

Abstract
Macrophages are critically involved in wound healing, from dampening inflammation to clearing cell debris and coordinating tissue repair. Within the wound, the complexity of macrophage function is increasingly recognized, with adverse outcomes when macrophages are inappropriately activated, such as in fibrosis or chronic non-healing wounds. Recent advances in in vivo and translational wound models, macrophage-specific deletions, and new technologies to distinguish macrophage subsets, have uncovered the vast spectrum of macrophage activation and effector functions. Here, we summarize the main players in wound healing macrophage activation and function, including cytokines, apoptotic cells, nucleotides and mechanical stimuli. We highlight recent studies demonstrating cooperation between these factors for optimal wound healing. Next, we describe recent technologies such as cell tracking and single cell RNA-seq, which have uncovered remarkable plasticity and heterogeneity in blood-derived or tissue-resident macrophages and discuss the implications for wound healing. Lastly, we evaluate macrophage dysfunction in aberrant wound healing that occurs in aging, diabetes and fibrosis. A better understanding of the longevity and plasticity of wound healing macrophages, and identification of unique macrophage subsets or specific effector molecules in wound healing, would shed light on the therapeutic potential of manipulating macrophage function for optimal wound healing.
Keywords: Macrophage, wound healing, M2 macrophage, resolving macrophage, Th2 cytokine, apoptotic cell
Introduction
Wound healing, which is the repair and restoration of tissue to homeostasis following injury caused by infection or mechanical trauma, occurs in three main stages: coagulation and inflammation; resolution of inflammation; and tissue vascularization and regeneration. These stages are common to all wounds, although the cell-types and secreted factors vary depending on the organ (e.g. skin, lung, liver, brain) and type of injury (e.g. burn, pathogen). In all wounds, macrophages are critical players, from contributing to the inflammation necessary to kill potential pathogens, to resolving inflammation once the pathogens are cleared, and initiating and maintaining tissue remodeling and regeneration.1 Based on these diverse contributions to wound healing, macrophages are broadly grouped into three activation types: first, the inflammatory macrophage for pathogen phagocytosis and killing; second, the resolving macrophage that removes dead cells and dampens inflammation; third, the tissue remodeling macrophage that instructs tissue repair. It is important to recognize that these activation phenotypes do not represent distinct macrophage subsets but likely a continuum of macrophage activation that changes according to cell ontogeny and environmental stimuli.2
This review will focus on the macrophages that contribute to the final stages of wound healing, specifically resolution of inflammation mediated by anti-inflammatory macrophages, and tissue repair, mediated by T helper type 2 (Th2) cytokine-activated ‘M2’ macrophages, also known as alternatively activated macrophages. We will first describe recently discovered pathways influencing the activation and function of these cells. Next, we will highlight the important influence of ontogeny and plasticity on the development of these wound healing macrophage subsets. Lastly, we will discuss how dysfunctional activation of these cells can contribute to disease. A better understanding of these wound healing macrophage subsets from activation to effector molecules, and whether they can be altered to improve wound healing, would have significant therapeutic benefit especially since deficient wound healing can be fatal.
Wound healing macrophage activation and function
Numerous soluble and cellular signals instruct macrophage activation for the final stages of wound healing: tissue remodeling and resolution of inflammation. These include: Th2 cytokines that mediate a tissue remodeling ‘M2’ program, and apoptotic cells that induce an anti-inflammatory macrophage phenotype. We will summarize the main features of these activation programs and how they act together to promote optimal wound healing. We will also discuss recently discovered factors that influence these macrophage activation programs.
Th2 Cytokines:
M2 macrophages are important players in tissue repair.1 M2 macrophages are activated by Th2 cytokines, such as IL-4 and IL-13, that are highly produced in allergic inflammation and helminth infection. For this reason, significant functional information on M2 macrophages has been acquired from helminth infection and allergy studies.3 These studies have provided insight into the M2-mediated effector pathways for wound healing. Indeed, helminths are macroscopic organisms that cause tissue injury, and the tissue remodeling that occurs in allergic responses shares similarities with the tissue repair stage in wound healing. In these models of tissue injury and inflammation, the Th2 cytokine response is mediated in a two-step process, as summarized in Figure 1. First, an insult to the barrier causes epithelial cells to release alarmins, including thymic stromal lymphopoietin (TSLP), IL-25 and IL-33.3 These in turn activate Th2 cytokine-producing innate cells, such as group 2 innate lymphoid cells, mast cells, basophils and eosinophils. The critical importance of M2 macrophages in mediating wound healing is demonstrated in numerous in vitro and in vivo studies. For instance, IL-4/IL-13-activated human THP-1 cells induced proliferation, collagen synthesis and α-smooth muscle actin (α-SMA) expression by human dermal fibroblasts (HDFs), in a co-culture assay.4 These M2-differentiated THP-1 cells also increased dermal fibroblast expression of α-SMA, a feature of myofibroblasts, indicating that fibroblasts were differentiated into myofibroblasts.4 Further, abrogation of IL-4Rα signaling in macrophages impaired wound repair in in vivo models of wound healing by skin punch biopsy, chemical-induced injury or invasive helminth infection-induced injury.5–7 Mechanistically, M2 macrophages initiate wound repair through numerous pathways including growth factors and matrix metalloproteinases (MMP), summarized in Figure 2 and in recent reviews.1 In addition, M2 macrophage-derived arginase 1 (Arg1) and RELMα (Retnla) are downstream effectors of wound healing in the skin and following helminth infection.3, 8 The mechanism by which macrophage-derived arginase promotes skin wound healing is likely two-fold: dampening inflammation, and promoting matrix deposition through its metabolism of L-Arginine. RELMα’s effector function in wound healing has recently been explored.9 RELMα activates the enzyme lysyl hydrolase 2 (Plod2), which mediates optimal collagen cross-linking.5, 10 Additionally, macrophage-derived RELMα was implicated in dampening lung inflammation and promoting tissue repair in a model of helminth infection-induced lung injury with rodent hookworm Nippostrongylus brasiliensis.11–13 In this model, gene expression analysis of RELMα-treated lung macrophages revealed that RELMα promoted expression of genes involved in extracellular matrix remodeling: matrix metalloproteinase 9 (Mmp9), integrin beta 1 (Itgb1), and junctional adhesion molecule A (F11r).
Figure 1. Wound healing macrophage activation.
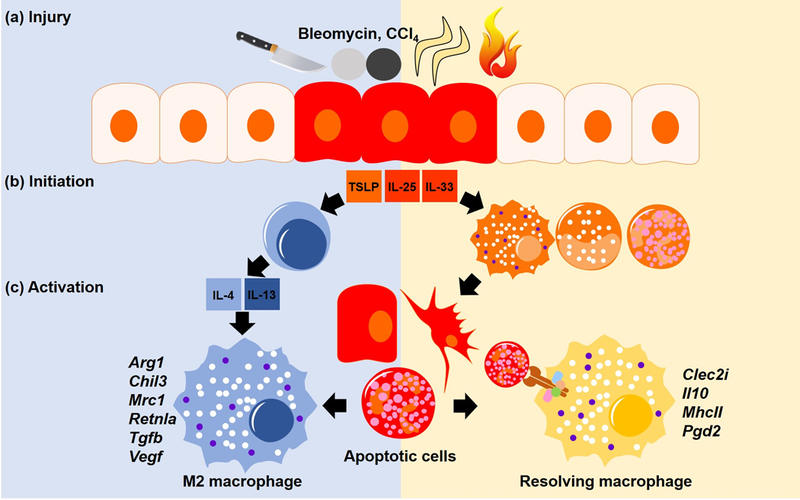
(a) Injury by cuts, chemicals (CCl4 and bleomycin), helminths or burn injury causes a breach in barrier. (b) The wound healing response is initiated by dying cells which release cytokines (TSLP, IL-25, and IL-33) that activate Th2 cytokine (IL-4/IL-13) producing cells (blue). Innate cells such as neutrophils (orange) are also recruited to kill invading pathogens, and apoptose once the challenge is resolved. (c) M2 macrophages (left) are activated by the Th2 cytokines. Equally important is the activation of resolving macrophages (right) which are activated by phagocytosis of the apoptotic cells resulting from the inflammation. Rather than distinct subsets, both M2 and resolving macrophages represent a continuum of macrophage activation that are influenced by both Th2 cytokines and apoptotic cells.
Figure 2. Macrophage enhancers and effectors in wound healing and fibrosis.
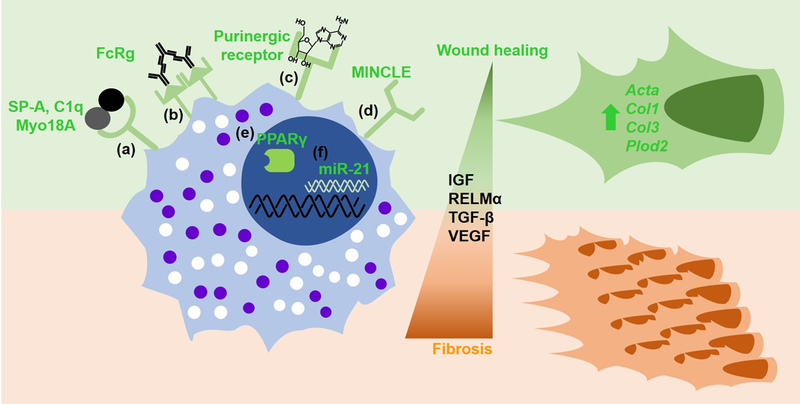
Wound healing macrophage activation is enhanced by the following surface markers: (a) signaling through the Myo18A receptor; (b) FcγR-mediated signaling by immune complexes; (c) ATP or adenosine binding to purinergic receptors; (d) Mincle surface expression; and intracellular factor; (e) nuclear receptor PPARγ; (f) micro RNA 21. These enhance macrophage effector function to promote wound healing (green), but if excessive, can lead to fibrosis (brown).
IL-4Rα signaling also stimulates tissue-resident macrophage proliferation, which can have the beneficial outcome of expanding and activating the effector macrophage population for wound healing.6, 14 Here, additional signals from the tissue environment promoted the IL-4-induced wound healing capacity of M2 macrophages. Specifically, the surfactant protein A (SP-A) produced in the lung acted through the receptor myosin 18A (Myo18A) to enhance M2 macrophage activation and lung wound healing following N. brasiliensis infection-induced injury. Interestingly, in the peritoneal cavity, resident macrophages did not respond to SP-A, but instead were activated by the complement protein C1q, which is structurally homologous to SP-A. The stimulatory effect of C1q on M2 macrophages, likely through Myo18a, was observed in several tissues including the peritoneal cavity, liver, spleen and adipose tissue. Functionally, C1q promoted M2 macrophage-mediated liver repair following infection with Listeria monocytogenes. Whether C1q affects resident tissue macrophage populations, or instead activates monocytes or macrophages recruited to the injury site from the blood or peritoneal cavity is unclear. Another study utilizing carbon tetrachloride (CCL4) treatment as a model of liver injury demonstrated that peritoneal macrophages could cross the mesothelium and penetrate into the injured liver tissue. These macrophages, originating from the peritoneal cavity, expressed M2 markers Arginase and RELMα, and exhibited a reparative function when recruited to the liver.15 While tissue-resident macrophages are likely more rapid responders to injury, monocytes recruited from the blood can also differentiate into M2 macrophages and contribute to tissue repair following Schistosoma mansoni infection-induced liver injury.16 This process was impaired in vitamin A deficient mice suggesting that dietary components are important for optimal M2 macrophage activation.
M2 macrophage-mediated killing of large extracellular helminths exhibits common features of tissue repair. In H. polygyrus infection M2 macrophages are recruited to the helminth, and produce factors to trap and kill the pathogen.17 The contributing factors that immobilize the worm include wound healing factors such as arginase 1. Understanding the activation pathway and effector molecules of M2 macrophages that kill H. polygyrus infection may therefore provide insight into new wound healing mechanisms. Mechanistically, recruitment and killing of H.polygyrus by M2 macrophages involved recognition of helminth antigen-antibody immune complexes and the production of CXCR2 ligands.18 Mice deficient in FcγR signaling and activation-induced cytidine deaminase (AID), which contributes to antibody maturation and class switching, exhibited impaired worm killing but also increased intestinal lesions suggesting defective wound repair. Further, at later timepoints, Fcrg−/−and Aid−/−mice showed severe peritonitis that might be attributed to defective lesion repair resulting in increased bacterial translocation. Investigation of the mechanism of wound repair in this helminth infection model also revealed the importance of an additional cell-type: the myofibroblast, which was activated CXCL2 and CXCL3, via CXCR2, and helminth antigens via dectin-2, to mediate wound closure potentially through expression of α-SMA. Of translational significance, CXCL3 from human monocyte-derived macrophages induced by Ascaris suum, the pig helminth closely related to the human parasite Ascaris lumbricoides, up-regulated wound healing by human myofibrobalsts. These data indicate that crosstalk between M2 macrophages and other cell-types is necessary for effective wound healing.
Apoptotic cells:
A critical step to wound healing is the clearance of apoptotic cells resulting from the inflammatory environment. This process is mediated by resolving, or resolution, macrophages. Resolving macrophages sense and phagocytose phostaphatidylserine (PtdSer)-exposed apopotic cells in a process called efferocytosis.19 Effective efferocytosis is dependent on the receptor tyrosine kinases Axl and Mertk. In addition to clearing dead cells, resolving macrophages contribute to wound healing and tissue homeostasis by producing anti-inflammatory molecules such as IL-10, and tissue remodeling growth factors such as TGF-β. Th2 cytokines and apoptotic cell engulfment were originally considered distinct signals, which activated M2 or resolving macrophages respectively, however, recent studies have uncovered synergism of these signals for optimal wound healing. Indeed, co-treatment of bone marrow-derived macrophages with apoptotic neutrophils and Th2 cytokines induced maximal expression of wound healing genes Retnla (RELMα), Chil3 (Ym1), Ear2 (Eosinophil associated, ribonuclease A family, member 2), and Fn1 (Fibronectin 1).19 The essential function of apopotic cell recognition for optimal M2 activation was demonstrated utilizing macrophages deficient in Axl and Mertk in N. brasiliensis infection as an in vivo model of M2 macrophage-dependent wound healing.19 Critically, Axl and Mertk functional effects on tissue repair were not restricted to the lung, as these proteins were required for upregulation of the anti-inflammatory and wound healing genes in macrophages in the damaged intestine and peritoneal cavity. It has previously been shown that SP-A induces efferocytosis while C1q activates MERTK expression20, Thus, in addition to M2 activation, SP-A and C1q might promote apoptotic cell sensing.21 Together, these studies implicate an inter-dependent, positive feedback loop whereby apoptotic cells and Th2 cytokines act together to promote macrophage-mediated wound healing.
Nucleotides:
Macrophage recognition of nucleotides, such as adenosine triphosphate (ATP) and their metabolites (e.g. adenosine), by P2 and P1 purinergic receptors respectively, influences their activation and recruitment.22, 23 For instance, ATP released by apoptotic cells promotes macrophage recruitment and efferocytosis.22 In a mouse model of traumatic brain injury, microglia chemotaxis towards the localized injury was dependent on extracellular ATP activation of the P2 receptor P2Y12.24 The P1 purinergic receptors that recognize adenosine also influence macrophage activation. Specifically, activation of the P1 receptors A2A and A2B promotes M2 macrophage activation,23 while the A3 receptor signaling induces an anti-inflammatory response.25 Consistent with a tissue protective role for the P1 adenosine receptors, A2A receptor-deficient mice exhibit extensive tissue damage and inflammation following concanavalin A or CCl4-induced liver injury as well as in response to endotoxic shock.26 One potential mechanism of A2A-mediated tissue protection may be through promoting macrophage production of IL-10.23 The A2B receptor, which is expressed on macrophages but also non-immune cells such as epithelial cells, also promoted M2 macrophage activation, and was tissue protective in helminth infection and endotoxin-induced lung injury.27, 28
MicroRNAs (miRs) also play an important role in macrophage-mediated wound healing by regulating efferocytosis-mediated suppression of the innate immune response. miR21 was upregulated in macrophages after efficient efferocytosis where it suppressed pro-inflammatory TNFα, and induced anti-inflammatory IL-10.29 The mechanism of action of miR21 included silencing of the signaling molecules PTEN, which contributes to NF-κB-induced inflammation and TNFα expression, and PDC4, which suppresses IL-10 expression. Together, these studies identify nucleotides as additional factors that regulate macrophage activation in injury and inflammation. Regulation of these signals in macrophages may be novel targets to promote wound healing.
Mechanical stimulus:
In addition to soluble factors or cells, tissue structure or physical cues in the extracellular matrix during wound healing also affects macrophage activation and behavior.30 For instance, macrophage elongation caused by migration in the fibrous tissue of a wound, rather than in healthy tissue or the vasculature, likely provides different mechanical cues. Recent studies suggest that these mechanical stimuli regulate macrophage polarization.31 In bone marrow-derived macrophage shape studies, M1 macrophages, activated by IFNγ and LPS, were flattened and round, while Th2 cytokine-activated M2 macrophages were elongated.31 Conversely, manipulating macrophage cell shape by micropatterning altered expression of macrophage phenotype markers. Cell elongation increased expression of M2 markers arginase-1, CD206, Ym1, characteristic of wound healing macrophages. Moreover, elongation augmented arginase-1 expression induced by IL-4/IL-13 but decreased inducible nitric oxide synthase (iNOS) expression caused by IFNγ/LPS, suggesting that cell elongation preferentially skews macrophages towards an M2 phenotype. The effect of cell elongation on macrophage polarization was mediated by actin and actin-associated contractility because pharmacological inhibition of actin and the actin signaling pathway abrogated up-regulation of arginase-1 expression by elongation but not by cytokine stimulation. Hence, polarization induced by changes in the extracellular matrix structure may promote wound healing through activation of genes required for tissue repair. In summary, we have highlighted in this section the complexity of factors that promote wound healing macrophages and have discussed main downstream macrophage effector pathways, outlined in Figure 1.
Wound healing macrophage heterogeneity and plasticity
We have summarized thus far multiple studies mapping the wound healing program of macrophages. Overall, there is a clear consensus that Th2 cytokines and apoptotic cells are key drivers of wound healing macrophages, but activation can be enhanced or modulated by other factors. Two active areas of research remain, of relevance to selective treatment strategies to target wound healing macrophages. First, investigation of the degree of heterogeneity in the wound healing macrophage subsets would allow for the optimal wound healing profile be selected and expanded. Second, evaluating the plasticity of the wound healing macrophage phenotype would guide therapeutic strategies to promote wound healing function. Specifically, are all macrophages able to differentiate into wound healing macrophages, are they long-lived, and can they change their phenotype dependent on the microenvironment? Recent research technologies including macrophage lineage tracking and single cell profiling have made it possible to start addressing these questions.
Wound healing macrophage heterogeneity:
While original macrophage studies grouped these cell-types into distinct non-overlapping subsets such as M1, M2 and regulatory macrophages, it is increasingly apparent that macrophage activation represents a spectrum of phenotypes dependent on the tissue microenvironment and cell lineage.32 Two main lineages of macrophages exist: tissue-resident macrophages originating from the embryonic precursors, and monocyte-derived macrophages originating from the bone marrow. The importance of these distinct macrophage lineages in wound healing is still undefined, however, tissue-resident macrophages are likely the first responders to wounds. Following injury, tissue-resident macrophages express adhesion molecules that recruit and guide multiple cell-types.33 Further, tissue-resident macrophages can replicate to increase their numbers, are highly M2 polarized in response to IL-4, and orchestrate the wound healing stages.34
Tissue-resident macrophages are long-lived and have the capacity for self-renewal. Indeed, fate-mapping and parabiosis studies revealed that tissue macrophages such as Kupffer cells, microglia, alveolar, and peritoneal macrophages are established prenatally and can be maintained independently of replenishment by blood monocytes under homeostatic conditions.35 In contrast, in the intestine, skin and heart, macrophages are replenished in the steady state by monocytes. The half-life of intestinal and dermal macrophages was estimated to range from 4 to 6 weeks, while those of the heart ranged from 8 to 12 weeks.36 In contrast to self-renewing tissue-resident macrophages, monocytes recruited to inflamed tissues cannot maintain themselves and die once inflammation resolves.35 Given the longevity of tissue-resident macrophages, they may consitute a better long-term target for wound healing compared to monocyte-derived macrophages.
Tissue-resident macrophage development is governed by distinct groups of transcription factors. For instance, GATA6 drives peritoneal macrophage differentiation, while PPARγ is involved in alveolar macrophage differentiation.37 With the advent of new technologies such as single cell RNA-seq (SC-RNA Seq), a more comprehensive identification of tissue macrophage subsets has been possible. In particular, the transcription factor zinc finger E-box binding homeobox 2 (ZEB2) was identified as a key determinant of tissue-specific macrophages in diverse organs including the liver, lung, spleen, brain, and colon.38 In ZEB2 deficient mice, there was alteration of different tissue-specific macrophage markers, highlighting the technical difficulty of identifying macrophages within tissues if using limited markers such as CD64 and F4/80.
To this end, a study using Single Cell Recognition, a reference-based computational tool that enables unbiased annotation of SC-RNA Seq, investigated macrophage heterogeneity in lung fibrosis.39 They characterized three distinct groups of macrophages, including alveolar macrophages (C1), interstitial macrophages (C3), and an intermediate cluster of cells (C2) in the lung. Both C2 and C3 were highly enriched in bleomycin-induced fibrosis, and C2 had high expression of genes from both C1 and C3, indicating that C2 is a transitional state between C1 and C3 in lung fibrosis. CX3CR1 was expressed in both C2 and C3, and these CX3CR1-lineage cells (CLCs) were increased after injury. MHC II expression was decreased, but expression of SiglecF, an alveolar macrophage marker, was increased in these cells, indicating that CLCs transit towards an alveolar macrophage identity. CLCs were in direct contact with fibroblasts expressing PDGF receptors. The ligand, PDGF, was induced in activated MHC IIhigh alveolar macrophages after injury, and conditioned media from MHC IIhigh alveolar macrophages mediated gap closure by fibroblasts better than media from MHClow alveolar macrophages. Last, deprivation of CLCs attenuated bleomycin-induced lung fibrosis, implying that CLCs provide trophic support for fibroblasts in the wound site.
Diverse macrophage populations were also identified by PrimeFlow, a flow-based technique which allows RNA cellular profiling of markers that may not have been detectable by antibodies.40 PrimeFlow analyses of in vitro and in vivo M2-activated macrophages revealed differential expression of the canonical M2 markers Arg1 and Retnla, with comparable frequencies of single and double positive subsets.40 Since both these genes contribute to wound healing, it is likely that targeting the double positive population would be of greatest therapeutic benefit. Heterogeneity in M2 macrophage subsets was also observed in the human peritoneum. Patients with peritoneal fibrosis caused by peritoneal dialysis exhibited heterogenous M2 macrophage populations with differential expression of CD163 and CD206.41 The functional relevance of these subsets in peritoneal fibrosis is unclear, however, high CD163 but not CD206 expression correlated with active peritonitis, and CD163-sorted macrophages produced chemokine CCL18 and promoted fibroblast proliferation.41 More research is needed to determine to what extent functional heterogeneity is influenced by macrophage origin versus subtle differences in the microenvironmental cues.
Wound healing macrophage plasticity:
As highlighted previously, macrophages can respond to a plethora of external cues leading to a wide spectrum of activation phenotypes. Whether these represent terminally differentiated cells, or whether these cells are plastic and can adapt their function according to new environmental cues is less clear. Additionally, transcriptional studies have suggested that the epigenetic landscape may govern macrophage polarization and plasticity.37 Within the ever-changing microenvironment of the wound, plasticity and longevity of the macrophages would be attractive features for therapeutic targeting allowing them to adapt their function to address the immediate needs within the wound. The monocyte/macrophage lineage is general recognized as a highly plastic lineage.2 Indeed, more specific research investigating macrophage plasticity between M1 and M2 macrophages does support a certain degree of plasticity in M2 macrophages. M2 macrophages that were differentiated following chronic helminth exposure could respond to M1 activating signals such as LPS and IFNγ.42 In an in vivo co-infection model, peritoneal M2 macrophages activated by H. polygyrus infection were able to change to antimicrobial nitric oxide-producing M1 macrophages capable of killing an attenuated Salmonella strain when challenged intraperitoneally.43 In contrast, Salmonella-induced M1 macrophages could not be repolarized by IL-4 treatment, indicating that M1 macrophage activation has a more restrictive effect on plasticity than M2 macrophage activation.43 These studies show that M2 macrophages have some ability to alter their phenotype in response to different stimuli. It can be inferred from these studies that wound healing M2 macrophage subsets may be equally plastic, however, further mechanistic investigation in wound healing models is needed.
As mentioned above, pro-inflammatory activated M1 macrophages kill pathogens in wounded tissues, on the other hand, M2 macrophages dampen these inflammatory responses, and these sequential steps are required for wound healing. AMP-activated protein kinase α1 subunit, a catalytic domain (AMPKα1) was proven to modulate M1/M2 macrophage polarization.44 Following cardiotoxin injection, AMPKα1 activity was increased in the macrophages recruited to the regenerating muscle. In both whole body and macrophage-specific Ampkα1−/−mice, there were more necrotic myofibers than regenerating myofibers following cardiotoxin injection, suggesting that AMPKα1 promotes muscle regeneration. Ampkα1−/−macrophages exhibit preferential expression of M1 macrophage markers (e.g. iNOS) over M2 markers (e.g. CD206, CD163, and TGFβ). Since AMPK senses the cellular energy level as ratios of ADP:ATP and AMP:ATP, this data supports the concept that energy sensing and metabolic activity dictates macrophage activation.45 Indeed, M2 macrophages exhibit high oxygen consumption rate, that is impaired in Ampkα1−/−mice. AICAR, a pharmacological AMPK activator, dampened M1 activation and enhanced M2 activation in wild-type but not Ampkα1−/−mice.44 Therefore, pharmacologically targeting metabolic pathways in wounds may influence macrophage plasticity and promote a wound healing macrophage subset.
Macrophage function in wound healing disorders
In the previous sections, we have provided evidence supporting the role of macrophages in the wound healing process. Consistent with their importance in wound healing, impaired or aberrant macrophages are key features in dysfunctional wounds. In this section, we will describe wound healing disorders that are mediated by macrophage dysfunction. These include aging and diabetes, which are associated with deficient wound healing macrophage activation. On the flipside, excessive wound healing macrophage activation is also detrimental. This is apparent in fibrotic disorders, which is mediated by uncontrolled M2 macrophage activation.
Aging:
Aging can result in immune dysfunction, including immunosenescence, defined as age-related impairments of the immune system,46 and ‘inflamm-aging’, defined as association of advanced age with chronic low-grade inflammation.47 Both these age-related immune disorders affect the wound healing process because they can result in impaired pathogen killing, or conversely, the inappropriate control of inflammation. For instance, monocyte-derived macrophages from elderly subjects produce less TNFα and IL-6 in response to Streptococcus pneumoniae, resulting in hampered bacterial killing compared to young subjects.48 Microarray analysis of LPS-stimulated macrophages revealed reduced immune response and signal transduction genes in older mice.49 The deficient activation of macrophages from aged mice could be attributed to decreased levels of p38 and c-Jun N terminal kinase, which are important for pro-inflammatory gene expression.50 In addition, A20 that inhibits NF-κB and MAPK signaling pathways was elevated in the lung and alveolar macrophages of aged mice, which caused reduced cytokine responses to bacteria.51 Stimulation of alveolar macrophages with TNFα increased A20 levels, implying the role of TNFα in inflamm-aging.
Conversely, levels of pro-resolving mediators, such as lipoxins, protectins, maresins, and the D-or E-series resolvins, are impaired in aged mice. Specifically, aged mice exhibit altered lipid biosynthesis dynamics with lower level of specialized pro-resolving mediators (SPMs) but higher levels of pro-inflammatory lipid mediators.52 The phagocytic ability of peritoneal macrophages is also decreased in aged mice, potentially due to exposure to elevated levels of IL-10 in the peritoneal cavity.46 Of significance to the wound healing process, peritoneal macrophages from aged mice are less able to phagocytose necrotic cells.53 In response to certain stimuli, however, macrophages from aged mice remained plastic and were able to respond to factors in their microenvironment, suggesting that macrophage dysfunction can be reversed. Indeed, aged macrophages responded to in vitro treatment with IFNγ as effectively as young macrophages, underscoring the importance of the microenvironment over intrinsic defects.54 Additionally, deficiencies in macrophage polarization in aged mice could be restored with exercise and diet changes.51, 55
Diabetes:
Deficient wound healing is also a severe and potentially fatal consequence of diabetes. The mechanisms by which diabetics suffer from unhealed chronic wounds are multi-factorial but do involve dysfunctional macrophage responses. In particular, macrophage activation via the nuclear receptor PPARγ is impaired in diabetes.56 PPARγ activation promotes wound healing by decreasing the expression of pro-inflammatory cytokines and increasing wound healing genes.56 The induction of PPARγ activity also leads to granulation tissue formation, angiogenesis, and collagen deposition that are key for wound repair. In the diabetic wound, the decreased PPARγ activity was caused by sustained production of IL-1β leading to inflammasome activation.56 This could be reversed by treatment of the wound with PPARγ agonists as a promising treatment strategy to promote wound healing macrophages.
Fibrosis:
Optimal wound healing is dependent on a highly regulated M2 macrophage response. While a deficient M2 macrophage response leads to impaired wound closure, excessive M2 macrophage activation causes scarred or fibrotic tissue.57 In particular, the Th2 cytokine IL-13 can drive pathologic fibrosis through excessive M2 macrophage activation. In models of helminth-induced fibrosis, the IL-13 driven inflammation and fibrosis were ameliorated with depletion of CD11b+ macrophages.57 Additionally, in bleomycin-induced pulmonary fibrosis, the signaling molecule IRAK-M was shown to enhance IL-13 production and fibrosis as IRAK-M−/−mice had reduced bleomycin-induced collagen accumulation in the lung.58 When lung fibroblasts were co-cultured with macrophages from bleomycin-treated IRAK-M−/−mice, collagen and α-SMA expression was reduced compared to wild type mice, suggesting that M2 macrophages were the downstream effectors of IRAK-M signaling in promoting pathologic fibrosis.
Mincle, a C-type lectin expressed on macrophages, was also identified as a mediator of fibrosis.59 A high-fat diet increased Mincle expression in macrophages in the crown-like structures (CLS) of the epididymal fat, which are a characteristic structure in obese adipose tissue. Mincle was preferentially expressed by CD11b+F4/80lo rather than CD11b+F4/80hi cells. These Mincle-expressing macrophages had higher CD11c and lower CD206 expression in line with previous data showing that it is classically activated macrophages that express Mincle.59 Mincle−/−mice were protected from hepatic steatosis and insulin resistance, associated with reduced interstitial fibrosis in epididymal fat tissue, α-SMA+ cells and myofibroblasts.
The beneficial or pathologic effect of M2 macrophages in fibrosis may be critically dependent on the timing. Using a model of liver fibrosis and spontaneous recovery, where mice were treated with CCL4 for several weeks then left untreated for the liver to recover, Weng et al. investigated the function of M2 macrophages.7 Intriguingly, macrophage-specific IL-4Rα−/−mice were protected from liver fibrosis progression during CCl4 treatment, but had delayed fibrosis reversal during the recovery phase. The phase-specific function of M2 macrophages was confirmed with an anti-sense IL-4Rα nucleotide at different timepoints, where it was shown that early activation of M2 macrophages promotes fibrosis, while at later timepoints, M2 macrophages speed up fibrosis reversal. These studies highlight the importance of tightly controlling macrophage activation to promote wound healing while circumventing pathologic fibrosis, summarized in Figure 2.
Conclusion
Macrophages are critical participants in the wound healing process and provide a useful therapeutic target for wound healing disorders. Indeed, dysfunctional macrophages are key features of delayed wound healing in aging and diabetes, or excessive wound healing in fibrosis. There is increasing evidence that macrophages are long-lived and plastic and can change their phenotype depending on external stimuli. Therefore, it may be possible to skew their function within the aberrant wound for improved outcomes. While both activating factors and downstream effectors of wound healing macrophages are well-defined, challenges to macrophage-specific wound healing strategies remain. These include the extensive spectrum and heterogeneity of the macrophage subsets, and the lack of understanding of the individual cues that can control this heterogeneity. New technologies targeting individual macrophage lineages and macrophage-derived molecules, as well phenotyping these subsets at the single cell level, provide the promising prospect that identification of an optimal wound healing macrophage program of therapeutic potential will soon be possible.
Acknowledgements:
The Nair lab is supported by the NIH (R21AI137830; R21AI135500). We thank Sarah Bobardt for critical review of the manuscript.
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- 1.Minutti CM, Knipper JA, Allen JE, Zaiss DM. Tissue-specific contribution of macrophages to wound healing. Semin Cell Dev Biol 2017; 61: 3–11. [DOI] [PubMed] [Google Scholar]
- 2.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol 2013; 13: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Z, Ding J, Ma Z, Iwashina T, Tredget EE. Alternatively activated macrophages derived from THP-1 cells promote the fibrogenic activities of human dermal fibroblasts. Wound Repair Regen 2017; 25: 377–388. [DOI] [PubMed] [Google Scholar]
- 5.Knipper JA, Willenborg S, Brinckmann J, et al. Interleukin-4 receptor α signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity 2015; 43: 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minutti CM, Jackson-Jones LH, García-Fojeda B, et al. Local amplifiers of IL-4Rα-mediated macrophage activation promote repair in lung and liver. Science 2017; 356: 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng SY, Wang X, Vijayan S, et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. EBioMedicine 2018; 29: 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol 2013; 133: 2461–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pine GM, Batugedara HM, Nair MG. Here, there and everywhere: Resistin-like molecules in infection, inflammation, and metabolic disorders. Cytokine 2018; 110: 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutherland TE, Rückerl D, Logan N, Duncan S, Wynn TA, Allen JE. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PLoS Pathog 2018; 14: e1007423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batugedara HM, Li J, Chen G, et al. Hematopoietic cell-derived RELMα regulates hookworm immunity through effects on macrophages. J Leukoc Biol 2018; 104: 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen G, Wang SH, Jang JC, Odegaard JI, Nair MG. Comparison of RELMα and RELMβ single-and double-gene-deficient mice reveals that RELMα expression dictates inflammation and worm expulsion in hookworm infection. Infect Immun 2016; 84: 1100–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nair MG, Herbert DR. Immune polarization by hookworms: taking cues from T helper type 2, type 2 innate lymphoid cells and alternatively activated macrophages. Immunology 2016; 148: 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011; 332: 1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 2016; 165: 668–678. [DOI] [PubMed] [Google Scholar]
- 16.Gundra UM, Girgis NM, Gonzalez MA, et al. Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat Immunol 2017; 18: 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esser-von Bieren J, Mosconi I, Guiet R, et al. Antibodies trap tissue migrating helminth larvae and prevent tissue damage by driving IL-4Rα-independent alternative differentiation of macrophages. PLoS Pathog 2013; 9: e1003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esser-von Bieren J, Volpe B, Sutherland DB, et al. Immune antibodies and helminth products drive CXCR2-dependent macrophage-myofibroblast crosstalk to promote intestinal repair. PLoS Pathog 2015; 11: e1004778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017; 356: 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 2016; 273: 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouchery T, Harris NL. Specific repair by discerning macrophages. Science 2017; 356: 1014. [DOI] [PubMed] [Google Scholar]
- 22.Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Csóka B, Selmeczy Z, Koscsó B, et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J 2012; 26: 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 2006; 9: 1512–1519. [DOI] [PubMed] [Google Scholar]
- 25.Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. Inhibition of TNF-alpha expression by adenosine: role of A3 adenosine receptors. J Immunol 1996; 156: 3435–3442. [PubMed] [Google Scholar]
- 26.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001; 414: 916–920. [DOI] [PubMed] [Google Scholar]
- 27.Schingnitz U, Hartmann K, Macmanus CF, et al. Signaling through the A2B adenosine receptor dampens endotoxin-induced acute lung injury. J Immunol 2010; 184: 5271–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel N, Wu W, Mishra PK, et al. A2B adenosine receptor induces protective antihelminth type 2 immune responses. Cell Host Microbe 2014; 15: 339–350. [DOI] [PubMed] [Google Scholar]
- 29.Das A, Ganesh K, Khanna S, Sen CK, Roy S. Engulfment of apoptotic cells by macrophages: a role of microRNA-21 in the resolution of wound inflammation. J Immunol 2014; 192: 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol 2010; 184: 1049–1061. [DOI] [PubMed] [Google Scholar]
- 31.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A 2013; 110: 17253–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity 2018; 49: 595–613. [DOI] [PubMed] [Google Scholar]
- 33.Kim ND, Luster AD. The role of tissue resident cells in neutrophil recruitment. Trends Immunol 2015; 36: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies LC, Rosas M, Jenkins SJ, et al. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat Commun 2013; 4: 1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013; 38: 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott CL, Henri S, Guilliams M. Mononuclear phagocytes of the intestine, the skin, and the lung. Immunol Rev 2014; 262: 9–24. [DOI] [PubMed] [Google Scholar]
- 37.T’Jonck W, Guilliams M, Bonnardel J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol 2018; 330: 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott CL, T’Jonck W, Martens L, et al. The transcription factor ZEB2 is required to maintain the tissue-specific identities of macrophages. Immunity 2018; 49: 312–325.e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aran D, Looney AP, Liu L, et al. Reference-based annotation of single-cell transcriptomes identifies a profibrotic macrophage niche after tissue injury. bioRxiv 2018.
- 40.Lai C, Stepniak D, Sias L, Funatake C. A sensitive flow cytometric method for multi-parametric analysis of microRNA, messenger RNA and protein in single cells. Methods 2018; 134–135: 136–148. [DOI] [PubMed]
- 41.Bellón T, Martínez V, Lucendo B, et al. Alternative activation of macrophages in human peritoneum: implications for peritoneal fibrosis. Nephrol Dial Transplant 2011; 26: 2995–3005. [DOI] [PubMed] [Google Scholar]
- 42.Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol 2009; 182: 3084–3094. [DOI] [PubMed] [Google Scholar]
- 43.Rückerl D, Campbell SM, Duncan S, et al. Macrophage origin limits functional plasticity in helminth-bacterial co-infection. PLoS Pathog 2017; 13: e1006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mounier R, Théret M, Arnold L, et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab 2013; 18: 251–264. [DOI] [PubMed] [Google Scholar]
- 45.Huang SC, Smith AM, Everts B, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 2016; 45: 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell 2014; 13: 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franceschi C, Bonafè M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000; 908: 244–254. [DOI] [PubMed] [Google Scholar]
- 48.Verschoor CP, Johnstone J, Loeb M, Bramson JL, Bowdish DM. Anti-pneumococcal deficits of monocyte-derived macrophages from the advanced-age, frail elderly and related impairments in PI3K-AKT signaling. Hum Immunol 2014; 75: 1192–1196. [DOI] [PubMed] [Google Scholar]
- 49.Chelvarajan RL, Liu Y, Popa D, et al. Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol 2006; 79: 1314–1327. [DOI] [PubMed] [Google Scholar]
- 50.Boehmer ED, Meehan MJ, Cutro BT, Kovacs EJ. Aging negatively skews macrophage TLR2-and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev 2005; 126: 1305–1313. [DOI] [PubMed] [Google Scholar]
- 51.Hinojosa CA, Akula Suresh Babu R, Rahman MM, Fernandes G, Boyd AR, Orihuela CJ. Elevated A20 contributes to age-dependent macrophage dysfunction in the lungs. Exp Gerontol 2014; 54: 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnardottir HH, Dalli J, Colas RA, Shinohara M, Serhan CN. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J Immunol 2014; 193: 4235–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi R, Totsuka S, Ishigami A, Kobayashi Y, Nagata K. Attenuated phagocytosis of secondary necrotic neutrophils by macrophages in aged and SMP30 knockout mice. Geriatr Gerontol Int 2016; 16: 135–142. [DOI] [PubMed] [Google Scholar]
- 54.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol 2005; 175: 342–349. [DOI] [PubMed] [Google Scholar]
- 55.Goh J, Ladiges WC. Exercise enhances wound healing and prevents cancer progression during aging by targeting macrophage polarity. Mech Ageing Dev 2014; 139: 41–48. [DOI] [PubMed] [Google Scholar]
- 56.Mirza RE, Fang MM, Novak ML, et al. Macrophage PPARγ and impaired wound healing in type 2 diabetes. J Pathol 2015; 236: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borthwick LA, Barron L, Hart KM, et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol 2016; 9: 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ballinger MN, Newstead MW, Zeng X, et al. IRAK-M promotes alternative macrophage activation and fibroproliferation in bleomycin-induced lung injury. J Immunol 2015; 194: 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ichioka M, Suganami T, Tsuda N, et al. Increased expression of macrophage-inducible C-type lectin in adipose tissue of obese mice and humans. Diabetes 2011; 60: 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]