

Cardiolipin induces conformational changes in complex I that modulate the accessibility of the quinone substrate to the enzyme.
Abstract
Cardiolipin modulates the activity of membrane-bound respiratory enzymes that catalyze biological energy transduction. The respiratory complex I functions as the primary redox-driven proton pump in mitochondrial and bacterial respiratory chains, and its activity is strongly enhanced by cardiolipin. However, despite recent advances in the structural biology of complex I, cardiolipin-specific interaction mechanisms currently remain unknown. On the basis of millisecond molecular simulations, we suggest that cardiolipin binds to proton-pumping subunits of complex I and induces global conformational changes that modulate the accessibility of the quinone substrate to the enzyme. Our findings provide key information on the coupling between complex I dynamics and activity and suggest how biological membranes modulate the structure and activity of proteins.
INTRODUCTION
Complex I (NADH:ubiquinone oxidoreductase) functions as a redox-driven proton pump in aerobic respiratory chains (1–3). With a molecular mass of ca. 1 MDa distributed among up to 45 subunits in eukaryotes (4–7), complex I is the largest, most intricate, and least understood enzyme of the respiratory chain. The over 100-Å-long hydrophilic domain of complex I catalyzes electron transfer between nicotinamide adenine dinucleotide (NADH) and quinone, and the released free energy is used for proton pumping across the 200-Å-wide membrane domain (Fig. 1) (8–10). The established proton motive force powers energy-requiring processes in the cell in the form of active transport and synthesis of adenosine triphosphate (ATP) (11, 12). The electron and proton transfer processes are fully coupled despite their large spatial separation. Although the overall mechanism of this process still remains unclear, molecular principles recently started to emerge, suggesting that the signal propagates by combined conformational, electrostatic, and hydration changes across the membrane domain (13–16).
Fig. 1. Structure and function of the respiratory complex I (PDB ID: 4HEA).
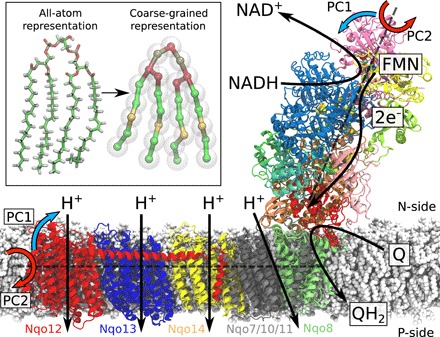
Electrons are transferred from the NADH/FMN (flavin mononucleotide) site to quinone (Q) via a tunneling wire comprising eight iron-sulfur centers. The Q reduction takes place at the interface between the hydrophilic and membrane domains and couples to the pumping of four protons across the membrane. The global bending (PC1, blue arrows) and twisting motions (PC2, red arrows) are shown along two principal axes (see text). Inset: Comparison of atomistic and coarse-grained molecular simulation models of a cardiolipin molecule.
In addition to the functional elements of the 14 core subunits (17), the activity of complex I strongly depends on cardiolipin (18–20), an anionic lipid that accounts for 20% of mitochondrial membranes. The activity of all mitochondrial energy-transducing enzymes depends on this lipid (21, 22), but unlike, e.g., FoF1–ATPase, where the lipid-protein interactions are transient (23, 24), complex I interacts with at least 10 tightly bound cardiolipin molecules that strongly modulate the enzymatic activity (25). Recent cryo–electron microscopy (cryo-EM) structures (6, 7) suggest that some cardiolipins are likely to bind between the proton-pumping membrane subunits, but the functional role of these lipids remains puzzling, considering that they are located up to 100 Å away from the quinone reduction site. The quinone site is located ca. 20 Å above the membrane plane, at the top of a narrow tunnel with an opening to the lipid membrane (Fig. 1).
To elucidate a molecular mechanism by which cardiolipin modulates the activity of complex I, we study here the dynamics of the bacterial enzyme in cardiolipin-containing membranes on millisecond time scales using a combination of coarse-grained and atomistic molecular dynamics (MD) simulation approaches (Fig. 1, inset).
RESULTS
Millisecond simulations identify cardiolipin binding sites
To probe the effect of cardiolipin on the structure and dynamics of complex I, we performed coarse-grained molecular dynamics (CG-MD) simulations on millisecond time scales with the bacterial complex I embedded in a lipid membrane containing 20% cardiolipin and compared these to simulations of complex I in POPC (palmitoyl-oleoyl phosphatidylcholine):POPE (palmitoyl-oleoyl phosphatidylethanolamine) membrane without cardiolipin (see Methods; table S1). The coarse-grained simulations suggest that complex I forms contacts with ca. 150 lipid molecules during the simulations (Fig. 2, A and B). However, in stark contrast to POPE and POPC (see Methods) that transiently interact with the protein, cardiolipin molecules form much stronger interactions with the membrane domain of complex I (Fig. 2C and movie S1). The Nqo8 subunit, which provides a hinge between the membrane domain and the hydrophilic domain, strongly interacts with 10 to 15 cardiolipin molecules (Fig. 2, A to C) that are also supported by atomistic MD simulation of the complex I hinge region (see Methods; fig. S1). We also observe prominent binding sites in the CG-MD models at the interface between the antiporter-like subunits, Nqo12/Nqo13 and Nqo13/Nqo14 (Fig. 2A and fig. S2). These sites are close to the broken transmembrane (TM) helix elements, TM12, which are involved in establishing proton channels across the membrane (15). The identified binding sites superimpose with cardiolipin molecules recently resolved from cryo-EM structure of mammalian complex I (Fig. 2A) (6, 7), suggesting that the cardiolipin interaction sites are conserved across different species, as in the other respiratory enzymes (22).
Fig. 2. Cardiolipin binding sites in complex I.
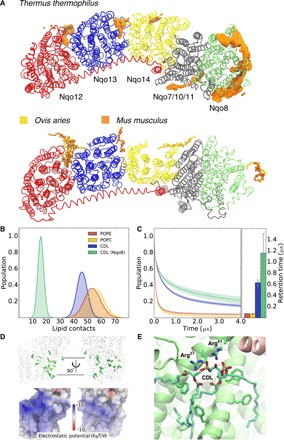
(A) Top: Cardiolipin (CDL) binding sites identified by CG-MD simulations of complex I from Thermus thermophilus. Bottom: Experimentally refined CDL molecules around cryo-EM structures of complex I from Ovis aries (6) (PDB ID: 5LNK) and Mus musculus (7) (PDB ID: 6G2J). Note that TM helices 1 to 3 of subunit Nqo14 (subunit in yellow, T. thermophilus) are not present in the homologous mammalian ND2 subunits. Both enzymes are viewed from the N-side of the membrane. The supernumerary subunits are omitted for visual clarity. (B) Statistics of lipid contacts with the membrane domain of complex I obtained from CG-MD simulations. (C) Autocorrelation function of the lipid binding dynamics. The average retention time for CDL is about six times higher than that for POPE or POPC, and ca. 12 times higher than that for Nqo8-bound CDL. (D) Top: Membrane-exposed phenylalanine residues in Nqo8. Bottom: Electrostatic potential map of complex I (front view). The color scale ranges from +10 kBT/e (blue, ca. +260 mV) to −10 kBT/e (red, ca. −260 mV). (E) Putative CDL binding sites in Nqo8, obtained from atomistic simulations, highlighting interactions with Arg37, Arg41, and phenylalanine residues.
The cardiolipin binding is induced by positively charged surface regions distributed around the membrane domain of complex I (Fig. 2D) and conserved across different complex I isoforms (fig. S3). In addition, phenylalanine residues interact with cardiolipin, particularly in the Nqo8 subunit (Fig. 2, D and E). Notably, phenylalanine residues provide important cardiolipin binding sites also in cytochrome c oxidase (21).
Cardiolipin binding affects complex I structure and dynamics
To analyze how cardiolipin affects the global dynamics of complex I, we projected out thermal fluctuations from the CG-MD simulation trajectories using principal components (PC) analysis (26). The analysis indicates that complex I undergoes large-scale bending (PC1) and twisting (PC2) motion around the hydrophilic and membrane domains with Nqo8 acting as a hinge (Fig. 1), consistent with global motions identified from previous network models and atomistic simulations of the bacterial complex I in a POPC membrane (27). The PC analysis suggests that the coupled twisting-bending motions (PC3) around the hydrophilic and membrane domains is strongly modulated by cardiolipin (Fig. 3A and fig. S4). This global motion displaces half of the membrane and hydrophilic domains (Fig. 3, A and B), and its origin can be traced to cardiolipin bound at the Nqo13/Nqo14 interface and at Nqo8 that induce a 0.4-nm structural shift between TM1 of Nqo8 and helix 1 of Nqo9 (Fig. 3C). These structural rearrangements may have important functional implications as they modulate the accessibility of the quinone to its binding site (see below).
Fig. 3. Effect of CDL on the dynamics of complex I.
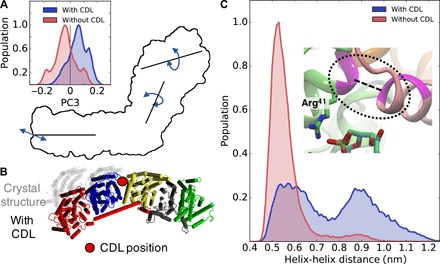
(A) The coupled twisting-bending motion (PC3) is strongly influenced by CDL binding to complex I. The crystal structure is taken as reference conformation for the PC. (B) Structural changes in the membrane domain (view from the N-side) induced by cardiolipin binding at the Nqo13/Nqo14 interface. (C) Cardiolipin induces conformational changes at the interface between Nqo8 and the hydrophilic domain. The figure shows the distance between helix TM1 of Nqo8 and helix 1 of Nqo9.
Cardiolipin modulates quinone access and channel formation dynamics
To probe how the quinone dynamics is affected by cardiolipin, we studied the global motions of complex I with and without a quinone molecule inside the quinone tunnel (see Methods; table S1). The quinone finds stable binding sites in the CG-MD simulations at the top (d ~ 0.5 to 0.7 nm), slightly below (d ~ 0.7 to 0.9 nm), and at the bottom part (d ~ 3.2 to 3.7 nm from Tyr87) of the cavity (Fig. 4, A and B). These binding sites are consistent with minima observed in free energy profiles of the quinone diffusion along the tunnel derived from a recent atomistic study of complex I in a POPC membrane on nanosecond time scales (28). We find that the enzyme undergoes a structural rearrangement in the CG-MD simulations along the bending mode when the quinone molecule moves from the lower edge of the tunnel opening (d ~ 3.2 to 4 nm in Fig. 4, A to C) to the upper binding site (d ~ 0.6 to 0.8 nm in Fig. 4, A to C). When the quinone passes a strongly bent kink region (d ~ 2 nm; Fig. 4B) (29) in the middle of the tunnel, we observe a structural rearrangement along the twisting mode that facilitates the substrate motions along its tunnel, indicated by a transition around d ~ 2 nm in Fig. 4A. In contrast, when the quinone is located at the lower edge of the tunnel around d ~ 3.2 to 4 nm, complex I samples conformations similar to those observed in simulations of the unbound (apo) state (Fig. 4A), suggesting that the initial quinone binding to complex I does not trigger large-scale conformational changes in the protein structure.
Fig. 4. CDL modulates quinone and complex I dynamics.
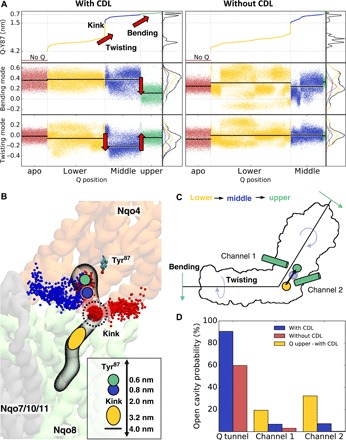
(A) Projection of complex I dynamics with and without CDL along the bending (PC1) and twisting (PC2) modes sorted by the quinone (Q) position along the tunnel. The quinone position is determined by the distance between the quinone headgroup and Tyr87. The quinone motion along the cavity is coupled to the complex I motion along the bending and twisting modes. (B) Quinone binding sites and channels connecting the quinone cavity with the N-side of the membrane. The upper binding sites (d ~ 0.6 to 0.8 nm from Tyr87) are shown as green and blue circles, the kink region is marked at d ~ 2.0 nm, and the lower binding sites (d ~ 3.2 to 4.0 nm) are shown as a yellow oval. Channel 1 (blue) is located near the Nqo7 loop region, and channel 2 (in red) is located at the interface between Nqo8 and Nqo9. Both channels are more stable in simulations with CDL and when quinone is bound at the upper site (d ~ 0.6 to 0.8 nm) (see also movie S2). Inset: Distance between Tyr87 and the kink region and different quinone binding sites. (C) Schematic representation of structural changes in complex I that couple to the quinone motion. (D) Probability of finding open cavities connected to the quinone binding site.
The activity of the mammalian complex I is regulated by the so-called active (A)–to–deactive (D) transition (30) that is also characterized by structural changes along these twisting and bending modes (5, 7, 27). Since we observe a strong correlation between the quinone position in the tunnel and these global motions (Fig. 4 and fig. S5), our findings suggest that quinone dynamics could also be modulated by the structural changes required for the A-D transition in the eukaryotic complex I.
In the absence of cardiolipin in the membrane, the quinone rarely visits the upper binding site, but it finds more stable binding poses at the edge of the tunnel (d ~ 2.7 to 3.6 nm; Fig. 4A). We find that complex I also undergoes similar global twisting and bending motions around the hydrophilic and membrane domains as in the cardiolipin-bound state (fig. S4A). However, in stark contrast to the latter, complex I now samples a configurational space that is very similar to that in the different quinone-bound states as well as that in the unbound (apo) state of the enzyme, with large fluctuations along the twisting and bending modes throughout the complete distance range (d ~ 0.6 to 4.2 nm; Fig. 4A).
This suggests that the twisting and bending motions are now decoupled from the quinone diffusion, indicating that cardiolipin alters the free energy landscape of the enzyme (Fig. 4A). In other words, the bound cardiolipin molecules seem to direct complex I to sample conformations that favor the quinone motion along its cavity and stabilize quinone binding. These findings thus provide a possible molecular explanation to how cardiolipin enhances complex I activity.
The bound cardiolipin enhances the stability of the quinone tunnel (Fig. 4D), in addition to two prominent cavities that are large enough for water molecules to enter from the negatively charged side of the membrane (N-side) to the quinone tunnel. Coarse-grained simulations usually have a tendency to overestimate protein-protein interactions (31), and they may therefore underestimate the probability of finding such channels.
One of these putative channels comprises several conserved residues (table S2) and leads from the N-side to the conserved His38 and Tyr87 residues of Nqo4 (Fig. 4C and movie S2), which function as proton donors upon reduction of quinone to quinol (QH2) (32). Reprotonation of these residues is thus a prerequisite for complex I to initiate the next catalytic cycle. This cavity is close to a loop region in Nqo7 that also undergoes conformational changes during the A-D transition in the mammalian complex I (5). It is important to note that a functional redox-driven pump requires tight gating of the protonation reactions to avoid leaks. A continuous water connectivity between the quinone reduction site and the N-side could thus compromise the proton-pumping machinery. To this end, we find that the stability of this putative channel depends on the position of the quinone in the tunnel (Fig. 4D), suggesting that opening of the channel could be controlled by the redox chemistry of the quinone site.
The second channel forms at the Nqo8/Nqo9 interface near TM1 (Fig. 4D). This channel also starts at the N-side of the membrane, but it leads to a unique kink region in the middle of the quinone tunnel. This region coincides with the putative second quinone binding site around d ~ 3.2 to 4.0 nm (Fig. 4C), to which the quinone molecule docks before exiting to the membrane [see above, and cf. also (28)]. The kink site could be relevant for the activation of the proton-pumping machinery in Nqo8, as this site involves many conserved residues that have been linked with human mitochondrial disorders. In simulations without cardiolipin, this channel remains strictly closed, whereas both cardiolipin and quinone bound to the active site strongly enhance opening of this channel (ca. 20 to 30% open channels with quinone and cardiolipin relative to 0.4 to 3% without cardiolipin; Fig. 4D).
DISCUSSION
We have identified here, on the basis of millisecond molecular simulations, how cardiolipin modulates the structure and dynamics of respiratory complex I. Our simulations indicate that cardiolipin binds at specific sites in the membrane domain of complex I, in particular around the Nqo8 subunit and at the interface between antiporter-like subunits Nqo12/Nqo13 and Nqo13/Nqo14. Some of the predicted cardiolipin binding sites are supported by recent cryo-EM structures of the mammalian enzyme (6, 7), whereas others remain to be experimentally validated. Our findings suggest that in addition to the conserved structural core features of complex I, the lipid binding sites are also conserved across different species.
Cardiolipin binding arises from a combination of electrostatic effects between the cardiolipin headgroup and the positively charged protein residues, as well as by dispersive interactions that are dominated by phenylalanine residues. The lipid binding modulates intersubunit contacts, which are important for the protonation signal propagation across the membrane domain (16). This suggests that cardiolipin binding could indirectly also affect the energetics of the proton pumping process. Cardiolipin molecules bound at Nqo8 might also stabilize the highly tilted TM helices of the subunit and affect the energetics of conformational changes linked with the protonation dynamics in this region.
In addition to these local conformational changes, our simulations suggested that complex I also undergoes global conformational changes along the bending and twisting modes that are strongly influenced by cardiolipin. These global motions were recently linked with the active-to-deactive transition (27). Our simulations indicate that bacterial complex I undergoes displacement along a similar structural motion when the quinone molecule moves along its tunnel. Recent studies suggest that the bacterial enzyme could also undergo a fast resting-to-active transition (33–35). In the absence of cardiolipin, the twisting and bending motions are decoupled from the quinone position along its tunnel, resulting in a lower accessibility of the quinone to its binding site. These findings might provide a molecular explanation to the regulatory role of cardiolipin in complex I: The lipid interactions modify the free energy landscape of complex I along the bending and twisting motions, which in turn modulate the quinone dynamics. Although detailed free energy calculations and atomistic simulations will be important to elucidate structural details of this effect, differences between the simulations with and without cardiolipin are systematic and robust and support that cardiolipin plays a central role in the regulation of the complex I dynamics.
We found that cardiolipin also strongly modulates the formation of two channels from the N-side of the membrane, one around the Nqo7 loop region that could provide protonation pathways for the quinone reduction site, and another channel at the Nqo8/Nqo9 interface that leads to a kink site of the quinone cavity, which could be functionally relevant for activating the pumping machinery (28). The functional role of these putative channels can be probed by site-directed mutagenesis experiments, to which our simulations provide important possible candidates (table S2).
In conclusion, our simulations identified how the lipid membrane may modulate global enzyme dynamics that in turn regulate the accessibility of substrates to the active site. These results provide a molecular understanding of complex I function and the role of cardiolipin in the regulation of this respiratory enzyme.
METHODS
General methodological overview
CG-MD simulations were used to study the interaction between cardiolipin and complex I up to millisecond time scales. The CG-MD trajectories were analyzed by PC analysis (26) and protein cavity search algorithms (36) to connect the large-scale motion with the quinone dynamics and the formation of channels leading to the quinone cavity. Cardiolipin binding around the Nqo8 region was validated by atomistic MD simulations to refine the molecular details of the cardiolipin-protein interactions. All CG-MD results were compared with global motions obtained from previous atomistic simulations and network models (27), as well as to atomistic simulations of the quinone dynamics (28).
CG-MD simulations
Coarse-grained simulation models of complex I were built from the x-ray crystal structure from Thermus thermophilus (PDB ID: 4HEA) (13). The protein was embedded in cardiolipin:POPE:POPC and POPE:POPC membranes with lipid ratios of 1:2:2 and 1:1, respectively, solvated using MARTINI water (37) and neutralized. The cardiolipin was modeled in its singly pronated state (Fig. 1B). CG-MD simulations were performed at T = 300 K using an NpT ensemble with a 20-fs time step, a thermostat with a coupling constant of τt = 1.0 ps (38), a semi-isotropic Parrinello-Rahman barostat (39) with a coupling constant of τp = 12.0 ps, and a compressibility of χ = 3.0 × 10−4 bar−1. Nonbonded interactions were treated with a dielectric constant of 15 and using a cutoff distance of 1.1 nm. The CG-MD simulations were performed in total for ca. 1.2 ms, with each individual trajectory at least 50 μs long. Simulation details are summarized in table S1. All CG-MD simulations were performed using Gromacs 2016.2 (40) and the MARTINI 2.2 force field (37, 41), and quinone parameters were obtained from the literature (42). Visual molecular dynamics (VMD) (43), Gromacs select tool, and PLUMED (44) were used for analysis and/or visualization, and the Backward script (45) was used to convert coarse-grained structures back to an atomistic representation. PyEMMA (46) was used to perform the PC analysis (26) on the coarse-grained simulation trajectories on the backbone atom level. Caver (36) was used with a probe radius of 2.25 Å to identify the channels that are shown in fig. S4.
Atomistic molecular simulation
Molecular models of the membrane domain of complex I were built from the x-ray crystal structure from T. thermophilus (PDB ID: 4HEA) (13). The system comprised subunits Nqo4/Nqo6/Nqo7, Nqo8 to Nqo11, Nqo14, and the C-terminal end of the HL helix (residues 569 to 605). The model was embedded in a lipid membrane with a cardiolipin:POPC:POPE ratio of 1:2:2 built using the CHARMM-GUI module (47), and solvated with TIP3P (48) water molecules. Sodium and chloride ions were added to neutralize the system with an ionic strength of ca. 100 mM. The total system comprised ca. 332,100 atoms. After minimization, relaxation, and equilibration using harmonic forces of 2 kcal mol−1 Å−2 on the protein backbone atoms, MD simulations without restraints were carried out for 0.5 μs at T = 310 K using a 2-fs time step, and the long-range electrostatics were treated using the Particle Mesh Ewald approach (49). The system was treated using the CHARMM36 force field (50) and the density functional theory–derived parameters for the redox-active cofactors. Classical MD simulations were performed with NAMD2 (51), and VMD (43) was used for visualization and analysis. The electrostatic potential calculations were performed using the Adaptive Poisson-Boltzmann Solver (52).
Supplementary Material
Acknowledgments
We are thankful for the computing time provided by SuperMuc at the Leibniz Rechenzentrum (pr27xu). Funding: This work received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program/grant agreement 715311. This work was also supported by the German Research Foundation. Author contributions: A.J., A.D.L., and V.R.I.K. designed the project; A.J. performed the coarse-grained simulations; A.D.L. performed the atomistic simulations; A.J., A.D.L., and V.R.I.K. analyzed the calculations; and V.R.I.K. wrote the manuscript with contributions from all the other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav1850/DC1
Fig. S1. Comparison of cardiolipin molecules bound to Nqo8 from coarse-grained and atomistic models.
Fig. S2. The structure of the membrane domain of complex I from T. thermophilus.
Fig. S3. Electrostatic potential surface of the bacterial and mammalian complex I.
Fig. S4. Global motion of complex I with and without cardiolipin.
Fig. S5. Projection of complex I dynamics with and without cardiolipin along the bending (PC1) and twisting (PC2) modes.
Table S1. Overview of all coarse-grained MD simulations.
Table S2. Residues within 3 Å of putative channels 1 and 2.
Movie S1. Cardiolipin binding to complex I from a coarse-grained simulation trajectory.
Movie S2. Cardiolipin-induced channel formation dynamics.
REFERENCES AND NOTES
- 1.Brandt U., Energy converting NADH: Quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75, 69–92 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Hirst J., Mitochondrial complex I. Annu. Rev. Biochem. 82, 551–575 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Sazanov L. A., A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Zickermann V., Wirth C., Nasiri H., Siegmund K., Schwalbe H., Hunte C., Brandt U., Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Zhu J., Vinothkumar K. R., Hirst J., Structure of mammalian respiratory complex I. Nature 536, 354–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiedorczuk K., Letts J. A., Degliesposti G., Kaszuba K., Skehel M., Sazanov L. A., Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agip A. A., Blaza J. N., Bridges H. R., Viscomi C., Rawson S., Muench S. P., Hirst J., Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones A. J. Y., Blaza J. N., Varghese F., Hirst J., Respiratory complex I in Bos taurus and Paracoccus denitrificans pumps four protons across the membrane for every NADH oxidized. J. Biol. Chem. 292, 4987–4995 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wikström M., Hummer G., Stoichiometry of proton translocation by respiratory complex I and its mechanistic implications. Proc. Natl. Acad. Sci. U.S.A. 109, 4431–4436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galkin A. S., Grivennikova V. G., Vinogradov A. D., →H+/2e stoichiometry in NADH-quinone reductase reactions catalyzed by bovine heart submitochondrial particles. FEBS Lett. 451, 157–161 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Mitchell P., Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148 (1961). [DOI] [PubMed] [Google Scholar]
- 12.Yoshida M., Muneyuki E., Hisabori T., ATP synthase — A marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2, 669–677 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Baradaran R., Berrisford J. M., Minhas G. S., Sazanov L. A., Crystal structure of the entire respiratory complex I. Nature 494, 443–448 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaila V. R. I., Long-range proton-coupled electron transfer in biological energy conversion: Towards mechanistic understanding of respiratory complex I. J. R. Soc. Interface 15, 20170916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Luca A., Gamiz-Hernandez A. P., Kaila V. R. I., Symmetry-related proton transfer pathways in respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, E6314–E6321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Luca A., Mühlbauer M. E., Saura P., Kaila V. R. I., How inter-subunit contacts in the membrane domain of complex I affect proton transfer energetics. Biochim. Biophys. Acta Bioenerg. 1859, 734–741 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirst J., Carroll J., Fearnley I. M., Shannon R. J., Walker J. E., The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta Bioenerg. 1604, 135–150 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Fry M., Green D. E., Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880 (1981). [PubMed] [Google Scholar]
- 19.Dröse S., Zwicker K., Brandt U., Full recovery of the NADH: Ubiquinone activity of complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica by the addition of phospholipids. Biochim. Biophys. Acta Bioenerg. 1556, 65–72 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Vos M., Geens A., Böhm C., Deaulmerie L., Swerts J., Rossi M., Craessaerts K., Leites E. P., Seibler P., Rakovic A., Lohnau T., de Strooper B., Fendt S.-M., Morais V. A., Klein C., Verstreken P., Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 216, 695–708 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnarez C., Marrink S. J., Periole X., Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Sci. Rep. 3, 1263 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnarez C., Mazat J.-P., Elezgaray J., Marrink S.-J., Periole X., Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 135, 3112–3120 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Mehdipour A. R., Hummer G., Cardiolipin puts the seal on ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 113, 8568–8570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan A. L., Robinson A. J., Walker J. E., Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Natl. Acad. Sci. U.S.A. 113, 8687–8692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharpley M. S., Shannon R. J., Draghi F., Hirst J., Interactions between phospholipids and NADH: Ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248 (2006). [DOI] [PubMed] [Google Scholar]
- 26.David C. C., Jacobs D. J., Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 1084, 193–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Luca A., Kaila V. R. I., Global collective motions in the mammalian and bacterial respiratory complex I. Biochim. Biophys. Acta Bioenerg. 1859, 326–332 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Warnau J., Sharma V., Gamiz-Hernandez A. P., Di Luca A., Haapanen O., Vattulainen I., Wikström M., Hummer G., Kaila V. R. I., Redox-Coupled Quinone Dynamics in the Respiratory Complex I. Proc. Natl. Acad. Sci. U.S.A. 115, E8413–E8420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fedor J. G., Jones A. J. Y., Di Luca A., Kaila V. R. I., Hirst J., Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 114, 12737–12742 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Babot M., Birch A., Labarbuta P., Galkin A., Characterisation of the active/de-active transition of mitochondrial complex I. Biochim. Biophys. Acta Bioenerg. 1837, 1083–1092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Javanainen M., Martinez-Seara H., Vattulainen I., Excessive aggregation of membrane proteins in the Martini model. PLOS ONE 12, e0187936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma V., Belevich G., Gamiz-Hernandez A. P., Róg T., Vattulainen I., Verkhovskaya M. L., Wikström M., Hummer G., Kaila V. R. I., Redox-induced activation of the proton pump in the respiratory complex I. Proc. Natl. Acad. Sci. U.S.A. 112, 11571–11576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belevich N., von Ballmoos C., Verkhovskaya M., Activation of proton translocation by respiratory complex I. Biochemistry 56, 5691–5697 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Belevich N., Verkhovskaya M., Resting state of respiratory complex I from Escherichia coli. FEBS Lett. 590, 1570–1575 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Belevich N., Belevich G., Chen Z., Sinha S. C., Verkhovskaya M., Activation of respiratory Complex I from Escherichia coli studied by fluorescent probes. Heliyon 3, e00224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., Damborsky J., CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLOS Comput. Biol. 8, e1002708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H., The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 40.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 41.de Jong D. H., Singh G., Bennett W. F. D., Arnarez C., Wassenaar T. A., Schäfer L. V., Periole X., Tieleman D. P., Marrink S. J., Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput. 9, 687–697 (2013). [DOI] [PubMed] [Google Scholar]
- 42.de Jong D. H., Liguori N., van den Berg T., Arnarez C., Periole X., Marrink S. J., Atomistic and coarse grain topologies for the cofactors associated with the photosystem II core complex. J. Phys. Chem. B 119, 7791–7803 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 44.Tribello G. A., Bonomi M., Branduardi D., Camilloni C., Bussi G., PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185, 604–613 (2014). [Google Scholar]
- 45.Wassenaar T. A., Pluhackova K., Böckmann R. A., Marrink S. J., Tieleman D. P., Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 10, 676–690 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Scherer M. K., Trendelkamp-Schroer B., Paul F., Pérez-Hernández G., Hoffmann M., Plattner N., Wehmeyer C., Prinz J.-H., Noé F., PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. J. Chem. Theory Comput. 11, 5525–5542 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Jo S., Kim T., Iyer V. G., Im W., CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L., Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- 49.Darden T., York D., Pedersen L., Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- 50.Best R. B., Zhu X., Shim J., Lopes P. E. M., Mittal J., Feig M., MacKerell A. D. Jr., Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav1850/DC1
Fig. S1. Comparison of cardiolipin molecules bound to Nqo8 from coarse-grained and atomistic models.
Fig. S2. The structure of the membrane domain of complex I from T. thermophilus.
Fig. S3. Electrostatic potential surface of the bacterial and mammalian complex I.
Fig. S4. Global motion of complex I with and without cardiolipin.
Fig. S5. Projection of complex I dynamics with and without cardiolipin along the bending (PC1) and twisting (PC2) modes.
Table S1. Overview of all coarse-grained MD simulations.
Table S2. Residues within 3 Å of putative channels 1 and 2.
Movie S1. Cardiolipin binding to complex I from a coarse-grained simulation trajectory.
Movie S2. Cardiolipin-induced channel formation dynamics.