

SUMMARY
Heterobifunctional molecule that recruits E3 ubiquitin ligases, such as cereblon, for targeted protein degradation is an emerging pharmacological strategy. A major unanswered question is how generally applicable this strategy is to all protein targets. In this study, we designed a multi-kinase degrader by conjugating a highly promiscuous kinase inhibitor with a cereblon-binding ligand, and used quantitative proteomics to discover 28 kinases, including BTK, PTK2, PTK2B, FLT3, AURKA, AURKB, TEC, ULK1, ITK and 9 members of the CDK family, as degradable. This set of kinases is only a fraction of the intracellular targets bound by the degrader, demonstrating that successful degradation requires more than target engagement. The results guided us to develop selective degraders for FLT3 and BTK, with potentials to improve disease treatment. Together, this study demonstrates an efficient approach to triage a gene family of interest to identify readily degradable targets for further studies and pre-clinical developments.
Keywords: Drug design, protein degradation, kinase, chemoproteomics, FLT3, BTK
Graphical Abstract

In brief
Heterobifunctional protein degrader is an emerging pharmacological strategy. Using a multi-kinase degrader, Huang et al. greatly expanded the number of known degradable kinases, and demonstrated an efficient approach to triage a gene family for actionable targets with potential therapeutic impacts.
INTRODUCTION
Networks of protein kinases serve as the major cellular signaling conduits in cells, and dysregulation of these pathways is common in human diseases, such as cancer and autoimmune diseases. To date, the Food and Drug Administration (FDA) has approved 35 small molecule kinase inhibitors for the treatment of cancer, rheumatoid arthritis and idiopathic pulmonary fibrosis (CenterWatch, 2017; Wu et al., 2015). The majority of these and other preclinical kinase inhibitors are ATP-competitive and block the ability of kinases to transfer phosphate to their substrates, while others bind outside the ATP-binding site and allosterically inhibit kinase activity.
Recently, rationally designed small molecule kinase degraders have emerged as an attractive pharmacological strategy for abrogating kinase functions. These heterobifunctional molecules act by binding a kinase target on one end and an E3 ubiquitin ligase on the other, thereby inducing ubiquitination of the target and subsequent proteosome-dependent degradation. Potential advantages of kinase degradation versus functional inhibition include prolonged cellular effects, especially for proteins with slow turnover rate, and the ability to abrogate non-enzyme-dependent, or so-called ‘scaffolding’ functions of the kinase. Kinase degradation may also prevent kinase upregulation, which is frequently observed as a resistance mechanism to conventional kinase inhibitors (Barouch-Bentov and Sauer, 2011). In addition, because degrader molecules can act catalytically to induce protein degradation (Bondeson et al., 2015), they may not require as high intracellular concentrations as are required by traditional ‘occupancy-based’ inhibitors to achieve an effect. Furthermore, any allosteric kinase binder that may or may not affect kinase function can be utilized for degrader design, opening up further possibilities to achieving selectivity and efficacy.
The concept of hijacking the ubiquitin proteosome system for targeted degradation was first exemplified by the Proteolysis Targeting Chimeras (PROTACs) (Sakamoto et al., 2001). However, the early iterations of PROTACs were unstable and poorly cell-penetrant due to the phosphopeptidic nature of the E3 ligase binders utilized. Subsequently, chemical ligands for the E3 ligase mouse double minute 2 homolog (MDM2) and cellular inhibitor of apoptosis protein 1 (cIAP1) were applied to improve the chemical properties of PROTACs (cIAP-mediated degraders were termed specific and non-genetic IAP-dependent protein erasers, or SNIPERs) (Itoh et al., 2010; Schneekloth et al., 2008). However, the effects of these degraders were still modest in cellular assays. Furthermore, the cIAP1 ligand bestatin has specificity issues (Orning et al., 1991; Umezawa et al., 1976) and causes cIAP1 degradation (Sekine et al., 2008), which further limits its utility. With recent advancement in the chemical properties of E3-binding ligands—specifically, the discovery and development of immunomodulatory imide drugs (IMiDs) (Ito et al., 2010) and von Hippel-Lindau (VHL) ligands (Buckley et al., 2012; Galdeano et al., 2014) as potent chemical binders of the cereblon (CRBN) and the VHL ubiquitin ligases, respectively—bifunctional degraders begin to demonstrate potency and in vivo efficacy that promise wide-spread applications in both discovery biology and small molecule therapeutics (Deshaies, 2015; Raina et al., 2016).
To date, VHL- and CRBN-mediated protein degraders have been developed for several targets, including: BRD4 (Lu et al., 2015; Winter et al., 2015; Zengerle et al., 2015), FKBP12 (Winter et al., 2015), BRD9 (Remillard et al., 2017) and for the kinases BCR-ABL (Lai et al., 2016), RIPK2 (Bondeson et al., 2015) and CDK9 (Olson et al., in press). Several reports described the importance of varying all three components of degrader molecules, namely the warhead, the linker and the E3 ligase ligand, to optimize degradation efficiency (Bondeson et al., 2015; Lai et al., 2016; Zengerle et al., 2015). Changes in the chemical composition may affect cellular uptake and stability, two key factors that affect the efficacy of bivalent degraders. Different warheads and linker lengths also affect the orientation in which the target and the E3 ligase can be brought into proximity, which may affect ubiquitination efficiency. Despite this knowledge, the medicinal chemistry efforts for protein degraders remain largely empirical, as there is limited mechanistic understanding for why certain linker compositions or combinations of warheads and E3 ligase ligands give rise to better degradation efficiency.
Recently, the crystal structure of the selective BRD4 degrader MZ1 sandwiched between the second bromodomain (BD) of BRD4 (BRD4BD2) and the VHL ligase complex provided molecular insights to the formation of ternary complex, the species that is essential for degrader-induce targeted ubiquitination. The crystal structure revealed unexpected molecular features including linker folding, linker-protein interactions as well as protein-protein interactions (Gadd et al., 2017). This degrader-induced protein-protein interaction, rather than just proximity between the target protein and an E3 ligase, mirror the actions of the IMiDs (Lu et al., 2014) and the splicing inhibitor sulfonamides (SPLAMs) (Han et al., 2017) at redirecting the substrate profile of an E3 ubiquitin ligase. Considering the protein-protein interactions in ternary complex formation (Gadd et al., 2017) and the reports describing improved target selectivity when an inhibitor is turned into a degrader (Lai et al., 2016; Zengerle et al., 2015), we hypothesized that protein targets are differentially susceptible to degradation mediated by a specific E3 ligase. Hence, we reasoned, in contrast to choosing a target of interest a priori for iterative rounds of medicinal chemistry efforts, there is great utility in surveying the “low-hanging fruits” of the degrader strategy for target prioritization.
In this study, we sought to develop a methodology that would allow us to broadly survey the susceptibility of kinases to rationally designed degraders. We developed a multi-kinase degrader based on a diaminopyrimidine scaffold and identified degradable targets using an unbiased, multiplexed quantitative proteomic approach employing tandem mass tag (TMT) reagents (McAlister et al., 2014). This approach allowed us to identify 28 kinases as readily degradable by CRBN in two leukemic cell lines, including therapeutically relevant kinases such as FLT3, BTK and CDK4/6. Informed by this data, we were able to prepare effective target-selective degraders of FLT3, and of BTK, based upon published selective ATP-competitive ligands with relative ease. This multi-targeted degrader strategy can be generally applied to rapidly enumerate readily degradable protein targets using promiscuous small molecule baits. These experiments are also the first steps to systematically understand the factors that may determine the susceptibility of protein targets to rationally designed degraders.
RESULTS
TL12-186 is a CRBN-dependent multi-kinase degrader
To develop a potential multi-kinase degrader, we elaborated a 2,4-diaminopyrimidine scaffold, which is a versatile ATP-site directed pharmacophore that has been exploited in numerous kinase inhibitors. In particular, we chose TAE684 as a starting point, as it is a well-characterized inhibitor of ALK that also exhibits potent binding to a number of additional kinase targets (Galkin et al., 2007). To further increase the promiscuity of binding to other kinases, we removed the 2-methoxy functionality from the 2-anilino hinge-binding segment, a known key selectivity determinant for this compound (Galkin et al., 2007). The resultant promiscuous kinase binder is termed TL13-87 (Figure 1A). An available crystal structure of ALK in complex with TAE684 (PDB: 2XB7) (Bossi et al., 2010) informed a site for polyethylene glycol (PEG) linker installation and conjugation with pomalidomide, a CRBN-recruiting IMiD analog, to generate TL12-186 (Figure 1A). We chose to focus on CRBN-mediated degradation over VHL-mediated degradation in this study because the former had demonstrated better degradation capabilities against ABL and BCR-ABL (Lai et al., 2016). To ensure that TL12-186 is still capable of binding to various kinases, we performed kinase-binding measurements against a panel of 468 kinases (KINOMEscan) (Karaman et al., 2008). Remarkably, TL12-186 displays >90% inhibition of 193 kinases at a screening concentration of 1 μM (Figure 1B, Table S1). We also confirmed that TL12-186 displays potent binding to CRBN (IC50 = 12 nM) using an AlphaScreen engagement assay (Figure 1C). A negative control of the degrader, TL13-27, was synthesize by replacing the glutarimide moiety on pomalidomide with a δ-lactam moiety (Figure 1A). The negative control TL13-27 demonstrated a similar kinome profile (Table S1) but lost much affinity for CRBN (IC50 = 5.5 μM, Figure 1C).
Figure 1. Rationally designed multi-kinase degrader TL12-186 presents CRBN-dependent pharmacology.
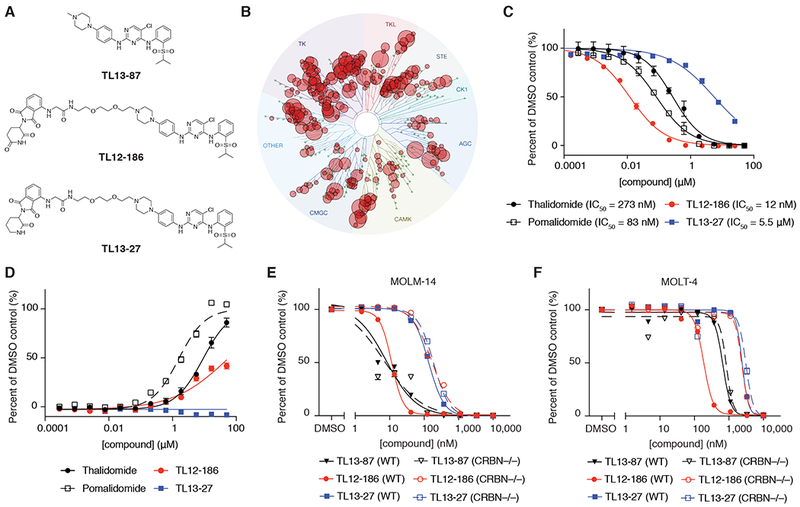
(A) Chemical structures of the multi-kinase degrader TL12-186, its parental kinase inhibitor TL13-87, and its negative control analog TL13-27.
(B) TREEspot visualization of the kinome selectivity profile of TL12-186 (1 μM). See also Table S1.
(C) CRBN-DDB1 engagement assay by AlphaScreen, testing the ability of thalidomide, pomalidomide, TL12-186 and TL13-27 at displacing biotinylated thalidomide for CRBN-DDB1 binding at indicated concentrations. Values represent quadruplicate means ± SD.
(D) Cellular CRBN engagement assay, testing the ability of thalidomide, pomalidomide, TL12-186 and TL13-27 at preventing CRBN-mediated degradation of FKBP12F36V-Nluc luciferase at indicated concentrations. Values represent triplicate means ± SD.
(E) 2-day proliferation assays of WT and CRBN−/− MOLM-14 cells treated with TL13-87, TL12-186 or TL13-27. Values represent duplicate means ± SD.
(F) 2-day proliferation assays of WT and CRBN−/− MOLT-4 cells treated with TL13-87, TL12-186 or TL13-27. Values represent duplicate means ± SD. For all plots within this figure, error bars shorter than the height of the symbols are not drawn.
As these high molecular weight bivalent molecules often suffer from poor cell penetration, we developed a dual-luciferase assay to evaluate intracellular CRBN binding. TL12-186 competitively prevented the degradation of FKBP12F36V-Nluc luciferase catalyzed by dTAG-7 (Erb et al., 2017), indicating its engagement with intracellular CRBN (Figure 1D). Consistent with the biochemical assay, the negative control compound TL13-27 could not engage with CRBN to rescue the degradation effect (Figure 1D).
To ensure that TL12-186 exhibits CRBN-dependent degrader functions, we compared its antiproliferative activity with TL13-87 and TL13-27 in WT and CRBN−/− cell lines. The parental kinase inhibitor, TL13-87, displayed more potent inhibition of cellular proliferation relative to the degrader molecules in CRBN−/− cells, likely reflecting the poorer permeability of the bivalent degraders (Figures 1E and 1F). However, only the degrader TL12-186 exhibited a CRBN-dependent pharmacological effect, displaying 13 to 15-fold more potent inhibition of survival of the WT cells than the CRBN−/− cells (Figures 1E and 1F).
Multiple kinases are degraded by TL12-186 in MOLM-14 and MOLT-4 cells
To identify the degradable targets of TL12-186, we employed a quantitative multiplexed proteomic approach to measure changes in protein abundance in an unbiased fashion (McAlister et al., 2014). We compared TL12-186 (100 nM) treatment to vehicle and TL13-27 (100 nM) controls in MOLM-14 cells. The non-CRBN-binding TL13-27 controlled for changes in protein abundance not mediated by the recruitment of CRBN. A 4-hour treatment time was chosen to capture the primary CRBN-dependent degradation response. We chose 100 nM as the treatment concentration because the maximal differential anti-proliferative effects were observed between WT and CRBN−/− MOLM-14 cells at this concentration (Figure 1E).
Upon TL12-186 treatment (see Differential Protein Abundance Analysis under STAR Methods), 14 out of 7559 identified proteins in MOLM-14 cells were significantly downregulated by at least 25% (false-discovery rate (FDR) adjusted P Value < 0.05). Notably, 12 out of the 13 degradable proteins (92.3%) were kinases: AAK1, AURKA, AURKB, BTK, CDK12, FLT3, FES, FER, PTK2, PTK2B, ULK1 and TEC (Figure 2A, Tables S2 and S3). This ratio is much higher than that of the total proteome (293 kinases out of 7559 identified proteins, 3.9%), consistent with TL12-186 acting as a kinase-targeting degrader.
Figure 2. Quantitative multiplexed proteomics revealed 28 kinases readily degradable by TL12-186 in two leukemic cell lines.
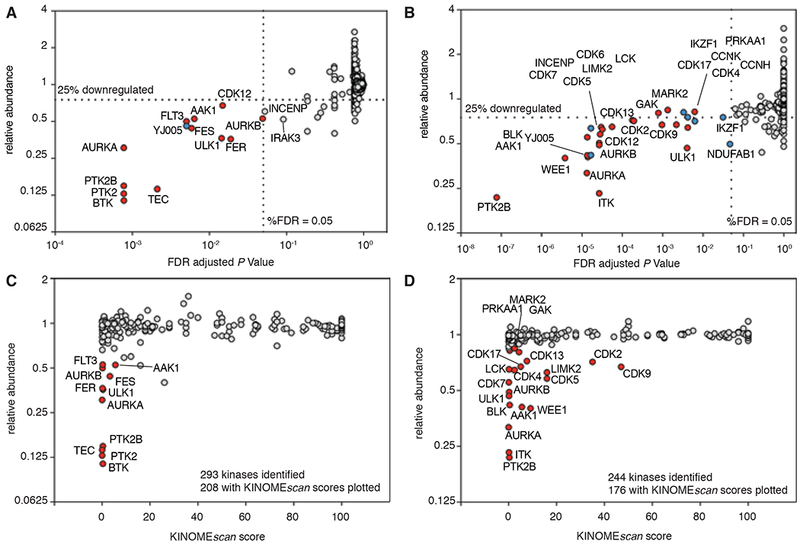
(A) Relative abundance of 7559 proteins in MOLM-14 cells when treated with TL12-186 (100 nM) for 4 hours, versus adjusted P Values (limma, duplicates, <5% FDR). Relative abundance was calculated using linear models for cells treated with TL12-186 compared to DMSO and TL13-27 controls. Red and blue circles represent significantly downregulated kinases and non-kinases, respectively. See also Tables S2 and S3.
(B) Relative abundance of 6609 proteins in MOLT-4 cells when treated with TL12-186 (100 nM) for 4 hours, versus adjusted P Values (limma, triplicates, <5% FDR). Otherwise, as in (A). See also Tables S4 and S5.
(C) Relative abundance, as in (A), of 208 kinases in MOLM-14 cells identified by mass spectrometry, versus their KINOMEscan scores. See also Tables S6 and S7.
(D) Relative abundance, as in (B), of 176 kinases in MOLT-4 identified by mass spectrometry, versus their KINOMEscan scores. See also Figure S1 and Tables S6.
To investigate the consistency of the degradable kinome by TL12-186 in a different cellular background, we performed a second proteomic analysis using MOLT-4 cells. In MOLT-4 cells, we found 23 out of 6609 identified proteins significantly downregulated by the same standard (Figure 2B, Tables S4 and S5). Five additional proteins were significantly downregulated (FDR adjusted P Value < 0.05), but to a lesser extent (0.75 < relative abundance < 0.85). Again, kinases were enriched among the degraded proteins (22 out of the 28, 78.6%): AAK1, AURKA, AURKB, BLK, CDK2, CDK4, CDK5, CDK6, CDK7, CDK9, CDK12, CDK13, CDK17, GAK, ITK, LCK, LIMK2, MARK2, PRKAA1, PTK2B, ULK1, and WEE1.
When comparing the repertoire of degraded kinases between the cell lines, MOLT-4 cells presented a longer list than MOLM-14 cells (22 kinases vs 12 kinases, Table 1). This was likely due to a higher replicate number in the MOLT-4 experiment, giving rise to better statistical power and fewer false negatives. Six kinases (PTK2B, AURKA, AURKB, AAK1, ULK1 and CDK12) were identified in both cell lines. Five and three degradable kinases were only identified in MOLM-14 cells (PTK2, BTK, TEC, FES and FER) and MOLT-4 cells (BLK, ITK and LCK), respectively. Among the 13 kinases that were significantly degraded (FDR adjusted P Value < 0.05) in MOLT-4 cells but not in MOLM-14 cells, 12 of them were reduced in abundance even though they did not meet the criteria for statistical significance (Table 1). FLT3 and LIMK2 were the only two kinases that showed discrepant degradability between the two cell lines. It is possible that FLT3 and LIMK2 were not degraded due to their lower protein levels in the respective cell lines, but further mechanistic investigation is needed to confirm the point.
Table 1.
Kinases significantly degraded by TL12-186 in MOLM-14 and/or MOLT-4 cells based on quantitative multiplexed proteomics
| MOLM-14 | MOLT-4 | ||||||
|---|---|---|---|---|---|---|---|
| Kinase | RAa | P Value | adjusted P Valueb | RAa | P Value | adjusted P Valueb | KINOMEscan scorec |
| PTK2B | 15% | 3.08E-07 | 7.76E-04 | 22% | 1.17E-11 | 7.71E-08 | 0.3 |
| AURKA | 30% | 2.49E-07 | 7.76E-04 | 32% | 5.97E-09 | 1.32E-05 | 0 |
| PTK2 | 13% | 2.84E-07 | 7.76E-04 | 0 | |||
| BTK | 11% | 4.12E-07 | 7.78E-04 | 0.3 | |||
| TEC | 14% | 1.40E-06 | 2.11E-03 | 0 | |||
| FLT3 | 50% | 4.41E-06 | 5.06E-03 | 99% | 9.52E-01 | 1.00E+00 | 0.15 |
| FES | 44% | 6.22E-06 | 5.88E-03 | 3.2 | |||
| AAK1 | 52% | 7.58E-06 | 6.36E-03 | 41% | 1.27E-08 | 1.40E-05 | 5.5 |
| ULK1 | 36% | 1.91E-05 | 1.44E-02 | 47% | 1.40E-05 | 4.02E-03 | 0.15 |
| CDK12 | 68% | 2.16E-05 | 1.48E-02 | 51% | 4.38E-08 | 2.67E-05 | |
| FER | 36% | 3.01E-05 | 1.90E-02 | 0.35 | |||
| AURKB | 53% | 8.42E-05 | 4.90E-02 | 49% | 4.45E-08 | 2.67E-05 | 0.25 |
| CDK13 | 80% | 4.17E-04 | 1.86E-01 | 72% | 4.28E-07 | 1.77E-04 | 7.6 |
| MARK2 | 73% | 4.74E-04 | 1.89E-01 | 84% | 4.03E-06 | 1.33E-03 | 2.4 |
| WEE1 | 59% | 7.71E-04 | 2.77E-01 | 40% | 1.15E-09 | 3.79E-06 | 9.1 |
| CDK7 | 66% | 8.88E-04 | 3.05E-01 | 55% | 1.01E-08 | 1.36E-05 | 0.1 |
| CDK17 | 68% | 1.88E-03 | 4.20E-01 | 67% | 6.94E-06 | 2.18E-03 | 5 |
| GAK | 83% | 1.68E-03 | 4.20E-01 | 81% | 2.13E-06 | 7.82E-04 | 4.3 |
| CDK5 | 74% | 1.74E-03 | 4.20E-01 | 58% | 5.13E-08 | 2.83E-05 | 16 |
| CDK6 | 85% | 2.01E-02 | 7.62E-01 | 65% | 5.88E-08 | 2.99E-05 | |
| CDK2 | 86% | 5.48E-03 | 7.62E-01 | 71% | 4.94E-07 | 1.92E-04 | 35 |
| CDK9 | 83% | 1.56E-02 | 7.62E-01 | 67% | 2.75E-06 | 9.56E-04 | 47 |
| CDK4 | 77% | 1.34E-02 | 7.62E-01 | 64% | 1.55E-05 | 4.19E-03 | 2.4 |
| PRKAA1 | 82% | 4.72E-03 | 7.62E-01 | 82% | 2.45E-05 | 6.22E-03 | 0.45 |
| LIMK2 | 110% | 4.80E-01 | 8.53E-01 | 63% | 6.80E-08 | 3.21E-05 | 16 |
| BLK | 42% | 1.03E-08 | 1.36E-05 | 0.45 | |||
| ITK | 23% | 3.69E-08 | 2.67E-05 | 0.15 | |||
| LCK | 65% | 1.27E-07 | 5.60E-05 | 0.15 | |||
RA stands for relative abundance.
Adjusted P Value represents P Value after correction for multiple-hypothesis testing using the Benjamini-Hochberg procedure.
TL12-186 was profiled at a concentration of 1 μM against a diverse panel of 468 kinases (390 non-mutants kinases). Scores are reported as a percent of the DMSO control (% control). Kinases without a score are not present in the KINOMEscan platform. The lower the score, the lower the Kd is likely to be, such that scores of zero represent strong binding to the kinase by the tested molecule.
Among the degraded non-kinase proteins, uncharacterized protein FLJ45252 was identified in both cell lines (Figures 2A and 2B). A few more non-kinase targets were identified in MOLT-4 cells, including INCENP, NDUFAB1, CCNH, CCNK, and IKZF1. The degradation of IKZF1, a known degradation target of the IMiDs (Kronke et al., 2014; Lu et al., 2014), suggests that pomalidomide can partially retain its IMiD functions despite linker attachment. We examined the presence of other known targets of pomalidomide and its derivatives in our proteomic studies, including IKZF3, casein kinase 1A1 (CK1α), and GSPT1 (Ito et al., 2010; Lu et al., 2014; Matyskiela et al., 2016). All these potential targets were detected (except for IKZF3 in MOLM-14 cells), but none were significantly degraded.
Engagement by TL12-186 does not guarantee degradation
When we compared the relative abundance of all identified kinases to their biochemical affinities towards TL12-186 based on KINOMEscan, we found that almost all degradable kinases are strongly bound by TL12-186, as expected (Figures 2C and 2D). The exceptions were CDK2 and CDK9, which appeared significantly degraded but are not strongly bound by TL12-186. The weak affinity of TL12-186 toward CDK2 and CDK9 is likely a false negative result due to a lack of cyclin partners in the binding assay. TL12-186 was confirmed to inhibit CDK2/cyclin A (IC50 = 73 nM) and CDK9/cyclin T1 (IC50 = 55 nM) using a biochemical substrate phosphorylation assay (Table S6). We found that many targets that are potently bound by TL12-186 are not degraded, suggesting that additional factors contribute to the degradability of kinases by TL12-186. Besides KINOMEscan, we further confirmed the binding of TL12-186 to a couple degraded and non-degraded kinases using biochemical assays optimized for each specific kinase target (Table S6).
To ensure that some non-degraded kinases are indeed engaged by TL12-186 in cells, we employed the KiNativ platform to assess the cellular kinase engagement profile of TL12-186. CRBN−/− MOLM-14 cells were treated with TL12-186 at 100 nM, 1 μM and 10 μM or DMSO for 4 hours, followed by cell lysis and labeling by the ATP-desthiobiotin probe. The CRBN−/− line was necessary for this experiment to prevent kinase degradation, which would complicate data interpretation. Two higher concentrations than 100 nM were selected because the interaction between kinases and reversible kinase binders may re-equilibrate after cell lysis, leading to an underestimation of kinase engagement. Competition between reversible kinase binders with the covalent ATP-desthiobiotin probe may also lead to an underestimation of engagement.
Only a handful of kinases were substantially engaged (>35%) by TL12-186 at 100 nM, including AURKA, AURKB, IRAK3, TEC and WEE1 (Table S7). All these kinases were detected and significantly degraded in our proteomic study (except for IRAK3, which was degraded but did not meet the criteria for significance). Meanwhile, many kinases that were significantly degraded, including BTK, FER and FES, were not strongly engaged at this concentration (Table S7). These results may support the idea that only partial target engagement is required to catalyze protein degradation. However, the KiNativ methodology may underestimate the degree of kinase engagement by the reversible TL12-186.
All kinases that were significantly degraded in MOLM-14 cells and detected by the KiNativ platform exhibited substantial engagement (>35%) when treated with 1 μM TL12-186 (Table S7). Thus, we think this is a reasonable concentration to assess cellular engagement of the non-degraded kinases. Indeed, many non-degraded kinases, including INSR, JAK1, JNK1, MST1, NEK9, SLK, TAK1 and TBK1, were substantially engaged by TL12-186, supporting the idea that target engagement by degrader molecules does not guarantee that the kinase will be degraded. To further support this conclusion, we examined how TL12-186 affects the protein levels of JAK1 and JAK2 when it inhibits IFN-γ-stimulated STAT1 phosphorylation mediated by JAK1/2. As expected, TL12-186 could inhibit STAT1 phosphorylation without causing JAK1/2 degradation (Figure S1).
Validation of CRBN-mediated kinase degradation induced by TL12-186 treatment
To ensure the reliability of our multiplexed proteomics study, we selected a subset of the identified degradable kinases for validation by Western blotting analysis. As expected, only TL12-186, but not its components pomalidomide or TL13-87, nor the negative control TL13-27, can cause the degradation of the 8 selected kinases in MOLM-14 cells (Figures 3A and S2A). For the kinases tested, the magnitude of degradation agreed well with our mass spectrometry-based analysis, with PTK2B, AURKA and BTK being strongly degraded, and FLT3, AURKB and CDK7 showing weaker responses (Figures 3A and S2A, Table 1). We were able to reverse the degradation effect of TL12-186 by pretreating MOLM-14 cells with carfilzomib, MLN4924, and pomalidomide, respectively, demonstrating that the degradation is dependent on the CRBN-mediated ubiquitin proteasome pathway (Figures 3B and S2B). Furthermore, we demonstrated that genetic loss of CRBN expression also negates the degradation effects of TL12-186, confirming the mechanism of action of the degrader (Figures 3C and S2C). We performed the same validation process in MOLT-4 cells, and confirmed that 10 of the kinases identified in the proteomic study are bona fide degradable targets mediated by CRBN and the proteosome (Figures 3D-F and S2D-F).
Figure 3. Western blotting validation of multi-kinase degradation induced by TL12-186, a CRBN-mediated proteosome-dependent process.
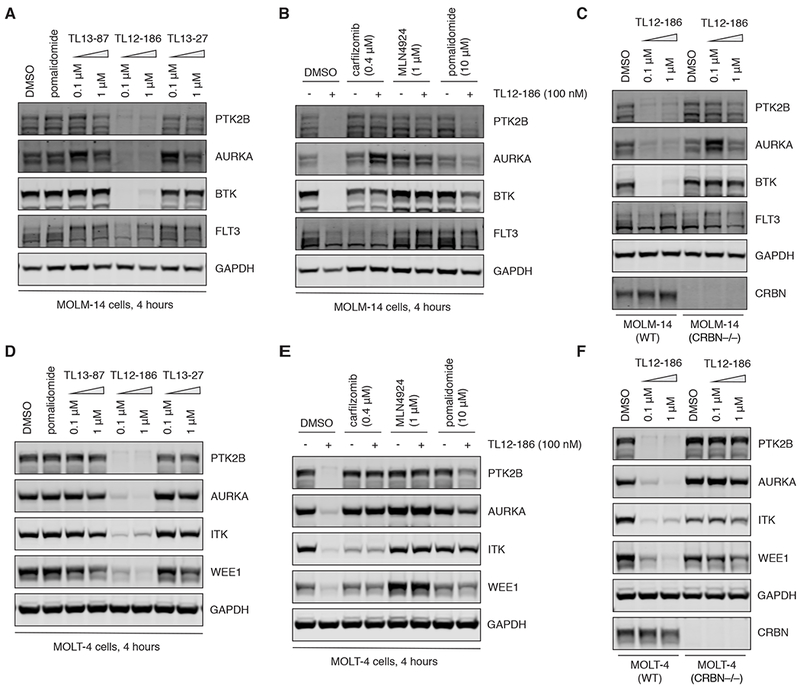
(A) Immunoblots for PTK2B, AURKA, BTK, FLT3 and GAPDH in MOLM-14 cells after 4-hour treatment of DMSO, pomalidomide, TL13-87, TL12-186 or TL13-27 at indicated concentrations. See also Figure S2A.
(B) Immunoblots for PTK2B, AURKA, BTK, FLT3 and GAPDH in MOLM-14 cells pre-treated with DMSO, carfilzomib, MLN4924 or pomalidomide for 4 hours, followed by 4-hour co-treatment with DMSO or TL12-186. See also Figure S2B.
(C) Immunoblots for PTK2B, AURKA, BTK, FLT3, GAPDH and CRBN in WT and CRBN−/− MOLM-14 cells after 4-hour treatment with DMSO or TL12-186. See also Figure S2C.
(D) Immunoblots for PTK2B, AURKA, ITK, WEE1 and GAPDH in MOLT-4 cells after the same treatment regimen described in (A). See also Figure S2D.
(E) Immunoblots for PTK2B, AURKA, ITK, WEE1 and GAPDH in MOLT-4 cells after the same treatment regimen described in (B). See also Figure S2E.
(F) Immunoblots for PTK2B, AURKA, ITK, WEE1, GAPDH and CRBN in WT and CRBN−/− MOLT-4 cells after 4-hour treatment with DMSO or TL12-186. See also Figure S2F.
We were unable to rescue the degradation of several kinase targets by carfilzomib because these targets were already downregulated upon treatment with carfilzomib, despite confirming CRBN dependence genetically. In particular, FLT3 in MOLM-14 cells, and ITK, ULK1, and CDK4/6 in MOLT-4 cells showed this trend. It was previously reported that FLT3-ITD is degraded upon proteosome inhibition due to upregulated autophagic flux (Larrue et al., 2016). We did not confirm whether autophagy is also responsible for downregulating these other kinases in MOLT-4 cells.
Development of FLT3-specific degrader
Among the degradable kinases discovered, FLT3 (particularly FLT3-ITD) was of great interest to us due to its well-credentialed role as an oncogenic driver in acute myelogenous leukemia (AML) (Smith et al., 2012). In approximately 30% of AML, FLT3 becomes constitutively activated due to point mutations (e.g. D835) or chromosomal translocations (e.g. FLT3-ITD) (Bacher et al., 2008; Levis, 2013). This has motivated the development of several clinical stage FLT3 kinase inhibitors including quizartinib (AC220) (Zarrinkar et al., 2009), crenolanib (Galanis et al., 2014; Zimmerman et al., 2013), and the recently approved midostaurin (Weisberg et al., 2002), among others (Wander et al., 2014). Using MOLM-14 cells, which harbor the FLT3-ITD translocation (Quentmeier et al., 2003), we confirmed the time-dependent degradation of FLT3 in response to TL12-186, which was apparent after 4-8 hours of treatment (Figure 4A). This response was slower than the observed degradation of AURKA, which occurred as early as 2 hours post treatment. We confirmed again the degradation effect is dependent on CRBN functions using MLN4924 and pomalidomide (Figure 4B). Since proteasome inhibition leads to paradoxical degradation of FLT3-ITD through autophagy (Larrue et al., 2016), we were unable to use carfilzomib to validate the dependency of FLT3 degradation on the proteasome as we did for AURKA and BTK (Figure 4B). We investigated the contribution of the lysosomal pathway for induced FLT3 degradation using bafilomycin A1 and chloroquine. Based on the increase in FLT3 levels upon autophagy inhibition, the lysosomal pathway is likely a constitutive pathway through which endogenous FLT3 proteins get turned over. However, autophagy inhibition cannot significantly rescue the degradation of FLT3 induced by TL12-186, suggesting that the lysosomal pathway is not primarily responsible for the induced FLT3 degradation (Figure 4B).
Figure 4. Design and characterization of AC220-based FLT3 degraders.
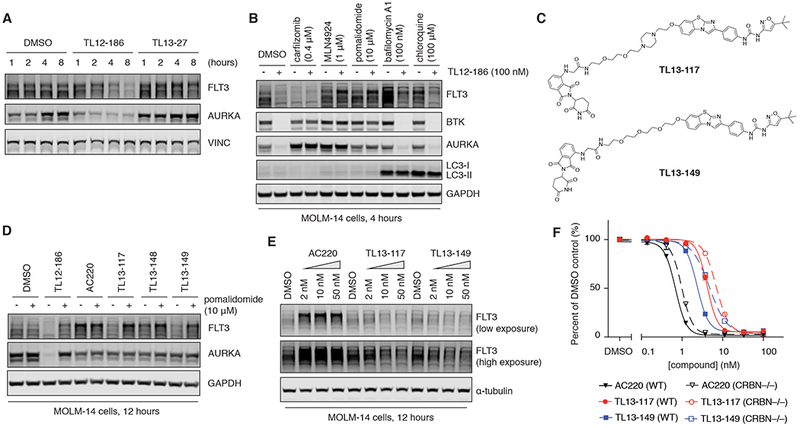
(A) Immunoblots for FLT3, AURKA and VINC in MOLM-14 cells treated with DMSO, TL12-186 (100 nM) or TL13-27 (100 nM) for the indicated amount of time.
(B) Immunoblots for FLT3, BTK, AURKA, LC3 and GAPDH in MOLM-14 cells pre-treated with DMSO, carfilzomib, MLN4924, pomalidomide, bafilomycin A1 or chloroquine for 4 hours, followed by 4-hour co-treatment with DMSO or TL12-186.
(C) Chemical structures of the two best AC220-based FLT3 degraders developed, TL13-117 and TL13-149. See also Figure S3A.
(D) Immunoblots for FLT3, AURKA, and GAPDH in MOLM-14 cells pre-treated with DMSO or pomalidomide for 4 hours, followed by 12-hour co-treatment with DMSO, TL12-186, AC220, TL13-117, TL13-148 or TL13-149 (all at 100 nM). See also Figures S3B and S3D.
(E) Immunoblots for FLT3 and α-tubulin in MOLM-14 cells treated with DMSO, AC220, TL13-117 or TL13-149 at indicated concentrations for 12 hours. See also Figure S3C and S3E.
(F) 2-day proliferation assays of WT and CRBN−/− MOLM-14 cells treated with AC220, TL13-117 or TL13-149. Values represent duplicate means ± SD. Error bars shorter than the height of the symbol are not drawn. See also Figure S3F.
We next sought to establish whether FLT3 could be efficiently degraded with a structurally distinct and more selective degrader molecule. We chose to build the degraders based on AC220, a well-characterized selective clinical-stage ATP-competitive FLT3 inhibitor (Zarrinkar et al., 2009). We used the available FLT3-AC220 crystal structure (PDB: 4XUF) to inform the site for installing a PEG linker, and 9 degraders with variable linker types and lengths were explored (Figures 4C and S3A). Unexpectedly, we observed that AC220 treatment in MOLM-14 cells led to a significant increase in FLT3 levels (Figures 4D and 4E), an effect that we postulate to occur via disruption of the control over FLT3 endocytosis and degradation. While several degraders demonstrated no effects on FLT3 degradation, likely due to suboptimal linker attachments and lengths, TL13-117 and TL13-149 caused FLT3 degradation to the endogenous levels or slightly below (Figures 4D, 4E, S3B and S3C). We confirmed the resultant FLT3 levels were the combined results of FLT3 upregulation and degradation by using pomalidomide to rescue the degradation effect (Figures 4D and S3B). Consistent with AC220 being a more selective kinase inhibitor relative to TL12-186, we observed that neither TL13-117 nor TL13-149 exhibited degradation of AURKA, BTK or PTK2B (Figures 4D and S3D). An extensive dose response analysis of TL13-117 revealed the hook effect, which is consistent with a ternary complex-mediated mechanism (Douglass et al., 2013), and demonstrated that concentrations between 10 and 100 nM resulted in the most efficient degradation (Figure S3E).
We next compared the antiproliferative properties of AC220 with the AC220-based degrader molecules and investigated whether they exhibited CRBN-dependent pharmacology. In both MOLM-14 and MV4-11 cells, AC220 exhibited the greatest anti-proliferative potency with an observed IC50 approximately 5-fold lower than TL13-117 and TL13-149 (Figures 4F and S3F). By comparing the sensitivity of WT and CRBN−/− cell lines to the degraders, we showed that the extent of FLT3 degradation achieved by TL13-117 and TL13-149 provided little improvement to their antiproliferative effects (Figures 4F and S3F).
Development of BTK-specific degrader
We next chose to develop selective degraders of BTK, a primary target of the covalent drug ibrutinib, which has demonstrated remarkable clinical efficacy in a variety of B cell-derived tumors including chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL) and Waldenström’s macroglobulinemia (WM) (Hendriks et al., 2014). Aberrant B-cell activities in several autoimmune diseases, including rheumatoid arthritis (Edwards et al., 2004), systemic lupus erythematosus (Navarra et al., 2011) and multiple sclerosis (Hauser et al., 2008), also motivate the development of new pharmacological strategies to block BTK functions.
First, we synthesized two distinct BTK degraders—one based on bosutinib, a cyanoquinoline BCR-ABL inhibitor that also inhibits the Src family and TEC family kinases, including BTK, and the other based on RN486, an aminopyridinone-based inhibitor that displays remarkable selectivity for BTK by occupying a selectivity pocket uniquely accessible in BTK (Lou et al., 2015; Remsing Rix et al., 2009). Again, available co-crystal structures of both compounds (PDB: 3UE4 and 4OTR) revealed the solvent exposed side of the inhibitor for linker installation. Both BTK degraders—CJH-005-067 (bosutinib-based) and DD-04-015 (RN486-based)—effectively degrade BTK after a 4-hour treatment (Figures 5A, 5B and S4A). An extensive dose response analysis of TL12-186 and DD-04-015 revealed that both degraders were most efficient at 100 nM (Figure S4B). We also confirmed that DD-04-015 selectively degrades BTK, but not other degradable targets of TL12-186, which is expected based on the selectivity of RN486 (Figure 5B).
Figure 5. Design and characterization of bosutinib and RN486-based BTK degraders.
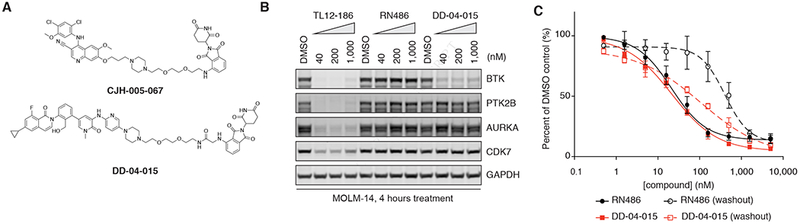
(A) Chemical structures of a bosutinib-based and an RN486-based BTK degrader, CJH-005-067 and DD-04-015, respectively.
(B) Immunoblots for BTK, PTK2B, AURKA, CDK7 and GAPDH in MOLM-14 cells treated with DMSO, RN486 or DD-04-015 at indicated concentrations for 4 hours. See also Figure S4.
(C) 3-day proliferation assays of TMD8 cells treated with RN486 or DD-04-015, with or without washout following 6 hours of treatment. Values represent triplicate means ± SD from one representative experiment. Error bars shorter than the height of the symbol are not drawn.
We next sought to determine whether the pharmacology of the degrader would be different from that of the conventional inhibitor. We compared the antiproliferative effects of DD-04-015 with RN486 on TMD8 cells, a diffuse large B-cell lymphoma line that is highly dependent on BTK activity for survival. After 3 days of treatment, we observed that RN486 and DD-04-015 demonstrated similar anti-proliferative potencies (Figure 5C). To evaluate the duration of response following inhibition versus degradation, we performed an experiment where cells were treated with RN486 or DD-04-015 for 6 hours, followed by compound washout. Cell viability following washout is lower in cells treated with DD-04-015 than those treated with RN486, which suggests that the degrader could exhibit prolonged pharmacodynamic effects, likely due to BTK degradation as its primary mode of action (Figure 5C). Taken together, selective BTK degraders such as DD-04-015 present a new pharmacological approach to treating BTK-dependent diseases.
DISCUSSION
The scope of the kinome that was investigated in this study was limited to the approximately 200 kinases targetable by TL12-186 and the kinases present in MOLM-14 and MOLT-4 cells. Furthermore, as warheads and linkers may also affect the efficiency of degraders (Bondeson et al., 2015; Lai et al., 2016; Zengerle et al., 2015), our study by no means defines the degradability of every kinase. Despite these caveats, we convincingly demonstrated how this strategy quickly expanded the number of kinases experimentally validated to be degradable, and enabled selection of targets for further optimization efforts.
Within the study, two systems were particularly useful for ensuring on-target mechanisms: (1) CRBN−/− cell lines and (2) negative degrader controls unable to bind CRBN. When combining the systems to examine the antiproliferative effects of TL12-186 versus TL13-27 in WT versus CRBN−/− cell lines, we demonstrated two important facts about the degraders. First, heterobifunctional degraders are less cell-permeable in comparison to its parental target binders. This is likely a result of their higher molecular weights, floppy structures, and possibly increased compound efflux. The lower intracellular concentration of the bivalent degrader molecules is also supported by our cell-based dual-luciferase assay, where thalidomide and pomalidomide exhibit more potent competition for intracellular CRBN than TL12-186. Second, the degraders can exhibit CRBN-dependent potentiation of their anti-proliferative effects due to degradation of essential genes. In certain cases, such as TL12-186 in MOLT-4 cells, multi-kinase degradation compensates for more than the loss of intracellular availability, leading to a stronger anti-proliferative effect than that of its parental inhibitor.
Interestingly, many degraded non-kinase proteins are associated with the degraded kinases. Specifically, INCENP is known to directly interact with Aurora B in the chromosomal passenger complex (Carmena et al., 2012). One possibility is that INCENP is less stable when its binding partner Aurora B is downregulated. It is also possible that INCENP is ubiquitinated due to its interaction and proximity to Aurora B. Two other components of the chromosomal passenger complex, borealin (CDCA8) and survivin (BIRC5), were unaffected. Borealin and survivin do not directly interact with Aurora B (Carmena et al., 2012), and the relative distance may explain why these two complex components are spared from degradation. As for uncharacterized protein FLJ45252, it is a putative protein encoded in the 3’UTR region of AAK1 kinase (Apweiler et al., 2004). Its identity and function have otherwise been poorly characterized, and it could be an isoform of AAK1 kinase, which would explain its degradability. CCNH and CCNK, similar to the case between INCENP and Aurora B, are perhaps degraded due to their interactions with CDK7 and CDK12, two degradable targets of TL12-186, respectively.
Based on the consistency of degradable kinases between the two cell lines, we postulate that the intrinsic degradability of a kinase is largely general across cell lines. However, differences may exist due to the variable proteomic compositions. It is possible that the proteins actively regulated by the ubiquitin-mediated degradation pathways are predisposed to the degrader strategy, as Aurora kinases, WEE1 and CDKs are among the degraded kinases we identified. We also consider the possibility that proteins endogenously regulated by the cullin-RING family of E3 ligases (CRL) are more susceptible to the CRBN-mediated degrader strategy as the E3 ligases are of the same class. For example, Aurora kinases (APC/C), ULK1 (Cul3-KLHL20) and WEE1 (SCFβ-TRCP) are all CRL substrates (Liu et al., 2016; Nguyen et al., 2005; Taguchi et al., 2002; Watanabe et al., 2004). Testing several other promiscuous degraders with different linker lengths and bifunctional ends in a variety of cell lines is necessary before we can examine these hypotheses. Factors such as protein half-life, stabilization by deubiquitinating enzymes (DUBs), distribution and density of surface lysines, compatibility of the molecular interface between the kinase and the ligase as well as cellular localization are all factors that deserve further investigation.
The success and relative ease of preparing selective FLT3 and BTK degraders using selective FLT3 and BTK binders support that our multi-kinase degrader approach indeed identifies the ‘low-hanging fruits’ of the degrader strategy. It also demonstrates an effective strategy to convert a promiscuous degrader to a selective one. Furthermore, none of the selective FLT3 or BTK degraders were derived from the 2,4-diaminopyrimidine scaffold, suggesting that the degradability of kinases are not necessarily sensitive to the scaffold of kinase binder employed, making our discoveries conveniently extensible.
Our evaluation of FLT3 degradation was complicated by an acute AC220-induced increase in FLT3 levels. A reversible increase of FLT3 level under prolonged treatment of FLT3 inhibitors was previously reported and attributed as a potential resistance mechanism (Weisberg et al., 2011). However, such short-term response has not been reported. Unfortunately, the extent of FLT3 degradation conferred by our best AC220-based FLT3 degraders did not translate to an improved anti-proliferative effect. One possible reason is that this increase in FLT3 levels is a dynamic signaling response that does not confer survival advantage. Another possibility is that we simply need a stronger degradation effect, one that brings FLT3 significantly below the endogenous level. Our future goal is to identify a more effective selective FLT3 degrader to study whether a FLT3 degrader confers longer-lasting antiproliferative response or prevents emergence of resistance.
The facile and strong degradation of BTK in BTK-expressing cell lines using degraders derived from three different scaffolds suggests that certain features of BTK make it especially susceptible to the degrader strategy. In fact, the TEC kinase family may all be especially susceptible to degradation based on our proteomics data, and it is of great importance to understand the reasons in future studies, which may involve crystallography and computational modeling.
In conclusion, our study demonstrates an efficient chemoproteomic strategy to survey the degradable kinome using a promiscuous kinase-directed heterobifunctional degrader. We quickly identified several well-validated therapeutic targets as tractable to this new mode of pharmacology, which may present advantages over traditional enzymatic inhibitors. Our study design can be applied beyond CRBN-mediated degraders to include the VHL-mediated degraders and any new E3 ligase recruiters. Furthermore, different promiscuous binders targeting other enzyme classes or domain-containing proteins can be investigated this way. We envision that a more comprehensive annotation of the degradability of the proteome may help elucidate the key factors that determine degradation efficiency, which is crucial for establishing the degrader strategy as a widely applicable and modular platform.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nathanael Gray (nathanael_gray@dfci.harvard.edu).
Experimental Model and Subject Details
Cell Lines
MOLM-14 and MV4-11 cells were authenticated using short tandem repeat (STR) profiling. Cell lines are routinely examined to be free of mycoplasma using the MycoAlert mycoplasma detection kit (Lonza, LT07-318). MOLM-14 (male), MOLT-4 (male) and MV4-11 (male) cells, and their CRBN−/− counterparts were maintained in RPMI medium 1640 (Cat#11875119; Thermo Fisher Scientific) supplemented with 10% FBS (Gibco) and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific). TMD8 (male) cells were maintained in IMDM media (Cat#12440061; Thermo Fisher Scientific) supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. 293FT (female) cells were maintained in DMEM media (Cat#11995073; Thermo Fisher Scientific) supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. All cell lines were cultured at 37 °C in a humidified chamber in the presence of 5% CO2.
Method Details
CRISPR-mediated CRBN Knockout
The CRBN gene-editing vectors were kindly provided by Dr. Georg Winter, which were generated by ligating linearized PX458 (BbsI-digested) with the annealed oligonucleotide pairs listed in KEY RESOURCES TABLE. MOLM-14 and MV4-11 cells were transfected with the CRBN-targeting CRISPR constructs using the Neon transfection system (1200V, 40ms, 1 pulse) (Thermo Fisher Scientific). GFP+ populations were single-cell sorted by FACS into 96-well plates. After 2-3 weeks, cell clones that grew up from each individual wells were expanded. CRBN-knockout clones were validated by both Western Blotting and PCR genotyping (Genewiz). The PCR primers used for genotyping are listed in KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Aurora A (D3E4Q) | Cell Signaling Technology | Cat#14475S; RRID: AB_2665504 |
| Rabbit polyclonal anti-Aurora B/AIM1 | Cell Signaling Technology | Cat#3094S; RRID: AB_10695307 |
| Rabbit monoclonal anti-BTK (D3H5) | Cell Signaling Technology | Cat#8547S; RRID: AB_10950506 |
| Rabbit monoclonal anti-CDK4 (D9G3E) | Cell Signaling Technology | Cat#12790S; RRID: AB_2631166 |
| Mouse monoclonal anti-CDK6 (DCS83) | Cell Signaling Technology | Cat#3136T; RRID: AB_2229289 |
| Mouse monoclonal anti-CDK7 (C-5) | Santa Cruz Biotechnology | Cat#sc-365075; RRID: AB_10845930 |
| Rabbit polyclonal anti-CRBN | Novus Biologicals | Cat#NBP191810; RRID: AB_11037820 |
| Mouse monoclonal anti-FER (5D2) | Cell Signaling Technology | Cat#4268S; RRID: AB_2278286 |
| Rabbit polyclonal anti-FLT3 (C-20) | Santa Cruz Biotechnology | Cat#sc-479; RRID: AB_631052 |
| Rabbit monoclonal anti-GAPDH (14C10) | Cell Signaling Technology | Cat#2118S; RRID: AB_561053 |
| Mouse monoclonal anti-ITK (2F12) | Cell Signaling Technology | Cat#2380S; RRID: AB_10692901 |
| Mouse monoclonal anti-JAK1 (D1T6W) | Cell Signaling Technology | Cat#50996S |
| Rabbit monoclonal anti-JAK2 (D2E12) | Cell Signaling Technology | Cat#3230S; RRID: AB_2128522 |
| Rabbit monoclonal anti-LCK (D88) | Cell Signaling Technology | Cat#2984T; RRID: AB_2136313 |
| Rabbit monoclonal anti-LC3A/B (D3U4C) | Cell Signaling Technology | Cat#12741T; RRID: AB_2617131 |
| Rabbit polyclonal anti-Pyk2 (H364) | Cell Signaling Technology | Cat#3090S; RRID: AB_2174095 |
| Mouse monoclonal anti-α-tubulin (DM1A) | Cell Signaling Technology | Cat#3873S; RRID: AB_1904178 |
| Mouse monoclonal anti-vinculin (V284) | Sigma-Aldrich | Cat#SAB4200080; RRID: AB_10604160 |
| Rabbit monoclonal anti-WEE1 (D10D2) | Cell Signaling Technology | Cat#13084T; RRID: AB_2713924 |
| Rabbit polyclonal anti-ULK1 (A705) | Cell Signaling Technology | Cat#4776S; RRID: AB_2212518 |
| Rabbit monoclonal anti-phospho-STAT1 (Y701) (58D6) | Cell Signaling Technology | Cat#9167S; RRID: AB_561284 |
| Rabbit monoclonal anti-STAT1 (D4Y6Z) | Cell Signaling Technology | Cat#14995S |
| Goat IRDye 680RD anti-Rabbit IgG (H+L), 0.5 mg | LI-COR Biosciences | Cat#926-68071; RRID: AB_10956166 |
| Goat IRDye 800CW anti-Mouse IgG (H+L), 0.5 mg | LI-COR Biosciences | Cat#926-32210; RRID: AB_621842 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete, Mini Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11836153001 |
| PhosSTOP Phosphatase Inhibitor Tablets | Sigma-Aldrich | Cat#04906837001 |
| recombinant human IFN-γ | R & D Systems | Cat#285-IF-100 |
| dTAG-7 | Erb et al., 2017 | N/A |
| MLN4924 | Active Biochem | Cat#A-1139 |
| pomalidomide | Selleckchem | Cat#S1567 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| AlphaScreen Streptavidin Donor beads | PerkinElmer | Cat#6760002 |
| Nickel Chelate AlphaLISA Acceptor beads | PerkinElmer | Cat#AL108 |
| Deposited Data | ||
| Multiplexed Proteomics data in MOLM-14 cells | This paper |
https://www.ebi.ac.uk/pride/archive/ PRIDE: PXD007851 |
| Multiplexed Proteomics data in MOLT-4 cells | This paper |
https://www.ebi.ac.uk/pride/archive/ PRIDE: PXD007861 |
| Original, unprocessed data | This paper | http://dx.doi.org/10.17632/wbbbfhg5bj.1 |
| Experimental Models: Cell Lines | ||
| Human: MOLM-14 cells (male) | Laboratory of Scott Armstrong | RRID:CVCL_7916 |
| Human: MOLT-4 cells (male) | Laboratory of James Bradner | RRID:CVCL_0013 |
| Human: MV4-11 cells (male) | Laboratory of Scott Armstrong | RRID:CVCL_0064 |
| Human: TMD8 cells (male) | Belfer Center for Applied Cancer Science | RRID:CVCL_A442 |
| Human: 293FT cells (female) | ATCC | RRID:CVCL_6911 |
| Truchoplusia ni: High Five cells | ThermoFisher Scientific | Cat#B85502; RRID: CVCL_C190 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| sgCRBN-494 Forward 5′-CACCGTAAACAGACATGGCCGGCGA-3′ | Laboratory of James Bradner | N/A |
| sgCRBN-494 Reverse 5′-AAACTCGCCGGCCATGTCTGTTTAC-3′ | Laboratory of James Bradner | N/A |
| sgCRBN-496 Forward 5′-CACCGATATGCCTATCGAGAAGAAC-3′ | Laboratory of James Bradner | N/A |
| sgCRBN-496 Reverse 5′-AAACGTTCTTCTCGATAGGCATATC-3′ | Laboratory of James Bradner | N/A |
| Genotyping primer Forward (for locus targeted by 494): 5′-CGTGAAGCAGCTTTCCGGTG-3′ | Laboratory of James Bradner | N/A |
| Genotyping primer Reverse (for locus targeted by 494): 5′-GCAGGGGACGGCGTTCTGTC-3′ | Laboratory of James Bradner | N/A |
| Genotyping primer Forward (for locus targeted by 496): 5′-GATGCATAGCAAGGTTGGAAACCAC-3′ | Laboratory of James Bradner | N/A |
| Genotyping primer Reverse (for locus targeted by 496): 5′-CAACGCAGCGAGACTCCATC-3′ | Laboratory of James Bradner | N/A |
| Recombinant DNA | ||
| Plasmid: pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene Plasmid #48138 |
| Plasmid: pFastBac vectors encoding CRBN and DDB1 | Fischer et al., 2014; Laboratory of Nicolas Thomä | N/A |
| Plasmid: Nluc/Fluc plasmid | Lu et al., 2015; Laboratory of Willian Kaelin | N/A |
| Software and Algorithms | ||
| R | R Core Team, 2016 | https://www.r-project.org/ |
| Limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| GraphPad Prism 7.0a | GraphPad Software, Inc. | http://www.graphpad.com/ |
| Other | ||
Immunoblotting
Cells were lysed with RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate, pH 7.4) supplemented with protease inhibitor cocktails (cOmplete, Roche) and phosphatase inhibitor cocktails (PhosSTOP, Roche) at 4°C for at least 30 minutes. C lear lysates were collected after max speed centrifugation at 4°C for 10 minutes, and were analyzed for protein concentration using Pierce BCA protein assay (Thermo Fisher Scientific). Lysate concentrations are normalized, mixed with 4X NuPAGE LDS sample buffer (supplemented with 10% fresh 2-mercaptoethanol; Thermo Fisher Scientific), and denatured at 95C for 10 minutes. Equal amount of p roteins (20–40 μg) was resolved by Bolt 4-12% Bis-Tris Plus gels, and then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% non-fat milk in TBS-T (TBS with 0.2% Tween-20), followed by incubation with primary antibodies at 4°C overnight. Next day, after washing with TBS-T, the membrane was incubated with fluorophore-conjugated secondary antibodies for 1 hour at room temperature. The membrane was then washed and scanned with an Odyssey Infrared scanner (Li-Cor Biosciences, Lincoln, NE).
CRBN-DDB1 Expression and Purification
pFastBac vectors encoding human CRBN (residues 1-442) and DDB1 (residues 1-1140) were a kind gift from Nicolas Thomä and Eric Fischer (Fischer et al., 2014).
Recombinant baculoviruses were prepared according to the manufacturer’s protocol. N-terminal His6-tagged DDB1 and CRBN were expressed in High Five cells (Invitrogen). For purification, cells were re-suspended in lysis buffer containing 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.25 mM TCEP (tris(2-carboxyethyl)phosphine), 1 mM PMSF, and 1 tablet/500 ml protease inhibitor cocktail (Sigma), and lysed by sonication. The lysate was clarified by ultracentrifugation and proteins purified using Ni-NTA affinity purification. Size exclusion chromatography (SEC) (HiLoad 16/60 Superdex 200; GE Healthcare) in 50 mM HEPES pH 7.4, 200 mM NaCl and 0.25 mM TCEP yielded proteins without visible contamination (Coomassie-stained SDS-PAGE). The purified protein was concentrated using “Amicon Ultra” spin concentration devices, flash frozen in liquid nitrogen and stored at −80°C until further use.
CRBN-DDB1 AlphaScreen Engagement Assay
Assays were performed with minimal modifications from the manufacturer’s protocol (PerkinElmer, USA). All reagents were diluted in 50 mM HEPES, 150 mM NaCl, 0.1% w/v BSA, 0.01% w/v Tween20, pH 7.5 and allowed to equilibrate to room temperature prior to addition to plates. After addition of Alpha beads to master solutions all subsequent steps were performed under low light conditions. A 2X solution of components with final concentrations of CRBN-DDB1 (see protein expression section) at 50 nM and 125 nM biotinylated-thalidomide (see bio-thal under the synthesis section) was added in 20 μL to 384-well plates (AlphaPlate-384, PerkinElmer, USA). Plates were spun down at 150 × g, 100 nL of compound in DMSO from stock plates were added by pin transfer using a Janus Workstation (PerkinElmer, USA). The streptavidin-coated donor beads and Ni-coated acceptor beads (both at 10 μg/ml final) were added as with the previous solution in a 2X, 20 μL volume. Following this addition, plates were sealed with foil to prevent light exposure and evaporation. The plates were spun down again at 150 × g. Plates were incubated at room temperature for 1 hour and then read on an Envision 2104 (PerkinElmer, USA) using the manufacturer’s protocol.
Cellular CRBN Engagement Assay
A lentiviral construct comprising firefly luciferase (Fluc) and FKBP12F36V fused to nanoluciferase (FKBP12F36V-Nluc) was used to produce virus and transduce 293FT cells. Nluc/Fluc plasmids were a kind gift from Dr. William Kaelin. For intracellular CRBN competition assays, cells were dispensed into white 384-well culture plates (Thermo) in 20 μL at 4000 cells / well. The cells were incubated overnight and pinned with 100 nL of competitor compound in DMSO followed by 100 nL of 16 uM dTAG-7 (Erb et al., 2017) using a JANUS workstation (PerkinElmer). Cells were incubated with compounds for 5.5 hours at 37 °C, 5% CO2 and then allowed to cool to room temperature for 30 minutes. Following incubation, 25 μL/well Fluc buffer (200 mM Tris, 15 mM MgSO4, 100 uM EDTA, 1 mM DTT, 1 mM ATP, 200 uM Coenzyme A, 400 uM D-Luciferin, 0.1% Trition X-100) was added to each plate, incubated for 15 minutes at room temperature, and then read on an Envision 2104 (PerkinElmer) for luminescence. 25 μL of Nluc buffer (25 mM Na4PPi, 10 mM NaOAc, 15 mM EDTA, 500 mM NaSO4, 500 mM NaCl, 16 uM coelenterazine, 50 uM 4-(6-methyl-1,3-benzothiazol-2-yl)aniline [sc-276812]) was then added to each well and the plate incubated for 15 minutes at room temperature before a second read. Nluc values were normalized by setting values from dTAG-7-only wells to 0% and those treated with DMSO to 100%. Data were further analyzed and plotted using GraphPad PRISM v6 and IC50 values were determined using the ‘log(agonist) vs normalized response –variable slope’ analysis module.
Cell Proliferation Assay
For 2-day proliferation assays of MOLM-14, MOLT-4, MV4-11 cells, and their CRBN−/− counterparts, 100 μL of 5 × 105 cells/mL cells were seeded per 96-well one day before compound treatment (in duplicates). For 3-day proliferation assay of TMD8 cells, 100 μL of 1 × 105 cells/mL cells were seeded per 96-well and treated with compounds at indicated concentrations (5 μM to 0.5 nM in triplicates) for 3 days. Washout assays were performed in the same way as the 3-day proliferation assay described above, except that after 6 hours of drug treatment, cells were washed 3 times with culture medium without drugs, and allowed to grow in culture medium without drugs for the remaining time. Relative cell numbers were assessed using CellTiter-Glo assay kits (Promega, G7571) as described in product manual by luminescence measurements on an Envision plate reader (PerkinElmer). IC50 values were determined using GraphPad Prism v6 nonlinear regression curve fit.
IFN-γ-stimulated STAT1 Phosphoryation
MOLT-4 cells were pre-treated with DMSO or TL12-186 for 3.5 hours, followed by IFN-γ (10 ng/mL; cat# 285-IF-100, R&D Systems) stimulation for 30 minutes. After a total of 4 hours, cells were pelleted by centrifugation at 130 × g for 3 minutes, washed with ice-cold PBS once, pelleted (130 × g for 3 minutes) again, and analyzed as described in the immunoblotting section.
Sample Preparation for Live-cell KiNativ
ATP and ADP acyl-nucleotide probes were synthesized as described previously (Patricelli et al., 2007). MOLM-14 cells were treated in duplicates with DMSO and in singlicates with TL12-186 (0.1 μM, 1.0 μM and 10 μM) for 4 hours. Cells were washed with ice-cold PBS once, pelleted, and snap-frozen with liquid nitrogen. The cell pellets were lysed by sonication in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton-X-100, phosphatase inhibitors (Cocktail II AG Scientific #P-1518)). After lysis, the samples were cleared by centrifugation, and the supernatant collected for probe labeling. For live cell treatment, 50 μL of a 10× aqueous solution of the desthiobiotin-ATP probe was added to each sample for a final probe concentration of 20 μM, and samples were incubated with probe for 10 minutes.
Samples were prepared for MS analysis as described previously (Patricelli et al., 2011). Briefly, probe-labeled lysates were denatured and reduced (6 M urea, 10 mM DTT, 65°C, 15 min), alkylated (40 mM iodoacetamide, 37°C, 30 min), and gel filtered (Biorad Econo-Pac® 10G) into 10 mM ammonium bicarbonate, 2 M urea, 5 mM methionine. The desalted protein mixture was digested with trypsin (0.015 mg/ml) for 1 hour at 37°C, and desthiobiotinylated peptides captured using 12.5 μl high-capacity streptavidin resin (Thermo Scientific). Captured peptides were then washed extensively, and probe-labeled peptides eluted from the streptavidin beads using two 35μl washes of a 50% CH3CN/water mixture containing 0.1% TFA at room temperature.
Sample Preparation for Proteomics Study
MOLM-14 or MOLT-4 cells were treated with DMSO, TL12-186, or TL13-27. MOLM-14 cells were treated in duplicates with DMSO, TL12-186 (0.1 μM and 1.0 μM) or TL13-27 (0.1 μM and 1.0 μM) for 4 hours. MOLT-4 cells were treated in triplicates with DMSO, TL12-186 (0.1 μM) or TL13-27 (0.1 μM) for 4 hours. Cells were washed with ice-cold PBS once, and lysed in 1 mL of lysis buffer (8 M urea, 1% SDS, 50 mM Tris pH8.5 supplemented with protease and phosphatase inhibitors). A micro-BCA assay (Pierce) was used to determine the protein concentration of each sample. A total of 1200 μg of proteins from each sample was depleted of abundant proteins using two Pierce Top 12 protein depletion spin columns. Eluted proteins were combined, and protein concentration was determined by micro-BCA. Proteins were precipitated on ice for 1 hour with 13% trichloroacetic acid (TCA), washed twice with acetone, and pellets were allowed to air dry. Proteins were resuspended in 6 M urea, 50 mM Tris pH 8.5, reduced with 5 mM dithiothreitol (DTT) at room temperature for 1 hour, and alkylated with 15 mM iodoacetamide in the dark at room temperature for 1 hour. Alkylation was quenched with DTT. Urea concentration was reduced to 4 M and proteins were digested with LysC (1:50; enzyme:protein) for 6 hours at 25°C. The Lys C digestion was diluted to 1 M urea, 50 mM Tris pH 8.5 and then digested with trypsin (1:100; enzyme:protein) overnight at 37°C. The reaction was quenched with 1% formic acid , subjected to C18 solid-phase extraction (Sep-Pak, Waters) and vacuum-centrifuged to near-dryness. Dried peptides were re-suspended in 200 mM EPPS pH 8.0. Peptide quantification was performed using the micro-BCA assay (Pierce). The same amount of peptide from each condition was labeled with tandem mass tag (TMT) reagent (1:4; peptide:TMT label) (Pierce). The 10-plex (only 9 labels were used for the MOLT-4 experiment) labeling reactions were performed for 2 hours at 25°C. Modification of tyro sine residue with TMT was reversed by the addition of 5% hydroxyl amine for 15 minutes at 25°C. The reaction was quenched with 0.5% trifluoroactic acid (TFA) and samples were combined at a 1:1:1:1:1:1:1:1:1:1 ratio. Mixed and labeled peptides were solubilized in buffer A (5% ACN, 50 mM ammonium bicarbonate, pH 8.0) and subjected to high-pH reverse-phase HPLC fractionation on an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm i.d., and 20 cm in length). Using an Agilent 1100 binary pump equipped with a degasser and a photodiode array (PDA) detector, a 50 min linear gradient from 18% to 45% acetonitrile in 50 mM ammonium bicarbonate pH 8 (flow rate of 0.8 mL/min) separated the peptide mixture into a total of 96 fractions. The 96 fractions were consolidated into 24 samples, acidified with 10% formic acid, and vacuum-dried. Each sample was re-dissolved with 5% formic acid/5% ACN, desalted via StageTip, dried via vacuum centrifugation, and reconstituted for LC-MS3 analysis.
Liquid Chromatography-MS3 Spectrometry
12 of the 24 peptide fractions from the high-pH reverse-phase fractionation (every other fraction) were analyzed with an LC-MS3 data collection strategy (McAlister et al., 2014) on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) equipped with a Proxeon Easy nLC 1000 for online sample handling and peptide separations. Approximately 5 μg of peptide resuspended in 5% formic acid + 5% acetonitrile was loaded onto a 100 μm inner diameter fused-silica micro capillary with a needle tip pulled to an internal diameter less than 5 μm. The column was packed in-house to a length of 35 cm with a C18 reverse-phase resin (GP118 resin 1.8 μm, 120 Å, Sepax Technologies). The peptides were separated using a 180 min linear gradient from 3% to 25% buffer B (100% acetonitrile (ACN) + 0.125% formic acid) equilibrated with buffer A (3% ACN + 0.125% formic acid) at a flow rate of 400 nL/min across the column. The scan sequence for the Fusion Orbitrap began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 400–1400 m/z scan range, AGC target 2 × 105, maximum injection time 100 ms, dynamic exclusion of 90 seconds). The “Top10” precursors was selected for MS2 analysis, which consisted of CID (quadrupole isolation set at 0.5 Da and ion trap analysis, AGC 8 × 103, NCE 35, maximum injection time 150 ms). The top ten precursors from each MS2 scan were selected for MS3 analysis (synchronous precursor selection), in which precursors were fragmented by HCD prior to Orbitrap analysis (NCE 55, max AGC 1 × 105, maximum injection time 150 ms, resolution 60,000.
For the KiNativ target engagement assay, samples were analyzed by LC-MS/MS as described (Patricelli et al., 2011). Briefly, samples were analyzed on Thermo LTQ ion trap mass spectrometers coupled with Agilent 1100 series micro-HPLC systems with autosamplers, essentially as described, using a custom target list comprising 352 unique kinase peptides that had been previously identified during the characterization of various in data dependent mode (Nomanbhoy et al., 2016; Patricelli et al., 2011).
General Information for Chemical Synthesis
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on Bruker Avance (400 MHz) or Bruker A500 (500 MHz), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5μM) using a gradient of 15-95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis. Details of chemical synthesis and the relevant NMR spectra are included in Method S1.
Quantification and Statistical Analysis
LC-MS3 Data Quantification and Analysis
A suite of in-house software tools were used to for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides as previously described. MS/MS spectra were searched against a Uniprot human database (February 2014) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 50 ppm, fragment ion mass tolerance of 1.0 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.162932 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (<200 summed signal-to-noise across 10 channels and <0.5 precursor isolation specificity).
For signal extraction/quantification of the KiNativ experiment, typically up to four ions were selected for based on their presence, intensity, and correlation to the reference MS/MS spectrum. The resulting chromatographic peaks from each run were then integrated and the integrated peak areas used to determine % inhibition values relative to control runs. For each peptide quantitated, the MS signal for the adult myocyte samples relative to the MS signal for the neonatal myocyte samples was expressed as fold-change using the following equation:
All data points were visually verified, as were all data points showing variability outside of normal limits. Significance of data points changing more than 2-fold were determined according to the Student T-test (one-tailed distribution, two-sample equal variance). In cases where scores <0.04, the fold-change was considered significant. In cases where the peptide signals in the adult myocyte samples were undetectable (noise), the fold-change value was preceded by “>“.
Differential Protein Abundance Analysis
In the first experiment (MOLM-14 cells) we included two concentrations for the degrader compounds (100 nM and 1 μM) to assess the optimal concentration for this experiment. When inspecting the data, we observed reduced degradation for most targets at 1 μM (known as the hook effect and common to heterobifunctional degraders; consistent with the dose response of TL12-186 on BTK shown in Figure S4B) and the 1-μM concentration points were therefore subsequently excluded from downstream analysis and not presented in detail. We normalized and scaled (as described below) the full dataset before excluding the 1-μM data points prior to statistical analysis.
All data analysis was carried out using the R statistical framework (R Core Team, 2016). Reporter ion intensities from all channels were normalized (normalization factors calculated by dividing the summed intensities of each channel by the maximum value of all channels, and subsequently applied to each reporter ion value) and scaled (scaling the sum of reporter ion intensities to 100 for each protein/peptide) using in house scripts. Proteins quantified with a minimum of 2 unique peptides were considered for downstream analysis. Log2-transformed, scaled and normalized reporter ion intensities were analyzed using a linear model approach implemented in the limma package (Ritchie et al., 2015), which allows modeling of both, the general changes induced by TL12-186 or TL13-27 relative to vehicle (DMSO), and the specific effect of TL12-186 on protein abundance. The resulting data was subjected to a moderated t-test to assess statistical significance also implemented in the limma package. In the moderated t-test, the standard errors are more robust calculated using an empirical Bayes method inferring information across all proteins. The nominal P Values are further corrected for multiple testing (termed FDR adjusted P Value in this study) by controlling for the false discovery rate (FDR), as proposed by Benjamini and Hochberg (Reiner et al., 2003). The “topTable” output from limma is provided as Table S3 and S5, and contains the values “logFC” (log2-transformed fold change of TL12-187 to DMSO/TL13-27 controls), “AveExpr” (not interpretable in the study design), “t” (moderated t-statistics), “P.Value” (nominal P Value), “adj.P.Val” (FDR adjusted P Value, corrected for multiple hypothesis testing) and “B” (B-statistics; log2-transformed odds that a protein is significantly decreased upon drug treatment). Additional columns appended to the table were “isKinase” (Boolean identity about whether the gene is a kinase) and “Kinomescan” (KINOMEscan score of TL12-186 on the associated kinase).
Statistical Analysis
Statistical tests and the associated error bars are identified in the corresponding figure legends. Typical replicate numbers describe the number of technical replicates analyzed in a single experiment. “Independent” replicate numbers describe the number of biological replicates, which were repeat experiments performed on different days. Data met the assumptions for all tests used.
Data and Software Availability
The mass spectrometry raw data files for quantitative multiplexed proteomics have been deposited in the PRIDE Archive under the accession numbers PXD007851 (MOLM-14) and PXD007861 (MOLT-4). Original data not included in supplemental tables are deposited to Mendeley Data under this link: http://dx.doi.org/10.17632/wbbbfhg5bj.1.
Supplementary Material
KINOMEscan kinase selectivity profiles of TL12-186 and TL13-27. Related to Figure 1B: Table S1
Raw data from the multiplexed proteomics study in MOLM-14 cells. Related to Figure 2A: Table S2
Statistical summary of the multiplexed proteomics study in MOLM-14. Related to Figure 2A: Table S3
Raw data from the multiplexed proteomics study in MOLT-4 cells. Related to Figure 2B: Table S4
Statistical summary of the multiplexed proteomics study in MOLT-4 cells. Related to Figure 2B: Table S5
KiNativ profiling of TL12-186 in CRBN−/− MOLM-14 cells. Related to Figure 2C: Table S7
SIGNIFICANCE.
Despite the promise that small molecule degraders can be developed for any protein of interest, we have demonstrated that there are considerable differences in the intrinsic “degradability” of different kinase targets. This study provides a road map to systemically survey susceptible targets in a gene family-focused fashion. By using a multi-targeted kinase degrader, we rapidly discovered degradable kinase targets of therapeutic importance and then demonstrated how more selective degraders can be efficiently generated.
Highlights.
Promiscuous degrader as a strategy to triage a gene family for tractable targets
Efficient degradation induced by degraders requires more than target engagement
Therapeutic targets BTK and FLT3 are susceptible to targeted degradation
A selective BTK degrader demonstrates prolonged pharmacodynamic effects
ACKNOWLEDGEMENTS
We thank Ryan C. Kunz and Rachel B. Rodrigues for performing the quantitative proteomics studies at the Thermo Fisher Scientific Center for Multiplexed Proteomics at Harvard Medical School. We thank Georg E. Winter for generating and providing the CRBN-targeting CRISPR vectors and CRBN−/− MOLT-4 cell line. We also thank Fleur M. Ferguson for critical reading of the manuscript. D.L.B. is a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2196-14). This work was supported by a grant of the Daegu-Gyeongbuk Medical Innovation Foundation New Drug Development Center R&D Project (DG15C011) for J.-H.C., E.K. and H.G.C. Financial support for this work was provided by NIH grant NCI R01CA214608 (E.S.F.). N.S.G. is a founder of C4 Therapeutics, which has licensed degrader related intellectual property from DFCI. J.E.B. is a Scientific Founder of Syros Pharmaceuticals, SHAPE Pharmaceuticals, Acetylon Pharmaceuticals, Tensha Therapeutics (now Roche) and C4 Therapeutics and is the inventor on IP licensed to these entities. J.E.B. is now an executive and shareholder in Novartis AG. E.S.F. is a member of the scientific advisory board of C4 Therapeutics and a consultant to Novartis AG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, et al. (2004). UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32, D115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacher U, Haferlach C, Kern W, Haferlach T, and Schnittger S (2008). Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood 111, 2527–2537. [DOI] [PubMed] [Google Scholar]
- Barouch-Bentov R, and Sauer K (2011). Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs 20, 153–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschelli DH, Ye F, Wang YD, Dutia M, Johnson SL, Wu B, Miller K, Powell DW, Yaczko D, Young M, et al. (2001). Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem 44, 3965–3977. [DOI] [PubMed] [Google Scholar]
- Bossi RT, Saccardo MB, Ardini E, Menichincheri M, Rusconi L, Magnaghi P, Orsini P, Avanzi N, Borgia AL, Nesi M, et al. (2010). Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 49, 6813–6825. [DOI] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, and Crews CM (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134, 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M, Wheelock M, Funabiki H, and Earnshaw WC (2012). The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 13, 789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CenterWatch (2017). FDA Approved Drugs. [Google Scholar]
- Deshaies RJ (2015). Protein degradation: Prime time for PROTACs. Nat Chem Biol 11, 634–635. [DOI] [PubMed] [Google Scholar]
- Douglass EF Jr., Miller CJ, Sparer G, Shapiro H, and Spiegel DA (2013). A comprehensive mathematical model for three-body binding equilibria. J Am Chem Soc 135, 6092–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, and Shaw T (2004). Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 350, 2572–2581. [DOI] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, et al. (2014). Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, and Ciulli A (2017). Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, and Levis M (2014). Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 123, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, Hewitt S, Dias DM, and Ciulli A (2014). Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem 57, 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, et al. (2007). Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A 104, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, and Nijhawan D (2017). Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, et al. (2008). B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358, 676–688. [DOI] [PubMed] [Google Scholar]
- Hendriks RW, Yuvaraj S, and Kil LP (2014). Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer 14, 219–232. [DOI] [PubMed] [Google Scholar]
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, and Handa H (2010). Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Ishikawa M, Naito M, and Hashimoto Y (2010). Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc 132, 5820–5826. [DOI] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, and Crews CM (2016). Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrue C, Saland E, Boutzen H, Vergez F, David M, Joffre C, Hospital MA, Tamburini J, Delabesse E, Manenti S, et al. (2016). Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 127, 882–892. [DOI] [PubMed] [Google Scholar]
- Levis M (2013). FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology Am Soc Hematol Educ Program 2013, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Lin YC, Chen YH, Chen CM, Pang LY, Chen HA, Wu PR, Lin MY, Jiang ST, Tsai TF, et al. (2016). Cul3-KLHL20 Ubiquitin Ligase Governs the Turnover of ULK1 and VPS34 Complexes to Control Autophagy Termination. Mol Cell 61, 84–97. [DOI] [PubMed] [Google Scholar]
- Lou Y, Han X, Kuglstatter A, Kondru RK, Sweeney ZK, Soth M, McIntosh J, Litman R, Suh J, Kocer B, et al. (2015). Structure-based drug design of RN486, a potent and selective Bruton’s tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. J Med Chem 58, 512–516. [DOI] [PubMed] [Google Scholar]
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong KK, Bradner JE, and Kaelin WG Jr. (2014). The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. (2015). Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, et al. (2016). A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, Leon MG, et al. (2011). Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 377, 721–731. [DOI] [PubMed] [Google Scholar]
- Nguyen HG, Chinnappan D, Urano T, and Ravid K (2005). Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol Cell Biol 25, 4977–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomanbhoy TK, Sharma G, Brown H, Wu J, Aban A, Vogeti S, Alemayehu S, Sykes M, Rosenblum JS, and Kozarich JW (2016). Chemoproteomic Evaluation of Target Engagement by the Cyclin-Dependent Kinase 4 and 6 Inhibitor Palbociclib Correlates with Cancer Cell Response. Biochemistry 55, 5434–5441. [DOI] [PubMed] [Google Scholar]
- Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, Zhang T, Kwiatkowski N, Boukhali M, Green J, et al. (in press). Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning L, Krivi G, and Fitzpatrick FA (1991). Leukotriene A4 hydrolase. Inhibition by bestatin and intrinsic aminopeptidase activity establish its functional resemblance to metallohydrolase enzymes. J Biol Chem 266, 1375–1378. [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. (2011). In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol 18, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, and Kozarich JW (2007). Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry 46, 350–358. [DOI] [PubMed] [Google Scholar]
- Quentmeier H, Reinhardt J, Zaborski M, and Drexler HG (2003). FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 17, 120–124. [DOI] [PubMed] [Google Scholar]
- R Core Team (2016). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, et al. (2016). PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, and Benjamini Y (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375. [DOI] [PubMed] [Google Scholar]
- Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastierdi S, Wuhr M, Dhe-Paganon S, Armstrong SA, et al. (2017). Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chem Int Ed Engl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remsing Rix LL, Rix U, Colinge J, Hantschel O, Bennett KL, Stranzl T, Muller A, Baumgartner C, Valent P, Augustin M, et al. (2009). Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 23, 477–485. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth AR, Pucheault M, Tae HS, and Crews CM (2008). Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett 18, 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K, Takubo K, Kikuchi R, Nishimoto M, Kitagawa M, Abe F, Nishikawa K, Tsuruo T, and Naito M (2008). Small molecules destabilize cIAP1 by activating auto-ubiquitylation. J Biol Chem 283, 8961–8968. [DOI] [PubMed] [Google Scholar]
- Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, et al. (2012). Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485, 260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi S, Honda K, Sugiura K, Yamaguchi A, Furukawa K, and Urano T (2002). Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett 519, 59–65. [DOI] [PubMed] [Google Scholar]
- Umezawa H, Aoyagi T, Suda H, Hamada M, and Takeuchi T (1976). Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J Antibiot (Tokyo) 29, 97–99. [DOI] [PubMed] [Google Scholar]
- Wander SA, Levis MJ, and Fathi AT (2014). The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol 5, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, and Osada H (2004). M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A 101, 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, and Griffin JD (2002). Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell 1, 433–443. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Ray A, Nelson E, Adamia S, Barrett R, Sattler M, Zhang C, Daley JF, Frank D, Fox E, et al. (2011). Reversible resistance induced by FLT3 inhibition: a novel resistance mechanism in mutant FLT3-expressing cells. PLoS One 6, e25351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, and Bradner JE (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, and Clausen MH (2015). FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 36, 422–439. [DOI] [PubMed] [Google Scholar]
- Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, et al. (2009). AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114, 2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, and Ciulli A (2015). Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, et al. (2013). Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood 122, 3607–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KINOMEscan kinase selectivity profiles of TL12-186 and TL13-27. Related to Figure 1B: Table S1
Raw data from the multiplexed proteomics study in MOLM-14 cells. Related to Figure 2A: Table S2
Statistical summary of the multiplexed proteomics study in MOLM-14. Related to Figure 2A: Table S3
Raw data from the multiplexed proteomics study in MOLT-4 cells. Related to Figure 2B: Table S4
Statistical summary of the multiplexed proteomics study in MOLT-4 cells. Related to Figure 2B: Table S5
KiNativ profiling of TL12-186 in CRBN−/− MOLM-14 cells. Related to Figure 2C: Table S7
Data Availability Statement
The mass spectrometry raw data files for quantitative multiplexed proteomics have been deposited in the PRIDE Archive under the accession numbers PXD007851 (MOLM-14) and PXD007861 (MOLT-4). Original data not included in supplemental tables are deposited to Mendeley Data under this link: http://dx.doi.org/10.17632/wbbbfhg5bj.1.