

Abstract
Porphyromonas gingivalis is a member of the dysbiotic oral microbiome and a “keystone pathogen” that causes severe periodontal disease, which is among the most prevalent infectious diseases. Part of the virulence factors secreted by P. gingivalis are the essential cysteine peptidases gingipain K (Kgp) and R (RgpA and RgpB), which account for 85% of the extracellular proteolytic activity of the pathogen and are thus prime targets for inhibition. We report the high-resolution (1.20 Å) complex structure of Kgp with KYT-36, a peptide-derived, potent, bioavailable and highly selective inhibitor, which is widely used for studies in vitro, in cells and in vivo. Sub-nanomolar inhibition of Kgp is achieved by tight binding to the active-site cleft, which is covered for its sub-sites S3 through S1’ under establishment of nine hydrophobic interactions, 14 hydrogen bonds and one salt bridge. In addition, an inhibitor carbonyl carbon that mimics the scissile carbonyl of substrates is pyramidalized and just 2.02 Å away from the catalytic nucleophile of Kgp, C477Sγ. Thus, the crystal structure emulates a reaction intermediate of the first nucleophilic attack during catalysis of cysteine peptidases. The present study sets the pace for the development of tailored next-generation drugs to tackle P. gingivalis.
Introduction
The human oral microbiome is extraordinarily diverse and includes phages, viruses, archaea, bacteria, fungi, and protozoa1. Bacteria are represented by ~1000 different species at 108–109 bacteria per mL saliva or mg dental plaque, which makes the oral microbiome second only to the colon microbiome in complexity2. Oral bacteria mainly belong to the phyla Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, Spirochaetes, Synergistetes and Tenericutes, and they divide into commensal and dysbiotic. While the former are usually beneficial to the host, the latter are associated with disease due to changes in microbiome composition and functional activities3. Dysbiotic bacteria are mostly Gram-negative and anaerobic, and they are responsible for two of the most widespread human diseases: dental caries and periodontal disease (PD). These are not classical infectious diseases originated by single pathogens but have polymicrobial origins and result from a combination of microbiota relationships, host susceptibility, and environmental factors, such as smoking and diet1,4. In particular, PD is the sixth most prevalent disabling health condition and affects an estimated ~750 million people worldwide5. It causes alveolar bone resorption, formation of deep periodontal pockets, and tooth loosening, and is epidemiologically associated with several systemic diseases including atherosclerosis, diabetes and cardiovascular conditions6. PD derives from an exacerbated inflammatory response to normal microbiota triggered by the presence of dysbiotic species including Aggregatibacter (formerly Actinobacillus) actinomycetemcomitans, Fusobacterium nucleatum, Prevotella intermedia, Treponema denticola, Tannerella forsythia and Porphyromonas gingivalis. For many years, the latter three species were englobed in the “red complex,” which substantially contributes to the subgingival biofilm and plaque and is intimately associated with severe forms of PD7. Among these species, P. gingivalis is a “keystone pathogen,” which converts other benign members of the biofilm into pathobionts and causes aggressive damage to periodontal tissues8. To this aim, it employs an armamentarium of virulence factors, which further contribute to pathogenesis by deregulating immune and inflammatory responses in the host.
P. gingivalis virulence factors include peptidases, which break down proteins within infected tissues, thus nourishing bacteria and facilitating their dissemination and host colonization9. Peptidases also dismantle host defenses and outcompete bacterial competitors within periodontal pockets10. The most relevant are the cysteine peptidases gingipain K (alias Kgp) and R (RgpA and RgpB), which cleave proteins and peptides after lysines and arginines, respectively11. They are translocated from the periplasm across the outer membrane layer to the extracellular space through a type-IX secretion system, which consists of at least 18 proteins, some of which are engaged in post-translational modification of cargo proteins12,13. The signal for translocation is a C-terminal domain conserved across cargos, which in RgpB adopts an immunoglobulin-like fold encompassing seven antiparallel β-strands organized in a β-sandwich14.
Gingipains are detected at concentrations exceeding 100 nM15 in gingival crevicular fluid from P. gingivalis-infected periodontitis sites, where they account for 85% of the total extracellular proteolytic activity of the bacterium16,17. Kgp, which is responsible for most of this activity18, is a 1723/1732-residue multidomain enzyme encompassing an N-terminal signal peptide, a pro-domain for latency, a caspase-like cysteine peptidase catalytic domain (CD), an immunoglobulin superfamily-like domain (IgSF), between three and five hemagglutinin-adhesion domains, and the C-terminal domain for type-IX secretion19. Kgp degrades connective tissue and plasma proteins, for example heme- and hemoglobin-transporting proteins, fibrinogen, fibronectin, plasma kallikrein, immunoglobulins, as well as peptidase inhibitors, thus causing vascular permeability and bleeding19,20. Kgp is indispensable for bacterial survival and the outcome of PD16,18, and has thus been hailed as a prime target for the development of novel drugs to treat PD19,21,22. This is of particular importance given that the current standard treatment of PD includes mechanical debridement and the widespread use of antibiotics and disinfectants, which have serious adverse effects due to toxicity and the development of bacterial resistance. Moreover, this treatment does not guarantee disease eradication23.
While a lot of effort has been dedicated lately to phylogenetic associations and meta-omics of the oral microbiome24,25, molecular and functional studies to discover valid biomarkers of oral pathophysiology, understand host–microbiome interactions, and develop novel drugs have largely been neglected1. An exception is the drug precursor candidate KYT-36 (Fig. 1), a peptide-derived, small-molecule inhibitor developed in 2004 in the laboratory of Kenji Yamamoto26. It is very specific for and potent against Kgp (Ki ≈ 10−10 M). Together with inhibitor KYT-1, which specifically tackles RgpA and RgpB, it strongly inhibited degradation of host proteins in culture supernatants and abolished thriving of P. gingivalis in cell cultures and in periodontal pockets in vivo. Moreover, it prevented Kgp-triggered vascular permeability in guinea pigs, i.e. demonstrating its efficacy against bacterial virulence in vivo, with no toxicity effects at the doses tested19. Based on these properties, the molecule and its derivatives are subject of patents by Cortexyme, Inc. for the therapeutic treatment of P. gingivalis (US20160096830A1, US2017014468A1 and WO2017201322A1) and by others (JP2010270061A and JP4982908B2). KYT-36 is currently distributed by at least four companies (Peptides International, www.pepnet.com; Pepta Nova, peptanova.de; MyBioSource, www.mybiosource.com; and Peptide Institute, www.peptide.co.jp) and has been used for years as the Kgp inhibitor of reference for studies in vitro, in cells and in vivo (see21,22,27 for examples).
Figure 1.
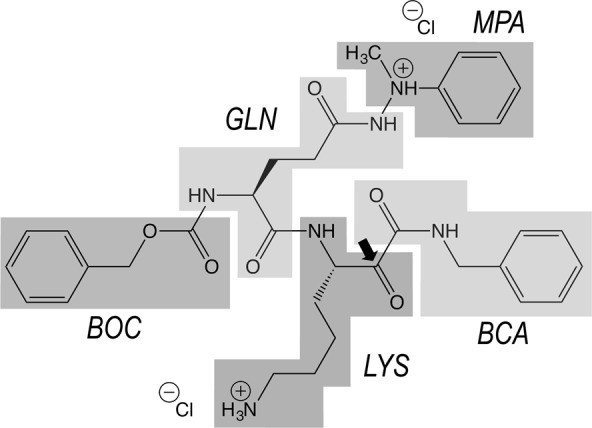
Chemical structure of KYT-36. The inhibitor, with IUPAC name benzyl-N-[(2S)-1-[[(3S)-7-amino-1-(benzylamino)-1,2-dioxoheptan-3-yl]amino]-5-(2-methyl-2-phenylhydrazinyl)-1,5-dioxopentan-2-yl]carbamate, consists of benzyloxycarbonyl (BOC), L-glutaminyl (GLN), methylphenylamino (MPA), L-lysinyl (LYS), and benzylcarbamoyl (BCA) moieties. A black arrow indicates the carbonyl mimicking the scissile carbonyl of a substrate. The molecular mass of the dichloride salt is 703.7 Da.
Whilst the efficacy of KYT-36 is well established, no information is available on its chemical mechanism of inhibition. This information is provided by three-dimensional structural studies, which are part of rational drug design strategies28,29. To this aim, we recently determined the crystal structure of the CD and IgSF domains of Kgp30 and of their zymogenic complex with the pro-domain31. These results revealed the mechanisms of action and latency of this peptidase. Here, we analyzed the crystal structure of Kgp from P. gingivalis strain W83 in complex with KYT-36 to very high resolution (1.20 Å). This is the first complex structure of the major proteolytic virulence factor of the periodontal pathogen reported with a drug or lead compound.
Results and Discussion
Structure of the Kgp catalytic domain
The Kgp fragment analyzed encompassed domains CD (residues D229-P600) and IgSF (K601-P683). Taken together, these domains form an elongated structure that resembles a tooth: the CD forms the crown with the cusp at its top, and the IgSF, which is a six-stranded antiparallel open β-barrel, shapes the root (see Fig. 2A). The CD is subdivided into an N-terminal subdomain (NSD; D229-K375) and a C-terminal subdomain (CSD; S376-P600), which are laterally attached to each other. Each of these subdomains is an α/β/α-sandwich consisting of a central β-sheet flanked by α-helices on either side. In NSD, the sheet is four-stranded and parallel; in CSD, it is six-stranded and parallel for all strands except the outermost strand at the interface with NSD, which is antiparallel to all other strands. In this way, the overall structure spans a central pseudo-continuous ten-stranded β-sheet. The NSD further contains two and three helices on either side of the sheet, respectively, plus an inserted β-ribbon and a calcium-binding site with structural functions. The CSD contains five and four helices on either side of the sheet, respectively, plus a β-ribbon and two sodium-binding sites. A second calcium site is found at the NSD-CSD interface. For further structural details on the general architecture of Kgp, see30.
Figure 2.
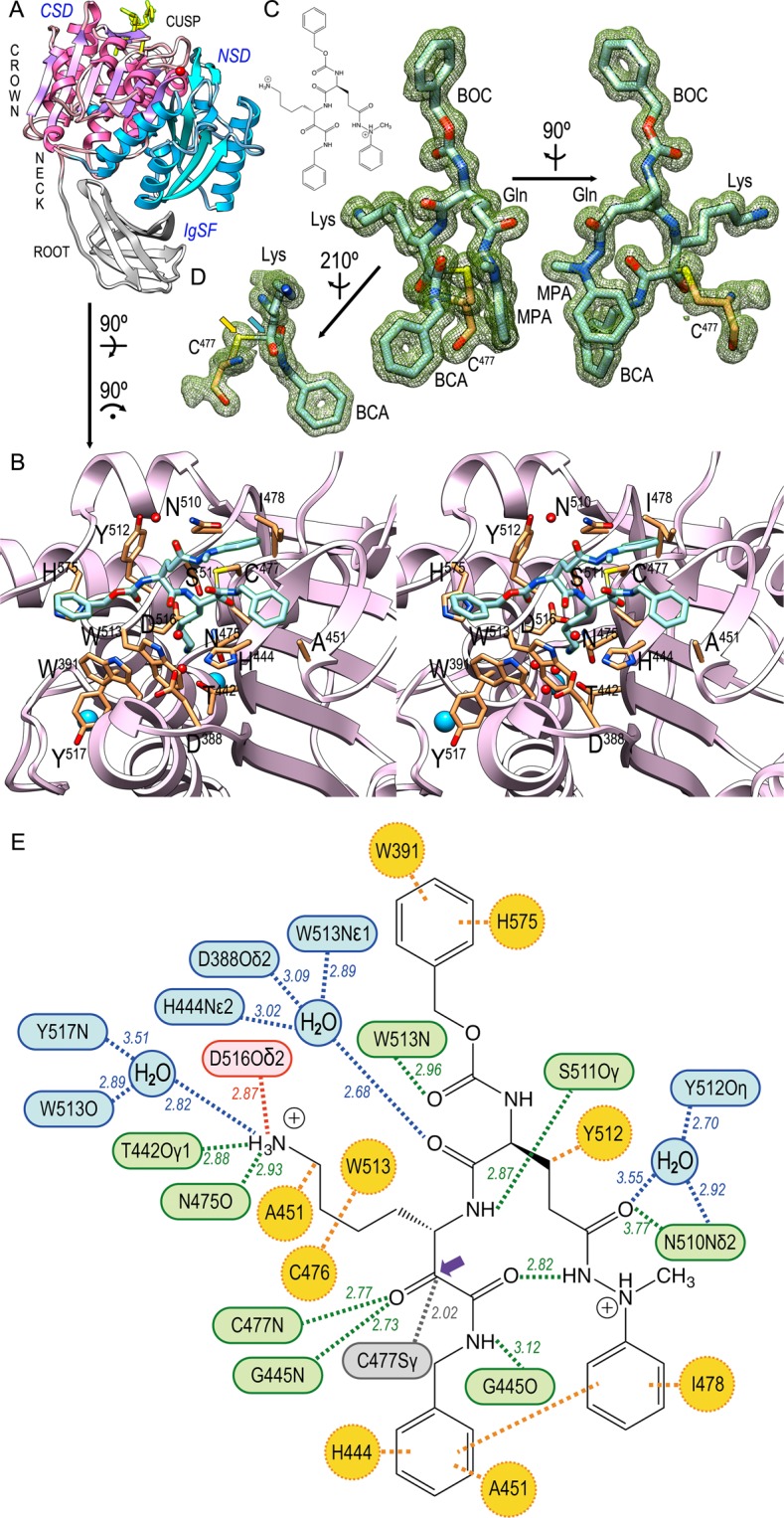
Interactions of the Kgp·KYT-36 complex. (A) Ribbon plot of Kgp, which mimics a tooth, whose crown encompasses the cusp in the top and consists of the NSD (blue ribbon) and CSD domains (magenta ribbon). Domain IgSF (grey ribbon) features the tooth root. KYT-36 is displayed as yellow sticks for reference. (B) Close-up of the tooth cusp encompassing the active site. The cleft runs from left (non-primed sub-sites) to right (primed sub-sites). Only the CSD is displayed as a plum ribbon for clarity. Kgp residues relevant for the complex are shown for their side chains (carbons in sandy brown) and labeled. The proposed catalytic triad is C477, H444 and D388 30. Solvent molecules and structural sodium cations are depicted as red and blue spheres, respectively. KYT-36 is shown as a stick model with carbons in light blue. (C) Structure of KYT-36 and Kgp catalytic residue C477 superposed with a (2mFobs-DFcalc)-type Fourier map contoured at 0.8σ (left) and after a 90°-rotation (right). The five moieties of the inhibitor (see Fig. 1) are labeled. The inset in the top left depicts the chemical structure of the inhibitor for reference. (D) Detail of (C, left) after reorientation depicting the pseudo-covalent bond (2.02 Å) between C477Sγ (yellow arrow) and the carbonyl carbon of LYS (blue arrow), which mimics the scissile carbonyl carbon of a substrate and is pyramidalized. (E) Scheme with the average distance values of direct (green) and solvent-mediated (blue) hydrogen bonds, salt bridges (red), hydrophobic interactions (orange), and the pseudo-covalent bond between the LYS carbonyl carbon (purple arrow) and catalytic C477Sγ (grey).
The active-site cleft of Kgp is found at the tooth cusp, on the CSD surface (Fig. 2A,B). As common in α/β-hydrolase-enzymes, residues engaged in substrate binding and catalysis come from loops that link strands of the central β-sheet on its the C-terminal edge32. As found in other cysteine peptidases33, Kgp probably contains a catalytic triad (C477, H444 and D388), which may form a charge-relay system for catalysis19,30,34. Atom C477Sγ acts as the nucleophile that attacks the scissile carbonyl carbon of substrates, which in a first step proceeds over a covalent tetrahedral reaction intermediate to an acyl-enzyme thioester complex with concomitant release of the amine reaction product35. In a second step, the covalent acyl-enzyme is hydrolyzed by a solvent molecule to release the acyl reaction product. In Kgp, substrates are bound with a lysine intruding the specificity pocket (sub-site S1) of the active-site cleft (for substrate and enzyme sub-site nomenclature, see36). The bottom of the specificity pocket leads to an internal water channel, which extends across the CSD to the opposite outer surface of the subdomain30.
The Kgp·KYT-36 complex
The complex structure was determined to very high resolution (1.20 Å; Fig. 2C,D), which enabled us to unambiguously assign the molecular determinants that cause sub-nanomolar inhibition of Kgp (Fig. 2B,E). KYT-36 is a L-peptide-derived molecule that mimics a substrate binding in extended conformation to cleft sub-sites S3, S2, S1 and S1’ (Fig. 2B). It can be divided into five moieties: BOC, GLN, MPA, LYS and BCA (Fig. 1).
The BOC benzyl group nestles in a hydrophobic pocket created by the side chains of H575 and W391, which together with Y512 and W513 create a shallow S3 sub-site in Kgp. The BOC carbonyl, which imitates the eponymous group of a substrate residue in position P3, is hydrogen-bonded to W513N (Fig. 2E). Downstream moiety GLN is in S2 and thus protrudes into the bulk solvent. Its side chain is folded towards the primed side of the cleft and its main-chain carbonyl establishes solvent-mediated hydrogen-bonds with H444Nε2, D388Oδ2, and W513Nε1. The aliphatic part of its side chain interacts with Y512 and its side-chain carboxamide performs an intramolecular hydrogen bond via the nitrogen with the carbonyl of the downstream BCA moiety. In addition, the carboxamide oxygen makes a direct and a solvent-mediated hydrogen bond with N510Nδ2 and Y512Oη, respectively. The aromatic phenyl ring of the MPA moiety hydrophobically interacts with I478—which explains why this residue has disallowed main-chain conformation angles—and, intramolecularly, with the benzyl group of the BCA moiety.
The LYS group of KYT-36 simulates a substrate residue in P1 and thus matches the specificity of the enzyme19. Its side chain penetrates the specificity pocket and its aliphatic part is pinched between W513, A451 and C476 through hydrophobic interactions. The terminal ε-amino group is tetrahedrally bound by D516Oδ2 through a salt bridge, by N475O and T442Oγ1 through direct hydrogen bonds, and by Y517N and W513O through hydrogen bonds mediated by a solvent molecule (Fig. 2E). The interactions made by T442 and H444 to bind the inhibitor also explain why intermediate residue A443 has disallowed main-chain conformation angles. Further downstream, the BCA moiety possibly occupies the S1’ sub-site and its amide nitrogen is hydrogen-bonded to G445O. In addition, the benzyl group establishes hydrophobic interactions with A451 and H444.
The LYS carbonyl emulates the scissile carbonyl of a substrate and its oxygen is tightly bound by C477N and G445N, which play the role of an oxyanion hole35 in Kgp to stabilize the tetrahedral reaction intermediate. The carbonyl carbon is just 2.02 Å apart (2.00 Å and 2.03 Å in the two Kgp molecules A and B found in the asymmetric unit of the crystal, respectively) from catalytic C477Sγ, which is roughly perpendicular to the carbon and its three bound atoms (Fig. 2D). This distance is larger than a standard aliphatic single C-S bond (1.82 Å;37) and the covalent bond found in the 1.75 Å-resolution structure of Kgp with a lysylmethyl group (1.84 Å; Protein Data Bank access code [PDB] 4RBM;30). However, the distance is shorter than that reported for reaction-intermediate mimics of serine endopeptidases in complex with protein inhibitors (2.6 Å for the complex between trypsin and bovine pancreatic trypsin inhibitor;38) and also than the sum of the van-der-Waals radii of carbon and sulfur (3.50 Å;39). In addition, the inhibitor carbonyl carbon is pyramidalized, i.e. not coplanar with its three bound atoms but shifted towards a tetrahedral configuration. This is reflected by angles C477Sγ-LYS(C)-LYS(O), C477Sγ-LYS(C)-LYS(Cα) and C477Sγ-LYS(C)-BCA(C) spanning on average 109.9°, 103.4° and 99.9°, respectively, instead of 90°. Thus, the present structure simulates a state immediately previous to formation of the tetrahedral reaction intermediate of the nucleophilic addition.
Conclusions
The complex structure of Kgp with its specific inhibitor KYT-36 revealed that the sub-nanomolar inhibition exerted by the inhibitor is based on 24 intermolecular interactions and the fact that the active-site cleft of the enzyme is blocked from sub-sites S3 to S1’. The side chain of inhibitor moiety LYS penetrates the S1 pocket like a substrate and makes four hydrogen bonds and a salt bridge, in addition to hydrophobic interactions with three protein residues.
The complex is also a valid model for the state preceding the formation of the tetrahedral reaction intermediate of the nucleophilic attack of C477Sγ onto the scissile carbonyl carbon during catalysis. This is reminiscent of structures of complexes between serine endopeptidases and protein inhibitors. In either case, the distances between the catalytic nucleophile and the scissile-carbonyl-carbon-mimic are larger than a regular bond but too short for a van-der-Waals interaction. Moreover, the carbon is pyramidalized, i.e. in a state preceding the tetrahedral intermediate.
Finally, the present data will foster the development of novel specific drugs against a major virulence factor of P. gingivalis, which may add to the locally-applied therapeutic agents currently used for PD as adjuncts to non-surgical therapy22. These adjuncts include doxycycline and minocycline, which are tetracycline antibiotics that inhibit host matrix metalloproteinases at doses low enough not to have antimicrobial activity. In this way, they do not select for antibiotic resistance within bacteria40. The potential of KYT-36 to contribute to such a development was demonstrated recently by KYT-41. This is a further development of KYT-36 and KYT-1, which potently and selectively blocks both Kgp (Ki = 2.7 × 10−10 M) and RgpA/B (Ki = 4.0 × 10−8 M), and shows therapeutic potential in guinea pig and dog models21.
Experimental Procedures
Protein production and complex formation
A Kgp construct spanning the CD and IgSF domains from P. gingivalis strain W83 (sequence D229-P683; UniProt (UP) entry Q51817) and a C-terminal His6-tag was purified from culture medium of P. gingivalis mutant strain ABM1 by affinity chromatography on Nickel-Sepharose beads as previously described41,42. The resulting sample was first incubated with Nα-tosyl-L-lysinylchloromethane (Sigma) prior to elution from the beads to avoid autolysis and then with excess of KYT-36 (purchased from Peptide International, KY, USA). The final complex was concentrated to ~10 mg/ml in 5 mM Tris·HCl pH 8, 150 mM sodium chloride, 0.02% sodium azide, 1 mM 1,4-dithiothreitol (DTT) for crystallization.
Crystallization and diffraction data collection
Crystallization assays were performed by the sitting-drop vapor diffusion method. Reservoir solutions were prepared with a Tecan robot and 100-nL crystallization drops were dispensed on 96 × 2-well MRC nanoplates (Innovadyne) by a Phoenix nanodrop robot (Art Robbins) or a Cartesian Microsys 4000 XL (Genomic Solutions) robot at the IBMB-IRB joint Automated Crystallography Platform at Barcelona Science Park. Plates were stored in Bruker steady-temperature crystal farms at 4 °C or 20 °C. Successful conditions were scaled up to the microliter range in 24-well Cryschem crystallization dishes (Hampton Research). The best Kgp·KYT-36 complex crystals were obtained at 20 °C with protein solution and 20% polyethylene glycol 8000, 0.1 M HEPES pH 7.5 as reservoir solution from 1 μL: 1 μL drops. Crystals were cryo-protected by immersion in harvesting solution containing reservoir solution plus 20% glycerol. Diffraction data were collected at 100 K from liquid-N2 flash cryo-cooled crystals (Oxford Cryosystems 700 series cryostream) on a Pilatus 6 M pixel detector (Dectris) at beam line XALOC43 of the ALBA synchrotron in Cerdanyola (Catalonia, Spain). These data were processed with programs XDS44 and XSCALE45, and transformed with XDSCONV to formats suitable for the CCP4 suite of programs46. Given that cell constants a and b were very similar (86.66 Å and 87.05 Å, respectively), initial indexation suggested a tetragonal setting. This was proven wrong during integration and merging of the reflections, which revealed that the crystals actually belonged to a primitive orthorhombic space group with two peptidase·inhibitor complexes per asymmetric unit.
Structure solution and refinement
The structure of Kgp·KYT-36 was solved by likelihood-scoring molecular replacement with the PHASER47 program using the coordinates of the protein part of Kgp crystallized in a different unit cell (PDB 4RBM;30) and diffraction data processed to 1.25 Å resolution. Two solutions were obtained at final Eulerian angles (α, β, γ, in °) 188.9, 71.0, 312.8 and 287.3, 71.3, 313.9; and fractional cell coordinates (x, y, z) 0.472, −0.103, 0.293 and −0.209, −0.295, 0.529, respectively. The initial values for the rotation/translation function Z-scores were 7.4/8.0 and 7.7/9.0, respectively, and the final log-likelihood gain was 62,873. These calculations revealed that P212121 was the correct space group. Subsequently, an automatic tracing step with ARP/wARP48 yielded a model, which was completed through successive rounds of manual model building with the COOT program49 and crystallographic refinement with the PHENIX50 and BUSTER/TNT51 programs, which included TLS refinement. In the final stages, anisotropic B-factor and alternate occupancy refinement was performed with BUSTER/TNT using data reprocessed to 1.20 Å resolution (see Table 1 for data processing statistics). The final model contained residues D229-G681, two calcium and two sodium cations, two unknown atoms/ions (UNK), and one KYT-36 moiety for each of the two Kgp molecules in the asymmetric unit. Further one DTT, six glycerols, two HEPES, and 1,580 solvent molecules completed the final model. The HEPES molecules were only tentatively assigned based on poor density and show two positions. Three residues of each molecule (A443, I478 and I576) were in disallowed regions of the Ramachandran plot but were unambiguously resolved in the final Fourier map. Three proline residues were found in cis conformation (P241, P424, and P453). Table 1 provides refinement and model validation statistics.
Table 1.
Crystallographic data.
| Dataset | Kgp·KYT-36 |
|---|---|
| Data processing | |
| Space group | P212121 |
| Cell constants (a, b, c, in Å) | 86.67, 87.05, 129.21 |
| Wavelength (Å) | 0.97949 |
| No. of measurements/unique reflections | 2,922,120/290,672 |
| Resolution range (Å) | 72.2 – 1.20 (1.27 – 1.20)a |
| Completeness (%) | 95.8 (75.2) |
| Rmerge | 0.065 (0.726) |
| Rmeas/CC1/2 | 0.068 (0.800)/1.000 (0.843) |
| <I/σ(I)> of unique reflections after merging | 19.6 (2.9) |
| B-Factor (Wilson) (Å2)/Aver. Multiplicity | 15.9/10.1 (5.2) |
| Structure refinement | |
| Resolution range used for refinement (Å) | 72.2 – 1.20 |
| No. of reflections used (test set) | 289,551 (1120) |
| Crystallographic Rfactor (free Rfactor) | 0.146 (0.148)b |
| No. of protein residues + atoms/solvent molecules/ligandsc | 906 + 7,048/1,580/2 K36, 4 Ca2+, 4 Na+, 4 UNK, 1 DTT, 6 GOL, 2 EPE |
| Correlation coefficient Fobs-Fcalc | 0.962b |
| Rmsd from target valuesb | |
| bonds (Å)/angles (°) | 0.012/1.17 |
| Average B-factors (Å2) (all// molec. A/B) | 16.3//13.2/13.3 |
| Overall anisotropic B-value (B11, B22, B33, in Å2) | 1.04, −3.67, 2.64 |
| All-atom contacts and geometry analysisd | |
| Protein residues in favored regions/outliers/all residues | 915e (97%)/6f/947e |
| Protein residues with outlying rotamers/bonds/angles/chirality/planarity | 2/0/0/0/0 |
| All-atom clashscore | 2.7 |
aData processing values in parenthesis are for the outermost resolution shell. bAccording to the final BUSTER/TNT refinement step. cK36, KYT-36; DTT, (2S,3S)-1,4-bis(sulfanyl)butane-2,3-diol; GOL, glycerol; UNK, unknown atoms/ions; and EPE, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES.). dwwPDB X-ray Structure Validation Report. eIncluding residues with atoms in two positions. fAll outliers are unambiguously resolved in the final Fourier map.
Once the structure was solved, the two molecules in the asymmetric unit were found to be related by a pure non-crystallographic twofold parallel to (1 1 0). Upon superposition of the protein moieties, the two inhibitor molecules perfectly matched and were engaged in crystal contacts with segment Y389-Q394 from the non-crystallographic symmetry mate. These contacts are very similar but not identical, which might actually have given rise to rupture of the tetragonal symmetry suggested by the indexation procedure.
Miscellaneous
Ideal coordinates and parameters for crystallographic refinement of KYT-36 were obtained from the PRODRG server52. Structure figures were prepared with the CHIMERA program53. The model was validated with the wwPDB Validation Server (https://www.wwpdb.org/validation;)54. The final coordinates of P. gingivalis Kgp·KYT-36 are deposited with the PDB at www.pdb.org (access code 6I9A).
Acknowledgements
We are grateful to Joan Pous and Xandra Kreplin from the joint IBMB/IRB Automated Crystallography Platform and to Roman Bonet from the Protein Purification Facility at IBMB for assistance. We further acknowledge the help of local contacts provided by the ALBA synchrotron. This study was supported in part by grants from US American (NIH/NIDCR R01 DE022597), Polish (National Science Center and Ministry of Science and Higher Education, Miniatura 2017/01/X/NZ1/01378, UMO-2015/199/N/NZ1/00322, UMO-2015/17/B/NZ1/00666, UMO-2016/21/B/NZ1/00292, and Mobility Plus 1306/MOB/IV/2015/0), Spanish (BFU2015-64487R and MDM-2014-0435), and Catalan (2017SGR3, and Fundació “La Marató de TV3” 201815) agencies. The Structural Biology Unit (www.sbu.csic.es) of IBMB is a “María de Maeztu” Unit of Excellence from the Spanish Ministry of Science, Innovation and Universities.
Author Contributions
F.X.G.R. and J.P. conceived the work; A.M.L., M.K. and B.P. produced and purified the protein; T.G., A.R.B. and F.X.G.R. performed crystallization and structural studies; F.X.G.R. and J.P. wrote the paper with contributions from all authors.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Tibisay Guevara and Arturo Rodríguez-Banqueri contributed equally.
Contributor Information
Jan Potempa, Email: jan.potempa@icloud.com.
F. Xavier Gomis-Rüth, Email: fxgr@ibmb.csic.es.
References
- 1.Wade WG. The oral microbiome in health and disease. Pharmacol. Res. 2013;69:137–143. doi: 10.1016/j.phrs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Human Microbiome Project, Structure, function and diversity of the healthy human microbiome. Nature486, 207–214 (2012). [DOI] [PMC free article] [PubMed]
- 3.Haenel H, Schmidt EF, Feldheim G. Fäkale Dysbiose im Säuglingsalter [Fecal dysbiosis in infancy] Z. Kinderheilkd. 1959;82:595–603. doi: 10.1007/BF00444718. [DOI] [PubMed] [Google Scholar]
- 4.Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol. Med. 2015;21:172–183. doi: 10.1016/j.molmed.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kassebaum NJ, et al. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J. Dent. Res. 2014;93:1045–1053. doi: 10.1177/0022034514552491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potempa J, Mydel P, Koziel J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017;13:606–620. doi: 10.1038/nrrheum.2017.132. [DOI] [PubMed] [Google Scholar]
- 7.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 8.Hajishengallis G, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell host & microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potempa J, Travis J. Porphyromonas gingivalis proteinases in periodontitis, a review. Acta Biochim. Pol. 1996;43:455–465. [PubMed] [Google Scholar]
- 10.Koziel J, Potempa J. Protease-armed bacteria in the skin. Cell Tissue Res. 2013;351:325–337. doi: 10.1007/s00441-012-1355-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potempa J, Sroka A, Imamura T, Travis J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: structure, function and assembly of multidomain protein complexes. Curr. Prot. Pept. Sci. 2003;4:397–407. doi: 10.2174/1389203033487036. [DOI] [PubMed] [Google Scholar]
- 12.Lasica AM, Ksiazek M, Madej M, Potempa J. The Type IX Secretion System (T9SS): highlights and recent insights into Its structure and function. Front. Cell. Infect. Microbiol. 2017;7:215. doi: 10.3389/fcimb.2017.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lauber, F., Deme, J. C., Lea, S. M. & Berks, B. C. Type 9 secretion system structures reveal a new protein transport mechanism. Nature564, 77–82 (2018). [DOI] [PMC free article] [PubMed]
- 14.de Diego I, et al. The outer-membrane export signal of Porphyromonas gingivalis type IX secretion system (T9SS) is a conserved C-terminal β-sandwich domain. Sci. Rep. 2016;6:23123. doi: 10.1038/srep23123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guentsch A, et al. Comparison of gingival crevicular fluid sampling methods in patients with severe chronic periodontitis. J. Periodontol. 2011;82:1051–1060. doi: 10.1902/jop.2011.100565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y, Nguyen KA, Potempa J. Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol. 2010;2000(54):15–44. doi: 10.1111/j.1600-0757.2010.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Potempa J, Pike R, Travis J. Titration and mapping of the active site of cysteine proteinases from Porphyromonas gingivalis (gingipains) using peptidyl chloromethanes. Biol. Chem. 1997;378:223–230. doi: 10.1515/bchm.1997.378.3-4.223. [DOI] [PubMed] [Google Scholar]
- 18.Pathirana RD, O’Brien-Simpson NM, Brammar GC, Slakeski N, Reynolds EC. Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect. Immun. 2007;75:1436–1442. doi: 10.1128/IAI.01627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pike, R. N. & Potempa, J. In Handbook of Proteolytic Enzymes Vol. 2 (eds N. D. Rawlings & G. Salvesen) 2337–2344 (Academic Press, 2013).
- 20.Brochu V, Grenier D, Nakayama K, Mayrand D. Acquisition of iron from human transferrin by Porphyromonas gingivalis: a role for Arg- and Lys-gingipain activities. Oral Microbiol. Immunol. 2001;16:79–87. doi: 10.1034/j.1399-302x.2001.016002079.x. [DOI] [PubMed] [Google Scholar]
- 21.Kataoka S, et al. A novel, potent dual inhibitor of Arg-gingipains and Lys-gingipain as a promising agent for periodontal disease therapy. FASEB J. 2014;28:3564–3578. doi: 10.1096/fj.14-252130. [DOI] [PubMed] [Google Scholar]
- 22.Hosn KN, Jefferson MM, Leding C, Shokouh-Amiri S, Thomas EL. Inhibitors of bacterial protease enzymes for periodontal therapy. Clin. Exp. Dent. Res. 2015;1:18–25. doi: 10.1002/cre2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haffajee AD, Socransky SS, Gunsolley JC. Systemic anti-infective periodontal therapy. A systematic review. Ann. Periodontol. 2003;8:115–181. doi: 10.1902/annals.2003.8.1.115. [DOI] [PubMed] [Google Scholar]
- 24.Dewhirst FE, et al. The human oral microbiome. J. Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gómez A, Nelson KE. The oral microbiome of children: development, disease, and implications beyond oral health. Microb. Ecol. 2017;73:492–503. doi: 10.1007/s00248-016-0854-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadowaki T, et al. Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol. Pharmacol. 2004;66:1599–1606. doi: 10.1124/mol.104.004366. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, et al. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017;7:11759. doi: 10.1038/s41598-017-12173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 29.Mittl PR, Grütter MG. Opportunities for structure-based design of protease-directed drugs. Curr. Opin. Struct. Biol. 2006;16:769–775. doi: 10.1016/j.sbi.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 30.de Diego I, et al. Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 2014;289:32291–32302. doi: 10.1074/jbc.M114.602052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pomowski A, et al. Structural insights unravel the zymogenic mechanism of the virulence factor gingipain K from Porphyromonas gingivalis, a causative agent of gum disease from the human oral microbiome. J. Biol. Chem. 2017;292:5724–5735. doi: 10.1074/jbc.M117.776724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ollis DL, et al. The α/β hydrolase fold. Prot. Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 33.Guarné A, et al. Structural and biochemical features distinguish the foot-and-mouth disease virus leader proteinase from other papain-like enzymes. J. Mol. Biol. 2000;302:1227–1240. doi: 10.1006/jmbi.2000.4115. [DOI] [PubMed] [Google Scholar]
- 34.Pavloff N, et al. Molecular cloning and characterization of Porphyromonas gingivalis lysine-specific gingipain. A new member of an emerging family of pathogenic bacterial cysteine proteinases. J. Biol. Chem. 1997;272:1595–1600. doi: 10.1074/jbc.272.3.1595. [DOI] [PubMed] [Google Scholar]
- 35.Polgár, L. In Handbook of Proteolytic Enzymes Vol. 2 (eds N. D. Rawlings & G. S. Salvesen) 1773–1784 (Academic Press, 2013).
- 36.Schechter I, Berger A. On the size of active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- 37.Sutton, L. E. E. Tables of interatomic distances and configuration in molecules and ions (Supplement 1956–1959). (The Chemical Society, 1965).
- 38.Huber R, Bode W. Structural basis of the activation and action of trypsin. Acc. Chem. Res. 1978;11:114–122. doi: 10.1021/ar50123a006. [DOI] [Google Scholar]
- 39.Bondi A. Van der Waals volumes and radii. J. Phys. Chem. 1964;68:441–451. doi: 10.1021/j100785a001. [DOI] [Google Scholar]
- 40.Caton J, Ryan ME. Clinical studies on the management of periodontal diseases utilizing subantimicrobial dose doxycycline (SDD) Pharmacol. Res. 2011;63:114–120. doi: 10.1016/j.phrs.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Potempa J, Nguyen KA. Purification and characterization of gingipains. Curr. Protoc. Prot. Sci. 2007;Suppl. 49:21.20.21–21.20.27. doi: 10.1002/0471140864.ps2120s49. [DOI] [PubMed] [Google Scholar]
- 42.Sztukowska M, et al. Disruption of gingipain oligomerization into non-covalent cell-surface attached complexes. Biol. Chem. 2012;393:971–977. doi: 10.1515/hsz-2012-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juanhuix J, et al. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 2014;21:679–689. doi: 10.1107/S160057751400825X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabsch W. XDS. Acta Crystallogr. sect. D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. sect. D. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. sect. D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. sect. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. sect. D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smart OS, et al. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. sect. D. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. sect. D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 53.Pettersen EF, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 54.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003;10:980–980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]