

Abstract
Rationale:
Cardiac myxoma is the most common cardiac neoplasm. Currently, there are not many reports on familial cardiac myxoma. Herein, we reported 2 first-degree relatives with left atrial myxoma.
Patient concerns:
A 20-year-old female was admitted in our hospital for lapsing into a coma for 24 hours, and was diagnosed with recurrent left atrial cardiac myxoma. The patient's father also had a history of cardiac myxoma.
Diagnosis:
The patient was diagnosed with left atrial myxoma using transthoracic echocardiography (TTE). Whole exome sequencing (WES) identified a p.Val164Aspfs (c.491-492delTG) mutation in the cAMP-dependent protein kinase A (PKA) regulatory (R) subunit 1 (PRKAR1A) gene for both the proband and her father, but not in her uncle and brother, who had not shown manifestation of cardiac myxoma by the time of this report.
Interventions:
The myxoma resection was performed following the standard procedure of open chest surgery.
Outcomes:
The tumor was successfully removed along with the tuberculum. The patient recovered well and was discharged home. No recurrence occurred during 1-year follow-up.
Lessons:
Our findings suggest that PRKAR1A mutation (c.491_492delTG) may be associated with cardiac myxoma, and genetic counseling and specific locus mutation tests may contribute to assessing the risk of cardiac myxoma.
Keywords: cardiac myxoma, diagnosis, PRKAR1A mutation, treatment
1. Introduction
Cardiac myxoma is the most common cardiac neoplasm that typically arises as an isolated mass in the left atrium.[1] In approximately 3% to 10% cases, it occurs as one of the manifestations of the Carney complex (CNC).[2] Patients with cardiac myxoma in the context of CNC are prone to develop other syndromic manifestations and have higher rates of tumor occurrence, and cardiac myxoma has been found with familial tendency.[2] Imaging materials, including transthoracic echocardiogram (TTE), cardiac computed tomogram (CT) and cardiac magnetic resonance images (MRI), are pivotal in the diagnosis, management guidance, and surveillance of cardiac myxoma.[3] Surgical excision of the cardiac myxomas is the established definitive treatment and is highly efficacious.[4] Previous reports on molecular etiology of CNC have identified that mutation of the cAMP-dependent protein kinase A (PKA) regulatory (R) subunit 1 (PRKAR1A) gene was associated with CNC.[1,5]
Currently, there are not many reports on familial cardiac myxoma. In this article, we report 2 first-degree relatives who were diagnosed with left atrial myxoma and have undergone surgical resection. Whole exome sequencing (WES) was conducted to identify the mutated genes related to cardiac myxoma. This article illustrates the diagnosis and management of a case of familial atrial myxoma, and highlights the responsibility of pathogenic gene variants for familial cardiac myxoma.
1.1. Case presentation
1.1.1. Ethical statement
This study has been approved by the Ethics Committee of our hospital (ethical approval number: 2017096). Informed written consent was obtained from the patient for publication of this case report and accompanying images.
1.2. Patient features and investigations
A 20-year-old female has a history of left atrial cardiac myxoma, which was diagnosed in 2015 and surgical resection was performed to remove the myxoma. In 2018, the patient was admitted in our hospital for lapsing into a coma for 1 day, and was diagnosed with recurrent left atrial cardiac myxoma (Fig. 1A). Physical examination did not reveal any yellow stain or bleeding spots on skin or mucous membrane of the whole body, and no thyroid nodule was found upon palpation. Diastolic murmur was present on cardiac auscultation; ultrasonography indicated the presence of an 18 × 18 × 47 mm mass at the left atrial septum.
Figure 1.
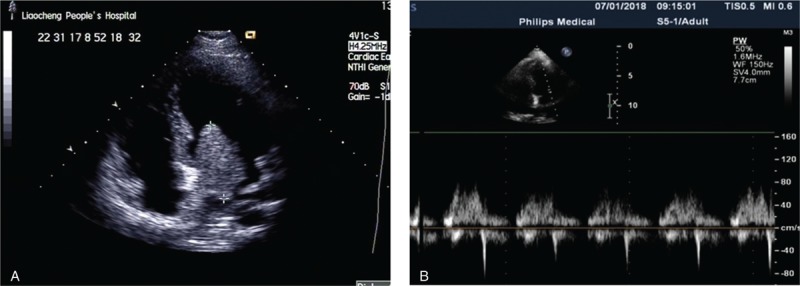
Left ventricular long axis view of transthoracic echocardiography (TTE) (A) before and (B) after the surgery.
1.3. Treatment
After routine examinations, myxoma resection was performed following the standard procedure of open chest surgery. Transverse aortotomy indicated the presence of a pedunculated myxoma mass of 3 × 2 × 3 cm at the left atrial septum. The tumor was successfully removed along with the tuberculum (Fig. 1B).
1.4. Outcome and follow up
After the surgery, the patient was transferred to the intensive care unit (ICU). The patient recovered well and was discharged home. No recurrence occurred during 1-year follow-up.
1.5. Family history
A family pedigree across 3 generations was constructed for the patient (Fig. 2). The proband's father (44 years old) was diagnosed with atrial cardiac myxoma and the mass has been surgically resected, therefore we speculated that the cardiac myxoma of the proband may be genetically related. The proband's uncle (38 years old) and brother (12 years old) did not show similar examination findings.
Figure 2.
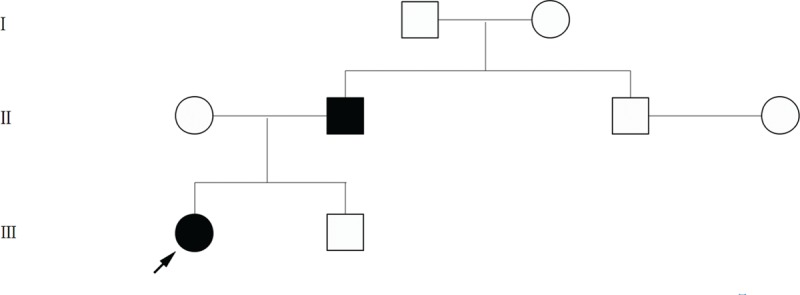
Pedigree of the family with cardiac myxoma. Arrow indicates the proband.
1.6. Whole exome sequencing (WES)
Peripheral blood specimens were collected for the patient, her father, uncle, and brother. Specimens of the patient's mother and grandparents were not able to be obtained. WES was performed to identify probable pathogenic variants responsible for familial cardiac myxoma. Bioinformatics analyses of WES identified a p.Val164Aspfs (c.491-492delTG) mutation in the PRKAR1A gene for both the patient and her father, but not in her uncle and brother. The results were further confirmed by Sanger sequencing (Fig. 3).
Figure 3.
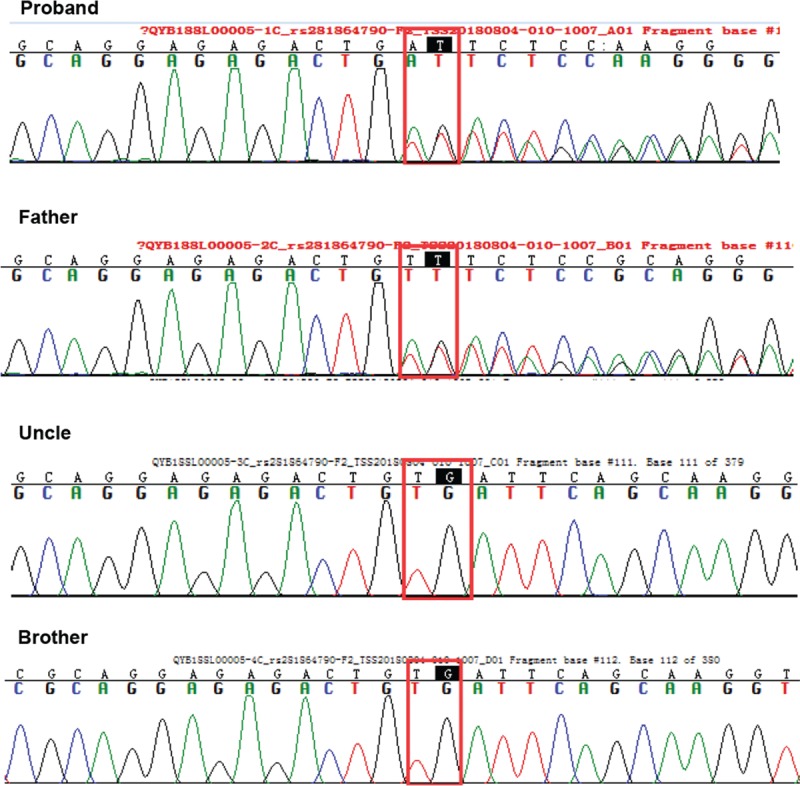
Gene mutation in the patients and his family. Both cardiac myxoma cases in the family (the proband and her father) carried the same PRKAR1A exon 5 mutation (c.491-492delTG), and her uncle and younger brother did not carry the mutation. PRKAR1A, protein kinase type I-alpha regulatory subunit.
2. Discussion
Myxomas are the most common primary cardiac tumor. It often occurs at 30 to 60 years old and the most common type is atrial myxoma. Mostly myxoma occurs without familiality, but a few cases showed a familial genetic tendency in a pattern of autosomal dominant inheritance. Recurrence of atrial myxomas is uncommon.[1,4] In this article, we reported 2 first-degree relatives with cardiac myxoma and the proband had recurrence 3 years after the first operation.
Mounting evidence indicated that loss of PRKAR1A expression may play a role in myxoma tumorigenesis. Studies on its molecular mechanism revealed that the regulatory subunit R1 binds to the catalytic subunits of protein kinase A, inactivating the holoenzyme PRKAR1A. Mutant-associated loss of PRKAR1A function may lead to increased cAMP-stimulated activity of protein kinase A, which may promote tumorigenesis.[6] More than 100 different mutations have been reported within the coding region of PRKAR1A.[7] Online PRKAR1A mutation database has been established by Horvath to facilitate the characterization of pathogenic variants.[8]PRKAR1A mutations have been detected in two-thirds of CNS-associated cardiac myxoma.[9]PRKAR1A appears to play a role in the development of both syndromic and non-syndromic cardiac myxoma.[2]
We identified a putative causal gene variant of PRKAR1A that included the deletion of TG nucleotides at the position 491–492 in 2 first-degree relatives with cardiac myxoma (the proband and her father), but not in her uncle and brother, who did not show sign of cardiac myxoma. This mutation was included in the Clinvar database as a pathogenic mutation (https://www.ncbi.nlm.nih.gov/clinvar/variation/12662/). Consistently, Wang et al identified a c.491-492delTG mutation in PRKAR1A in a recurrent left atrial myxoma family.[10] The TG deletion is a frameshift mutation of PRKAR1A, which leads to the premature termination of polypeptide chain synthesis and increased cAMP-stimulated kinase activity, which may initialize tumorigenesis.[11]
Familial atrial myxoma is usually inherited as an autosomal dominant trait, which means PRKAR1A variation carriers inherit the mutation from their parents, and their siblings and offsprings have a 50% chance of carrying the same mutation. Therefore, it is recommended that the first- and second-degree relatives of patients undergo genetic counseling and specific locus mutation tests to assess the risk of myxomatosis. Appropriate genetic test may detect mutant carriers who have high risk of atrial mysoma and should be closely monitored by TTE, therefore set the stage for early detection and early application of effective therapy.
3. Conclusions
This article presents a familial case of 2 first-degree relatives with cardiac myxoma. The proband was diagnosed with recurrent left atrial myxoma and underwent surgical resection. WES analysis identified the c.491-492delTG mutation in PRKAR1A in the 2 patients (proband and her father), but not in her uncle and brother, who had not shown manifestation of cardiac myxoma as of this article drafting. Our findings suggest that PRKAR1A mutation (c.491_492delTG) may be associated with cardiac myxoma, and genetic consulting and specific locus mutation tests may contribute to assessing the risk of cardiac myxoma.
Author contributions
Investigation: Shengjun Ma, Wei Liu, Anqi Zhang, Li Pan, Wenqiang Tang, Bo Jiang, Fengju Wang, Bo Fu.
Project administration: Shengjun Ma.
Supervision: Shengjun Ma, Bo Fu.
Validation: Shuangfeng Chen.
Writing – original draft: Bo Fu.
Writing – review & editing: Wei Liu, Anqi Zhang.
Footnotes
Abbreviations: CNC = carney complex, CT = computed tomogram, ICU = intensive care unit, MRI = magnetic resonance images, PKA = cAMP-dependent protein kinase A, TTE = transthoracic echocardiogram, WES = whole exome sequencing.
This study was funded by National Nature Science Foundation of China (Grant no. 81702884), China Postdoctoral Science Foundation (Grant no. 2017M612290), Natural Science Foundation of Shandong Province (Grant no. ZR2016HB17), and Medicine and Health Science Technology Foundation of Shandong Province (Grant no. 2015WS0381).
This study has been approved by the Ethics Committee of our hospital (ethical approval number: 2017096). Informed written consent was obtained from the patient for publication of this case report and accompanying images.
The authors confirm that there are no conflicts of interest.
References
- [1].Jain S, Maleszewski JJ, Stephenson CR, et al. Current diagnosis and management of cardiac myxomas. Expert Rev Cardiovasc Ther 2015;13:369–75. [DOI] [PubMed] [Google Scholar]
- [2].Maleszewski JJ, Larsen BT, Kip NS, et al. PRKAR1A in the development of cardiac myxoma: a study of 110 cases including isolated and syndromic tumors. Am J Surg Pathol 2014;38:1079–87. [DOI] [PubMed] [Google Scholar]
- [3].Bernatchez J, Gaudreault V, Vincent G, et al. Left atrial myxoma presenting as an embolic shower: a case report and review of literature. Ann Vasc Surg 2018;53:266 e13–20. [DOI] [PubMed] [Google Scholar]
- [4].Shah IK, Dearani JA, Daly RC, et al. Cardiac myxomas: a 50-year experience with resection and analysis of risk factors for recurrence. Ann Thorac Surg 2015;100:495–500. [DOI] [PubMed] [Google Scholar]
- [5].Massobrio L, Nasti S, Martinuzzi C, et al. Mutation analysis of PRKAR1A gene in a patient with atrial myxoma. Clin Lab 2016;62:731–4. [DOI] [PubMed] [Google Scholar]
- [6].Imai Y, Taketani T, Maemura K, et al. Genetic analysis in a patient with recurrent cardiac myxoma and endocrinopathy. Circ J 2005;69:994–5. [DOI] [PubMed] [Google Scholar]
- [7].Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009;94:2085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Horvath A, Bertherat J, Groussin L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 2010;31:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhang YL, Wang XC, Yu W, et al. A case of familial Carney complex. Arch Iran Med 2015;18:324–8. [PubMed] [Google Scholar]
- [10].Wang L, Wang Q, Zhou Y, et al. Recurrent left atrial myxoma in Carney complex: a case report of a familial pedigree. Medicine 2018;97:e0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000;26:89–92. [DOI] [PubMed] [Google Scholar]