

Abstract
Rationale:
The case with congenital macular coloboma and cataract was rarely reported, and the pathogenic gene of the disease is still not clear. Moreover, it is difficult to improve the visual acuity of the eye with this disease.
Patient concerns:
An 11-year-old boy presented low visual acuity and horizontal nystagmus in both eyes. Ophthalmologic examination showed the patient with bilateral congenital coloboma and cataract. The visual acuity of the patient improved slightly after cataract surgery. Heterozygous mutations of frizzled-4 (FZD4) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2) were identified by next-generation sequencing in this case.
Diagnosis:
Congenital macular coloboma and cataract of both eyes.
Interventions:
We performed the standard phacoemulsification and intraocular lens implantation on both eyes of the patient for the treatment of congenital cataract, and then followed up the fundus lesions regularly.
Outcomes:
Cataract surgery may improve the visual acuity of the eyes with congenital macular coloboma and cataract at some degree, but the vision of this patient was still very poor postoperatively. Furthermore, the heterozygous mutations of FZD4 and NOD2 were found in this patient.
Lessons:
Cataract surgery may improve the visual acuity of the eyes with congenital macular coloboma and cataract at some degree, and heterozygous mutations of FZD4 and NOD2 may be involved in the occurrence of congenital macular coloboma and cataract.
Keywords: congenital cataract, gene mutation, intrauterine infection, macular coloboma, nystagmus
1. Introduction
Congenital coloboma is a relatively rare condition involving 0.5–0.7/10,000 births,[1] which was thought to result from the failure of fusion of the optic fissure that normally occurs at 5 to 7 weeks.[2] It is usually sporadic, although autosomal dominant or other inheritance patterns may be followed. The abnormality of development resulted by intrauterine inflammation was considered to the main causes of macular coloboma.[2,3] However, the genetic changes were also reported in some literature recently.[4,5]
Congenital cataract is a major cause of induced blindness in children. Genetic factors as the main reason of congenital cataract affected approximately 50% of all cases.[6] Autosomal dominant transmission seems to be the most frequent in all 3 types of Mendelian inheritance. Furthermore, congenital infections, multiple anomalies, and syndromes were also involved in the development of congenital cataract.[6–8] Here we first reported a case with congenital coloboma and cataract in both eyes, and nystagmus also occurred in him due to the existing ocular abnormality.
2. Case report
2.1. Patients and clinical investigations
An 11-year-old male presented with blurred vision in both eyes from childhood. There was no contributory medical or family history, and it is non-consanguineous marriage for his parents. No syndromes or associations were detected in this child and ocular examination of family members revealed no abnormalities. Uncorrected (UCVA) and best-corrected visual acuity (BCVA) of this child were performed using the standard 20 feet Snellen acuity chart. External eye exams including eye movements and reverse afferent pupillary defect test were also done. Anterior segment photograph was obtained using a BX 900 Slit Lamp (Haag-Streit, Bern, Switzerland). Intraocular pressure (IOP) was measured with non-contact tonometer (Topcon Corporation, Tokyo, Japan). On postoperative 1 month, fundus photographs were carried out using a TRC-NW300 Retinal Camera (Topcon Corporation). Peripheral retinal examination was done with an indirect ophthalmoscope and 3 mirror contact lens, and macular zone of the patients was examined by optical coherence tomography (OCT) (Heidelberg Engineering, Inc, Heidelberg, Germany) and multifocal electroretinography (mfERG) (Roland Consult, Inc, Keltern, Germany). The patient had also undergone fundus fluorescein angiography (FFA) (Heidelberg Engineering, Inc, Heidelberg, Germany) to determine whether the retinal vascular abnormalities were present. Physical examinations were performed to exclude systemic diseases before cataract surgery. Venous blood samples from this patient and his unaffected family members were collected. All of the ocular examinations were performed by 2 academic experienced ophthalmologists. All experimental protocols were carried out according to the guidelines approved by the Ethics Committee of Weifang Eye Hospital and in accordance with the Declaration of Helsinki. Informed written consent was obtained from the patient for publication of this case report and accompanying images.
2.2. Surgery
The standard phacoemulsification was performed on both eyes of the patient. After general combined with topical anesthesia, a 1-mm side port clear corneal incision was made, and a 2.2 mm clear cornea incision was created at the position 90 degrees to the right of the side port incision. Then, viscoelastic (Discovisc, Alcon) was injected into the anterior chamber continuous curvilinear capsulorhexis was performed manually, followed by phacoemulsification with Centurion cataract surgery platform (Alcon, Fort Worth, Texas) using divide and conquer technique, bimanual irrigation, and aspiration of cortical remnants, foldable IOL implantation and evacuation of viscoelastic. After surgery, compound neomycin sulfate eye drops, gatifloxacin eye drops, and diclofenac sodium eye drops were used 4 times per day. Brinzolamide eye drops were given 3 times a day, when the patient showed high IOP.
2.3. Target capture and next-generation sequencing
A capture panel of the genes related to congenital cataract and retinal diseases was previously designed and assessed by our group. The capture panel, which covered all exons, included 4500 genes causing common inherited eye diseases that have been previously reported.
Next-generation sequencing was performed, and the methods were similar to the previous report.[5] Briefly, genomic DNA from peripheral blood leucocytes was extracted using the QIAamp DNABlood Midi Kit (Qiagen, Hilden, Germany). Then the genomic DNA was sheared using Covaris Ultra Sonicator (Covaris, Inc, MA). The DNA library was constructed using an Illumina Truseq DNA Sample Preparation Kit (Illumina, San Diego, CA) following the manufacturer's protocol and amplified using precapture LM-PCR. Agilent 1000 Chip (Agilent Technologies, Inc, CA) was used for library quality assessment. The amplified library was then hybridized for exon regions using a SeqCap EZ human Exome kit v2.0 (Roche, Inc, Basel, Switzerland) according to the User's Guide (Joy Orient, Inc, Beijing, China). The library was then washed and amplified using post-capture LM-PCR. Agilent DNA 1000Chip was used to assess quality, concentration, and size distribution of the capture library, and qPCR was used to measure enrichment and quality. The exon enriched DNA library was sequenced on the Illumina Hiseq2500 Analyzers (Illumina, Inc, CA) according to the manufacturer's protocol for 150-bp paired-end reads. The raw imagine file was processed using bcl2fastq (Illumina, Inc, CA) for base calls, which generated the raw data. Only genotypes with quality scores ≥20 were included for further data analysis. Pindel analysis software and Samtools were used to analyze SNPs and indels relative to the reference sequence. The variant analysis was filtered based on their frequencies (MAF < 0.05) in the databases of the EXOme Affregation Consortium, the Exom sequencing project, or 1000 human genome dataset. Nonsynonymous changes were then evaluated using the SIFT software and Polyphen software. All genes were analyzed for the association of putative dominant and recessive disease-causing variants with patients’ phenotypes.
Mutations were further confirmed using PCR and Sanger sequencing on the DNA samples of proband and his parents. The PCR products were sequenced on an ABI 3730XL DNA Sequencer (Thermo Fisher Scientific, Inc, MA). The sequences were analyzed with the DNASTAR software to confirm the mutations discovered from all the samples.
2.4. Outcomes
Examination of the patient revealed bilateral horizontal nystagmus. Before surgery, his UCVA and BCVA were 20/500 and 20/200 in the right eye, respectively, and visual acuity of the left eye was 20/400 and could not be corrected. Refractive correction in diopters (dpt) was −8.50 dpt −2.00 ×5 for the right eye and −10.00 dpt −2.00 × 05 for the left eye with spectacles. The IOP was 23 millimeter of mercury (mm Hg) in the right eye and 20 mm Hg in the left eye. Cornea, anterior chamber, and iris were normal. The lens opacities were categorized into C0N1P3 according to the lens opacities classification system II (Fig. 1). On postoperative 1 week, the fundus examination after mydriasis showed that a bilateral excavated macular coloboma about 1.5 × 1.5 disc diameter in size of the right eye and 2 × 2 disc diameter in size of the left eye. The boundary of the defect is legible. The large choroidal vessels were visible at the base and some pigment clumps located in the area around the defect in both eyes. The optic discs of both eyes were pale color and the cup to disc ratio was about 0.5:1 (Fig. 2). OCT revealed that the thickness of retina became thinner than normal in macular region (Fig. 3), and no significant the peak value was found by mfERG (Fig. 4). Based on the typical clinical features and ophthalmologic examination, the patient was diagnosed to congenital pigmented macular coloboma in combination with congenital cataract in both eyes.
Figure 1.

Anterior segment photograph showed lens opacities in both eyes.
Figure 2.

Fundus photographs of the patient.
Figure 3.

OCT and FFA imagine of the macular region from both eyes of the patient. FFA = fundus fluorescein angiography, OCT = optical coherence tomography.
Figure 4.

mfERG showed that no significant the peak value was found in both eyes. mfERG = multifocal electroretinography.
On postoperative 1 day, IOP of the patient was 28 and 29 mm Hg in the right and left eyes, respectively, and it returned to normal after using brinzolamide eye drops. UCVA was 20/100 in both eyes, and BCVA was 20/70 in both eyes.
2.5. Mutation analysis
A heterozygous frizzled-4 (FZD4) mutation c.205C>T (p.H69Y) and a heterozygous nucleotide-binding oligomerization domain-containing protein 2 (NOD2) mutation c. 1118G>A (p.R373H) were identified in the affected case. FZD4 mutation was also present as a heterozygous state in the patient's mother with normal eyes, but NOD2 mutation was not found in his parents (Fig. 5).
Figure 5.
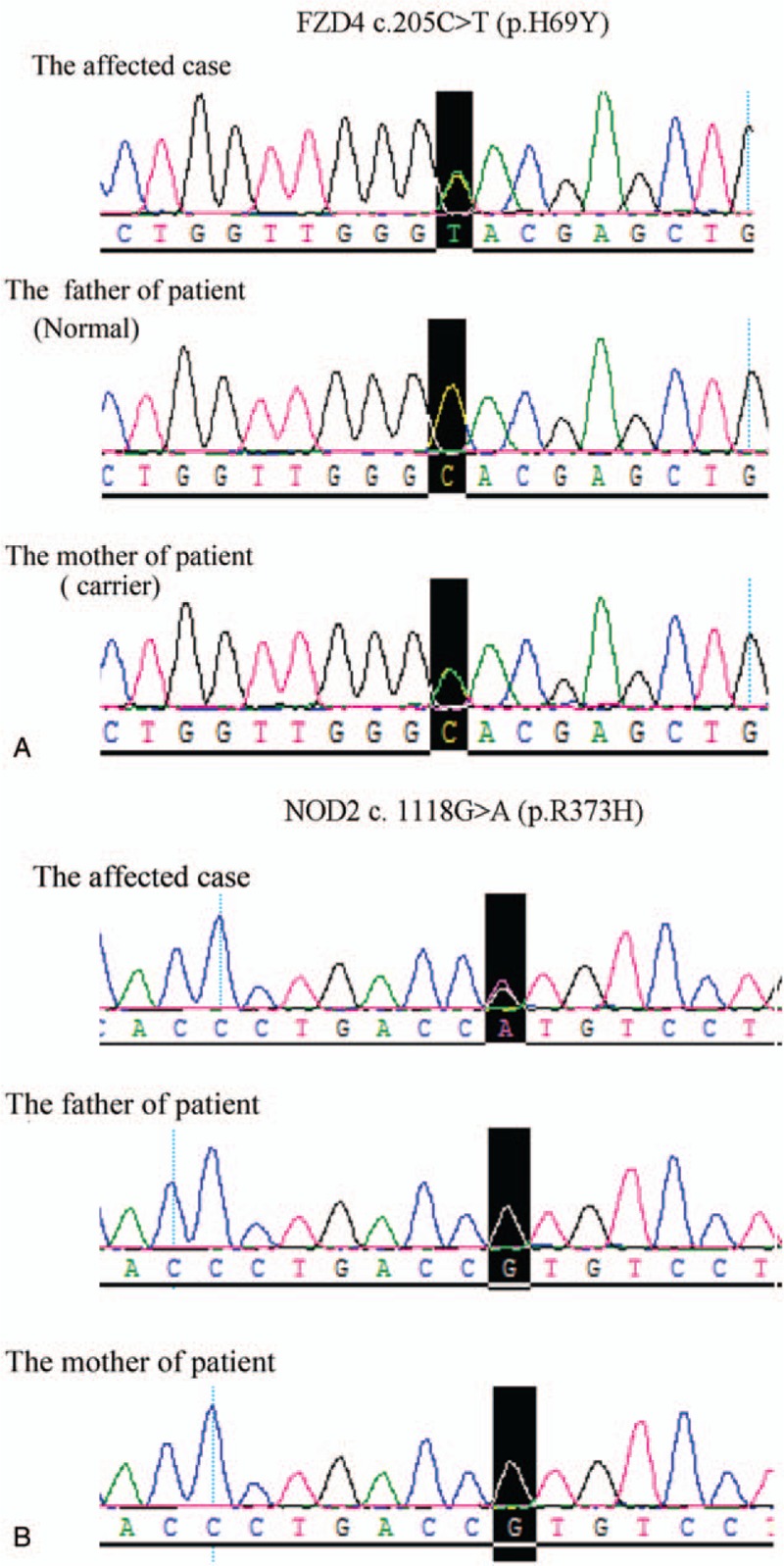
A heterozygous FZD4 mutation 205C>T (p.H69Y) and a heterozygous NOD2 mutation c. 1118G>A (p.R373H) were identified in the affected case. FZD4 = frizzled-4, NOD2 = nucleotide-binding oligomerization domain-containing protein 2.
3. Discussion
Mann[9] classified macular coloboma into 3 types as follow:
-
(1)
pigmented macular coloboma;
-
(2)
nonpigmented macular coloboma;
-
(3)
macular coloboma associated with abnormal vessels.
For the pigmented macular coloboma, it shows a more or less circular patch at the macular area and is covered with dense irregularly arranged masses of pigment. The large choroidal vessels could be seen deep to the pigment. For the nonpigmented macular coloboma, it is round or oval patch with punched-out appearance. The base is pearly white and slightly hollowed out. There were no choroidal and retinal vessels across the area of the defect. For the third type of macular coloboma, the retinal vascular anomaly may apparently take the form either of an abnormal anastomosis of choroidal vessels in the patch or of a vessel passing forwards from the coloboma into the vitreous. Two hypotheses were proposed for the causes of macular coloboma. The first is genetic factors. The inheritance pattern for most of the patients was autosomal dominant, and a few of patients were thought to be inherited in an autosomal recessive manner. The combination of macular coloboma with other systemic diseases, such as apical dystrophy of the hands and feet, achondroplasia, and retinitis pigmentosa, and so on, was commonly reported in these patients.[4,10,11] The second reason for this disease was considered to be an intrauterine infection.[3]
Previous studies reported that heterozygous bestrophin 1 and regulating synaptic membrane exocytosis 1 mutation and homozygous DHX38 mutation had been identified in the patients with bilateral macular coloboma.[4,5] However, the mutations of these genes were not detected in this study. Heterozygous FZD4 and NOD2 mutations were found in the patient. FZD4 was reported to take part in the regulation of retinal angiogenesis.[12] FZD4 mutation had been found in the patients with familial exudative vitreoretinopathy[12–14] and caused a failure of peripheral retinal vascularization. Retinal vascular abnormalities were also detected by FFA in the patient reported in this study, but which located on macular regions of his both eyes rather than peripheral retina. Nevertheless, the patient's mother with same mutation of FZD4 did not show any ocular abnormalities, which leaded us to speculate that other factors might also play some roles in this disease. NOD2 mutation had been reported to be the cause of the Blau syndrome, which showed congenital cataract, glaucoma, uveitis, bilateral band keratopathy, arteritis, Crohn disease, and so on.[15,16] This patient has congental cataract in his both eyes, and IOP of his right eye was also a little higher than normal. But other symptoms, such as uveitis, bilateral band keratopathy, arteritis, and Crohn disease, were not present in the patient. Due to nystagmus, field of vision could not be performed. Therefore, it could not be sure whether he is suffering from glaucoma. We speculated that the heterozygous FZD4 and NOD2 mutations might play some role in the disease of this patient, but it still needs further verification in our future study.
Intrauterine infection was also thought to be one of the causes of macular coloboma.[3] It also was described the concomitance between macular coloboma and other retinochoroidal diseases, such as retinitis pigmentosa, Leber amaurosis, and retinal dystrophy, and so on.[17,18] The infection of toxoplasma and Zika virus was reported to be in association with macular coloboma.[3,7] Intrauterine infection of Zika virus can cause congenital macular coloboma and cataract in infants.[7] However, other abnormalities, for example, cerebral calcifications, malformations of cortical development, hypoplasia of the cerebellum and brain stem, ventriculomegaly, lissencephaly, iris coloboma, and lens subluxation, were not observed in this patient. Furthermore, there was no family history and systemic diseases for patient. The etiology of this case is still unknown, but the presence of bilateral macular coloboma and cataract together with negative family history and other diseases lead us to infer that intrauterine infection might also be a reason that caused the diseases of this patient.
In summary, this study was the first to report the case that showed bilateral congenital macular coloboma, cataract, and nystagmus, and the mutations of FZD4 and NOD2 were identified in this patient, which may be the cause of these ocular abnormalities. Moreover, we also found that cataract surgery may improve the visual acuity of the eyes with macular coloboma and cataract at some degree.
Acknowledgments
The authors are grateful to the patient and his family.
Author contributions
Data curation: Peng Wu, Canwei Zhang.
Formal analysis: Peng Wu.
Funding acquisition: Canwei Zhang, Yaqin Jiang.
Investigation: Canwei Zhang, Peng Wu, Jing Gao, Yaqin Jiang.
Project administration: Yaqin Jiang.
Resources: Yaqin Jiang.
Supervision: Jing Gao, Yaqin Jiang.
Validation: Canwei Zhang, Peng Wu, Luping Wang, Jing Gao, Xudong Huang, Yaqin Jiang.
Writing - original draft: Canwei Zhang.
Writing – review and editing: Peng Wu, Luping Wang, Jing Gao, Xudong Huang, Yaqin Jiang.
Footnotes
Abbreviations: BCVA = best-corrected visual acuity, FFA = fundus fluorescein angiography, FZD4 = frizzled-4, IOP = intraocular pressure, mfERG = multifocal electroretinography, NOD2 = nucleotide-binding oligomerization domain-containing protein 2, OCT = optical coherence tomography, UCVA = uncorrected visual acuity.
CZ and PW are authors equally contributed to the work.
This study was supported by the Science and Technology Development Plan Project of Weifang (grant number 2018YX042).
The authors report no conflicts of interest.
References
- [1].Hornby SJ, Adolph S, Gilbert CE, et al. Visual acuity in children with coloboma: clinical features and a new phenotypic classification system. Ophthalmology 2000;107:511–20. [DOI] [PubMed] [Google Scholar]
- [2].Varghese M, Kavalakatt JA, Pandey S, et al. Macular coloboma. Oman J Ophthalmol 2016;9:67–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chen MS, Yang CH, Huang JS. Bilateral macular coloboma and pigmented paravenous retinochoroidal atrophy. Br J Ophthalmol 1992;76:250–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ajmal M, Khan MI, Neveling K, et al. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet 2014;51:444–8. [DOI] [PubMed] [Google Scholar]
- [5].Li T, Lin Y, Gao H, et al. Two heterozygous mutations identified in one Chinese patient with bilateral macular coloboma. Mol Med Rep 2017;16:2505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pichi F, Lembo A, Serafino M, et al. Genetics of congenital cataract. Dev Ophthalmol 2016;57:1–4. [DOI] [PubMed] [Google Scholar]
- [7].Guevara JG, Agarwal-Sinha S. Ocular abnormalities in congenital Zika syndrome: a case report, and review of the literature. J Med Case Rep 2018;12:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tasdemir S, Sahin I, Cayir A, et al. Vici syndrome in siblings born to consanguineous parents. Am J Med Genet A 2016;170A:220–5. [DOI] [PubMed] [Google Scholar]
- [9].Mann IC. On certain abnormal conditions of the macular region usually classed as colobomata. Br J Ophthalmol 1927;11:99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thompson EM, Baraitser M. Sorsby syndrome: a report on further generations of the original family. J Med Genet 1988;25:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ahoor MH, Amizadeh Y, Sorkhabi R. Achondroplasia and macular coloboma. Middle East Afr J Ophthalmol 2015;22:522–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Robitaille J, MacDonald ML, Kaykas A, et al. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet 2002;32:326–30. [DOI] [PubMed] [Google Scholar]
- [13].Tang M, Ding X, Li J, et al. Novel mutations in FZD4 and phenotype-genotype correlation in Chinese patients with familial exudative vitreoretinopathy. Mol Vis 2016;22:917–32. [PMC free article] [PubMed] [Google Scholar]
- [14].Khan AO, Lenzner S, Bolz HJ. A family harboring homozygous FZD4 deletion supports the existence of recessive FZD4-related familial exudative vitreoretinopathy. Ophthalmic Genet 2017;38:380–2. [DOI] [PubMed] [Google Scholar]
- [15].Pillai P, Sobrin L. Blau syndrome-associated uveitis and the NOD2 gene. Semin Ophthalmol 2013;28:327–32. [DOI] [PubMed] [Google Scholar]
- [16].Snyers B, Dahan K. Blau syndrome associated with a CARD15/NOD2 mutation. Am J Ophthalmol 2006;142:1089–92. [DOI] [PubMed] [Google Scholar]
- [17].Parmeggiani F, Milan E, Costagliola C, et al. Macular coloboma in siblings affected by different phenotypes of retinitis pigmentosa. Eye (Lond) 2004;18:421–8. [DOI] [PubMed] [Google Scholar]
- [18].Heckenlively JR, Foxman SG, Parelhoff ES. Retinal dystrophy and macular coloboma. Doc Ophthalmol 1988;68:257–71. [DOI] [PubMed] [Google Scholar]