.1016/j.ijcard.2011.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seidl MD, Stein J, Hamer S, Pluteanu F, Scholz B, Wardelmann E, Huge A, Witten A, Stoll M, Hammer E, Völker U, Müller FU. Characterization of the genetic program linked to the development of atrial fibrillation in CREM-IbΔC-X mice. Circ Arrhythm Electrophysiol. 2017;10:e005075. doi: 10.1161/CIRCEP.117.005075. [DOI] [PubMed] [Google Scholar]
- 11.Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. doi: 10.1038/nrm3072. doi: 10.1038/nrm3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tenbrock K, Juang YT, Leukert N, Roth J, Tsokos GC. The transcriptional repressor cAMP response element modulator alpha interacts with histone deacetylase 1 to repress promoter activity. J Immunol. 2006;177:6159–6164. doi: 10.4049/jimmunol.177.9.6159. [DOI] [PubMed] [Google Scholar]
- 13.Kee HJ, Kook H. Roles and targets of class I and IIa histone deacetylases in cardiac hypertrophy. J Biomed Biotechnol. 2011;2011:928326. doi: 10.1155/2011/928326. doi: 10.1155/2011/928326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stratton MS, McKinsey TA. Epigenetic regulation of cardiac fibrosis. J Mol Cell Cardiol. 2016;92:206–213. doi: 10.1016/j.yjmcc.2016.02.011. doi: 10.1016/j.yjmcc.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKinsey TA. Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. J Mol Cell Cardiol. 2011;51:491–496. doi: 10.1016/j.yjmcc.2010.11.009. doi: 10.1016/j.yjmcc.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 16.McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–319. doi: 10.1146/annurev-pharmtox-010611-134712. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- 17.Lkhagva B, Kao YH, Chen YC, Chao TF, Chen SA, Chen YJ. Targeting histone deacetylases: a novel therapeutic strategy for atrial fibrillation. Eur J Pharmacol. 2016;781:250–257. doi: 10.1016/j.ejphar.2016.04.034. doi: 10.1016/j.ejphar.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 18.Zhang D, Hu X, Henning RH, Brundel BJ. Keeping up the balance: role of HDACs in cardiac proteostasis and therapeutic implications for atrial fibrillation. Cardiovasc Res. 2016;109:519–526. doi: 10.1093/cvr/cvv265. doi: 10.1093/cvr/cvv265. [DOI] [PubMed] [Google Scholar]
- 19.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64:1079–1086. doi: 10.1158/0008-5472.can-03-0799. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Levin MD, Petrenko NB, Lu MM, Wang T, Yuan LJ, Stout AL, Epstein JA, Patel VV. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J Mol Cell Cardiol. 2008;45:715–723. doi: 10.1016/j.yjmcc.2008.08.015. doi: 10.1016/j.yjmcc.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulte JS, Fehrmann E, Tekook MA, Kranick D, Fels B, Li N, Wehrens XH, Heinick A, Seidl MD, Schmitz W, Müller FU. Cardiac expression of the CREM repressor isoform CREM-IbΔC-X in mice leads to arrhythmogenic alterations in ventricular cardiomyocytes. Basic Res Cardiol. 2016;111:15. doi: 10.1007/s00395-016-0532-y. doi: 10.1007/s00395-016-0532-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulte JS, Seidl MD, Nunes F, Freese C, Schneider M, Schmitz W, Müller FU. CREB critically regulates action potential shape and duration in the adult mouse ventricle. Am J Physiol Heart Circ Physiol. 2012;302:H1998–H2007. doi: 10.1152/ajpheart.00057.2011. doi: 10.1152/ajpheart.00057.2011. [DOI] [PubMed] [Google Scholar]
- 24.Bögeholz N, Pauls P, Kaese S, Schulte JS, Lemoine MD, Dechering DG, Frommeyer G, Goldhaber JI, Seidl MD, Kirchhefer U, Eckardt L, Müller FU, Pott C. Triggered activity in atrial myocytes is influenced by Na+/Ca2+ exchanger activity in genetically altered mice. J Mol Cell Cardiol. 2016;101:106–115. doi: 10.1016/j.yjmcc.2016.11.004. doi: 10.1016/j.yjmcc.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishtala K, Phong TQ, Steil L, Sauter M, Salazar MG, Kandolf R, Felix SB, Völker U, Klingel K, Hammer E. Proteomic analyses of age related changes in A.BY/SnJ mouse hearts. Proteome Sci. 2013;11:29. doi: 10.1186/1477-5956-11-29. doi: 10.1186/1477-5956-11-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bukowska A, Felgendreher M, Scholz B, Wolke C, Schulte JS, Fehrmann E, Wardelmann E, Seidl MD, Lendeckel U, Himmler K, Gardemann A, Goette A, Müller FU. CREM-transgene mice: An animal model of atrial fibrillation and thrombogenesis. Thromb Res. 2018;163:172–179. doi: 10.1016/j.thromres.2017.07.033. doi: 10.1016/j.thromres.2017.07.033. [DOI] [PubMed] [Google Scholar]
- 27.Zona C, Avoli M. Effects induced by the antiepileptic drug valproic acid upon the ionic currents recorded in rat neocortical neurons in cell culture. Exp Brain Res. 1990;81:313–317. doi: 10.1007/BF00228121. [DOI] [PubMed] [Google Scholar]
- 28.Krusche CA, Holthöfer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol. 2011;106:617–633. doi: 10.1007/s00395-011-0175-y. doi: 10.1007/s00395-011-0175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reilly SN, Liu X, Carnicer R, Recalde A, Muszkiewicz A, Jayaram R, Carena MC, Wijesurendra R, Stefanini M, Surdo NC, Lomas O, Ratnatunga C, Sayeed R, Krasopoulos G, Rajakumar T, Bueno-Orovio A, Verheule S, Fulga TA, Rodriguez B, Schotten U, Casadei B. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci Transl Med. 2016;8:340ra74. doi: 10.1126/scitranslmed.aac4296. doi: 10.1126/scitranslmed.aac4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shirokova N, Niggli E. Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies. J Mol Cell Cardiol. 2013;58:217–224. doi: 10.1016/j.yjmcc.2012.12.009. doi: 10.1016/j.yjmcc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gudbjartsson DF, Holm H, Sulem P, Masson G, Oddsson A, Magnusson OT, Saemundsdottir J, Helgadottir HT, Helgason H, Johannsdottir H, Gretarsdottir S, Gudjonsson SA, Njølstad I, Løchen ML, Baum L, Ma RC, Sigfusson G, Kong A, Thorgeirsson G, Sverrisson JT, Thorsteinsdottir U, Stefansson K, Arnar DO. A frameshift deletion in the sarcomere gene MYL4 causes early-onset familial atrial fibrillation. Eur Heart J. 2017;38:27–34. doi: 10.1093/eurheartj/ehw379. doi: 10.1093/eurheartj/ehw379. [DOI] [PubMed] [Google Scholar]
- 32.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kourliouros A, Yin X, Didangelos A, Hosseini MT, Valencia O, Mayr M, Jahangiri M. Substrate modifications precede the development of atrial fibrillation after cardiac surgery: a proteomic study. Ann Thorac Surg. 2011;92:104–110. doi: 10.1016/j.athoracsur.2011.03.071. doi: 10.1016/j.athoracsur.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 34.Barth AS, Merk S, Arnoldi E, Zwermann L, Kloos P, Gebauer M, Steinmeyer K, Bleich M, Kääb S, Hinterseer M, Kartmann H, Kreuzer E, Dugas M, Steinbeck G, Nabauer M. Reprogramming of the human atrial transcriptome in permanent atrial fibrillation: expression of a ventricular-like genomic signature. Circ Res. 2005;96:1022–1029. doi: 10.1161/01.RES.0000165480.82737.33. doi: 10.1161/01.RES.0000165480.82737.33. [DOI] [PubMed] [Google Scholar]
- 35.Spronk HM, De Jong AM, Verheule S, De Boer HC, Maass AH, Lau DH, Rienstra M, van Hunnik A, Kuiper M, Lumeij S, Zeemering S, Linz D, Kamphuisen PW, Ten Cate H, Crijns HJ, Van Gelder IC, van Zonneveld AJ, Schotten U. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur Heart J. 2017;38:38–50. doi: 10.1093/eurheartj/ehw119. doi: 10.1093/eurheartj/ehw119. [DOI] [PubMed] [Google Scholar]
- 36.Roldán V, Marín F, Muiña B, Torregrosa JM, Hernández-Romero D, Valdés M, Vicente V, Lip GY. Plasma von Willebrand factor levels are an independent risk factor for adverse events including mortality and major bleeding in anticoagulated atrial fibrillation patients. J Am Coll Cardiol. 2011;57:2496–2504. doi: 10.1016/j.jacc.2010.12.033. doi: 10.1016/j.jacc.2010.12.033. [DOI] [PubMed] [Google Scholar]
- 37.Bauer A, McDonald AD, Donahue JK. Pathophysiological findings in a model of persistent atrial fibrillation and severe congestive heart failure. Cardiovasc Res. 2004;61:764–770. doi: 10.1016/j.cardiores.2003.12.013. doi: 10.1016/j.cardiores.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 38.Ausma J, Litjens N, Lenders MH, Duimel H, Mast F, Wouters L, Ramaekers F, Allessie M, Borgers M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J Mol Cell Cardiol. 2001;33:2083–2094. doi: 10.1006/jmcc.2001.1472. doi: 10.1006/jmcc.2001.1472. [DOI] [PubMed] [Google Scholar]
- 39.Habibi M, Samiei S, Ambale Venkatesh B, Opdahl A, Helle-Valle TM, Zareian M, Almeida ALC, Choi E-Y, Wu C, Alonso A, Heckbert SR, Bluemke DA, Lima JAC. Cardiac magnetic resonance-measured left atrial volume and function and incident atrial fibrillation: results from MESA (Multi-Ethnic Study of Atherosclerosis). Circ Cardiovasc Imaging. 2016;9:e004299. doi: 10.1161/CIRCIMAGING.115.004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E. Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol. 2015;66:943–959. doi: 10.1016/j.jacc.2015.06.1313. doi: 10.1016/j.jacc.2015.06.1313. [DOI] [PubMed] [Google Scholar]
- 41.Thijssen VL, Ausma J, Liu GS, Allessie MA, van Eys GJ, Borgers M. Structural changes of atrial myocardium during chronic atrial fibrillation. Cardiovasc Pathol. 2000;9:17–28. doi: 10.1016/s1054-8807(99)00038-1. [DOI] [PubMed] [Google Scholar]
- 42.Kirchhof P, Eckardt L, Franz MR, Mönnig G, Loh P, Wedekind H, Schulze-Bahr E, Breithardt G, Haverkamp W. Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1027–1033. doi: 10.1046/j.1540-8167.2003.03165.x. [DOI] [PubMed] [Google Scholar]
- 43.Thompson SA, Copeland CR, Reich DH, Tung L. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation. 2011;123:2083–2093. doi: 10.1161/CIRCULATIONAHA.110.015057. doi: 10.1161/CIRCULATIONAHA.110.015057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd, Ross J, Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harada M, Melka J, Sobue Y, Nattel S. Metabolic considerations in atrial fibrillation- Mechanistic insights and therapeutic opportunities. Circ J. 2017;81:1749–1757. doi: 10.1253/circj.CJ-17-1058. doi: 10.1253/circj.CJ-17-1058. [DOI] [PubMed] [Google Scholar]
- 46.Gore-Panter SR, Rennison JH, van Wagoner DR. Genetic-genomic insights into the metabolic determinants of spontaneous atrial fibrillation. Circ Arrhythm Electrophysiol. 2017;10:e005636. doi: 10.1161/CIRCEP.117.005636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jalife J, Kaur K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc Med. 2015;25:475–484. doi: 10.1016/j.tcm.2014.12.015. doi: 10.1016/j.tcm.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamoto Y, Chaves A, Chen J, Kelley R, Jones K, Weed HG, Gardner KL, Gangi L, Yamaguchi M, Klomkleaw W, Nakayama T, Hamlin RL, Carnes C, Altschuld R, Bauer J, Hai T. Transgenic mice with cardiac-specific expression of activating transcription factor 3, a stress-inducible gene, have conduction abnormalities and contractile dysfunction. Am J Pathol. 2001;159:639–650. doi: 10.1016/S0002-9440(10)61735-X. doi: 10.1016/S0002-9440(10)61735-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. doi: 10.1172/JCI2950. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kehat I, Heinrich R, Ben-Izhak O, Miyazaki H, Gutkind JS, Aronheim A. Inhibition of basic leucine zipper transcription is a major mediator of atrial dilatation. Cardiovasc Res. 2006;70:543–554. doi: 10.1016/j.cardiores.2006.02.018. doi: 10.1016/j.cardiores.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 51.Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci. 2007;64:2090–2103. doi: 10.1007/s00018-007-7079-x. doi: 10.1007/s00018-007-7079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- 53.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.French JA, Pedley TA. Clinical practice. Initial management of epilepsy. N Engl J Med. 2008;359:166–176. doi: 10.1056/NEJMcp0801738. doi: 10.1056/NEJMcp0801738. [DOI] [PubMed] [Google Scholar]
- 55.Eyal S, Yagen B, Sobol E, Altschuler Y, Shmuel M, Bialer M. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia. 2004;45:737–744. doi: 10.1111/j.0013-9580.2004.00104.x. doi: 10.1111/j.0013-9580.2004.00104.x. [DOI] [PubMed] [Google Scholar]
- 56.Kim SW, Hooker JM, Otto N, Win K, Muench L, Shea C, Carter P, King P, Reid AE, Volkow ND, Fowler JS. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl Med Biol. 2013;40:912–918. doi: 10.1016/j.nucmedbio.2013.06.007. doi: 10.1016/j.nucmedbio.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang D, Hu X, Li J, Hoogstra-Berends F, Zhuang Q, Esteban MA, de Groot N, Henning RH, Brundel BJJM. Converse role of class I and class IIa HDACs in the progression of atrial fibrillation. J Mol Cell Cardiol. 2018;125:39–49. doi: 10.1016/j.yjmcc.2018.09.010. doi: 10.1016/j.yjmcc.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 58.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–358. doi: 10.1161/CIRCULATIONAHA.113.005300. doi: 10.1161/CIRCULATIONAHA.113.005300. [DOI] [PubMed] [Google Scholar]
- 59.Ooi JY, Tuano NK, Rafehi H, Gao XM, Ziemann M, Du XJ, El-Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics. 2015;10:418–430. doi: 10.1080/15592294.2015.1024406. doi: 10.1080/15592294.2015.1024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112:863–871. doi: 10.1172/JCI19137. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seki M, LaCanna R, Powers JC, Vrakas C, Liu F, Berretta R, Chacko G, Holten J, Jadiya P, Wang T, Arkles JS, Copper JM, Houser SR, Huang J, Patel VV, Recchia FA. Class I histone deacetylase inhibition for the treatment of sustained atrial fibrillation. J Pharmacol Exp Ther. 2016;358:441–449. doi: 10.1124/jpet.116.234591. doi: 10.1124/jpet.116.234591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schuetze KB, Stratton MS, Blakeslee WW, Wempe MF, Wagner FF, Holson EB, Kuo YM, Andrews AJ, Gilbert TM, Hooker JM, McKinsey TA. Overlapping and divergent actions of structurally distinct histone deacetylase inhibitors in cardiac fibroblasts. J Pharmacol Exp Ther. 2017;361:140–150. doi: 10.1124/jpet.116.237701. doi: 10.1124/jpet.116.237701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Gao F, Tang Y, Xiao J, Li C, Ouyang Y, Hou Y. Valproic acid regulates Ang II-induced pericyte-myofibroblast trans-differentiation via MAPK/ERK pathway. Am J Transl Res. 2018;10:1976–1989. [PMC free article] [PubMed] [Google Scholar]
- 64.Larsson P, Bergh N, Lu E, Ulfhammer E, Magnusson M, Wåhlander K, Karlsson L, Jern S. Histone deacetylase inhibitors stimulate tissue-type plasminogen activator production in vascular endothelial cells. J Thromb Thrombolysis. 2013;35:185–192. doi: 10.1007/s11239-012-0831-6. doi: 10.1007/s11239-012-0831-6. [DOI] [PubMed] [Google Scholar]
- 65.Larsson P, Alwis I, Niego B, Sashindranath M, Fogelstrand P, Wu MC, Glise L, Magnusson M, Daglas M, Bergh N, Jackson SP, Medcalf RL, Jern S. Valproic acid selectively increases vascular endothelial tissue-type plasminogen activator production and reduces thrombus formation in the mouse. J Thromb Haemost. 2016;14:2496–2508. doi: 10.1111/jth.13527. doi: 10.1111/jth.13527. [DOI] [PubMed] [Google Scholar]
- 66.Avery LB, Bumpus NN. Valproic acid is a novel activator of AMP-activated protein kinase and decreases liver mass, hepatic fat accumulation, and serum glucose in obese mice. Mol Pharmacol. 2014;85:1–10. doi: 10.1124/mol.113.089755. doi: 10.1124/mol.113.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, Yin XM, Dong Z. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018;9:322. doi: 10.1038/s41419-018-0374-7. doi: 10.1038/s41419-018-0374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harada M, Nattel SN, Nattel S. AMP-activated protein kinase: potential role in cardiac electrophysiology and arrhythmias. Circ Arrhythm Electrophysiol. 2012;5:860–867. doi: 10.1161/CIRCEP.112.972265. doi: 10.1161/CIRCEP.112.972265. [DOI] [PubMed] [Google Scholar]
- 69.Choi YJ, Lee KY, Jung SH, Kim HS, Shim G, Kim MG, Oh YK, Oh SH, Jun DW, Lee BH. Activation of AMPK by berberine induces hepatic lipid accumulation by upregulation of fatty acid translocase CD36 in mice. Toxicol Appl Pharmacol. 2017;316:74–82. doi: 10.1016/j.taap.2016.12.019. doi: 10.1016/j.taap.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 70.Lugenbiel P, Govorov K, Rahm AK, Wieder T, Gramlich D, Syren P, Weiberg N, Seyler C, Katus HA, Thomas D. Inhibition of histone deacetylases induces K+ channel remodeling and action potential prolongation in HL-1 atrial cardiomyocytes. Cell Physiol Biochem. 2018;49:65–77. doi: 10.1159/000492840. doi: 10.1159/000492840. [DOI] [PubMed] [Google Scholar]
- 71.Ruiz-Giménez J, Sánchez-Alvarez JC, Cañadillas-Hidalgo F, Serrano-Castro PJ Andalusian Epilepsy Society. Antiepileptic treatment in patients with epilepsy and other comorbidities. Seizure. 2010;19:375–382. doi: 10.1016/j.seizure.2010.05.008. doi: 10.1016/j.seizure.2010.05.008. [DOI] [PubMed] [Google Scholar]