

Summary
Mitochondrial DNA (mtDNA) segregation associated with donor-recipient mtDNA mismatch in mitochondria replacement therapy leads to unknown risks. Here, to explore whether matching mtDNA haplotypes contributes to ameliorating segregation, we reproduced various degrees of heteroplasmic mice with three single nucleotide polymorphisms to monitor segregation severity. “Segregation” presented in tissues of heteroplasmic mice containing low-level donor mtDNA heteroplasmy, and disappeared as donor mtDNA heteroplasmy levels ascended. Meanwhile, we found that distribution of donor mtDNA among the blastomeres of preimplantation embryos from the heteroplasmic mice shared the same tendency as that in adult tissues. Statistical analysis showed that no selective replication of donor mtDNA occurred during lifespan. Tracking donor mtDNA distribution showed that uneven distribution of donor mtDNA among embryonic blastomeres gradually became even as donor mtDNA heteroplasmy increased, indicating that the “segregation” in tissues was inherited from the uneven distribution. Our finding suggested that donor-recipient mtDNA matching could circumvent segregation in mitochondria replacement therapy.
Subject Areas: Biological Sciences, Developmental Genetics, Genetics
Graphical Abstract
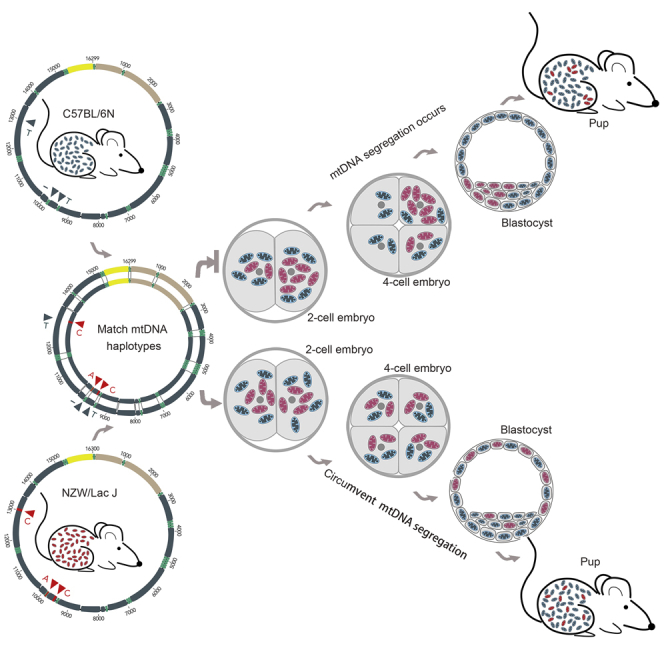
Highlights
-
•
Matching mitochondrial DNA haplotypes make the nucleus treat different mtDNA the same
-
•
Similar mtDNA haplotypes prevents tissue-specific segregation bias
-
•
Low level of mtDNA heteroplasmy results in uneven inheritance rather than segregation
Biological Sciences; Developmental Genetics; Genetics
Introduction
Mitochondrial replacement has the potential to reduce the transmission of inherited mitochondrial diseases (Wang et al., 2014, Paull et al., 2013, Graven et al., 2010, Tachibana et al., 2009). However, mitochondrial replacement will inevitably result in trace levels of heteroplasmy (Paull et al., 2013, Graven et al., 2010, Tachibana et al., 2009). Although such trace levels of heteroplasmy do not exceed a pathogenic threshold, if the pathogenic maternal mitochondrial DNA (mtDNA) is given a selective advantage, it is possible that it achieves dominance and manifests a pathogenic phenotype through the process of segregation, a common phenomenon in tissues of patients with mitochondrial disease (Frederiksen et al., 2006, Nishizuka et al., 1998). Recent studies of human mitochondrial replacement show sharp drifts of pathogenic mtDNA haplotype on a cellular level (Hyslop et al., 2016, Kang et al., 2016, Yamada et al., 2016), indicating that the nuclear genome preferentially regulates the replication and segregation of the native pathogenic mitochondria. Thus the requirement of a functional match between donor and recipient mtDNA, as well as between the mtDNA and nuclear genome, is of the utmost importance in clinical applications of mitochondria replacement (Latorre-Pellicer et al., 2016, Reinhardt et al., 2013, Sharpley et al., 2012; St John and Campbell, 2010). Previous study suggested that the segregation of donor mtDNA in mitochondria replacement could be alleviated if the donor mtDNA haplotype matches with the recipient mtDNA haplotype (Latorre-Pellicer et al., 2016, Røyrvik et al., 2016).
Obviously, the optimal recipient mitochondria would have the same haplotype as the donor. However, it is impossible to have two identical haplotypes of mtDNA in humans (He et al., 2010). However, the shorter the genetic distance between haplotypes, the less pronounced is the segregation bias. Therefore, exploring haplotype matching between donor and recipient mtDNA is particularly important for eliminating segregation of donor mtDNA. As tissue-specific segregation of different mtDNA genotypes is also common in heteroplasmic mice created from ooplasm or nuclear transfer, various heteroplasmic mice models were used to elucidate the mechanisms for controlling segregation of different mtDNA genotypes (Jokinen et al., 2015, Neupane et al., 2015, Sato et al., 2007, Battersby et al., 2005, Takeda et al., 2000, Jenuth et al., 1997). Among these models, heteroplasmic mice constituted by NZB and C57BL/6N were the dominant heteroplasmic models with 106 single nucleotide polymorphism (SNP) differences. In addition, Burgstaller et al. found highly significant positive correlation between individual tissue-specific segregation and mtDNA genetic distance in specific heteroplasmic mice generated using wild mice in Europe and C57BL/6N, where the wild mice display a spectrum of genetic distance (18, 86, 107, and 416 SNP differences at mtDNA) with C57BL/6N (Burgstaller et al., 2014a). Common segregation present in those heteroplasmic mice indicated that none of the models above underwent haplotype matching, leading to the absence of related progress in segregation.
Here we therefore intend to shorten the distance between the donor and recipient mtDNA haplotypes to explore whether matching mtDNA haplotypes between donor and recipient mtDNA can circumvent the segregation bias toward donor mtDNA in tissues of mitochondria replacement mice (Figures S1A and S1B). We had established a specific model of heteroplasmic mice from NZW/Lac J and B6D2F1 (C57/BL6×DBA) using mitochondria replacement from our past study (Wang et al., 2014). The mtDNA genotypes of NZW strain differ from those of C57 strain at only three SNPs, making them ideal models for matching mtDNA haplotypes. To study the effect of matching mtDNA haplotypes on the tissue segregation, we reared offspring of the heteroplasmic mice to test whether matching mtDNA haplotypes between “donor” and “recipient” can circumvent the segregation of donor mtDNA (Figure S1C).
Results
Tissue-Specific “Segregation” Gradually Disappeared with the Increase of Donor mtDNA Mean Heteroplasmy in Adult Mice
To explore whether matching mtDNA haplotypes can circumvent the segregation bias, we first sought to test whether the segregation occurred in different tissues from heteroplasmic mice with 3 SNP difference (Table S1), which was derived as described using mitochondria replacement technique (Wang et al., 2014). To avoid the potential impact of heteroplasmy level and trauma from mitochondria replacement manipulation on segregation behavior, heteroplasmic mice from the mitochondria replacement founder were used, which possessed naturally inherited levels of heteroplasmy. Pyrosequencing, which has a 1% detection threshold, 100% sensitivity, and 100% specificity (Hyslop et al., 2016, Wang et al., 2014, Blakely et al., 2013, White et al., 2005) (Figure S2), was adopted to measure the level of donor mtDNA heteroplasmy with primary and second primers (Table S2, also see Transparent Methods). The donor mtDNA heteroplasmy was measured with pyrosequencing in 16 tissues from 37 heteroplasmic mice that were sacrificed as adults (6–8 months old) (Figure 1A). The mean heteroplasmy level of each adult displaces the natural range (from 1.86% to 38.13%) (Figure 1B and Table S3). The heteroplasmic value of 16 tissues showed similar regional distribution (p > 0.05) (Figure 1C), which initially indicated that segregation of donor mtDNA does not appear in different tissues of the heteroplasmic mice with 3 SNP difference. After incorporation of different tissues into the corresponding germ layer, it was found that there was no significant difference in donor mtDNA heteroplasmy between the three germ layers (p > 0.05) (Figure 1D). Then we compared the distribution of donor mtDNA in individual tissues of each adult mouse using normalized variance (V′(h)) calculation as a way to measure the dispersion of donor mtDNA heteroplasmy. A drastic dispersion of donor mtDNA presented in individual tissues of adult mice with lesser than 10% donor mtDNA, evidenced by the greater V′(h) (r = −0.53, p < 0.001) (Figure 1E). However, a much narrower distribution with few deviations appeared in individual tissues of adult mice with 10%–20%, or greater than 20%, donor mtDNA, supported by the low V′(h) (Figure 1E).
Figure 1.

Tissue-Specific Segregation Disappeared with the Increase of Donor mtDNA Mean Heteroplasmy in Adult Mice
(A) Schematic model of heteroplasmic mice and dissected tissues in this study.
(B) Heteroplasmy of donor mtDNA in individual tissues from heteroplasmic mice with different heteroplasmic levels.
(C) Comparison of heteroplasmic distribution in 16 tissues from 37 mice (p > 0.05, Friedman test). Data are represented as scatterplot with mean ± SD.
(D) Comparison of mean heteroplasmy levels in the ectoderm, endoderm, and mesoderm (p > 0.05, Mann-Whitney test). Data are represented as mean ± SD.
(E) Negative correlation between heteroplasmic levels (green) and V′(h) (red) in 16 tissues of each mouse (r = −0.53, p < 0.001, Spearman correlation test). Error bars indicate SD, with the mean value.
See also Table S3.
Distribution of Donor mtDNA in the Blastomeres of Preimplantation Embryos Shared the Same Tendency as that in Adult Tissues
It is known that no net replication of mtDNA takes place before embryo implantation. Therefore each subsequent cell division reduces the amount of mtDNA within the daughter cells by about 50% (Carling et al., 2011). Next, to explore whether “segregation” actually occurs at lower levels of heteroplasmy in adult tissues, we observed the distribution of the two different mtDNA genotypes in each blastomere of embryos at 2-cell, 4-cell, and 8-cell stages from the heteroplasmic mice. As the adult tissues of the heteroplasmic mice, embryos at 2-cell, 4-cell, and 8-cell stages maintained a natural distribution value, ranging from 1.95 to 39.63, 1.81 to 54.24, and 2.57 to 51.91, respectively (Figures 2A–2C, Tables S4–S6). Similar to adult tissues, we observed that distribution of donor mtDNA heteroplasmy in each blastomere became less diverse as the mean levels of donor mtDNA heteroplasmy gradually increased in 2-, 4-, and 8-cell embryos, respectively. It was witnessed that V′(h) values present a negative correlation with the mean heteroplasmy of embryos at 2-, 4-, and 8-cell stages (r = −0.45, p < 0.05 for 2-cell stage; r = −0.58, p < 0.0001 for 4-cell stage; r = −0.48, p < 0.005 for 8-cell stage) (Figure 2D).
Figure 2.

Distribution of Donor mtDNA in the Blastomeres of Pre-implantation Embryos Shared the Same Tendency as that in Adult Tissues
(A) Schematic model of embryos and dissected blastomeres of embryos at 2-cell stage. Heteroplasmy distribution of donor mtDNA in blastomeres of embryos at 2-cell stage from heteroplasmic mice.
(B) Schematic model of embryos and dissected blastomeres of embryos at 4-cell stage. Heteroplasmy distribution of donor mtDNA in blastomeres of embryos at 4-cell stage from heteroplasmic mice.
(C) Schematic model of embryos and dissected blastomeres of embryos at 8-cell stage. Heteroplasmy distribution of donor mtDNA in blastomeres of embryos at 8-cell stage from heteroplasmic mice.
(D) Negative correlation between donor mtDNA heteroplasmy (green) and V′(h) (red) values in blastomeres of embryos at 2, 4, and 8-cell stage (r = −0.45, p < 0.05 for 2-cell stage; r = −0.58, p < 0.0001 for 4-cell stage; r = −0.48, p < 0.005 for 8-cell stage. Spearman correlation test).
Error bars indicate SD, with the mean value. See also Tables S4–S6.
No Selective Replication of Donor mtDNA Took Place during the Progressive Cleavage across Developmental Stages
To relate the similar distribution trend of donor mtDNA seen in adult tissues with those in blastomeres of cleaving embryos, we compared the dispersion of donor mtDNA from 2-cell with that from adult stage using several statistical comparison methods. We first observed the spread trends from 2-cell to adult stage. The spread of donor mtDNA heteroplasmy between daughter blastomeres within each embryo, calculated as the heteroplasmic range and V′(h) values, increased gradually from the 2-cell through the 4-cell to 8-cell groups (p < 0.05) (Figures 3A–3D). These phenomena are consistent with recent studies' findings, which showed increasing cell-to-cell heteroplasmy variability through early embryonic cleavages (Lee et al., 2012, Johnston et al., 2015, Johnston and Jones, 2015, Johnston and Jones, 2016). However, the spread trend returned to the original level as that of 2-cell embryos as development progresses to adult stage (Figures 3A–3D). Then we further found that adult tissues share a similar distribution of heteroplasmy with early embryos at 2-, 4-, and 8-cell stages (p > 0.05), calculated as mean heteroplasmy (Figure 3E), frequency distribution (Figure 3F), and cumulative probability distribution (Figure 3G). Thus, from the spread trends and the distribution, it can be deduced that the distribution of donor mtDNA in adult tissues depends on the distribution present in early embryos, suggesting that no selective replication of donor mtDNA took place during the progressive cleavage from the 2- to 4- to 8-cell stages extending to adult.
Figure 3.

No selective replication of donor mtDNA took place during the progressive cleavage across developmental stages
(A and B) (A) Spread of donor mtDNA heteroplasmy from embryonic blastomere to adult tissue expressed by heteroplasmy range (maximum or minimum heteroplasmy value minus the mean median of heteroplasmy value). The mean median is defined as half of the sum of maximum and minimum values. Light bar = maximum − median = maximum − = ; dark bar = (B) V′(h) values of embryonic blastomeres and adult tissue.
(C) Comparison of the spread ranges among embryos at 2-, 4-, and 8-cell stage and adults. Data are represented as mean ± SD.
(D) Comparison of V′(h) values among embryos at 2-, 4-, and 8-cell stage and adults. Different letters indicate p values < 0.05 (Mann-Whitney test); error bars indicate SD. See also Tables S3–S6.
(E) Comparison of mean heteroplasmy values of embryos at 2-, 4-, and 8-cell stage and adult tissues (p > 0.05, Mann-Whitney test). Data are represented as scatterplot with mean ± SD.
(F) Frequency histogram of the donor mtDNA heteroplasmy of embryos at 2-, 4-, and 8-cell stage and adult tissues.
(G) Cumulative probability distribution functions for the heteroplasmy of embryos at 2-, 4-, and 8-cell stage and adult tissues. (p > 0.05, Kolmogorov-Smirnov test).
See also Tables S3–S6.
Tracking Donor mtDNA Distribution Exhibited that Low Level of Donor mtDNA Heteroplasmy Resulted in Its Uneven Inheritance during Early Embryonic Cleavage
To explore why donor mtDNA deviation occur in tissues and blastomeres in the <10% group, we generated heteroplasmic oocytes to observe the distribution of donor mtDNA via spindle-chromosome complex transfer (spindle transfer) (Figure 4A) (Wang et al., 2014). Briefly, the donor mitochondria were labeled with 250 nM MitoTracker Red. Then spindle transfer was performed between the stained oocytes (donor) and unstained oocytes (recipient) (Figures 4A and 4B and Video S1). Differing amounts of donor mtDNA were fused into an enucleated recipient oocyte, resulting in varying levels of heteroplasmy (<10% and >10%; here we only use >10% as past results showed no significant difference between the 10% to 20% and the >20% groups). The levels of heteroplasmy were calculated from the volume ratio (on average 10%:90%) by measuring the diameters of karyoplasts (carrying donor mtDNA). After the oocytes were fertilized in vitro and development proceeded, red mitochondria distribution was monitored in individual blastomeres of embryos at the 2-, 4-, and 8-cell stages and the blastocyst. A distinct correlation was observed between the distribution of donor mitochondria and the level of heteroplasmy. For <10% group, uneven and even configurations of red mitochondria distribution were found in the preimplantation embryos from 2-cell to blastocyst stage. In the uneven group, the number of red mitochondria in each blastomere varied significantly under confocal microscope, with no red mitochondria in several of the blastomeres (Figures 4B and S3–S6). By contrast, almost equal numbers of red granules were distributed in each blastomere in the even group (Figures 4B and S3–S6). However, in cells with higher levels of heteroplasmy (>10%), we observed only even distribution of stained mitochondria, with cells portraying close to equal levels of heteroplasmy (Figures 4B and S3–S6). Furthermore, statistical comparison found that there were significant differences between <10% uneven and <10% even or >10% groups for donor mtDNA distribution in each blastomere of embryos at 2-, 4-, and 8-cell stages and blastocysts (Figures 4C and S3–S6 and Tables S7–S10). On the contrary, there were no obvious differences between <10% even and >10% groups for the distribution in each blastomere of embryos at 2-, 4-, and 8-cell stages and blastocysts (Figures 4C and S3–S6 and Tables S7–S10). This suggests that disproportionate variance increase can arise from partitioning noise with low mitochondrial volumes.
Figure 4.

Tracking Donor mtDNA Distribution in Pre-implantation Embryos Exhibited that Low Level of Heteroplasmy Led to Its Uneven Inheritance during Cleavage
(A) Schematic model showing how differing amounts of donor mitochondria were transferred into the recipient oocytes, resulting in the formation of the <10% heteroplasmy group and the >10% heteroplasmy group (here we only use >10% as past results showed no significant difference between the 10%–20% and >20% group). Depicts the two possible outcomes for each group: even distribution or uneven distribution.
(B) The spindle transfer manipulation that resulted into the two groups of differing heteroplasmy (images were taken with Nikon TE, 2000 microscope, 40X); the two separate groups of heteroplasmic oocytes with different heteroplasmy of donor mtDNA; the final outcome of the two groups, with the <10% group displaying both even and uneven distributions of donor mtDNA in pre-implantation development; the >10% group only portraying an even distribution in pre-implantation development (images were taken with Leica confocal scanning microscope, 63X). The images of 2-cell in <10% uneven and >10% even, and blastocyst in <10% uneven and <10% even were also used for measuring fluorescence intensity in Figures S3 and S6, respectively.
(C) Statistical comparison of MitoTracker Red distribution in each blastomere of embryos at 2-, 4-, and 8-cell stage and blastocysts. UE, uneven; E, even. Data are represented as scatterplot with mean ± SD. Asterisks indicated significant differences (*p < 0.05, **p < 0.01, Mann-Whitney test). Scale bar, 40 μm.
See also Video S1, Figures S3–S6 and Tables S7–S10.
Discussion
As we know, segregation of mutant mtDNA is a universal event during individual development (Burgstaller et al., 2014b). The coexistence of two kinds of mitochondria and its mtDNA may have fatal consequences for the development of offspring (Schon et al., 2012). Mitochondrial replacement technology will inevitably lead to the coexistence of two kinds of mitochondria and mtDNA, so the public has been worried about the potential safety risks since its conception. This study demonstrated that matching the haplotypes of the donor and recipient mtDNA has the potential to circumvent segregation bias and prevent the occurrence of mitochondrial diseases.
When heteroplasmic values are close to the detection limit, technical variability will likely be mixed in with the biological variability and may contribute to “segregation” as well. However, the spread trend of mtDNA heteroplasmy in preimplantation embryo was consistent with recent studies (Johnston et al., 2015, Lee et al., 2012), suggesing that heteroplasmic values from pyrosequencing are reliable in our study. Our results showed that uneven inheritance donor mtDNA in embryonic blastomeres rather than selective replication of donor mtDNA causes the “segregation” in adult tissues of heteroplasmic mice with low level of heteroplasmy. The spread trend of donor mtDNA heteroplasmy and V′(h) values increases from 2-cell, through 3- to 4-cell, to 6- to 8-cell blastomeres, evidenced by how the spread trend of 8-cell is greater in turn than that of 4-cell and 2-cell blastomeres. This phenomenon is consistent with the results of Johnston et al. and Lee et al. studies (Johnston et al., 2015, Lee et al., 2012). Recent studies showed that mitochondrial concentration is controlled in the stochastic partition between cell division (Das Neves et al., 2010, Jajoo et al., 2016). Thus the spread trend could be attributed to how donor mitochondria are randomly assigned into two daughter blastomeres during mitosis of preimplantation embryos, resulting in much higher uneven inheritance of mtDNA in each blastomere when cleavage frequency increased, due to how they are stochastically partitioned during cell division. However, the spread trend in adult tissues almost resumes the initial spread range of the first embryonic cleavage in this study (Figures 3A–3D). As development progressed to adult stage, each tissue may be developed from more than one blastomere of an embryo at 8-cell stage, thus leading to the spread trend initialization, which indicated that no selective replication of donor mtDNA took place during the progressive cleavage across developmental stages.
Burgstaller et al. found that mtDNA segregation in heteroplasmic tissues is common in vivo and may be modulated by haplotype differences (Burgstaller et al., 2014a). Based upon their data (See Methods-Mathematical Analysis section), we created a mathematical model and formula that describes the relationship between the proliferation rate of donor mtDNA and genetic distance. From the formula we can deduce that when genetic distance (d) of haplotype differences is equal to or less than 9 SNPs, the expected level of segregation, albeit with substantial uncertainty, drops to zero (Figure S7). Thus, in our study, no segregation is present in tissue of heteroplasmic mice containing two mtDNA genotypes of NZW strain and C57 strain, as the mtDNA genotypes differ at only three SNPs. As mtDNA segregation is controlled by the nuclear genome (Agaronyan et al., 2015, Battersby et al., 2003), our results suggest that haplotype matching matches foreign mitochondrial and nuclear DNA as well, so that the nucleus treats similar mtDNA sequence the same, thus preventing segregation.
As we know, mtDNA point mutation causes a variety of different phenotypes in humans. The effect of point mutations on segregation in different tissues during lifetime is still enigmatic. Segregation of some mtDNA mutations, such as 8993T > G, yield no tissue segregation (White et al., 1999), whereas for others, such as 3243A > G, segregation varies drastically among tissues (Frederiksen et al., 2006). The latter seems to violate our results and speculation from Burgstaller's data, in which differences less than 9 SNPs could circumvent segregation of mutant mitochondria (Figure S7). Owing to a rapid mutation rate over the human lifetime, a number of novel mtDNA mutations, which constitutes mtDNA polymorphisms, were detected in both pathogenic mtDNA carriers' and healthy donors' oocytes (Kang et al., 2016). Owing to how genetic distance between two random selected people will differ at 100 SNPs (Røyrvik et al., 2016), previous studies found that polymorphisms can grant a replicative advantage (Burgstaller et al., 2014b, Kang et al., 2016). It has been demonstrated that some mtDNA point mutations, such as 3394C variant, may either be deleterious or beneficial depending on its haplogroup and environmental context (Ji et al., 2012), suggesting the segregation of point mutations associated with the polymorphisms. We hypothesize that original polymorphisms and novel variations may interact with pathogenic point mutation to constitute a network with nuclear DNA to regulate mutant and non-mutant mtDNA proliferation. Thus, multiple factors should be combined and taken into account for the point mutations as well as other mutants on segregation in further study.
In summary, mitochondrial segregation inevitably occurs in offspring from mitochondrial replacement manipulation if no haplotype matching has been conducted. This study indicates that genetic similarity between donor and recipient mtDNA has the potential to circumvent the segregation bias toward pathogenic maternal mtDNA in tissues of mitochondria replacement offspring. Thus our results recommend that mtDNA haplotype matching should be undertaken between the donor and recipient, as it could “fool” the nucleus into treating the donated mtDNA and the native pathogenic mtDNA the same, thereby eliminating any proliferative advantage, and circumvent any segregation bias and prevent the onset of mitochondrial diseases.
Limitations of the Study
Although this study has demonstrated that matching mtDNA haplotypes could circumvent the tissue segregation of mutant mitochondria in heteroplasmic mice with 3 SNP difference, more spectra (such as 3–18 SNPs) of mitochondrial genetic differences between two kinds of mice should be conducted to clearly address the minimum distance that can circumvent tissue segregation. Furthermore, screening key SNP loci or regulatory networks, which is associated with mtDNA replication and proliferation in nuclear and mtDNA sequences, may make it easier to find a suitable recipient donation and prevent the occurrence of mitochondrial diseases.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (81471512 and 31871506 to H.S.).
Author Contributions
H.S. and X.D. supervised and designed the experiments. H.S. and J.P. manipulated mitochondrial donation. J.P. and C.L. performed staining and confocal analysis. L.W. detected heteroplasmy level of samples. Y.Z. did data statistics. Z.M. supervised mice. H.S., C.L., and J.P. prepared the figures and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.03.002.
Contributor Information
Xi Dong, Email: dong.xi@zs-hospital.sh.cn.
Hongying Sha, Email: shahongying@fudan.edu.cn.
Supplemental Information
References
- Agaronyan K., Morozov Y.I., Anikin M., Temiakov D. Replication-transcription switch in human mitochondria. Science. 2015;347:548–551. doi: 10.1126/science.aaa0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battersby B.J., Loredo-Osti J.C., Shoubridge E.A. Nuclear genetic control of mitochondrial DNA segregation. Nat. Genet. 2003;33:183–186. doi: 10.1038/ng1073. [DOI] [PubMed] [Google Scholar]
- Battersby B.J., Redpath M.E., Shoubridge E.A. Mitochondrial DNA segregation in hematopoietic lineages does not depend on MHC presentation of mitochondrially encoded peptides. Hum. Mol. Genet. 2005;14:2587–2594. doi: 10.1093/hmg/ddi293. [DOI] [PubMed] [Google Scholar]
- Blakely E.L., Yarham J.W., Alston C.L., Craig K., Poulton J., Brierley C., Park S.M., Dean A., Xuereb J.H., Anderson K.N. Pathogenic mitochondrial tRNA point mutations: nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum. Mutat. 2013;34:1260–1268. doi: 10.1002/humu.22358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller J.P., Johnston I.G., Jones N.S., Albrechtová J., Kolbe T., Vogl C., Futschik A., Mayrhofer C., Klein D., Sabitzer S. mtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 2014;7:2031–2041. doi: 10.1016/j.celrep.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgstaller J.P., Johnston I.G., Poulton J. Mitochondrial DNA disease and developmental implications for reproductive strategies. Mol. Hum. Reprod. 2014;21:11–22. doi: 10.1093/molehr/gau090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling P.J., Cree L.M., Chinnery P.F. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion. 2011;11:686–692. doi: 10.1016/j.mito.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Das Neves R.P., Jones N.S., Andreu L., Gupta R., Enver T., Iborra F.J. Connecting variability in global transcription rate to mitochondrial variability. PLoS Biol. 2010;8:e1000560. doi: 10.1371/journal.pbio.1000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen A.L., Andersen P.H., Kyvik K.O., Jeppesen T.D., Vissing J., Schwartz M. Tissue specific distribution of the 3243A->G mtDNA mutation. J. Med. Genet. 2006;43:671–677. doi: 10.1136/jmg.2005.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graven L., Tuppen H.A., Greggains G.D., Harbottle S.J., Murphy J.L., Cree L.M., Murdoch A.P., Chinnery P.F., Taylor R.W., Lightowlers R.N. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y., Wu J., Dressman D.C., Iacobuzio-Donahue C., Markowitz S.D., Velculescu V.E., Diaz L.A., Jr., Kinzler K.W., Vogelstein B., Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyslop L.A., Blakeley P., Craven L., Richardson J., Fogarty N.M., Fragouli E., Lamb M., Wamaitha S.E., Prathalingam N., Zhang Q. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature. 2016;534:383–386. doi: 10.1038/nature18303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jajoo R., Jung Y., Huh D., Viana M.P., Rafelski S.M., Springer M., Paulsson J. Accurate concentration control of mitochondria and nucleoids. Science. 2016;351:169–172. doi: 10.1126/science.aaa8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuth J.P., Peterson A.C., Shoubridge E.A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- Ji F., Sharpley M.S., Derbeneva O., Alves L.S., Qian P., Wang Y., Chalkia D., Lvova M., Xu J., Yao W. Mitochondrial DNA variant associated with Leber hereditary optic neuropathy and high-altitude Tibetans. Proc. Natl. Acad. Sci. U S A. 2012;109:7391–7396. doi: 10.1073/pnas.1202484109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston I.G., Jones N.S. Closed-form stochastic solutions for non-equilibrium dynamics and inheritance of cellular components over many cell divisions. Proc. Math. Phys. Eng. Sci. 2015;471:20150050. doi: 10.1098/rspa.2015.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston I.G., Jones N.S. Evolution of cell-to-cell variability in stochastic, controlled, heteroplasmic mtDNA populations. Am. J. Hum. Genet. 2016;99:1150–1162. doi: 10.1016/j.ajhg.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston I.G., Burgstaller J.P., Havlicek V., Kolbe T., Rülicke T., Brem G., Poulton J., Jones N.S. Stochastic modelling, bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism. Elife. 2015;4:e07464. doi: 10.7554/eLife.07464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R., Marttinen P., Stewart J.B., Dear T.N., Battersby B.J. Tissue-specific modulation of mitochondrial DNA segregation by a defect in mitochondrial division. Hum. Mol. Genet. 2015;25:706–714. doi: 10.1093/hmg/ddv508. [DOI] [PubMed] [Google Scholar]
- Kang E., Wu J., Gutierrez N.M., Koski A., Tippner-Hedges R., Agaronyan K., Platero-Luengo A., Martinez-Redondo P., Ma H., Lee Y. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540:270–275. doi: 10.1038/nature20592. [DOI] [PubMed] [Google Scholar]
- Latorre-Pellicer A., Moreno-Loshuertos R., Lechuga-Vieco A.V., Sánchez-Cabo F., Torroja C., Acín-Pérez R., Calvo E., Aix E., González-Guerra A., Logan A. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–565. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- Lee H.S., Ma H., Juanes R.C., Tachibana M., Sparman M., Woodward J., Ramsey C., Xu J., Kang E.J., Amato P. Rapid mitochondrial DNA segregation in primate preimplantation embryos precedes somatic and germline bottleneck. Cell. Rep. 2012;1:506–515. doi: 10.1016/j.celrep.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane J., Ghimire S., Vandewoestyne M., Lu Y., Gerris J., Van Coster R., Deroo T., Deforce D., Vansteelandt S., De Sutter P. Cellular heterogeneity in the level of mtDNA heteroplasmy in mouse embryonic stem cells. Cell Rep. 2015;13:1304–1309. doi: 10.1016/j.celrep.2015.10.019. [DOI] [PubMed] [Google Scholar]
- Nishizuka S., Tamura G., Goto Y., Murayama K., Konno T., Hakozaki M., Nonaka I., Tohgi H., Satodate R. Tissue-specific involvement of multiple mitochondrial DNA deletions in familial mitochondrial myopathy. Biochem. Biophys. Res. Commun. 1998;247:24–27. doi: 10.1006/bbrc.1998.8709. [DOI] [PubMed] [Google Scholar]
- Paull D., Emmanuele V., Weiss K.A., Treff N., Stewart L., Hua H., Zimmer M., Kahler D.J., Goland R.S., Noggle S.A. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–637. doi: 10.1038/nature11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt K., Dowling D.K., Morrow E.H. Mitochondrial replacement, evolution, and the clinic. Science. 2013;341:1345–1346. doi: 10.1126/science.1237146. [DOI] [PubMed] [Google Scholar]
- Røyrvik E.C., Burgstaller J.P., Johnston I.G. mtDNA diversity in human populations highlights the merit of haplotype matching in gene therapies. Mol. Hum. Reprod. 2016;22:809–817. doi: 10.1093/molehr/gaw062. [DOI] [PubMed] [Google Scholar]
- Sato A., Nakada K., Shitara H., Kasahara A., Yonekawa H., Hayashi J. Deletion-mutant mtDNA increases in somatic tissues but decreases in female germ cells with age. Genetics. 2007;177:2031–2037. doi: 10.1534/genetics.107.081026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon E.A., DiMauro S., Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012;13:878. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpley M.S., Marciniak C., Eckel-Mahan K., McManus M., Crimi M., Waymire K., Lin C.S., Masubuchi S., Friend N., Koike M. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. doi: 10.1016/j.cell.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John J.C., Campbell K.H. The battle to prevent the transmission of mitochondrial DNA disease: is karyoplast transfer the answer? Gene Ther. 2010;17:147–149. doi: 10.1038/gt.2009.164. [DOI] [PubMed] [Google Scholar]
- Tachibana M., Sparman M., Sritanaudomchai H., Ma H., Clepper L., Woodward J., Li Y., Ramsey C., Kolotushkina O., Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Takahashi S., Onishi A., Hanada H., Imai H. Replicative advantage and tissue-specific segregation of RR mitochondrial DNA between C57bl/6 and RR heteroplasmic mice. Genetics. 2000;155:777–783. doi: 10.1093/genetics/155.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Sha H., Ji D., Zhang H.L., Chen D., Cao Y., Zhu J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157:1591–1604. doi: 10.1016/j.cell.2014.04.042. [DOI] [PubMed] [Google Scholar]
- White H.E., Durston V.J., Seller A., Fratter C., Harvey J.F., Cross N.C. Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet. Test. 2005;9:190–199. doi: 10.1089/gte.2005.9.190. [DOI] [PubMed] [Google Scholar]
- White S.L., Collins V.R., Wolfe R., Cleary M.A., Shanske S., DiMauro S., Dahl H.H., Thorburn D.R. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am. J. Hum. Genet. 1999;65:474–482. doi: 10.1086/302488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M., Emmanuele V., Sanchez-Quintero M.J., Sun B., Lallos G., Paull D., Zimmer M., Pagett S., Prosser R.W., Sauer M.V. Genetic drift can compromise mitochondrial replacement by nuclear transfer in human oocytes. Cell Stem Cell. 2016;18:749–754. doi: 10.1016/j.stem.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.