

Abstract
Aims
Understanding the spectrum of disease, symptom burden and natural history are essential for the management of children with hypertrophic cardiomyopathy (HCM). The effect of changing screening practices over time has not previously been studied. This study describes the clinical characteristics and outcomes of childhood HCM over four decades in a well-characterized United Kingdom cohort.
Methods and results
Six hundred and eighty-seven patients with HCM presented at a median age of 5.2 years (range 0–16). Aetiology was: non-syndromic (n = 433, 63%), RASopathy (n = 126, 18.3%), Friedreich’s ataxia (n = 59, 8.6%) or inborn errors of metabolism (IEM) (n = 64, 9%). In infants (n = 159, 23%) underlying aetiology was more commonly a RASopathy (42% vs. 11.2%, P < 0.0001) or IEM (18.9% vs. 6.4% P < 0.0001). In those with familial disease, median age of presentation was higher (11 years vs. 6 years, P < 0.0001), 141 (58%) presented <12 years. Freedom from death or transplantation was 90.6% (87.9–92.7%) at 5 years (1.5 per 100 patient years) with no era effect. Mortality was most frequently sudden cardiac death (SCD) (n = 20, 2.9%). Children diagnosed during infancy or with an IEM had a worse prognosis (5-year survival 80.5% or 66.4%). Arrhythmic events occurred at a rate of 1.2 per 100 patient years and were more likely in non-syndromic patients (n = 51, 88%).
Conclusion
This national study describes a heterogeneous disease whose outcomes depend on the age of presentation and aetiology. Overall mortality and SCD rates have not changed over time, but they remain higher than in adults with HCM, with events occurring in syndromic and non-syndromic patients.
Keywords: Hypertrophic cardiomyopathy, United Kingdom, Survival, Aetiology
See page 994 for the editorial comment on this article (doi: 10.1093/eurheartj/ehy889)
Introduction
Hypertrophic cardiomyopathy (HCM) is the second commonest cardiomyopathy during childhood, with an estimated annual incidence of 0.24–0.47 per 100 000.1–3 The disease in most children is caused by mutations in the cardiac sarcomere protein genes,4 but the aetiology is more heterogeneous than that seen in adults, and includes inborn errors of metabolism (IEM), neuromuscular disorders, and malformation syndromes.5–7
Understanding the spectrum of disease, symptom burden and outcomes are essential for the management of children with HCM. North American2,5 and Australian3,8,9 registry studies have provided valuable information on the epidemiology, aetiology and survival for childhood HCM, as well as morphological descriptions of the clinical phenotype at presentation.7 Long-term prognosis appears to depend on the age of presentation and aetiology.5–7 However, the effect of changing screening practices on the age of presentation, aetiology, and survival in childhood HCM has not been assessed. A detailed understanding of disease-specific causes of mortality and the incidence of outcomes other than mortality would enable a more individualized approach to patient care. This study sought to describe the clinical characteristics and outcomes of childhood HCM over four decades in a well-characterized United Kingdom (UK) cohort.
Methods
A retrospective, longitudinal multi-centre cohort of children diagnosed with HCM in the UK between 1980 and 2017 was formed. Data were available from 13 out of 14 paediatric cardiac centres.
Eligibility criteria
Patients aged 16 years or younger meeting the diagnostic criteria for HCM were eligible. This included patients with phenocopies of sarcomeric HCM (e.g. RASopathies, IEM, or neuromuscular diseases). The diagnosis of HCM was made if left ventricular wall thickness was greater than two standard deviations above the body surface area-corrected population mean (≥Z score +2), which could not be solely explained by abnormal loading conditions.10 Eligible patients were identified by the principle investigator at each site using multiple sources, including medical databases and echocardiography log books.
Data collection
A single researcher (G.N.) visited each site to assist with data collection and confirm diagnostic classification. Anonymised, non-invasive clinical data were collected, including: demographics; aetiology; symptoms; medical therapy; family history; resting and ambulatory electrocardiography (ECG); 2D, Doppler, and colour transthoracic echocardiography; and exercise testing. Data were collected from baseline evaluation and last clinical review.
Aetiology was classified as non-syndromic in the absence of a diagnosis of a RASopathy syndrome (Noonan or other malformation syndrome), neuromuscular disease (including Friedreich’s ataxia), or inborn error of metabolism. Patients with mutations in the cardiac sarcomere protein genes or in other HCM-causing genes were included in the non-syndromic group.
Clinical investigations
Non-sustained ventricular tachycardia (NSVT) was defined as three or more consecutive ventricular beats at a rate of greater than 120 b.p.m. with a duration of less than 30 s on ambulatory ECG recordings.11 An abnormal blood pressure (BP) response to exercise was defined as the failure of systolic BP to rise by ≥20 mmHg (flat BP response) or a fall in BP during exercise (hypotensive BP response).11,12
Echocardiographic measurements were made according to current guidelines.13 Specifically, maximal left ventricular wall thickness (MLVWT) was measured in the parasternal long-axis or parasternal short-axis views (2D or M-Mode) at end diastole. Left atrial diameter measurements were made in the parasternal long-axis view (2D or M-Mode) at end systole. Left ventricular outflow tract (LVOT) obstruction was defined as an instantaneous peak Doppler LVOT pressure gradient ≥30 mmHg at rest.11 A haemodynamically significant gradient was considered to be an instantaneous peak Doppler gradient ≥50 mmHg.14
Outcomes
The primary patient outcomes, taken from last clinic appointment, were: sudden cardiac death (SCD) as previously defined15 or equivalent event [appropriate implantable cardioverter-defibrillator (ICD) therapy, aborted cardiac arrest, or sustained ventricular tachycardia (VT)]; heart failure-related death; cardiac transplantation; or non-cardiac death. Outcomes were assessed by experienced cardiologists at each site. Patients were classified as lost to follow-up if last clinical review was more than 3 years ago. In order to investigate possible era effects, patients were grouped according to the decade in which they were first diagnosed (1980–1989; 1990–1999; 2000–2009; 2010–2017).
Statistical analysis
Body surface area was calculated from height and weight.16 Maximal left ventricular wall thickness and left atrial diameter absolute measurements are expressed in millimetres and as Z scores relative to the distribution of measurements vs. body surface area in normal children.17 Normally distributed continuous variables are described as mean ± standard deviation with two or three group comparisons conducted using Student t-test and analysis of variance (ANOVA), respectively. Skewed data are described as median [interquartile range (IQR)] with two or three group comparisons performed using Wilcoxon rank sum and Kruskal–Wallis tests, respectively. The distributions of categorical variables were compared using the χ2 test or Fisher’s exact test. Two-sample proportion tests were performed to compare groups within variables of interest. To account for multiple comparisons, the Bonferroni correction was applied. A significance level of 0.05 was used for all comparisons.
Estimates of survival were obtained using the Kaplan–Meier product limit method. Endpoints were all-cause mortality or cardiac transplantation, and a composite outcome of a major arrhythmic event (SCD, aborted cardiac arrest, sustained VT, or appropriate ICD therapy). Group differences in survival were assessed using the log rank test. Statistical analysis was performed using StataCorp. 2015. Stata Statistical Software: Release 14. College Station, TX, USA: StataCorp LP.
Ethics
Ethics committee approval was obtained at each participating site. The study complies with the principles of Good Clinical Practice and the Declaration of Helsinki. G.N. and J.P.K. had access to all data and final responsibility for submission of the manuscript. All investigators have agreed to the manuscript as written.
Results
Six hundred and eighty-seven patients diagnosed with HCM aged 16 years or younger were identified. Median age at presentation was 5.2 years (IQR 2.3–9.9 years; range 0–16 years). One hundred and fifty-nine patients (23%) presented under the age of 1 year and 314 (46%) during pre-adolescent years (1–12 years old) (Figure 1A). Four hundred and thirty-four patients (63%) were male.
Figure 1.

Age of presentation in years with childhood hypertrophic cardiomyopathy; (A) by family history, (B) by aetiology. Patients presenting in infancy are more likely to have a diagnosis of a RASopathy (χ2P < 0.001) or IEM (χ2P < 0.001).
Aetiology
Data on aetiology are shown in Table 1 and Supplementary material online, Table S1. Four hundred and thirty-three patients (63%) had non-syndromic HCM. Data on genetic testing strategy were not available. Pathogenic mutations in sarcomeric protein genes were reported in 100 (23%), most commonly in MYH7 (n = 50) and MYBPC3 (n = 39) (Table 2); twelve patients had more than one pathogenic mutation identified. One hundred and twenty-six patients (18.3%) had a RASopathy syndrome, 64 (9.3%) had an IEM and 59 (8.6%) Friedreich’s ataxia (Supplementary material online, Table S2). The number of patients with an underlying IEM or Friedreich’s ataxia increased over time (Supplementary material online, Table S3). Patients with Friedreich’s ataxia presented later in childhood [mean age of 10.4 years (range 4–16)], although four patients were diagnosed below the age of 7 years (Figure 1B).
Table 1.
Clinical and demographic characteristics at diagnosis by age of diagnosis
| Whole cohort (no. of patients (%)) | <1 year (no. of patients (%)) | >1 year (no. of patients (%)) | P-value | ||
|---|---|---|---|---|---|
| Gender | Male | 434 (63.2%) | 102 (64.2%) | 332 (62.9%) | 0.77* |
| Aetiology | Non-syndromic | 433 (63%) | 62 (39%) | 371 (70.3%) | <0.0001 |
| Noonan syndrome (or RASopathies) | 126 (18.3%) | 67 (42%) | 59 (11.2%) | <0.0001 | |
| Friedreich’s ataxia | 59 (8.6%) | 59 (11.2%) | |||
| Other neuromuscular syndrome | 5 (0.7%) | 5 (1%) | |||
| Inborn error of metabolism | 64 (9.3%) | 30 (18.9%) | 34 (6.4%) | <0.0001 | |
| Family history of HCM (n = 680) | 242 (35.6%) | 26 (16.4%) | 216 (41.5%) | <0.0001* | |
| Family history of SCD (n = 682) | 50 (7.3%) | 3 (1.9%) | 47 (9%) | 0.039* | |
| NYHA/Ross at presentation (n = 684) | I | 516 (75.4%) | 94 (59.5%) | 422 (80.2%) | <0.0001 |
| II | 133 (19.4%) | 42 (26.6%) | 91 (17.3%) | 0.12 | |
| III | 29 (4.2%) | 17 (10.8%) | 12 (2.3%) | <0.0001 | |
| IV | 6 (0.9%) | 5 (3.2%) | 1 (0.2%) | 0.267 | |
| Cause of mortality (n = 75) | SCD | 20 (2.9%) | 2 (1.3%) | 18 (3.4%) | |
| CCF | 12 (1.7%) | 10 (6.3%) | 2 (0.4%) | ||
| Other CV | 12 (1.7%) | 8 (5%) | 4 (0.8%) | ||
| Non-CV | 17 (2.5%) | 12 (7.5%) | 5 (0.9%) | ||
| Unknown | 14 (2%) | 3 (1.9%) | 11 (2.1%) | ||
Data expressed as n (%). Total number of patients is 687 unless otherwise stated. All comparisons are made using a two-sample proportion test unless otherwise stated.
Comparisons were made using χ2 test.
Table 2.
Genes in which pathogenic mutations were identified in patients with non-syndromic hypertrophic cardiomyopathy
| Gene | Number of patients | |
|---|---|---|
| Sarcomeric protein gene | 100 | |
| MYH7 | 50 | |
| MYBPC3 | 39 | |
| TPM1 | 4 | |
| TNNT2 | 3 | |
| TTN | 2 | |
| TNNI3 | 2 | |
| Non-sarcomeric gene | 7 | |
| DES | 3 | |
| PRKAG2 | 3 | |
| JPH2 | 1 | |
| Variant of unknown significance (VUS) | 10 | |
Family history
Two hundred and forty-two patients (35.6%) had a first-degree relative with HCM based on family history; of these, 214 (88%) had non-syndromic HCM. The median age of presentation was higher compared to those without a family history [11 years (5–13) vs. 6 years (0–12), P < 0.0001] and they were less likely to present during infancy (Table 1 and Figure 1A). One hundred and forty-one patients (58%) with a family history presented in pre-adolescent years (<12 years). Fifty patients (7.3%) had a family history of SCD in a first-degree relative.
Symptoms at presentation
Five hundred and sixteen patients (75.4%) were asymptomatic for heart failure symptoms at presentation [New York Heart Association (NYHA)/Ross functional Class 1]. Thirty-five patients (5.1%) had symptoms of congestive cardiac failure (CCF) (NYHA/Ross > 2). Heart failure symptoms were more common in patients presenting in infancy (Table 1). Thirty-eight patients (5.6%) had a history of unexplained syncope at presentation. Twenty-four patients (3.5%) presented following an aborted SCD or out of hospital arrest.
Investigations at baseline
Maximal left ventricular wall thickness on 2D echocardiography at baseline ranged from 4 to 48 mm (median 13 mm, IQR 11–18 mm, n = 645), with median Z score +10.1 (IQR +3.6 to +34.5). Left ventricular outflow tract gradient at baseline was documented in 604 (88%) patients; one hundred and sixty patients (26.5%) had LVOT obstruction and 32 patients (5.3%) had a gradient >90 mmHg (Table 3).
Table 3.
Results of baseline investigations
| No. of patients (%) | |||
|---|---|---|---|
| Echocardiographic data at diagnosis (n = 677) | LVMWT (mm) [median (IQR)] | 13 (11–18) | |
| LVMWT (Z score) [median (IQR)] | +10.1 (3.6–37) | ||
| Left atrial diameter (mm) [mean (SD)] [n = 352] | 29 (9.2) | ||
| Left ventricular outflow gradient at rest (mmHg) [n = 604] | <30 | 444 (64.6%) | |
| 30–50 | 47 (6.8%) | ||
| 50–90 | 81 (11.8%) | ||
| >90 | 32 (12.1%) | ||
| Ambulatory ECG performed (n = 483) | Yes | NSVT | 7 (1.4%) |
| No NSVT | 476 (98.6%) | ||
| No | 204 (29%) | ||
| Exercise test performeda (n = 220) | Non-syndromic | 196 (45%) | |
| RASopathy | 12 (10%) | ||
| Friedreich ataxia | 7 (12%) | ||
| Inborn error of metabolism | 5 (8%) | ||
| Blood pressure response to exercisea | Normal (>20 mmHg rise in systolic BP) | 126 (57%) | |
| Flat (<20 mmHg rise in systolic BP) | 68 (31%) | ||
| Hypotensive (fall in systolic BP) | 16 (7%) | ||
Data expressed as number (%). Total number of patients is 687 unless otherwise stated.
SD, standard deviation.
Exercise test in patients ≥6 years.
Of 483 patients (71%) with 24-h ambulatory ECG at baseline, NSVT was detected in 7 (1.4%). An exercise test was performed in 220 out of 412 patients (53%) aged 6 years and above. Blood pressure response to exercise was classified as abnormal in 84 patients (38%), of which 23 (27%) had LVOT obstruction at rest and 37 (44%) were on beta-blocker therapy. No patient had documented arrhythmias during exercise.
Outcomes
Median length of follow-up was 5.2 years (IQR 2.3–9.9 years). 529 (77%) patients remained under the age of 18 years at last follow-up. Ninety-eight patients (14.3%) were classified as lost to follow-up, of which 41 (42%) had been transitioned to adult care. one hundred and thirty-five patients (20%) underwent ICD implantation for primary (n = 108, 80%) or secondary (n = 27, 10%) prevention.
Mortality
Six hundred and twelve patients (89.1%) were alive at last clinical follow-up. Seventy-five patients (10.9%) died: SCD in 20 (2.9%), non-cardiovascular (CV) in 17 (2.5%), CCF in 12 (1.8%), other CV causes in 12 (1.8%), and unknown in 14 (2.3%). Sixteen patients (80%) who died suddenly were aged under 18 years at the time of death. Ten patients (1.5%) underwent cardiac transplantation. Survival free from death or transplantation was 95.6% [95% confidence interval (95% CI) 93.7–96.9%] at 12 months and 86.3% (95% CI 82.6–89.2) at 10 years (Figure 2A). No difference in survival was seen by era of presentation (Supplementary material online, Figure S1). Children diagnosed during infancy or with an IEM had a worse prognosis, with survival at 12 months of 83.3% (95% CI 76.5–88.3%, P < 0.0001) or 82.2% (95% CI 70.2–89.8%) respectively (Figure 2B,C and Table 4). Cause of death differed by underlying aetiology (Supplementary material online, Table S1) and age of diagnosis (Table 1).
Figure 2.
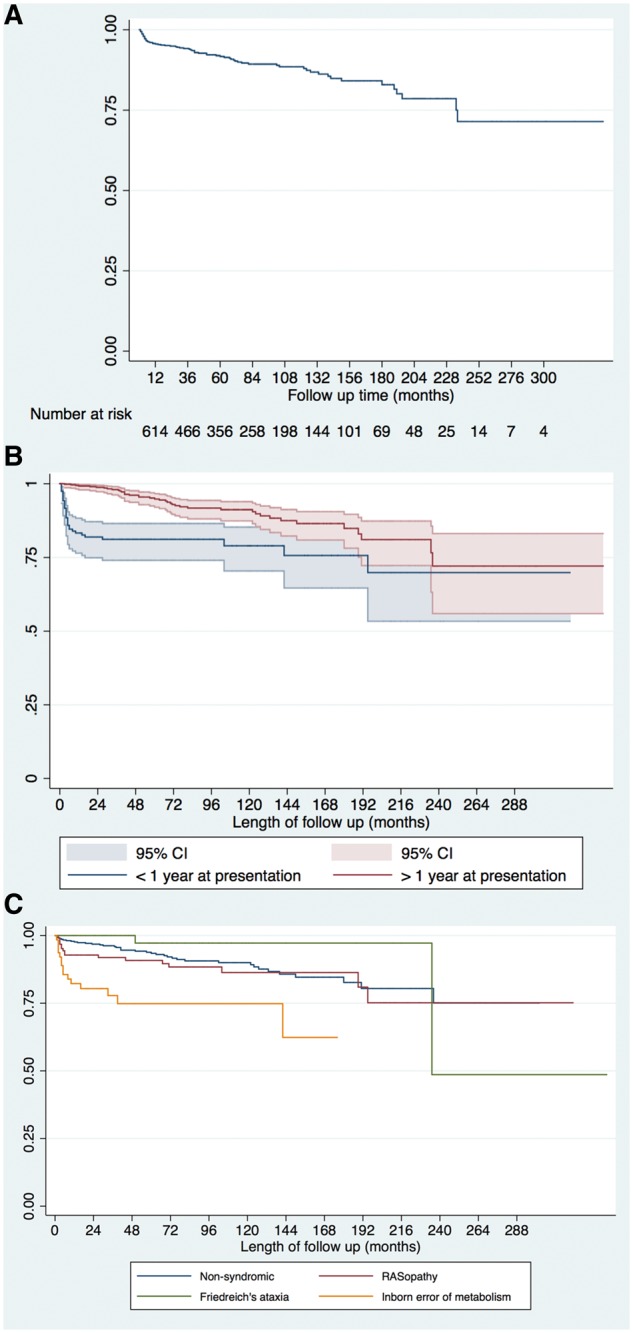
The Kaplan–Meier curves for survival free from all-cause mortality or cardiac transplantation; (A) for whole cohort; (B) stratified by age of presentation, Log rank test P < 0.0001. 95% CI are shown. (C) Stratified by aetiology, Log rank test <0.0001.
Table 4.
Survival rates from time of presentation to death or heart transplantation
| Survival after HCM diagnosis, % (95% confidence interval) |
|||||
|---|---|---|---|---|---|
| 1 year | 5 year | 10 year | Log rank analysis | ||
| Whole cohort | 95.6% (93.7–96.9) | 90.6% (87.9–92.7) | 86.3% (82.6–89.2) | ||
| Age at presentation | <1 year (n = 159) | 83.3% (76.5–88.3) | 80.5% (73.2–85.9) | 78.3% (69.7–84.7) | <0.0001 |
| >1 year (n = 528) | 99.2% (97.9–99.7) | 93.6% (90.6–95.6) | 88.6% (84.3–91.7) | ||
| Aetiology | Non-syndromic (n = 433) | 97.6% (95.7–98.7) | 92.7% (89.4–95) | 87.5% (82.8–91.1) | <0.0001 |
| RASopathy (n = 126) | 92.5% (86.1–96) | 90.5% (83.4–95.6) | 85.9% (76.7–91.7) | ||
| Inborn error of metabolism (n = 64) | 82.2% (70.2–89.8) | 66.4% (50.2–78.3) | 66.4% (50.2–78.3) | ||
| Friedreich’s ataxia (n = 59) | 100 | 97.1% (80.9–99.6) | 97.1% (80.9–99.6) | ||
Survival rates, using the Kaplan–Meier method, with subgroup analysis based on age at diagnosis and aetiology. Patient numbers are indicated (n).
Major arrhythmic events:
Fifty-eight patients (8.4%) had an arrhythmic event [SCD in 20 (34%); resuscitated cardiac arrest in 11 (19%); sustained VT in 8 (14%); appropriate ICD discharge in 19 (33%)] during follow-up, with an event rate of 1.2 per 100 patient years at risk. Freedom from arrhythmic event was 98% (95% CI 97.3–99.3) at 1 year, 92.9% (95% CI 90.3–94.9) at 5 years and 89.6% (95% CI 86.1–92.2%) at 10 years. Arrhythmic events occurred in 51 patients (88%) with non-syndromic HCM, 5 (9%) with a RASopathy, and 2 (3%) with an IEM (Table 5). No patients with neuromuscular disease (e.g. Friedreich’s ataxia) had an arrhythmic event. Nine patients (7.6%) who had been transitioned had arrhythmic events (SCD n = 4, appropriate ICD discharge n = 5).
Table 5.
Clinical characteristic of patients with inborn error of metabolism or malformation syndrome and sudden cardiac death
| Diagnosis | Age at presentation | Symptoms at presentation | Phenotype | History of arrhythmias | Interventions | SCD or equivalent event | |
|---|---|---|---|---|---|---|---|
| Patient 1 | Danon’s disease | 13 years | Unexplained syncope |
|
Nil | ICD implanted aged 14 years | Sustained VT aged 16 years |
| Patient 2 | Glycogen storage disease | 3 months | NYHA 1 |
|
Nil | Reveal device implanted aged 14 (unexplained syncope aged 13) | SCD—reveal device documented VT + fine VF aged 14 |
| Patient 3 | Noonan’s | 13 years | NYHA 2 |
|
NSVT aged 16 years | Nil | SCD aged 18 years |
| Patient 4 | Noonan’s | 11 years | NYHA 1 |
|
NSVT aged 15 years | Primary prevention ICD implanted aged 16 years | Appropriate ICD therapy aged 16 years |
| Patient 5 | Noonan’s | 9 days | NYHA 2 |
|
Nil | Myectomy aged 5 years | SCD aged 8 years |
| Patient 6 | Other RASopathy | 2 years | NYHA 1 |
|
NSVT aged 9 years | Nil | Aborted cardiac arrest aged 10 years |
| Patient 7 | Noonan’s | 3 months | NYHA 3 |
|
Nil | Nil | Sustained VT aged 14 months |
ASH, asymmetric hypertrophy; BVH, biventricular hypertrophy; LVMWT, left ventricular maximal wall thickness; RVOTO, right ventricular outflow tract obstruction; VF, ventricular fibrillatrion.
Discussion
This study describes the clinical characteristics and outcomes of a national cohort of 687 childhood HCM patients, representing one of the largest studies in paediatric HCM, and the largest systematic study from Europe. Important novel findings include the detection of a phenotype in 58% of pre-adolescents with a first-degree family history of HCM and a change in the prevalence of syndromic HCM, particularly IEM, over time. Although the overall mortality and rates of SCD are lower than reported in early paediatric studies, they are nevertheless higher than seen in adults, where SCD rates are 0.81% per year. The cause of mortality differed by underlying aetiology, although arrhythmic events occurred in syndromic as well as non-syndromic patients.
Comparison with previous studies
Current understanding of the aetiology and clinical characteristics of childhood HCM is primarily derived from a North American registry, which described 855 patients under the age of 18 years.5 The present study describes a cohort of patients under 16 years at diagnosis with a comparable length of follow-up [although recruited from a wider time period (1980–2017 vs. 1990–2009)], which provides a useful comparison of the clinical presentations and outcomes of childhood HCM in different healthcare systems. In agreement with the North American study, we report that the underlying aetiology of childhood HCM is heterogeneous; although the most common aetiology was non-syndromic, 37% had syndromic disease which was more common in infant-onset disease. More patients with an inborn error of metabolism were included in the most recent era (2010 onwards, n = 41) compared to earlier time periods (1990–1999, n = 1), which is likely to be due to systematic screening of patients with metabolic disease for cardiac disease. In the present study, only 23% of non-syndromic patients had a confirmed pathogenic mutation in a cardiac sarcomere protein gene. This is almost certainly an underestimate of the true prevalence as previous studies have shown that most cases of HCM are caused by sarcomeric mutations, even in young children.4
This study contains a higher number of non-syndromic patients with familial disease compared to the North American registry (n = 214, 49% vs. n = 109, 17%), of which over half were diagnosed with a phenotype in pre-adolescence. The age of presentation for familial disease is determined both by disease-specific and healthcare system factors, such as the age at which family screening started. Patients with more severe disease may therefore present at a younger age with symptoms whereas screening may identify children with a milder subclinical disease. It is not known if these patients were diagnosed through screening or in response to symptoms, but it is noteworthy that the majority were asymptomatic at presentation. Current guidelines recommend that routine family clinical screening is not started before the age of 1011 or 12 years,18 which may in part explain the peak in adolescence previously reported with patients’ first clinical assessment occurring in adolescent years. The age of presentation for familial disease is not described in other registry studies, but the findings from this study suggest that the age-related penetrance of HCM is highly variable and that consideration should be given to commencing screening at an earlier age.
Outcomes of paediatric hypertrophic cardiomyopathy
This study shows that most children with HCM have a relatively good prognosis, with overall mortality or transplantation rates of 1.5 per 100 patient years at risk. Our data confirm previous reports5,8 that underlying aetiology has a significant impact on outcomes. Five-year survival was much lower for patients with an IEM (66%), compared to those with a diagnosis of a RASopathy syndrome (90.5%) or Friedreich’s ataxia (97%). These survival rates are higher than those previously reported in the North American registry,5 which likely reflects the variability in phenotype and outcomes that are seen. The cause of death differed depending on the underlying aetiology with non-cardiac deaths more common in patients with an IEM (n = 8, 12.5%) or RASopathy (n = 5, 4%). This highlights the multi-systemic nature of such conditions where cardiac involvement is only one aspect of the clinical phenotype and may not determine long-term outcomes. Also in agreement with previous reports,5,8 presentation at a young age was associated with worse outcomes. This finding may be partly explained by age-related difference in aetiology. However, it may also be a surrogate marker for disease severity as patients with more severe disease may be expected to present earlier in life and with worse outcomes. The phenotype and progression of familial hypertrophic cardiomyopathy in childhood, as well as the impact of clinical screening at a younger age, is largely unknown and warrants further investigation.
No era effect was seen on survival, although the earliest era (1980–1989) contained significantly smaller numbers of patients (Supplementary material online, Table S3 and Figure S1), suggesting that the findings are an accurate reflection of the prognosis of childhood HCM. The explanation for this is likely multifactorial. The medical management of children with HCM has not significantly changed over the study period. However, ICDs have become widely used and shown to be effective at terminating malignant arrhythmias19–21 although at the expense of a higher incidence of complications compared to adult populations. Our data would support that ICDs are effective at terminating malignant arrhythmias as over time the proportion of patients experiencing SCD decreased whilst those with an aborted SCD/appropriate ICD therapy increased. Current guidelines recommend implantation of a primary prevention ICD in children with two or more risk factors for SCD.21 Nonetheless, challenges remain in identifying patients at highest risk for an arrhythmic event as evidenced by patients experiencing SCD/aborted arrests whilst under clinical follow-up and the low number of appropriate ICD therapies (14%). The higher number of infants with an inborn error of metabolism or RASopathy in recent years, who are known to have a worse prognosis, could also contribute to the lack of difference in survival seen between eras.
Arrhythmic events occurred at a rate of 1.2 per 100 patient years at risk. While this is significantly lower than initial reports,22–24 it remains higher than in adult cohorts (0.81% per year).25,26 Interestingly, seven of the patients who died suddenly had phenocopies of sarcomeric HCM, groups of patients that are traditionally regarded as having a low risk for an arrhythmic event. One of these patients had Danon syndrome, which is recognized to have a high risk of SCD,27 however five had a diagnosis of a RASopathy syndrome and one had a glycogen storage disorder. This highlights the need for a systematic assessment of arrhythmic risk in all patients with HCM, regardless of the underlying aetiology. It is noteworthy that there were no arrhythmic events in the 59 children with Friedreich’s ataxia, suggesting that patients with Friedreich’s ataxia may be at lower risk of ventricular arrhythmias. The finding that arrhythmic events occurred after transition to adult care confirms recent reports28 that arrhythmic risk in this group of patients is not limited to childhood and adolescence meaning ongoing surveillance is required.
Limitations
This study is limited by inherent problems of retrospective studies, in particular missing or incomplete data. As no prospective database exists in the UK, all patients diagnosed with HCM over the study period (1980–2016) may not have been included. The number of patients by calendar year of diagnosis increased over time (Supplementary material online, Table S3). This may be due to more complete identification of affected patients. However, it may also represent a change in clinical practice, with earlier screening of affected families and routine cardiac assessment for patients with multi-systemic syndromes or diagnoses. Fourteen percentage of patients were classified as lost to follow-up (last clinical review more than 3 years ago) of whom 42% had been transitioned to adult care. Although the mortality rate is unlikely to be affected by this missing data due to nationally recorded death data in the UK National Health Service (NHS), other outcomes, such as arrhythmic events, could be underestimated. As patients were recruited from a number of centres, variations in clinical assessment and management between sites are inevitable. However, this is also a strength of the study, as it reflects accurately the clinical management of patients in the UK, both historically and in the current era.
Take home figure.
Kaplan-Meier curves for survival free from all cause mortality or cardiac transplantation. Aetiology is important for outcome with patients with an inborn error of metabolism having a worse prognosis most commonly secondary to congestive cardiac failure (log rank test <0.0001).
Conclusions
This study is the first European multi-centre investigation of paediatric HCM. The results show that, during childhood, HCM is a heterogeneous disease in terms of its age of presentation, aetiology, and outcomes. Important novel findings include the detection of a phenotype in pre-adolescents with a family history of HCM and a change in the prevalence of syndromic HCM, particularly IEM, over time. Although in most children survival is good (freedom from death or transplantation was 90.6% at 5 years), SCD is the most common cause of mortality, occurring at a rate of 1.2 per 100 patient years at risk. Patients diagnosed in infancy or with an inborn error of metabolism have a worse prognosis, most commonly secondary to CCF or non-CV death, but arrhythmic events are also seen in these patient groups. An improved understanding of the relationship between aetiology, phenotype, and outcomes will facilitate the systematic study of risk factors for adverse events in this diverse patient group.
Supplementary Material
{kind=link}
Funding
British Heart Foundation (FS/16/72/32270 to G.N. and J.P.K.); Max’s Foundation and Great Ormond Street Hospital Children’s Charity to E.F. and J.P.K. Part of this research was carried out at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health which is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of interest: none declared.
References
- 1. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O.. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol 1997;146:385–393. [DOI] [PubMed] [Google Scholar]
- 2. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, Colan SD.. The incidence of paediatric cardiomyopathy in two regions of the United States. N Engl J Med 2003;348:1647–1655. [DOI] [PubMed] [Google Scholar]
- 3. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, Weintraub RG.. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med 2003;348:1639–1646. [DOI] [PubMed] [Google Scholar]
- 4. Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM.. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009;2:436–441. [DOI] [PubMed] [Google Scholar]
- 5. Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA.. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Paediatric Cardiomyopathy Registry. Circulation 2007;115:773–781. [DOI] [PubMed] [Google Scholar]
- 6. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, Towbin JA, Connuck DM, Messere JE, Lipshultz SE.. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Paediatric Cardiomyopathy Registry. Am Heart J 2012;164:442–448. [DOI] [PubMed] [Google Scholar]
- 7. Lipshultz SE, Orav EJ, Wilkinson JD, Towbin JA, Messere JE, Lowe AM, Sleeper LA, Cox GF, Hsu DT, Canter CE, Hunter JA, Colan SD.. Risk stratification at diagnosis for children with hypertrophic cardiomyopathy: an analysis of data from the Paediatric Cardiomyopathy Registry. Lancet 2013;382:1889–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nugent AW, Daubeney PE, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG.. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation 2005;112:1332–1338. [DOI] [PubMed] [Google Scholar]
- 9. Alexander PMA, Nugent AW, Daubeney PEF, Lee KJ, Sleeper LA, Schuster T, Turner C, Davis AM, Semsarian C, Colan SD, Robertson T, Ramsay J, Justo R, Sholler GF, King I, Weintraub RG.. Long-term outcomes of hypertrophic cardiomyopathy diagnosed during childhood: results from a national population-based study. Circulation 2018. [DOI] [PubMed] [Google Scholar]
- 10. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A.. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2007;29:270–276. [DOI] [PubMed] [Google Scholar]
- 11. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H.. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 12. Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ.. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997;96:2987–2991. [DOI] [PubMed] [Google Scholar]
- 13. Yang YJ, Fan CM, Yuan JQ, Wang SY, Song YH, Qiao SB, You SJ, Wang ZM, Duan FJ, Li YS.. Effectiveness of alcohol septal ablation versus transaortic extended myectomy in hypertrophic cardiomyopathy with midventricular obstruction. J Interv Cardiol 2016;29:619–627. [DOI] [PubMed] [Google Scholar]
- 14. Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, Rakowski H, Williams WG.. Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis 1985;28:1–83. [DOI] [PubMed] [Google Scholar]
- 15. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ.. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000;36:2212–2218. [DOI] [PubMed] [Google Scholar]
- 16. Du Bois D, Du Bois EF.. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition 1989;5:303–311; discussion 312–313. [PubMed] [Google Scholar]
- 17. Lopez L, Colan S, Stylianou M, Granger S, Trachtenberg F, Frommelt P, Pearson G, Camarda J, Cnota J, Cohen M, Dragulescu A, Frommelt M, Garuba O, Johnson T, Lai W, Mahgerefteh J, Pignatelli R, Prakash A, Sachdeva R, Soriano B, Soslow J, Spurney C, Srivastava S, Taylor C, Thankavel P, van der Velde M, Minich L.. Relationship of echocardiographic Z scores adjusted for body surface area to age, sex, race, and ethnicity: the paediatric heart network normal echocardiogram database. Circ Cardiovasc Imaging 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW, Jacobs AK, Smith SC, Anderson JL, Albert NM, Buller CE, Creager MA, Ettinger SM, Guyton RA, Halperin JL, Hochman JS, Krumholz HM, Kushner FG, Nishimura RA, Ohman EM, Page RL, Stevenson WG, Tarkington LG, Yancy CW.. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011;142:e153–e203. [DOI] [PubMed] [Google Scholar]
- 19. Kaski JP, Tome Esteban MT, Lowe M, Sporton S, Rees P, Deanfield JE, McKenna WJ, Elliott PM.. Outcomes after implantable cardioverter-defibrillator treatment in children with hypertrophic cardiomyopathy. Heart 2007;93:372–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maron BJ, Spirito P, Ackerman MJ, Casey SA, Semsarian C, Estes NAM, Shannon KM, Ashley EA, Day SM, Pacileo G, Formisano F, Devoto E, Anastasakis A, Bos JM, Woo A, Autore C, Pass RH, Boriani G, Garberich RF, Almquist AK, Russell MW, Boni L, Berger S, Maron MS, Link MS.. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol 2013;61:1527–1535. [DOI] [PubMed] [Google Scholar]
- 21. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ.. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 22. McKenna W, Deanfield J, Faruqui A, England D, Oakley C, Goodwin J.. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol 1981;47:532–538. [DOI] [PubMed] [Google Scholar]
- 23. McKenna WJ, Deanfield JE.. Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 1984;59:971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ostman-Smith I, Wettrell G, Keeton B, Holmgren D, Ergander U, Gould S, Bowker C, Verdicchio M.. Age- and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur Heart J 2008;29:1160–1167. [DOI] [PubMed] [Google Scholar]
- 25. O'Mahony C, Tome-Esteban M, Lambiase PD, Pantazis A, Dickie S, McKenna WJ, Elliott PM.. A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/American Heart Association risk stratification and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart 2013;99:534–541. [DOI] [PubMed] [Google Scholar]
- 26. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F.. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000;102:858–864. [DOI] [PubMed] [Google Scholar]
- 27. Maron BJ, Roberts WC, Arad M, Haas TS, Spirito P, Wright GB, Almquist AK, Baffa JM, Saul JP, Ho CY, Seidman J, Seidman CE.. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009;301:1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maurizi N, Passantino S, Spaziani G, Girolami F, Arretini A, Targetti M, Pollini I, Tomberli A, Pradella S, Calabri GB, Vinattieri V, Bertaccini B, Leone O, De Simone L, Rapezzi C, Marchionni N, Cecchi F, Favilli S, Olivotto I.. Long-term outcomes of paediatric-onset hypertrophic cardiomyopathy and age-specific risk factors for lethal arrhythmic events. JAMA Cardiol 2018;3:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.