

Abstract
Carbonaceous nanomaterials represent a significant portion of ultra-fine airborne particulate matter, and iron is the most abundant transition metal in air particles. Owing to their high surface area and atmospheric oxidation, carbon nanoparticles (CNP) are enriched with surface carbonyl functional groups and act as a host for metals and small molecules. Using a synthetic model, concentration-dependent changes in the chemical speciation of iron adsorbed on oxidized carbon surfaces were investigated by a combination of X-ray and electron microscopic and spectroscopic methods. Carbon K-edge absorption spectra demonstrated that the CNP surface was enriched with carboxylic acid groups after chemical oxidation but that microporosity was unchanged. Oxidized CNP showed a high affinity for sorption of Fe(III) from solution (75–95% uptake) and spectroscopic measurements confirmed a 3+ oxidation state of Fe on CNP irrespective of surface loading. The bonding of adsorbed Fe(III) at variable loadings was determined by iron K-edge X-ray absorption spectroscopy. At low loadings (3 and 10 μmol Fe/m2 CNP), mononuclear Fe was octahedrally coordinated to oxygen atoms of carboxylate groups. As Fe surface coverage increased (21 and 31 μmol Fe/m2 CNP), Fe-Fe backscatters were observed at interatomic distances indicating iron (oxy)hydroxide particle formation on CNP. Electron-donating surface carboxylate groups on CNP coordinated and stabilized mononuclear Fe(III). Saturation of high-affinity sites may have promoted hydroxide particle nucleation at higher loading, demonstrating that the chemical form of reactive metal ions may change with surface concentration and degree of CNP surface oxidation. Model systems such as those discussed here, with controlled surface properties and known chemical speciation of adsorbed metals, are needed to establish structure-activity models for toxicity assessments of environmentally relevant nanoparticles.
Graphical Abstract
Iron speciation on oxidized carbon nanoparticles changes with surface concentration
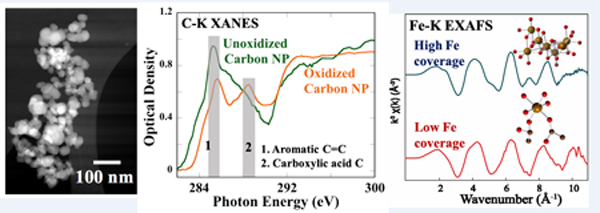
Introduction
Inhalation of ultrafine particulate matter poses several chronic and acute health impacts.1 Health risks associated with particulate matter of aerodynamic diameters of a few micrometers (PM2.5, PM10) and the risks of acute lung inflammation to carcinogenicity have been assessed.2,3 Because of their smaller size and high reactive surface area, the purportedly greater health impacts of nanometer-sized particulate matter (ultrafine fraction, <0.1 μm) have more recently been the focus of studies,4 although their damaging effects were noted over twenty years ago.5 Toxicity studies of engineered nanomaterials support the hypothesis that ultrafine particles have higher cellular uptake in model systems, although their uptake mechanisms are not well established.6,7 Many biological studies of nanoparticulates of anthropogenic and geogenic origin are aimed at understanding mechanisms of toxicity using representative synthetic particle systems, rather than natural materials, because of their availability and purity. Atmospheric oxidation, UV exposure, and deposition of organic molecules and metals on particles affect their surface composition, morphology, and physiochemical nature. The majority of toxicity studies, however, use model particles of a single composition (e.g., carbon or SiO2), often without modification, which fails to capture the impact of compositional mixtures and surface alterations typical of airborne particulate matter.
Carbon, in the form of nanoparticles from high temperature combustion and automobile emissions, is of specific interest because it is a major component of ultrafine particulate pollution in air.8 Carbon particulates found as sub-μm sized aggregates of smaller graphitic spheres (10–40 nm)9 are a common carrier of transition metals,10 polyaromatic hydrocarbons (PAH),11 and quinones.12 Carbon nanoparticles (CNP) are commercially synthesized by high temperature aromatization of fatty acids or fossil fuels. Ambient CNP are characterized by being highly heterogeneous in size and chemical composition compared to synthetic CNP. Surface oxygen functional groups (~2–10% oxygen) are generated on CNP by photochemical ozonolysis and other hydroxyl radical-mediated reactions.13 For example, Takahoma et al 14 examined atmospheric CNP (0.5–1 μm) using Scanning Transmission X-ray Microscopy (STXM) and carbon K-edge X-ray Absorption Near-edge Structure (XANES) spectroscopy, and confirmed the presence of carboxylic acid groups on the exterior of particles. Oxidation of carbon surfaces induces changes in physiochemical properties of CNP specifically because of electrostatic and steric interactions offered by carboxylic acid groups. Due to the large surface area and negative charge on carboxylate groups, ambient CNP is a potential carrier for cations. Metals, metal ions, and/or metal oxides are often chemisorbed or physiosorbed on ambient CNP. Analysis of ultrafine particles collected from several locations showed that iron is the most abundant transition metal present in atmospheric particles.15,16,17 The presence of hematite, magnetite, maghemite, and metallic iron phases were reported in urban PM (0.1 to 2.5 μm), which is mainly from industrial or transportation sources.18,19,20
The cytotoxicity and inflammation of iron associated with CNP have been investigated previously. For example, a prior study demonstrated that the presence of Fe(III) (100 μM) and Printex 90 (15 μg/mL), a commonly used synthetic surrogate for CNP, in cell culture media increased oxidative stress signals by 3–4 times compared to CNP alone.21 However, oxidation of the fluorescent reporter dye is often misinterpreted in studies as representing the generation of oxygen-derived oxidizing species, but is actually a measure of iron-dependent dye oxidation.22,23 Waldman et al.23 investigated the inflammatory responses of human macrophages and epithelial cells from exposure to hydrothermally synthesized CNP that contained 3–7 % surface iron by mass, as measured by X-ray photoelectron spectroscopy (XPS) and elemental analysis from scanning electron microscopy (Fe(II)/Fe(III) = 3/7). At nontoxic particle doses (0–25 μg/cm2), they found that iron-containing particles induced cytokine production about 5–10 times more than that of CNP without iron. They attributed the inflammatory response from these particles to the presence of Fe(II) that generated hydroxyl radicals via Fenton chemistry.24 We previously demonstrated the effect of surface-adsorbed iron on engineered silica micro/nano particles to induce inflammatory responses using human-derived THP-1 macrophages.25,26 In contrast, a recent study noted that catalytic iron impurities in carbon nanotubes had a negligible role in the cytotoxicity induced by the particles.27 Differences in cell response from exposure to iron-CNP observed in prior studies suggest that iron oxidation state, chemical form, and bonding to nanocarbon surfaces influences the biochemical pathway and response mechanism.
Here, we designed and characterized a synthetic CNP system involving surface oxidation and iron deposition intended to simulate an atmospheric particle system. We investigated surface oxidation of synthetic CNP by strong acid as a reasonable modification to match the surface chemistry and physiochemical properties of ambient particles. The chemistry of surface-adsorbed iron on CNP and changes in iron speciation with surface loading under environmentally relevant conditions have not been previously investigated. In this study, the nature of the carbon surface and chemical speciation and structure of surface-adsorbed iron were examined from bulk to nanoscale dimensions using electron and X-ray spectro-microscopy techniques to produce well-characterized particles with known surface properties and iron speciation.
Materials
The sources and properties of four different synthetic CNP are listed in Table 1. Potassium iron (III) trisoxalate (K3Fe(C2O4)3 or K3Fe(ox)3), trace metal grade HNO3 and H2O2 (30% v/v in water) were purchased from Fisher Scientific. FeCl3.6H2O, hematite (α-Fe2O3), and iron(III) acetylacetonate (Fe(acac)3) were received from Sigma Aldrich (St. Louis, MO, USA). Amorphous hydrous ferric oxide (HFO: Fe(OH)3. nH2O) was synthesized by following the reported method.28 Stainless steel nanoparticles were purchased from US Research Nanomaterials, Inc. (Houston, TX, USA). All washing and dilutions were done in milli-Q (18 mΩ.cm) pure water.
Table 1.
Properties of carbon nanoparticles.
Methods
Oxidation of CNP and Fe Sorption:
Carbon nanoparticles were oxidized using concentrated nitric acid treatment.29 One gram CNP was refluxed with 10 mL trace metal-free concentrated HNO3 at ~80°C for ~12 h. Excess acid was removed by centrifugation at 14000 rpm for 90 minutes, and the pellet was washed and separated by centrifugation. The pellet was washed further until the pH of the dispersion was near neutral. To remove the larger particles the suspension was centrifuged at 3000 rpm for one hour and the pellet was discarded. The final concentration was calculated by taking the dry weight (60°C, oven) of a known volume of the dispersion. The resulting suspensions were stable for 2–3 months in water at room temperature without visible precipitation. Deposition of iron onto the oxidized surfaces of CNP was performed according to a published protocol with minor modifications.30 All the sorption experiments were carried out ultrapure water (18.2 mΩ cm) and no buffers were used to avoid any interference from other ions. A calculated amount of FeCl3 solution (1 mM, pH ~ 3) was added to the CNP dispersion to reach a final [Fe(III)] concentration from 0.5 to 1000 μM in N2 purged water. After equilibrating the CNP dispersion (pH ~ 4–7) with Fe(III) solutions for 12 h on a rotating shaker (40 rpm), pH was adjusted slowly to 7–7.5 using dilute NaOH to avoid precipitation of ferric hydroxide, followed by another 12 h equilibration. Next, the dispersion was centrifuged at 14000 rpm for 90 minutes to separate the insoluble particles, and washed with MilliQ water and centrifuged for three more times. The final pellet was kept in an oven at ~40°C until completely dried. No visual precipitation of iron oxide particles was observed under these conditions.
Inductively Coupled Plasma - Optical Emission Spectrometry (ICP-OES) analysis:
The amount of iron adsorbed onto CNP was determined by total sample digestion and measurement of total iron; uptake from solution was calculated by difference. For digestion, CNP samples (~50 mg) were reacted in Teflon tubes with 4 mL H2O2 (30% v/v) for 12 h and then 4 mL trace metal-free conc. HNO3 was added. Samples were digested by microwave (Anton Paar Multiwave 3000; 50 min, 160 °C, 18 atm) to ensure the complete dissolution of iron from the particles.31 Digested samples were transferred to 50 mL Falcon tubes followed by rinsing the Teflon vessels three times with water. Filtered samples were diluted to 10 mL (5% HNO3) and measured for total Fe by ICP-OES (Perkin-Elmer Optima 5300 DV). Calibration standards for Fe were prepared from a purchased standard solution (Invitrogen, Carlsbad, CA, USA) and a linear calibration plot (R2 = 0.99) was obtained from 5 to 5000 ppb. The method detection limit for Fe was measured as 3 ppb, and the relative standard deviation (RSD) of the measurements were 0.3 to 5%. Total Fe concentration was normalized to sample particle surface area using the measured BET surface area.
Transmission Electron Microscopy (TEM):
Morphology and size distribution of the CNP were examined by a JEOL JEM-2011 transmission electron microscope (Peabody, MA, USA) equipped with a LaB6 filament, operated at 200 kV accelerating voltage. A dilute solution of CNP (~ 20 μg/mL, 2 μL) was dropped onto a holey carbon-coated Cu-mesh grid (400 mesh, Ted Pella, Redding, CA, USA) and dried under vacuum (>12 hours) immediately prior to imaging. Scanning transmission electron microscopy (STEM) and elemental mapping using energy-dispersive X-ray spectroscopy (EDXS) were performed in a Philips CM200/FEG instrument operated at 200 kV and equipped with an Oxford INCA detector with energy resolution of 136 eV for Mn K-alpha radiation. Electron energy-loss spectroscopy (EELS) of the Fe L-edge was performed at 0.2 eV/channel dispersion using a Tridiem® Gatan imaging filter (GIF) on an F20 UT Tecnai microscope (200 kV), equipped with high-angle annular dark-field (HAADF) and bright-field (BF) detectors. The spectral resolution of EELS was 0.5 eV, measured from the full width at half-maximum (FWHM) of the zero-loss peak. The EELS collection and convergence angle were 42 mrad and 12 mrad, respectively. The continuum background of the EELS spectra was subtracted by following Egerton’s power law method over a ~25 eV range in the pre-edge region (~50 eV below the edge).32 For acquisition of carbon maps and EELS, particles in a hole are preferred to avoid background from the holey carbon film. Energy-loss spectra from several regions were collected, aligned, and summed for comparison with the reference compounds.
Scanning Transmission X-ray Microscopy (STXM):
Synchrotron X-ray STXM studies were done at the Canadian Light Source (CLS Beamline 10ID-1, Saskatoon, Canada) and the Advanced Light Source (Beamline 5.3.2.2, Berkeley, CA) designed for soft X-ray microscopy at ambient conditions. Samples were prepared by drop-casting the nanoparticle solution (~ 20 μg/mL) onto a 0.5×0.5 mm silicon nitride membrane (100 nm thick, Norcada, Edmonton, Canada). Data were acquired in transmission X-ray mode using a ~30 nm spot size under He atmosphere (1/6 atm, room temperature). X-ray absorption spectra were obtained by subtracting the intensity of a particle-free region on the membrane (I0) from particle intensity (I) in the same image (expressed as optical density = log(I0/I)). I0 were collected from minimum absorbing regions (pixels) in the STXM image at the absorption edge, which showed maximum contrast. The spectral data were analyzed using aXis2000 and MANTiS 2.3.02 programs available online.33,34 The images were realigned before calculating the full spectra from the stack plots. All pixels in the absorbing regions are integrated to obtain the spectra. Cluster analysis was used to group image stacks with similar spectroscopic signatures. The cluster identification procedure uses principal component analysis (PCA) to find the pixels with similar weightings of the eigenspectra. The recommended method from the developers for cluster analysis is followed here to identify the statistically significant regions in the image. The estimations are started to identify about 6 different regions in the image and the number of possible regions are reduced after manually inspecting the spectral features.
Synchrotron X-ray Spectroscopy (XAS): Microfocused and bulk:
X-ray absorption spectra at the Fe K-edge were collected on iron-coated CNP at ambient conditions using synchrotron X-ray radiation (beam current 500 mA) at the Stanford Synchrotron Radiation Lightsource (SSRL). For microfocused X-ray fluorescence (XRF) and X-ray absorption near-edge structure (XANES) data collection, a particle suspension (10 mg/mL) was air-dried on a Teflon filter (>12 h) and mounted on a sample holder. Data were collected at beamline 2–3 at room temperature using a 2×2 μm spot size, achieved by a Kirkpatrick-Baez mirror system. Iron-containing particles were located by Fe XRF mapping over a large area (~350 × 350 μm, 25 s dwell time). XANES spectra at individual spots were collected using a fluorescence detector (Vortex 2X), and three scans were collected and averaged manually.
Iron K-edge extended X-ray absorption fine structure (EXAFS) spectra were collected in bulk mode on beamline 4–1 with a Si(220) monochromator crystal and a beam size of 1×4 or 1×2 mm. Samples were in Al sample holders, sealed with sulfur-free tape, and held in a liquid nitrogen cryostat during data collection. Energy was calibrated using an Fe metal foil with the first inflection on the absorption edge set to 7112.0 eV. Spectra were collected in fluorescence using a solid-state Ge detector (Canberra) with a Mn filter and Soller slits, to reduce background noise. Reference compounds (Fe(III) acetylacetonate, (Fe(acac)3) and potassium Fe(III) oxalate, (K3Fe(ox)3), and hydrous ferric oxide (HFO) were diluted with a calculated amount of amorphous boron nitride powder to have 10% transmittance of the X-ray beam. Spectra were collected at liquid N2 temperature using a fluorescence PIPS detector. SIXpack35 and Athena/Artemis36 software packages were used for data processing, background subtraction, normalization, and spectral fits. Theoretical phase shift and amplitude functions were calculated for scattering paths in Fe(acac)3, K3Fe(ox)3, and goethite (α-FeO(OH)), which was used to fit the experimental amorphous hydrous ferric oxide (HFO) spectrum, using FEFF 8.237 using interatomic distances from known crystal structures. Non-linear least-squares fits to the experimental spectra of the reference compounds were used to estimate Debye-Waller factors (σ2) for absorber-backscatterer pairs of known number of backscatterers (N). The experimental EXAFS spectra of CNP/Fe were fit using Fe-O and Fe-C scattering paths from Fe(III) acetyl acetonate with interatomic distances (R) and number of backscatterers (N) varied, and σ2 fixed for each path; energy shift (ΔE0) was linked as a single adjustable parameter for all shells. Error estimates of fitted parameters are: R – ±0.0.1 Å, N and σ2 – ±15% for the first coordination shell. For outer shells, R – ±0.0.2 Å, N and σ2 - ±25%. 38,39
Results
Aggregate Size and Surface Area of CNP
The effect of oxidation on aggregate stability and size distribution of aqueous CNP dispersions was examined by measuring particle hydrodynamic size (Figure 1A). All CNP types formed stable aqueous suspensions after HNO3 treatment. The mean hydrodynamic size of oxidized CNP was about 170–400 nm for all types, whereas the unoxidized particles could not be measured by dynamic light scattering (DLS) due to poor dispersity. Note that DLS measured the size of aggregated particles but not the diameter of individual carbon spheres (20–50 nm).
Figure 1.
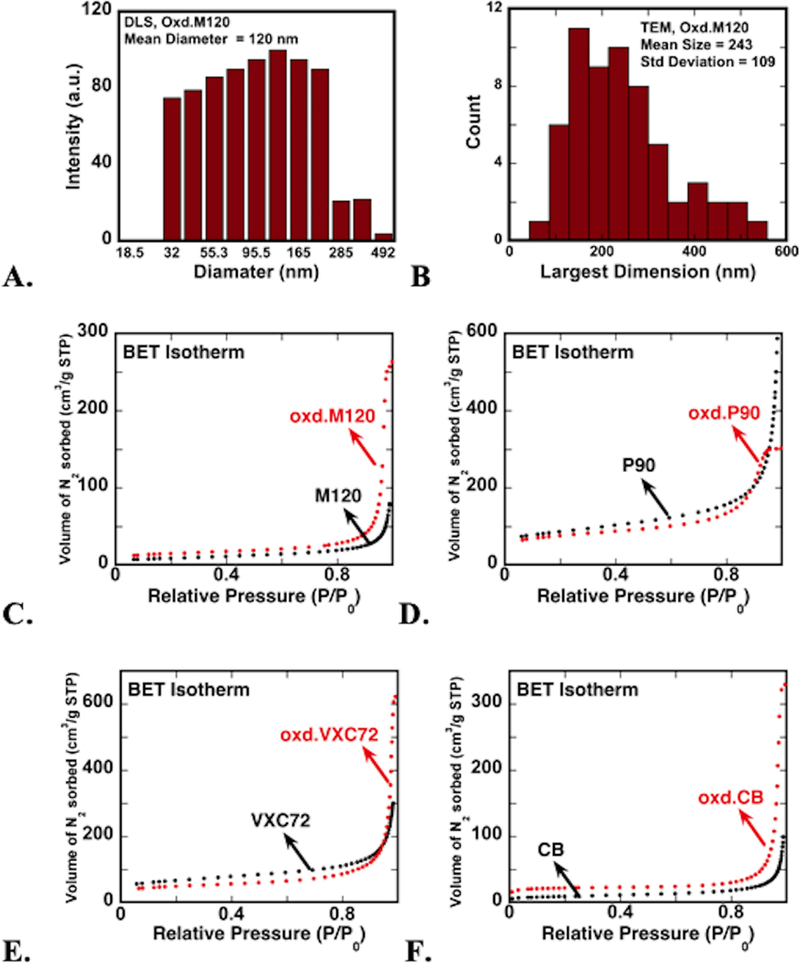
a. size distribution of oxidized m120 cnp dispersed in deionized water from dls measurement. b. distribution of largest dimension of oxidized m120 aggregates measured from tem images. c-f. nitrogen gas adsorption isotherms (at 77 k) of carbon nanoparticles before (black dots) and after (red dots) particle surface oxidation for four synthetic cnp (see table 1). see table s1 for numerical results of isotherm analysis.
Changes in surface area and microporosity of CNP after oxidation were examined from N2 adsorption measurements. Table 1 summarizes the surface properties determined by fitting N2 adsorption data to BET and BJH models. Both oxidized and untreated CNP resulted in type II-like isotherms corresponding to a nonporous surface (Figure 1.C-F).40 The isotherm data suggests that overall surface properties of the particles were not affected strongly by oxidation, but changes in specific surface area and pore dimensions were observed in BET and BJH results. The surface areas of M120 and CB CNP were increased from ~30 m2/g to ~50 m2/g after nitric acid oxidation, probably due to the disintegration of aggregated and agglomerated particles. This increase corresponded with an increase in average pore width from ~30 nm to ~40 nm (SI Table S1). In contrast, the surface areas of P90 (~300 m2/g) and VXC72 CNP (~230 m2/g) decreased after oxidation (265 and 170 m2/g, respectively). The surface area decrease was probably due to a reduction of average pore width from 17 to 11 nm for P90 particles after oxidation. Interestingly, the average pore width of VXC72 particles showed a three-times increase after oxidation (from 12 to 36 nm). In VXC72, the particles are vulcanized carbon particles, hence the increase in pore size can be attributed to the loss of sulfur from the carbon surface by nitric acid oxidation. The decrease in surface area may be attributed to the loss of free space (intraparticle-voids) in the agglomerate after particle dispersion during oxidation. Analyses of N2 adsorption showed that CNP surfaces lack microporosity and therefore reaction with iron solutions would coat iron onto the surfaces, rather than trapping it in pores.
Particle Size and Morphology of CNP
Transmission electron microscopy (TEM) images of all unoxidized particles showed highly aggregated μm-sized clusters, whereas oxidized CNP were discrete aggregates of particles about 100–300 nm in size (Figure 2). The size and shape of the primary particles did not change after oxidative treatment. The statistical distribution of aggregate size (largest dimension was measured) was estimated to be 250(±110) nm from TEM images (Figure 1.B). The aggregate size of dried particles is expected to be smaller than the hydrated aggregate particle size measured from DLS experiments. The size distribution of oxidized CNP was below ~500 nm, which is in the size range of ambient CNP observed in microscopy images in other studies.13
Figure 2.

a-d. representative tem images of the four different cnp after surface oxidation showing primary spheres (~15–50 nm) merged to form submicron-size aggregates.
Surface Chemistry of Oxidized CNP
Among the four CNP types, M120 showed homogenous 30–50 nm spherical primary spheres (low neck formation) in the TEM images and was selected for in-depth characterization of surface chemistry. Particle imaging with STXM and C and O K-edge XANES was used to identify the functional group distribution on carbon surfaces before and after oxidation. Significant differences were observed in C XANES spectra in STXM studies after CNP oxidation. The particles showed the highest contrast at the absorption energy for C (285.2 eV, Figure 3.A & B) compared to the pre-absorbing energy range, confirming the location of carbon-rich areas. and XANES from 270 to 320 eV showed characteristic absorption features corresponding to C bonds. Oxidized M120 showed spectral signatures for aromatic carbon (sp2) at 285.5 eV, carboxylic acid or carboxylate groups at 288.5 eV and carbon σ* resonance at about 295 eV (Figure 3.C). Unoxidized M120 particles showed aromatic carbon resonance at 285.2 eV, similar to the oxidized CNP, but the carboxylic acid absorbance feature was absent. Characteristic absorption peaks for alcohol, phenols, and ketones at 286.5–286.7 eV41 were not observed in the XANES and suggest that the surface is enriched mostly by carboxylic groups, although a minor fraction of other functional groups cannot be ruled out. Note that the aromatic C absorbance (at 285 eV) shifted about 0.2 eV higher after oxidation, indicating an increase in sp3 carbon bonds.42 To further confirm the oxygenation of CNP, O K-edge (1s) XANES spectra exhibited peaks at 531.5 eV (C=O), and 540 eV (σ* resonance) associated with oxygen functional groups (Figure 3.D). Poor absorption of unoxidized particles at O K-edge was also noticed. A cluster analysis of the STXM data of oxidized M120 CNP at the C K-edge was used to spatially identify regions of similar absorbance intensity, based on the eigen score of the locations from the PCA analysis. Both strongly absorbing regions (Figure 3.E, blue area) and regions of lower absorbance (Figure 3.E, green region) had identical spectral absorbance features (Figure 3.F) implying a uniform chemical distribution at the sub-micrometer level. A weak spectrum from the red region is due to the averaging of background pixels and weakly absorbing particle clusters in this region during clustering of pixels with eigen scores within specific limits (red is the minimum score region). Similarly, C XANES of oxidized P90 particles showed spectra similar to oxidized M120, suggesting that the oxidation procedure had the same effect on different types of CNP (Figure 3 G & H).
Figure 3.
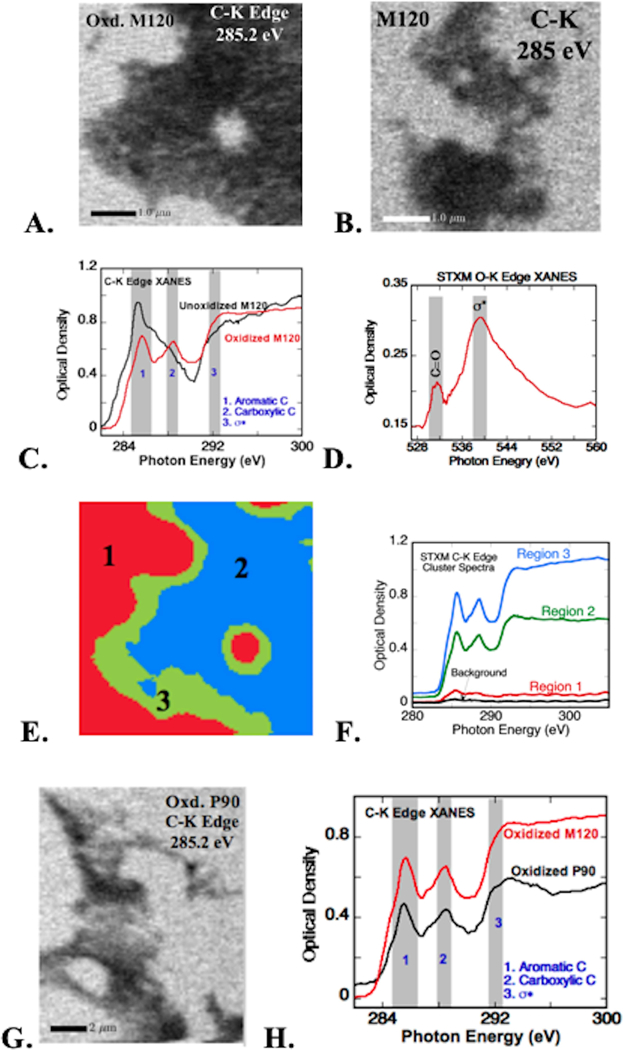
stxm images of a. oxidized m120 cnp and b. unoxidized (pristine) m120 at the carbon k absorption edge. c. carbon k-edge xanes before and after oxidation of m120 cnp (spectra collected from absorbing (dark) regions in a and b); shaded bands indicate the energy range of c absorption for aromatic and carboxylic acid groups, and ω* resonance. d. oxygen xanes collected at the dark region in a show features of c=o (531.5 ev) and ω* resonances (540 ev). e. cluster analysis of the stxm image in a showing spectroscopically distinguishable regions. f. carbon xanes spectra corresponding to the clustered regions in e (color in the image matches with spectra). g. stxm image of oxidized printex 90 (p90) cnp at c-k absorption edge. h. carbon xanes of oxidized p90 from absorbing regions in g; spectral ranges same as those marked in b.
In support of the STXM analyses, STEM images and electron energy-loss spectra (STEM-EELS) of the C K-edge were collected from CNP (Figure 4). EELS of oxidized M120 CNP showed strong π* and σ* resonance attributed to sp2 carbons, but the energy resolution of EELS was lower than that of STXM and spectral features were not as well resolved (Figure 4B). Elemental carbon and oxygen maps (from STEM-EDS) of oxidized M120 particles confirmed surface oxidation at an individual particle level (Figure 4C). Quantitative analysis of STEM-EDS maps showed 80–85% C and 13–18% O as major components. Therefore, both STXM and STEM-EDS data confirmed the generation of oxygen functional groups on CNP surfaces after nitric acid treatment.
Figure 4.
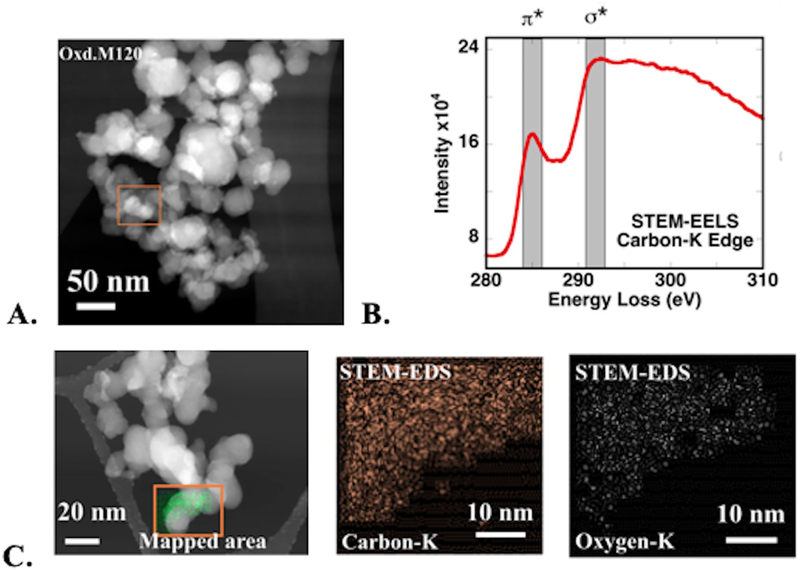
a. stem image of oxidized m120 cnp. b. carbon eels from the box marked in a; shaded bands show the energy range for π* and * resonances. c. stem-eds fluorescence maps showing the distribution of c and o atoms in the box marked in the stem image.
Iron Adsorption on CNP Surfaces
The ability of oxidized CNP surfaces to adsorb Fe(III) was examined in bulk uptake experiments for all four CNP samples. The amount of iron coated on oxidized CNP was controlled by changing the solution concentration of Fe(III) ([Fe]) and pH (Figure 5). The amount of adsorbed iron measured on CNP was proportional to the solution concentration, indicating a high surface affinity for Fe(III). For comparison, Fe(III) adsorption to unoxidized M120 CNP (SI Figure S1A) at low concentrations were performed, which shows similar adsorption behavior at low concentrations (>50 μM, but tend to saturate at about 6 μmol/m2. Iron sorption was linear with respect to solution concentration and about ~95% of iron in solution was adsorbed by all CNP types up to 200 μM [Fe]. Experiments with oxidized M120 CNP at higher solution iron concentrations (500 μM and 1000 μM), showed non-linearity with respect to surface concentration and about 65% of iron in solution was adsorbed to CNP at 1000 μM [Fe] (Figure 5). The adsorption data for M120/Fe was fitted to Freundlich adsorption model (Figure S1B). Langmuir model fits showed linear behavior, suggesting was the adsorption behavior is well below the plateaus.
Figure 5.
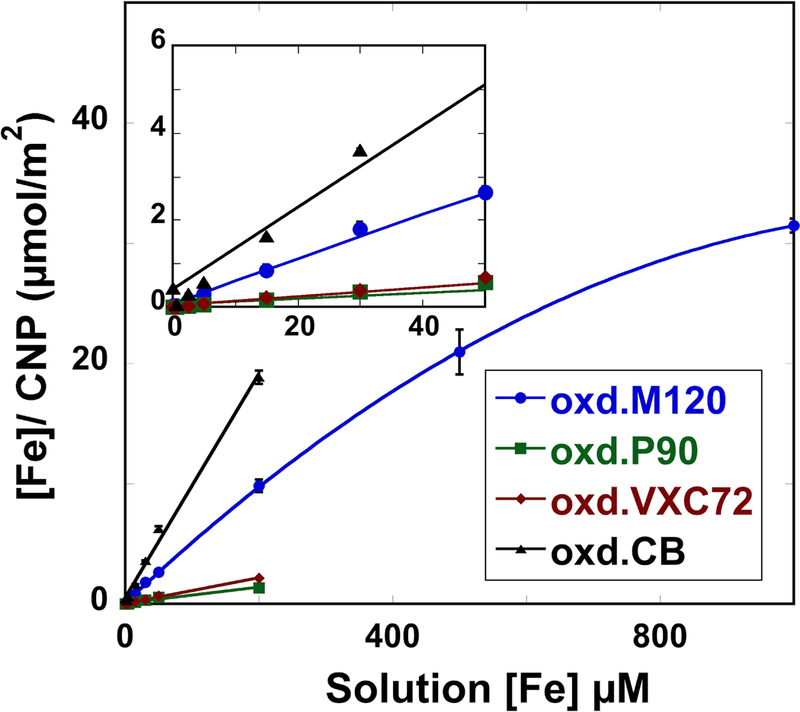
total iron concentration ([fe]) of fe-reacted carbon nanoparticles (cnp) compared to total [fe] measured in reactant solutions for all four cnp at low solution concentrations (inset) up to [fe] = 200 μm; oxidized m120 particles were also studied at [fe] = 500 μm and 1000 μm. iron associated with cnp was measured by particle digestion and fe mass was normalized to bulk particle bet surface area. iron concentrations of uncoated cnp were below detection. inset shows sorption at low fe(iii) concentrations.
Examination by STEM of one sample after iron coating at a surface coverage of 10 μmol / m2 (M120–10Fe) showed that particle morphology did not change compared with uncoated particles (Figure 6). Elemental mapping by STEM/EDS showed the presence of trace iron on oxidized CNP particles, although the amount was too low for quantitative analysis (Figure 6B). These observations suggest that Fe was dispersed on CNP surfaces and did not substantially change CNP bulk surface properties.
Figure 6.

oxidized and fe-coated cnp sample m120/10fe. tem bright-field images. b. stem dark-field image and elemental maps of c, o, and fe from eds.
Speciation of Iron on CNP Surfaces
Iron L-edge EELS and XANES.
Oxidized M120 CNP was selected to examine the chemical speciation and bonding of iron as a function of surface loading by analysis of both Fe L-edge (STEM-EELS and STXM-XANES) and K-edge (synchrotron XAS) spectra. Spatially resolved STEM-EELS and STXM-XANES studies were performed for the sample at 10 μmol/m2 Fe/CNP (M120–10Fe) surface coverage. Typically, iron L-edge EELS shows two energy loss peaks (L3 at ~710 eV and L2 at ~720 eV) that are an indicator of the oxidation state of iron. Compared with reference standards of known oxidation state using EELS, the energy position and intensity of Fe L-edge features of M120/200Fe were similar to hematite (α-Fe2O3) and HFO (am-Fe(OH)3) spectra acquired under similar conditions, indicative of Fe(III)-oxygen bonding, and different from reference compound spectra containing Fe(II) and Fe(0) (Figure 7A). Analysis of M120/200Fe using STXM showed the presence of iron on CNP surfaces using the difference in absorption below and above the Fe L-edge (Figure 7B). Compared with the same reference compounds used for EELS, the STXM-XANES spectrum of the sample was more similar to HFO and hematite spectra in the energy and intensity of the absorption peaks compared with the peak position and shape of Fe(II) and Fe(0) reference compound spectra, which were considerably different (Figure 7B). Note that the spatial resolution of STXM is much lower (~30 nm), and that the XANES spectrum was averaged over a greater area (~6 × 6 μm), compared with STEM-EELS (~0.5 nm resolution, spectrum over ~20 nm area). However, the energy resolution of L-edge spectra was higher for STXM (~0.2 eV) than for EELS (~0.5 eV), and spectra showed fine-scale absorption features that were not resolved in EELS. Both sets of data, with different spatial and energy resolution, indicated that Fe on CNP surfaces maintained its Fe(III) oxidation state and was most similar to Fe(III) coordinated by oxygen atoms.
Figure 7.
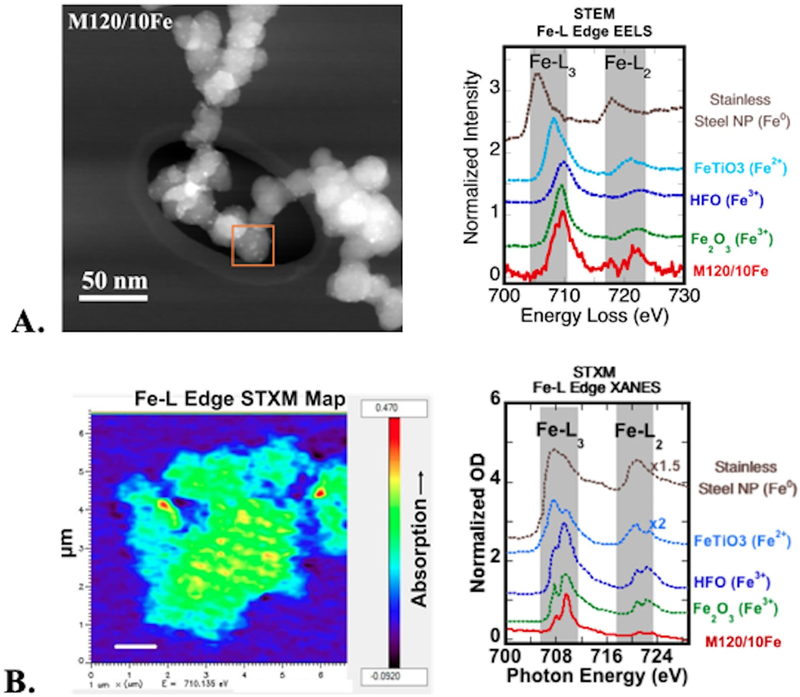
oxidized and fe-coated cnp sample m120/10fe. a. stem-dark field image and corresponding fe l-edge eels compared with fe reference compounds of different oxidation state. b. stxm fe l-edge energy difference map (pre-edge map at 700 ev subtracted from maximum absorption map at 710 ev). high-absorbing regions appear in red. iron l-edge xanes spectra from the map averaged over the whole aggregate, compared with the same fe reference compounds used for eels.
Iron K-edge XANES and EXAFS.
To further investigate the coordination environment of Fe on CNP surfaces, Fe K-edge XAS, including XANES and quantitative EXAFS analysis, was done on samples over a range of iron surface coverages. The chemical environment of adsorbed iron was compared to that of three representative Fe(III) reference compounds with bonding environments similar to that of the CNP/Fe samples (Table 2). At surface coverages of 3 and 10 μmol/m2 (M120–3Fe and M120–10Fe), bulk XANES were nearly identical, indicating a similar coordination environment (Figure 8A). For M120–10Fe, the bulk XANES spectrum was compared with microfocused XANES collected with a 2 μm spot size. Both were identical, indicating homogeneity of the microscale chemical environment of Fe coating on CNP. Moreover, the XANES features were similar to that of the reference compound Fe(acac)3. As the iron coating increased to ~21 μmol/m2 (M120–21Fe), XANES features showed noticeable differences from that of M120–10Fe, indicating a change in the coordination environment of iron. The XANES features were nearly identical for the next higher iron coating, ~31 μmol/m2 (M120–31Fe). Both M120–21Fe and M120–31Fe XANES showed a shoulder peak ~7150 eV similar to the spectrum of HFO, which is indicative of iron (hydr)oxide formation (Figure 8A). Similar to Fe spectra at lower surface coverage, microfocused XANES of M120–31Fe was identical to the bulk spectrum.
Table 2.
Fit parameters of Fe K-edge EXAFS over k = 2–11 Å−1.
| EXAFS Fit Results | Crystal Structure Data [g] |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reference Spectra | Path | N[a] | R (Å) [b] |
σ2 (Å2) [c] |
Eo [d] (eV) |
R- Factor[e] |
Red. γ2 [f] |
N | R (Å) |
| Iron(III) acetylacetonate | Fe-O | 6 | 2.00 | 0.0040 | 3.62 | 0.0208 | 481 | 6 | 1.992 |
| Fe-C | 6 | 2.98 | 0.0035 | 4 2 |
2.945 2.970 |
||||
| Fe-C | 3 | 3.28 | 0.0035 | 2 1 |
3.308 3.358 |
||||
| Fe-C | 7 | 4.40 | 0.0033 | 5 2 |
4.299 4.328 |
||||
| Fe-O-C | 8 | 4.32 | 0.0090 | 8 | 4.318 | ||||
| Potassium iron (III) oxalate | Fe-O | 6 | 2.02 | 0.0061 | 3.16 | 0.0512 | 212 | 6 | 2.023 |
| Fe-C | 6 | 2.83 | 0.0088 | 6 | 2.832 | ||||
| Fe-K | 2 | 3.88 | 0.0013 | 2 | 3.770 | ||||
| Fe-O | 6 | 3.91 | 0.0090 | 6 | 4.026 | ||||
| Fe-C-O | 12 | 4.14 | 0.0120 | 12 | 4.040 | ||||
| Hydrous ferric oxide (HFO) | Fe-O | 6 | 1.98 | 0.0131 | 0.00 | 0.0277 | 40.7 | ||
| Fe-Fe | 2.5 | 3.04 | 0.0128 | ||||||
| Fe-Fe | 1.6 | 3.45 | 0.0096 | ||||||
| Fe-coated M120 | Path | N[a] | R (Å) [b] | σ2 (Å2) [c] | Eo [d] (eV) | R-Factor[e] | Red. γ2[f] | ||
| 3 μmol Fe/m2 | Fe-O | 6 | 1.99 | 0.0077 | 2.00 | 0.0308 | 83.8 | ||
| Fe-C | 1.3 | 2.92 | 0.0035 | ||||||
|
10 μmol Fe/m2 |
Fe-O | 6 | 2.01 | 0.0074 | 3.62 | 0.0184 | 27.1 | ||
| Fe-C | 1.9 | 2.99 | 0.0035 | ||||||
| Fe-C | 3.5 | 4.36 | 0.0033 | ||||||
| 21 μmol Fe/m2 | Fe-O | 3 | 1.95 | 0.0117 | 0.747 | 0.0989 | 161 | ||
| Fe-O | 3 | 2.02 | 0.0117 | ||||||
| Fe-C | 1.2 | 2.77 | 0.0070 | ||||||
| Fe-Fe | 1.8 | 3.02 | 0.0128 | ||||||
| Fe-Fe | 1.2 | 3.42 | 0.0096 | ||||||
| 31 μmol Fe/m2 | Fe-O | 6 | 1.98 | 0.0144 | 0.00 | 0.0454 | 117 | ||
| Fe-Fe | 2.2 | 3.03 | 0.0128 | ||||||
| Fe-Fe | 1.3 | 3.43 | 0.0096 | ||||||
Effective coordination number for single-scattering path; fixed for all paths in reference compounds; for CNP samples, fixed at N=6 for all first shell Fe-O and varied for all other paths. For triangular multiple-scattering paths (Fe-C-O), N refers to the total number of scattering legs (= 3 here) that sum to distance R in the structure.
Interatomic distance of scattering path; varied for all the paths.
Debye-Waller parameter; varied for all paths in reference compounds (with N fixed); for CNP samples, varied for first shell Fe-O paths (N=6) and fixed on values determined from reference compounds for all other the paths.
Difference in Fermi energy level (k = 0 initially set to 7125 eV); varied as a single parameter for all shells.
R-factor is the normalized sum of the squared residuals of the fit (R = ∑(data-fit)2 / ∑data2); represents the relative error of the fit and data.
Reduced γ2 is a statistical goodness-of-fit parameter equal to (F-factor) / ( # of points - # of variables).
Figure 8.
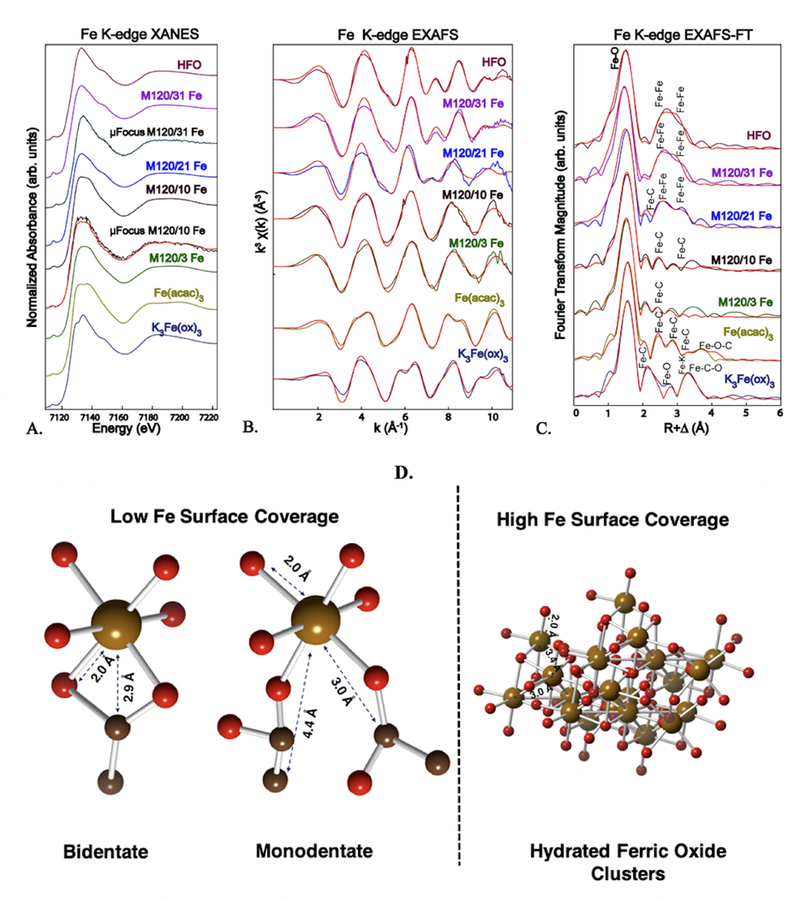
iron k-edge x-ray absorption spectra of oxidized and fe-coated cnp (m120) as a function of fe surface coverage compared with fe(iii) reference spectra (hydrous ferric oxide: hfo; fe(iii) acetylacetonate: fe(acac)3; potassium fe(iii) oxalate: k3fe(ox)3. a. normalized xanes bulk spectra and two microfocused spectra (μfocus, collected with a 2 μm spot size in black and smoothened spectrum in red) for comparison. b. k3-weighed exafs spectra and shell-by-shell best fits (in red). c. fourier transforms of exafs shown in b. d. chemical bonding of fe at low and high surface coverages based exafs fit results (given in table 1). atoms: fe: tan; o: red; c: black.
To evaluate the chemical structural environment of iron on CNP, bulk EXAFS were analyzed for bond distances (R) and coordination numbers (N) in Fe/CNP samples and reference compounds. The reference compounds K3Fe(ox)3 and Fe(acac)3 are monomeric octahedral Fe(III) complexes with Fe-O bond distances at ~2.0 Å (Table 2). Both complexes have three bidentate dicarbonyl ligands bonding with the Fe center. Due to the vicinal carboxylate groups in the oxalate ligand, 5-membered rings are formed with Fe in K3Fe(ox)3 (SI Figure S2B). The acetylacetonate ligand has a carbon atom (-CH2-) separating the carbonyl groups, leading to a 6-membered ring geometry with the Fe center (SI Figure S2A). Fits to the experimental EXAFS spectra were consistent with distances calculated from their known crystal structures (Figure 8; EXAFS and FT spectra of the model compounds with deconvoluted paths are presented in SI Figure S2.). Second-shell Fe-C atoms in Fe(acac)3 were fit at a longer R than K3Fe(ox)3 due to the expansion of the cyclic structure from a 5- to a 6- membered ring (Table 1). In Fe(acac)3, Fe-C backscattering from the –CH2 group separating the carbonyl groups was fit at R=3.28 Å (N=3), and this path was absent in K3Fe(ox)3, as expected. Due to the cyclic geometries, multiple-scattering paths were present in both complexes that corresponded to Fe-C-O or Fe-O-C triangular paths. The EXAFS spectrum of HFO (amorphous) was fit using Fe-O and Fe-Fe paths taken from crystalline goethite (α-FeO(OH)),43 following prior studies.36,37 The HFO EXAFS spectrum was fit assuming octahedral coordination by oxygen atoms (R = 1.98 Å) and two shells of more distant Fe atoms (Table 2).
The spectral similarities and differences noted in the XANES region of the spectra are reflected in EXAFS. The EXAFS spectra of Fe/CNP were dominated by the Fe-O scattering of the first coordination shell, with fitted interatomic Fe-O ranging from R = 1.93 to 2.01 Å (SI Figure S3, Table 2), which is consistent with octahedral coordination of Fe(III) by oxygen atoms for all samples. Resolving the second shell is critical to identifying differences in bonding of Fe on CNP surfaces. At the lowest Fe coating (3 μmol/m2), C backscattering atoms were fit at average R = 2.92 Å and N = 1.3. At the next highest Fe loading (~10 μmol/m2), C atoms were fit at average R = 2.99 Å and N = 1.86. In addition, a third shell of C atoms was fit at R = 4.36 Å and N = 3.5. For these two Fe/CNP spectra at lower Fe surface coatings, the atom geometries around Fe were more similar to that of Fe(acac)3 than to K3Fe(ox)3. In particular, Fe-C backscattering was fit at R = 2.92 and 2.99 Å compared with R = 2.98 Å for Fe(acac)3. As the Fe loading increased (21 μmol/m2), two shells of second-neighbor Fe-Fe backscattering were observed in addition to Fe-C backscattering (Figure 8, Table 2). This spectrum has both Fe(acac)3 and HFO signatures, but the strong Fe-Fe backscattering dominates over Fe-C backscattering. At the highest Fe surface coverage (31 μmol/m2), the EXAFS spectrum was fit with only second-neighbor Fe-Fe backscattering shells and fit results were very similar to those determined for the HFO reference spectrum. The SEM images of M120/31Fe show clusters or islands of Fe coating on CNP at nanoscale resolution (SI Figure S4).
Discussion
Morphology and Surface Properties of CNP
Reactions on carbon surfaces in the atmosphere create functional groups that change their physio-chemical properties.12 Highly reactive oxidative radicals in the gas phase generate carboxylic acid or carboxylate, carbonyl, and hydroxyl groups on carbon surfaces.44 In this study, oxidation of CNP was simulated using a chemical oxidant in solution. Strong oxidation with nitric acid did not affect the bulk surface properties of the CNP to a large extent. Evidence for nanopores was not observed in electron micrographs, which is consist with the type-II isotherms (nonporous surfaces) determined from N2 adsorption. Among the four types of CNP, both increases and decreases in bulk surface area were observed. Increases in available surface area after de-aggregation increased the measured surface area of CNP, but decreases in void space with decrease in aggregate size caused reduction in total surface area.45 When primary particle size was 30–100 nm, bulk surface area increased after oxidation, and when individual particle size was smaller (15–30 nm), reduction of voids decreased the overall surface area due to less particle aggregation. TEM images confirmed that after oxidation, the aggregates have fewer numbers of particles and voids compared to unoxidized CNP. Also, no primary particle porosity was observed in HR-TEM images, so the observed pore widths must come from the voids.
Primary spheroids (20–50 nm) of CNP aggregate to form coalesced particles that are either physically bound or merged (neck formation). This characteristic shape is referred as the ‘aciniform’ morphology that resembles ‘a cluster of grapes’.46 The introduction of ionizable, charged carboxylic acid groups reduced inner particle interactions and thereby reduced agglomeration. Hydrogen bonding with water molecules facilitated the solvation of the surface and resulted in a highly stable aqueous CNP dispersion with 100–300 nm aggregate size. An oxidation-induced, 6–7 days long transformation from a hydrophobic to hydrophilic surface was previously noted for ambient CNP.42 This slow transition suggests that the initial atmospheric oxidation process does not appreciably improve the wettability of the particles and as oxidation progresses, highly polar, negatively charged carboxylate groups (at neutral pH) are generated.42
As shown by STXM and C XANES, carboxylic acid or carboxylate groups dominated on CNP surfaces after nitric acid treatment and followed by the removal of the acid residues, compared with unoxidized CNP spectra that showed only a strong aromatic carbon resonance absorption. Other carbonyl or hydroxyl groups were not detected from C XANES after oxidation. Atmospherically aged black carbon particles likely have diverse surface functional groups that vary with the aging time, but prior studies of particles collected from different locations showed that particle exteriors were enriched with carboxylic acid or carboxylate groups.13,47 This could be due to the secondary oxidation of aldehyde and hydroxyl to carboxylic acid.42 The polarity of surface carboxylate groups on CNP helped to disperse CNP in aqueous solutions (near neutral pH) and maintain particle dispersion. Nitric acid oxidation treatment of engineered graphitic surfaces are routinely done for toxicity assays of carbon nanotubes and graphene.48 Similar acid oxidation treatments can be easily executed to generate model CNP with carboxylate as the dominant type of surface functional group for toxicity assessments.
Chemical Environment of Iron on CNP Surfaces
Carbon nanoparticles are a key transporter of iron in the air, where the association of iron occurs either during its formation from biomass or fossil fuels, or by association with iron-bearing particles and chemical alteration in a hydrated atmosphere.49 The chemical environment of iron on CNP surfaces can be changed by repeated wetting and drying, and by the presence of acids in which iron solubility is higher.50 Global iron concentrations in air vary from 0.001 to 10 μg/m3.51 Considering an average PM concentration of 100 μg/m3, average iron can be up to 10% of the total PM. The maximum iron surface coverage (M120/31Fe) in our experiments was about 3% by weight of CNP and therefore representative of average iron levels in PM. In this study, iron deposition onto CNP from solution and drying mimicked hydrated atmospheric processing from acid to near-neutral pH and provided information about the affinity of abundant and soluble Fe(III) for CNP surfaces. In PM, organic molecules are typically complex mixtures of different compounds with surface carboxylate groups originating from combustion and secondary oxidation processes,52,53 which provide sorption sites for iron. In our model system, Fe L- and K- edge absorption spectra (XANES and EELS) at different spatial resolutions demonstrated that surface iron maintained its Fe(III) oxidation state (at least 95% of total Fe)39 and was not reduced to lower oxidation states (see SI Figure S5). Mapping by STEM-EDS showed weak and dispersed iron fluorescence on particle surfaces. However, the species of Fe(III) on CNP was found to be dependent on the iron surface concentration (μmoles of Fe per unit area of CNP). At low Fe loadings (3 and 10 μmol/m2), Fe(III) existed mostly as a mononuclear coordinated complex with surface carbon atoms at an average Fe-C distance of ~2.9 Å. This Fe-C distance suggests that chelating carboxylate groups on average were not forming a 5-membered ring geometry with Fe(III) as seen in K3Fe(ox)3 (Fe-C, R = 2.83 Å). To form a such pentagonal geometry with Fe(III) in the complex, carboxylate groups must be on two neighboring carbon atoms (vicinal), which is less probable on randomly oxidized CNP surfaces. The observed Fe-C distance was closer to that of Fe(acac)3, where Fe(III) was part of a 6-membered ring geometry (Figure 8). Multiple scattering paths were not prominent in the sample spectra, contrary to the model systems, which suggests that the carboxylate ligands form dangling, monodentate bonds with Fe(III). To complete the octahedron, the rest of the oxygen atoms must be donated by water or hydroxide ions (Figure 8D). Absence of Fe-Fe backscattering in the EXAFS at low iron surface coverages rules out the formation of polynuclear Fe-complexes or nanoparticles. The average number of backscattering C atoms at ~2.9 Å was 1.3 to 1.9. Therefore, a plausible geometry of Fe(III) on CNP is a mononuclear Fe(III) complex coordinated to O atoms from a combination of monodentate and bidentate carboxylate ligands on CNP. The affinity of carboxylate groups for Fe(III) is predictable from hard-soft acid base theory (HSAB), where Fe(III) is listed as hard acid and carboxylate (–COO-) is a hard base. If reduction of ferric to ferrous iron occurs, the interactions could be weaker because Fe(II) is a moderate acid species in HSAB scale. Photo-reduction of Fe(III) to Fe(II) complexed to aqueous low molecular weight organic matter, coupled with oxalate or malonate radical formation, are also known, where iron (hydro)oxides were unreactive.54 Such photochemical conversion is possible in complexed Fe(III) in CNP, which may increase the availability of reactive Fe(II) in carbon-rich PM.52
At higher surface loadings on CNP, iron formed structures that were very similar to amorphous HFO. Iron hydroxide-like particles were observed on CNP surfaces at 21–31 μmol Fe/m2 surface coverage at neutral pH, in addition to evidence for Fe mononuclear complexes. These surface loadings correspond to solution iron concentrations (500 μM and 1000 μM) in which uptake behavior from solution changed from linear to non-linear with increasing concentration in sorption experiments (Figure 5). The transition from surface-coordinated mononuclear Fe(III) to Fe(III) hydroxide clusters with higher iron surface loading implies that the chemical environment of Fe(III) on CNP depends on the number and proximity of available carboxylate ligands on the surface. When surface sites are available on oxidized CNP, the high affinity of iron for electron-donating oxygen functional groups is favorable over iron oxide formation. When high affinity coordination sites are filled with Fe, the coordinated Fe sites may act as nucleation centers on CNP, and iron hydroxide or oxyhydroxide clusters are formed. A similar mechanism was previously discussed for Fe(III) chelation to dissolved organic matter (DOM) in solution, where at lower Fe(III) concentrations insoluble complexes were formed, whereas at a higher Fe(III) concentration, a mixture of a coordinated phase and oxyhydroxides were observed55
Studies of airborne particulate matter (PM) show that iron is present mostly as Fe(III) and predominately as an oxide in non-urban and non-industrial settings. However, the identity and relative abundance of specific mineral or amorphous phases varies considerably among studies depending on the method of characterization, particle size fraction, and mixture of PM sources. Crystalline iron oxide phases such as goethite (α-FeO(OH)) and hematite (α-Fe2O3), and iron structurally substituted in phyllosilicate minerals, are most often associated with geogenic PM, particularly in larger size fractions.56,57 Iron from combustion sources, including coal, transportation exhaust, biofuels, biomass, and industrial incineration is assumed to be associated with carbonaceous materials or with silica in the case of coal combustion products.58,59 Non-combustion iron sources include tire, brake, and road, wear and metal abrasion products, which may include Fe(0) and various Fe(II) or Fe(III) oxides and sulfates.47,60 Thus, the form of iron on particle surfaces may vary from chelated Fe(III) to particulate Fe oxide, which are likely to have different cellular and inflammatory responses in biological studies.
Conclusions
Chemical oxidation and adsorption of iron on carbon nanoparticle (CNP) surfaces to simulate hydrated atmospheric processes showed oxidation of sp2 carbon atoms and formation of surface carboxylate functional groups for Fe(III) binding and nucleation. Detailed spectroscopic investigations revealed that Fe(III) was bonded to oxidized CNP surfaces and maintained its 3+ oxidation state. Analyses of EXAFS spectra showed that the local bonding environment of Fe(III) depended on surface coverage on CNP. At lower loadings, mononuclear Fe(III) complexes were preferentially formed. As the concentration of Fe(III) on CNP increased, Fe clusters similar in local structure to amorphous hydrous ferric oxide (HFO) were formed. The driving force for a transition from mononuclear Fe(III) to iron hydroxide could be the high affinity of Fe(III) to electron-donating oxygens from CNP surfaces, compared to edge- and corner-shared bonding of Fe(III) octahedra found in HFO. Thus, the nature of Fe associated with atmospherically processed particles can vary with the degree of surface oxidation of carbon and surface concentration of soluble iron. Other factors that may influence the speciation of Fe on CNP are the chemical composition of the iron source, temperature, presence of other chemical species (metals, cations, anions, organic molecules), UV irradiation, aging time, and pH conditions in the atmosphere.
The surface properties of CNP significantly affect their aggregation size, degree of hydrophobicity, and ability to bind inorganic species and organic compounds to their surfaces. The form of surface iron associated with CNP can lead to either coordinated Fe(III) or a hydrous ferric oxide phase, each of which is likely to have a different biochemical response in toxicity studies. Previous studies using THP-1 macrophages showed that Fe on the surface of silica induced lipid peroxidation and disruption, increased cytokine production (TNF-α and IL-1β), and subsequently elevated phase II gene expression.61 In order to link specific biochemical signaling pathways to molecular scale mechanisms, investigation of the response of macrophages and other cells to different chemical forms of Fe on the same nanoparticle surface should be examined. Engineered nanoparticles with controlled physiochemical properties and known chemical speciation are needed to establish these structure-activity relationships in biological response assessments of environmentally relevant particles.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grant ES023864. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. Thanks to Paul Aronstein, Dr. Godwin Nwosu, Matthew Robinson and Benjamin Lash for assistance with data collection and analysis. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02–05CH11231. Special thanks to Dr. Adam Hitchcock’s group for their generous help at ALS. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231. Part of the STXM data were acquired at CLS beamline10ID-1 at the CLS, which is supported by the NSERC, the CIHR, the NRC, and the University of Saskatchewan. Special thanks to Dr. Jian Wang for his support and suggestions at the CLS. We thank the UC Merced IMF and Mike Dunlap for the support in TEM studies. We acknowledge the UC Merced EAL and Liying Zhao for ICP-OES analysis. We extend our gratitude to Prof. Wei-Chun Chin for letting us use the DLS facility.
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
References
- 1.Fuzzi S, Baltensperger U, Carslaw K, Decesari S, Denier H Gon Van Der, Facchini M, Fowler D, Koren I, Langford B, Lohmann U, Nemitz E, Pandis S, Riipinen I, Rudich Y, Schaap M, Slowik JG, Spracklen DV, Vignati E, Wild M, Williams M, and Gilardoni S, Particulate matter, air quality and climate: lessons learned and future needs, Atmospheric Chem. Phys, 2015, 15, 8217–8299. [Google Scholar]
- 2.Seaton A, Godden D, MacNee W and Donaldson K, Particulate air pollution and acute health effects, Lancet, 1995, 345, 176–178. [DOI] [PubMed] [Google Scholar]
- 3.Pope CA III and Dockery DW, Health effects of fine particulate air pollution: lines that connect, J. Air Waste Manag. Assoc., 2006, 56, 709–742. [DOI] [PubMed] [Google Scholar]
- 4.Kim KH, Kabir E and Kabir S, A review on the human health impact of airborne particulate matter, Environ. Int, 2015, 74, 136–143. [DOI] [PubMed] [Google Scholar]
- 5.Oberdörster G, Celein RM, Ferin J and Weiss B, Association of particulate air pollution and acute mortality: Involvement of ultrafine particles? Inhal. Toxicol., 1995, 7, 111–124. [DOI] [PubMed] [Google Scholar]
- 6.Nel A, Xia T, Mädler L and Li N, Toxic potential of materials at the nanolevel, Science, 2006, 311, 622–627. [DOI] [PubMed] [Google Scholar]
- 7.Sharifi S, Behzadi S, Laurent S, Forrest ML, Stroeve P and Mahmoudi M, Toxicity of nanomaterials, Chem. Soc. Rev, 2012, 41, 2323–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar P, Morawska L, Birmili W, Paasonen P, Hu M, Kulmala M, Harrison RM, Norford L and Britter R, Ultrafine particles in cities, Environ. Int. 2014, 66, 1–10. [DOI] [PubMed] [Google Scholar]
- 9.Kittelson DB, Engines and nanoparticles: a review, J. Aerosol Sci, 1998, 29, 575–588. [Google Scholar]
- 10.Li W, Wang T, Zhou S, Lee S, Huang Y, Gao Y and Wang W, Microscopic observation of metal-containing particles from Chinese continental outflow observed from a non-industrial site, Environ. Sci. Technol, 2013, 47, 9124–9131. [DOI] [PubMed] [Google Scholar]
- 11.Jonker MT and Koelmans AA, Extraction of polycyclic aromatic hydrocarbons from soot and sediment: solvent evaluation and implications for sorption mechanism, Environ. Sci. Technol, 2002, 36, 4107–4113. [DOI] [PubMed] [Google Scholar]
- 12.Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C and Nel A, Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity, Environ. Health Perspect, 2004, 112, 1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maria SF, Russell LM, Gilles MK and Myneni SC, Organic aerosol growth mechanisms and their climate-forcing implications, Science, 2004, 306, 1921–1924. [DOI] [PubMed] [Google Scholar]
- 14.Takahama S, Liu S and Russell L, Coatings and clusters of carboxylic acids in carbon‐containing atmospheric particles from spectromicroscopy and their implications for cloud‐nucleating and optical properties, J. Geophys. Res. Atmos., 2010, 115. [Google Scholar]
- 15.Moffet RC, Furutani H, Rödel TC, Henn TR, Sprau PO, Laskin A, Uematsu M and Gilles MK, Iron speciation and mixing in single aerosol particles from the Asian continental outflow, J. Geophys. Res. Atmos., 2012, 117. [Google Scholar]
- 16.Deboudt K, Gloter A, Mussi A and Flament P, Red‐ox speciation and mixing state of iron in individual African dust particles, J. Geophys. Res. Atmos, 2012, 117. [Google Scholar]
- 17.Johansen AM, Siefert RL and Hoffmann MR, Chemical composition of aerosols collected over the tropical North Atlantic Ocean, J. Geophys. Res. Atmos, 2000, 105, 15277–15312. [Google Scholar]
- 18.Sanderson P, Su S, Chang I, Saborit JD, Kepaptsoglou D, Weber R and Harrison RM, Characterisation of iron-rich atmospheric submicrometre particles in the roadside environment, Atmos. Environ, 2016, 140, 167–175. [Google Scholar]
- 19.Cartledge BT and Majestic BJ, Metal concentrations and soluble iron speciation in fine particulate matter from light rail activity in the Denver-Metropolitan area, Atmos. Pollut. Res, 2015, 6, 495–502. [Google Scholar]
- 20.Muxworthy A, Schmidbauer E and Petersen N, Magnetic properties and Mössbauer spectra of urban atmospheric particulate matter: a case study from Munich, Germany, Geophys. J. Int, 2002, 150, 558–570. [Google Scholar]
- 21.Wilson MR, Lightbody JH, Donaldson K, Sales J and Stone V, Interactions between ultrafine particles and transition metals in vivo and in vitro, Toxicol. Appl. Pharmacol, 2002, 184, 172–179. [DOI] [PubMed] [Google Scholar]
- 22.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts II LJ and Ischiropoulos H, Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations, Free Radic. Biol. Med, 2012, 52, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forman HJ and Finch CE, A critical review of assays for hazardous components of air pollution, Free Radic. Biol. Med, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldman WJ, Kristovich R, Knight DA and Dutta PK, Inflammatory properties of iron-containing carbon nanoparticles, Chem. Res. Toxicol, 2007, 20, 1149–1154. [DOI] [PubMed] [Google Scholar]
- 25.Premasekharan G, Nguyen K, Contreras J, Ramon V, Leppert VJ and Forman HJ, Iron-mediated lipid peroxidation and lipid raft disruption in low-dose silica-induced macrophage cytokine production, Free Radic. Biol. Med, 2011, 51, 1184–1194. [DOI] [PubMed] [Google Scholar]
- 26.Premshekharan G, Nguyen K, Zhang H, Forman HJ and Leppert VJ, Low dose inflammatory potential of silica particles in human-derived THP-1 macrophage cell culture studies–Mechanism and effects of particle size and iron, Chem. Biol. Interact, 2017, 272, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Visalli G, Facciolà A, Iannazzo D, Piperno A, Pistone A and Di Pietro A, The role of the iron catalyst in the toxicity of multi-walled carbon nanotubes (MWCNTs), J. Trace Elem. Med. Biol, 2017, 43, 153–160. [DOI] [PubMed] [Google Scholar]
- 28.Schwertmann U, and Cornell RM, Iron oxides in the laboratory: Preparation and characterization 2nd Edition, Wiley-VCH Editions, Weinhein, Germany, 2000. [Google Scholar]
- 29.Moreno-Castilla C, Ferro-Garcia M, Joly J, Bautista-Toledo I, Carrasco-Marin F and Rivera-Utrilla J, Activated carbon surface modifications by nitric acid, hydrogen peroxide, and ammonium peroxydisulfate treatments, Langmuir, 1995, 11, 4386–4392. [Google Scholar]
- 30.Ghio AJ, Kennedy TP, Whorton AR, Crumbliss AL, Hatch GE and Hoidal JR, Role of surface complexed iron in oxidant generation and lung inflammation induced by silicates, Am. J. Physiol. Lung Cell. Mol. Physiol, 1992, 263, L511–L518. [DOI] [PubMed] [Google Scholar]
- 31.Lepri FG, Borges DL, Araujo RG, Welz B, Wendler F, Krieg M and Becker-Ross H, Determination of heavy metals in activated charcoals and carbon black for Lyocell fiber production using direct solid sampling high-resolution continuum source graphite furnace atomic absorption and inductively coupled plasma optical emission spectrometry, Talanta, 2010, 81, 980–987. [DOI] [PubMed] [Google Scholar]
- 32.Egerton R and Malac M, Improved background-fitting algorithms for ionization edges in electron energy-loss spectra, Ultramicroscopy, 2002, 92, 47–56. [DOI] [PubMed] [Google Scholar]
- 33.Hitchcock A, Hitchcock P, Jacobsen C, Zimba C, Loo B, Rotenberg E, Denlinger J and Kneedler R, aXis 2000—Analysis of X-ray Images and Spectra, McMaster University: Hamilton, ON, Canada, 2012. [Google Scholar]
- 34.Lerotic M, Mak R, Wirick S, Meirer F and Jacobsen C, MANTiS: a program for the analysis of X-ray spectromicroscopy data, J. Synchrotron Radiat, 2014, 21, 1206–1212. [DOI] [PubMed] [Google Scholar]
- 35.Webb S, SIXpack: a graphical user interface for XAS analysis using IFEFFIT, Phys. Scr, 2005, 2005, 1011. [Google Scholar]
- 36.Ravel B and Newville M, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12, 537–541. [DOI] [PubMed] [Google Scholar]
- 37.Ravel B, 2001. EXAFS analysis with FEFF and FEFFIT. University of Washington 2, 31. [Google Scholar]
- 38.Root RA, Dixit S, Campbell KM, Jew AD, Hering JG and O’Day PA, Arsenic sequestration by sorption processes in high-iron sediments, Geochim. Cosmochim. Acta, 2007, 71, 5782–5803. [Google Scholar]
- 39.O’Day PA, Rivera N Jr, Root R and Carroll SA, X-ray absorption spectroscopic study of Fe reference compounds for the analysis of natural sediments, Am. Mineral, 2004, 89, 572–585. [Google Scholar]
- 40.Sing K, The use of nitrogen adsorption for the characterisation of porous materials, Colloids Surf. A, 2001, 187, 3–9. [Google Scholar]
- 41.Yabuta H, Uesugi M, Naraoka H, Ito M, Kilcoyne ALD, Sandford AA, Kitajima F, Mita H, Takano Y, Yada T, Karouji Y, Ishibashi Y, Okada T, Abe M, X-ray absorption near edge structure spectroscopic study of Hayabusa category 3 carbonaceous particles. Earth, Planets and Space 2014, 66, 156. [Google Scholar]
- 42.Vander Wal RL, Bryg VM and Hays MD, XPS analysis of combustion aerosols for chemical composition, surface chemistry, and carbon chemical state, Anal. Chem, 2011, 83, 1924–1930. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Lu R, Downs RT and Costin G, Goethite, α-FeO (OH), from single-crystal data, Acta Cryst. E, 2006, 62, i250–i252. [Google Scholar]
- 44.George I and Abbatt J, Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem, 2010, 2, 713. [DOI] [PubMed] [Google Scholar]
- 45.Lahaye J, Ehrburger-Dolle F,, Mechanisms of carbon black formation. Correlation with the morphology of aggregates. Carbon 1994, 32, 1319–1324. [Google Scholar]
- 46.Watson AY and Valberg PA, Carbon black and soot: two different substances, AIHAJ, 2001, 62, 218–228. [DOI] [PubMed] [Google Scholar]
- 47.Russell LM, Maria SF and Myneni SC, Mapping organic coatings on atmospheric particles, Geophys.l Res. Lett., 2002, 29. [Google Scholar]
- 48.Tchoul MN, Ford WT, Lolli G, Resasco DE and Arepalli S, Effect of mild nitric acid oxidation on dispersability, size, and structure of single-walled carbon nanotubes, Chem. Mater, 2007, 19, 5765–5772. [Google Scholar]
- 49.Sanderson P, Su S, Chang I, Saborit JD, Kepaptsoglou D, Weber R and Harrison RM, Characterisation of iron-rich atmospheric submicrometre particles in the roadside environment, Atmos. Environ, 2016, 140, 167–175. [Google Scholar]
- 50.Luo C, Mahowald N, Bond T, Chuang P, Artaxo P, Siefert R, Chen Y and Schauer J, Combustion iron distribution and deposition, Global Biogeochem. Cycles, 2008, 22. [Google Scholar]
- 51.Wang R, Balkanski Y, Boucher O, Bopp L, Chappell A, Ciais P, Hauglustaine D, Peñuelas J, Tao S, Sources, transport and deposition of iron in the global atmosphere. Atmos. Chem. and Phys. 2015, 15, 6247–6270. [Google Scholar]
- 52.Lin L, Lee ML, Eatough DJ, Review of recent advances in detection of organic markers in fine particulate matter and their use for source apportionment. J. Air Waste Manag. Assoc. 2010, 60, 3–25. [DOI] [PubMed] [Google Scholar]
- 53.Cheung KL, Ntziachristos L, Tzamkiozis T, Schauer JJ, Samaras Z, Moore KF and Sioutas C, Aerosol. Sci. Technol, 2010, 44, 500–513. [Google Scholar]
- 54.Wang Z, Chen C, Ma W and Zhao J, Photochemical coupling of iron redox reactions and transformation of low-molecular-weight organic matter, J. Phys. Chem. Lett., 2012, 3, 2044–2051. [Google Scholar]
- 55.Chen C, Dynes JJ, Wang J and Sparks DL, Properties of Fe-organic matter associations via coprecipitation versus adsorption, Environ. Sci. Technol, 2014, 48, 13751–13759. [DOI] [PubMed] [Google Scholar]
- 56.Cwiertny DM, Baltrusaitis J, Hunter GJ, Laskin A, Scherer MM and Grassian VH, Characterization and acid‐mobilization study of iron‐containing mineral dust source materials, J. Geophys. Res. Atmos., 2008, 113. [Google Scholar]
- 57.Formenti P, Caquineau S, Chevaillier S, Klaver A, Desboeufs K, Rajot JL, Belin S and Briois V, Dominance of goethite over hematite in iron oxides of mineral dust from Western Africa: Quantitative partitioning by X‐ray absorption spectroscopy, J. Geophys. Res. Atmos., 2014, 119. [Google Scholar]
- 58.Oakes M, Ingall E, Lai B, Shafer M, Hays M, Liu Z, Russell A and Weber R, Iron solubility related to particle sulfur content in source emission and ambient fine particles, Environ. Sci. Technol, 2012, 46, 6637–6644. [DOI] [PubMed] [Google Scholar]
- 59.Li W, Xu L, Liu X, Zhang J, Lin Y, Yao X, Gao H, Zhang D, Chen J and Wang W, Air pollution–aerosol interactions produce more bioavailable iron for ocean ecosystems, Sci. Adv, 2017, 3, e1601749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moreno T, Querol X, Martins V, Minguillón M, Reche C, Ku L, Eun H, Ahn K, Capdevila M and de Miguel E, Formation and alteration of airborne particles in the subway environment, Environ. Sci. Process. Impact, 2017, 19, 59–64. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H; Zhou L; Yuen J; Birkner N; Leppert V; O’Day PA; Forman HJ Delayed Nrf2-regulated antioxidant gene induction in response to silica nanoparticles. Free Radic. Biol. Med, 2017, 108, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu W, Miser DE, Chan WG and Hajaligol MR, HRTEM investigation of some commercially available furnace carbon blacks, Carbon, 2004, 42, 1841–1845. [Google Scholar]
- 63.Lu S and Chung D, Viscoelastic behavior of carbon black and its relationship with the aggregate size, Carbon, 2013, 60, 346–355. [Google Scholar]
- 64.Jacobsen NR, White PA, Gingerich J, Møller P, Saber AT, Douglas GR, Vogel U and Wallin H, Mutation spectrum in FE1‐MUTATMMouse lung epithelial cells exposed to nanoparticulate carbon black, Environ. Mol. Mutagen, 2011, 52, 331–337. [DOI] [PubMed] [Google Scholar]
- 65.Warheit DB, What is currently known about the health risks related to carbon nanotube exposures?, Carbon, 2006, 44, 1064–1069. [Google Scholar]
- 66.Iball J and Morgan C, A refinement of the crystal structure of ferric acetylacetonate, Acta Crystallogr. 1967, 23, 239–244. [Google Scholar]
- 67.Saritha A, Raju B, Ramachary M, Raghavaiah P and Hussain K, Synthesis, crystal structure and characterization of chiral, three-dimensional anhydrous potassium tris (oxalato) ferrate (III), Physica B, 2012, 407, 4208–4213. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.