

Abstract
Aims
Myocardial fibrosis is associated with profound changes in ventricular architecture and geometry, resulting in diminished cardiac function. There is currently no information on the role of the delta-like homologue 1 (Dlk1) in the regulation of the fibrotic response. Here, we investigated whether Dlk1 is involved in cardiac fibroblast-to-myofibroblast differentiation and regulates myocardial fibrosis and explored the molecular mechanism underpinning its effects in this process.
Methods and results
Using Dlk1-knockout mice and adenoviral gene delivery, we demonstrate that overexpression of Dlk1 in cardio-fibroblasts resulted in inhibition of fibroblast proliferation and differentiation into myofibroblasts. This process is mediated by TGF-β1 signalling, since isolated fibroblasts lacking Dlk1 exhibited a higher activation of the TGF-β1/Smad-3 pathway at baseline, leading to an earlier acquisition of a myofibroblast phenotype. Likewise, Dlk1-null mice displayed increased TGF-β1/Smad3 cardiac activity, resulting in infiltration/accumulation of myofibroblasts, induction and deposition of extra-domain A-fibronectin isoform and collagen, and activation of pro-fibrotic markers. Furthermore, these profibrotic events were associated with disrupted myofibril integrity, myocyte hypertrophy, and cardiac dysfunction. Interestingly, Dlk1 expression was down-regulated in ischaemic human and porcine heart tissues. Mechanistically, miR-370 mediated Dlk1’s regulation of cardiac fibroblast–myofibroblast differentiation by directly targeting TGFβ-R2/Smad-3 signalling, while the Dlk1 canonical target, Notch pathway, does not seem to play a role in this process.
Conclusion
These findings are the first to demonstrate an inhibitory role of Dlk1 of cardiac fibroblast-to-myofibroblast differentiation by interfering with TGFβ/Smad-3 signalling in the myocardium. Given the deleterious effects of continuous activation of this pathway, we propose Dlk1 as a new potential candidate for therapy in cases where aberrant TGFβ signalling leads to chronic fibrosis.
Keywords: Cardiac fibrosis, Fibroblast–myofibroblast transdifferentiation, Dlk1, miR-370, TGF-β signalling
See page 979 for the editorial comment on this article (doi: 10.1093/eurheartj/ehy307)
Translational perspective
Myocardial fibrosis is a major determinant of clinical outcomes in patients with heart failure. In fact, accumulating experimental and clinical evidences suggest that emergence of myofibroblasts and early activation of pro-fibrotic signalling pathways occur before adverse ventricular remodelling occurs, and prior to heart failure progression. However, the molecular factors that control progression of the fibrotic response are still unclear. In this regard, delta-like homologue-1 (Dlk1) is demonstrated to negatively regulate cardiac fibroblast-to-myofibroblast differentiation and control myocardial fibrosis. These novel anti-fibrogenic properties of Dlk1 epitomize a potential intervention strategy in cases where aberrant signalling leads to chronic cardiac fibrosis.
Introduction
Cardiac fibrosis, characterized by excess deposition of extracellular matrix (ECM) and myofibroblast accumulation, is an integral feature of the remodelling of the failing heart.1,2 Cardiac fibroblasts, accurately estimated to comprise <20% of the total cell population in the adult murine heart,3 play a critical role in the repair and remodelling of the heart that occurs following myocardial infarction (MI) because of their exceptional plasticity to undergo conversions into myofibroblasts. Myofibroblasts, typically not present in the healthy heart, are highly specialized cells that regulate ECM turnover and remodel tissue due to their contractile capacity. Following an infarction insult, myofibroblasts initially promote the formation of a protective fibrotic scar that prevents wall rupture; however, their persistent presence in the heart can lead to chronic cardiac fibrosis and subsequent dysfunction.3 Secreted TGF-β1 and activation of Smad-dependent canonical pathway upon injury is the main inducer of ECM production and fibroblast-to-myofibroblast conversion.4 Current overwhelming evidence indicates that activation of the Smad2/3 cascade, more specifically Smad-3, is essential for the development of cardiac fibrosis; as such, TGF-β1/Smad-3 signalling is activated in the border zone of healing infarcts leading to fibrotic remodelling of the infarcted ventricle.4,5
Delta-like homologue 1 (Dlk1) is a paternally imprinted gene that encodes a transmembrane protein belonging to the epidermal growth factor (EGF)-like family, which includes Notch receptors and their ligands.6 Delta-like homologue 1 is alternatively spliced, with transcripts encoding a large, soluble secreted isoform containing a tumour necrosis factor converting enzyme (TACE) cleavage site, and membrane-bound isoform lacking the protease cleavage region.7 Delta-like homologue 1 has been shown to play a critical role in controlling cell differentiation processes including adipogenesis, muscular and neuronal differentiation, bone differentiation, and haematopoiesis throughout embryonic and adulthood.8,9 Intriguingly, several studies have documented different biological roles for Dlk1 variants; for instance soluble Dlk1 inhibits adipocyte differentiation,10 whereas membrane-bound Dlk1 regulates neural stem cell number via a mechanism that requires soluble Dlk1.11
Currently there is no information on the role of Dlk1 in the heart. In the present study, we found that Dlk1 is a critical determinant of fibroblast-to-myofibroblast differentiation as deletion of Dlk1 leads to down-regulation of miR-370 and increased TGFβ/Smad3 pro-fibrotic activity, resulting in myofibroblast infiltration, increased ECM deposition and cardiac dysfunction. Restoration of Dlk1 or miR-370 expression suppressed TGFβ/Smad-3 activation. To our knowledge, these findings are the first to demonstrate an inhibitory role of Dlk1 in the process of cardiac fibroblast-to-myofibroblast conversion by interfering with TGFβ/Smad-3 signalling in the myocardium.
Methods
An extended methods section is provided in Supplementary material online.
Animals and echocardiography
The Mount Sinai Institutional Animal Care and Use Committees approved handling of animals in accordance with the ‘Principles of Laboratory Animal Care by the National Society for Medical research and the Guide for the Care and Use of Laboratory Animals’ (NIH Publication No. 86-23, revised 1996).
Homozygous male Dlk1−/− mice (mix background SvJ129xC57BL/6)6 and wild types were anesthetized with intraperitoneal ketamine (100 μg/g) for echocardiographic analysis at 7, 13, and 19 weeks of age using GE Vivid 7.
Isolation of mouse cardiac myocytes, cardiac fibroblasts, adenoviral infection, and microRNA mimics and anti-miRs transfection
Cardiomyocytes and fibroblasts were enzymatically isolated from wild-type and Dlk1−/− mice hearts and processed as detailed in the extended methods section in Supplementary material online.
Cell proliferation assays
Cardiac fibroblasts were seeded in a 96 multi-well plate at a density of 2000 cells/well. Incorporation of BrdU was assessed at 16, 20, and 24 h following manufactures’ instructions (Roche). Cell proliferation was assayed in the presence of 10% FBS.
Results
Cardiac myocytes and fibroblasts express different delta-like homologue 1 isoforms
We first evaluated the Dlk1 expression pattern in the heart. We found that isolated cardiac fibroblasts and cardiomyocytes express different Dlk1 isoforms with fibroblasts displaying higher expression levels. Using primers designed to amplify the protease encoding site-only present in soluble/cleaved-producing Dlk1 mRNA isoforms-and primers designed against exons 4 and 5 of Dlk1 to detect all variants, we identified membrane-bound encoding Dlk1 variants as the most abundant in cardiomyocytes (Figure 1A), whereas fibroblasts expressed the larger Dlk1 mRNA variant, encoding for a secreted protein as the predominant isoform (Figure 1B). Supporting these results, an antibody against Dlk1 identified all isoforms based on their different molecular weights (Figure 1C).
Figure 1.

Cardiac cells express different delta-like homologue 1 (Dlk1) isoforms with delta-like homologue 1 expression decreasing in fibroblasts differentiating into myofibroblasts. Real time RT-PCR of total delta-like homologue 1 isoform (A) and delta-like homologue 1 soluble/cleaved (B) encoding mRNA variants from cardiac myocytes (CM) and fibroblasts (CF). (C) Representative western blot of delta-like homologue 1 reveals bands of different molecular weights corresponding to delta-like homologue 1 isoforms in cardiac myocytes and fibroblasts. GAPDH is a loading control. (D) Representative images of mouse cardiac fibroblasts maintained in 1% or 10% serum showing the effect on morphology and stress fibre expression (F-actin, red). (E) Western blot for α-smooth muscle actin, Smemb and delta-like homologue 1; GAPDH is a loading control. Graphs represent delta-like homologue 1 mRNA expression and protein densitometric analysis of delta-like homologue 1. (F) Representative images from non-treated (Cont) and treated (TGF-β1) fibroblasts cultured without serum. Top panel displays the change of fibroblast morphology after 24 h with TGF-β1. Middle (α-smooth muscle actin, green) and lower (merged) panels show an enhanced α-smooth muscle actin expression in TGF-β1-treated fibroblasts. (G) qRT-PCR of α-smooth muscle actin, fibronectin, collagen 1, connective tissue growth factor (CTGF), and delta-like homologue 1 in non-treated and TGF-β1-treated fibroblasts. Scale bars: 40 µm.
Lack of delta-like homologue 1 promotes fibroblast to myofibroblast differentiation in vitro
Since Dlk1 plays a critical role in the differentiation of various cell types,8,9 we hypothesized that Dlk1 will be implicated in the process of cardiac fibroblast-to-myofibroblast conversion, a crucial mechanism during the remodelling of the heart after MI. Previous reports have shown that cardio-fibroblasts spontaneously differentiate into myofibroblasts under standard culture conditions12; therefore, we performed our studies with no-passage fibroblasts maintained in 1% serum. Using these conditions, the fibroblastic phenotype was retained, as indicated by the low presence of stress fibres and low expression of α-smooth muscle actin (αSMA) and non-muscle myosin heavy chain (Smemb), along with the maintenance of the elongated shape characteristic of fibroblasts (Figure 1D, E). Indeed, we found that Dlk1 expression significantly decreased when fibroblasts, exposed to 10% serum, differentiate into myofibroblasts, identified as large cells expressing stress fibres and αSMA (Figure 1D, E). TGF-β1 is a potent inducer of ECM production and fibroblast-to-myofibroblast differentiation. We therefore treated fibroblasts with TGF-β1 for 24-h and, as expected, we observed a drastic change in cell morphology and up-regulation of αSMA, collagen-1a, fibronectin, and connective tissue growth factor (CTGF), all major markers of myofibroblasts (Figure 1F, G). Similarly, TGF-β1-treated fibroblasts (i.e. myofibroblasts) showed a significant down-regulation of Dlk1 expression compared to non-treated fibroblasts (Figure 1G).
To further confirm Dlk1 association with fibroblast-to-myofibroblast conversion, we used cardiac fibroblasts isolated from Dlk1-knockout mice. Delta-like homologue 1-deficient fibroblasts exhibited an early increase in cell size, appearance of stress fibres and increased αSMA expression compared to wild-type fibroblasts, even under low serum conditions (Figure 2A). Of notice, we observed significant traces of αSMA that have been incorporated into the stress fibres in Dlk1-deficient fibroblasts, indicating that these cells were already differentiated into myofibroblasts.12 On the contrary, wild-type fibroblasts still display a more elongated phenotype, reduced αSMA expression and absence of stress fibres (Figure 2A). Furthermore, in the presence of 10% serum, Dlk1-null fibroblasts displayed a remarkable further increase in cell size, a well-organized polymerized actin cytoskeleton (Figure 2B), and lower proliferative capacity (Figure 2C), all features of fully differentiated myofibroblasts.12 Taken together, these data indicate that the absence of Dlk1 induces profound changes in fibroblasts related to the acquisition of an early myofibroblast phenotype in vitro.
Figure 2.

Lack of delta-like homologue 1 promotes fibroblast-to-myofibroblast conversion in cardiac fibroblasts. (A) Representative images exhibiting elongated wild-type fibroblasts vs. large, polygonal-shaped delta-like homologue 1-null fibroblasts cultured in the presence of 1% serum (left panel). Immunostaining of α-smooth muscle actin (middle panels, green) and F-actin (right panels, red) in wild-type and delta-like homologue 1-null fibroblasts with α-smooth muscle actin staining quantification (right panel; n = 5 different staining with 10–25 cells/field). Arrows point to co-localization of α-smooth muscle actin and stress fibres. (B) Images from wild-type and delta-like homologue 1-null fibroblasts cultured in the presence of 10% serum with fibroblasts lacking delta-like homologue 1 appearing large with a better organized actin cytoskeleton. Right panel shows cells at higher magnification with size quantification (n = 16 different fields with 5–10 cells/field. (C) Cell proliferation analysis assessed by incorporation of BrdU at 16, 20, and 24 h from wild-type and delta-like homologue 1-null fibroblasts. Scale bars: (A) 40 µm; (B) 40 µm (left images), 25 µm (right images).
To determine whether activated fibroblasts expressing Dlk1 required down-regulation of Dlk1 expression to differentiate into myofibroblasts, we overexpressed Dlk1 in fibroblasts using an adenovirus encoding the large soluble Dlk1 isoform (Ad.Dlk1). As shown in Supplementary material online, Figure S1A, non-infected and Ad.GFP-infected fibroblasts spontaneously differentiated into myofibroblasts in the presence of 10% serum, as indicated by the polygonal shape of the cells and expression of αSMA and Smemb. However, Ad.Dlk1-infected fibroblasts retained a more elongated phenotype and reduced αSMA and Smemb expression, suggesting that Dlk1 expression partially prevented differentiation into myofibroblasts under these conditions in vitro. Moreover, isolated Dlk1-null fibroblasts almost immediately differentiated to myofibroblasts (within 1–2 days), and thus they become insensitive to the overexpression of Ad.Dlk1 (Supplementary material online, Figure S1B). This observation is in agreement with a previous report claiming that fully differentiated myofibroblasts have reached an irreversible steady-state insensitive to changes in external factors.12
TGF-β1 signalling is enhanced in delta-like homologue 1-null fibroblasts in vitro
To obtain insight on how Dlk1 stimulates fibroblast-to-myofibroblast differentiation, we examined the effect of Dlk1 deletion on TGF-β1-induced myofibroblast phenotype. TGF-β1 increased myofibroblasts cell size (Figure 3A) and enhanced the expression of αSMA, lysyl oxidase (LOX), CTGF, and tissue inhibitor of metalloproteinase-1 (TIMP-1) in fibroblasts lacking Dlk1 compared to wild-type cells (Figure 3B). Interestingly, even non-TGF-β1-treated Dlk1-null cells displayed higher expression of αSMA, LOX, and TIMP-1 compared to non-treated wild-type fibroblasts (Figure 3B), suggesting increased basal level of active TGF-β1 signalling in Dlk1-null fibroblasts. Since TGF-β1 pro-fibrotic effects are largely attributed to Smad3-mediated signalling, we determined TGF-β1 activation of Smad3 phosphorylation. Interestingly, whereas phosphorylation in wild-type cells reached its maximal level by 30 min following TGF-β1 stimulation, Smad3 in Dlk1-null fibroblasts was further phosphorylated and reached a significantly higher level compared to wild-type cells after 90 min of TGF-β1 incubation (Figure 3C). Taken together, these data suggest that Dlk1 negatively regulates the TGF-β1/Smad3 pathway.
Figure 3.

Deletion of delta-like homologue 1 leads to activation of TGF-β1 signalling. (A) Representative images of wild-type and delta-like homologue 1-null fibroblasts ± TGF-β1 for 24 h (10 ng/mL). Scale bars: 40 µm. (B) qRT-PCR for TIMP-1, α-smooth muscle actin, connective tissue growth factor, and lysyl oxidase from wild-type and delta-like homologue 1-null fibroblasts ± TGF-β1 (upper panel) with representative western blots of α-smooth muscle actin and lysyl oxidase (lower panel). The numbers indicate α-smooth muscle actin and lysyl oxidase densitometric analyses normalized to tubulin and GAPDH loading, respectively. (C) Western blot analysis of phosphorylated Smad-3 in wild-type and delta-like homologue 1-null fibroblasts ±TGF-β1 for 30 and 90 min. Time 0 shows basal levels of phospho-Smad3. Graph represents the ratio of phospho-Smad3/total Smad3 at 0, 30, and 90 min.
Delta-like homologue 1-null mice exhibit hyperactivation of the TGF-β1/Smad3 pathway and extra-domain A-fibronectin splicing in vivo
We then investigated if Dlk1 deficiency also promotes fibroblast-to-myofibroblast differentiation in vivo. As shown in Figure 4A, TGF-β1 activation, reflected by Smad3 phosphorylation, was enhanced in cardiac tissues from Dlk1-null mice compared to wild types. Furthermore, confocal immunohistochemistry revealed the presence of phospho-Smad3 nuclear localization (i.e. activation) in myocytes (white arrows) and fibroblasts (yellow arrows) from mice lacking Dlk1 (Figure 4A). TGF-β1 is a potent inducer of fibronectin extra-domain A (EDA-FN) splicing, a necessary ECM mediator of TGF-β1 promotion of fibroblast differentiation.13,14 Accordingly, immunoblot analysis of Dlk1-null cardiac lysates showed a remarkable up-regulation of EDA-FN expression compared to wild-type lysates (Figure 4B). Intriguingly, confocal images of ventricular tissue demonstrated deposition of EDA-FN around cardiomyocytes in mice lacking Dlk1, whereas no EDA-FN was found in wild types (Figure 4B). Moreover, co-immunostaining with αSMA antibody to localize myofibroblasts, revealed the presence of myofibroblasts within an EDA-FN-rich extracellular network (Figure 4B, lower right panel). Supporting these results, expression of mTOR and SF2, upstream molecules involved in EDA-FN splicing14 (Figure 4C), and expression of αSMA (Figure 4D) are enhanced in Dlk1-null mice. Altogether, these findings point to an important role of Dlk1 in the process of fibroblast differentiation in vitro and in vivo by interfering with TGF-β1 signalling.
Figure 4.

Deletion of delta-like homologue 1 (Dlk1) promotes TGF-β1/Smad3 activation and induction of extra-domain A-fibronectin in mice. (A) (upper panel) Phosphorylated Smad-3 immunostaining (green) in wild-type and delta-like homologue 1-null myocardium. WGA (magenta) stains membrane/extracellular matrix. White arrows indicate cardiomyocyte Smad-3 activation. Yellow arrows point to non-myocyte (i.e. fibroblast) Smad-3 activation. Representative western blots and ratio quantification of phosphorylated Smad-3/total Smad-3 in heart lysates (lower panel). (B) Extra-domain A-fibronectin immunostaining (red) in wild-type and delta-like homologue 1-null cardiac tissues. Double immunostaining for extra-domain A (red) and α-smooth muscle actin (green) identified a group of α-smooth muscle actin-positive cells (i.e. myofibroblasts) embedded in an extra-domain A-rich extracellular matrix (lower panel, right image). (C) Western blots and ratio quantification of extra-domain A fibronectin, α-smooth muscle actin, mTOR, and SF2 from wild-type and delta-like homologue 1-null hearts. Calsequestrin, actin, and Ponceau Red are loading controls. (D) qRT-PCR showing up-regulation of α-smooth muscle actin mRNA in delta-like homologue 1-null hearts. Scale bars: (A) 20 µm; (B) 40 µm (left panel), 20 µm (right panel). qRT-PCR, quantitative real time polymerase chain reaction; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; TGF, Transforming growth factor; WGA, Wheat germ agglutinin; MMP9, Matrix metallopeptidase 9.
Delta-like homologue 1 deletion promotes collagen deposition, myocyte hypertrophy, and impairs heart function
Excess ECM deposition leads to fibrotic disorders and, eventually, cardiac dysfunction.2,5,15 We assessed left ventricular (LV) function and dimensions in Dlk1-deficient mice and age-matched wild types by serial echocardiography at 7, 13, and 19 weeks of age. Delta-like homologue 1-null mice demonstrated clear posterior LV wall thinning and a substantial LV chamber dilatation, and exhibited significantly reduced contractility compared to wild-type mice, as assessed by the fractional shortening (FS%) which, surprisingly, did not worsen over time (Figure 5A and Table 1). Gross cardiac examination indicated modest heart size increase in Dlk1-null mice and post-mortem cross-sectional areas analysis revealed increased cardiomyocyte size (Figure 5B; Supplementary material online, Figure S2) along enhanced foetal cardiac stress gene expression compared to wild types (Supplementary material online, Figure S2).
Figure 5.

Ablation of delta-like homologue 1 leads to extracellular matrix remodelling and cardiac dysfunction. (A) Representative H&E staining of whole hearts and raw tracings of M-mode echocardiography of 13 week-old wild-type and Dlk1−/− mice. Measurements of left ventricular fractional shortening and left ventricular end-diastolic/systolic diameter are shown. (B) Representative epifluorescence images of left ventricular histological sections stained with actinin (cardiomyocytes) and DAPI (nuclei). Top panel represents longitudinal section; lower panel shows cross-section. White drawing indicates myocyte sizes. Scale bar: 20 μm. (C) Representative Masson’s trichrome-stained left ventricular sections and quantification of fibrosis area from wild-type (WT) and Dlk1−/− mice at 13 weeks. (D) qRT-PCR mRNA expression of the different pro-fibrotic genes indicated normalized to 18S RNA and expressed as fold change vs. wild type.
Table 1.
Left ventricular (LV) function and dimensions were measured by serial echocardiography at 7, 13, and 19 weeks in wild-type (WT) and delta-like homologue 1-null (KO) mice
| 7 weeks old |
13 weeks old |
19 weeks old |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | KO | P | WT | KO | P | WT | KO | P | |
| (n = 9) | (n = 10) | (n = 9) | (n = 10) | (n = 9) | (n = 9)a | ||||
| IVSd (mm) | 0.97 ± 0.03 | 0.91 ± 0.14 | 0.3078 | 1.00 ± 0.05 | 0.94 ± 0.13 | 0.1452 | 1.04 ± 0.07 | 0.90 ± 0.13 | 0.0378 |
| LVIDd (mm) | 2.94 ± 0.22 | 3.29 ± 0.37 | 0.0270 | 3.08 ± 0.18 | 3.7 ± 0.42 | 0.0011 | 3.24 ± 0.16 | 3.67 ± 0.22 | 0.0006 |
| LVPWd (mm) | 1.06 ± 0.04 | 1.09 ± 0.10 | 0.1055 | 1.07 ± 0.04 | 0.96 ± 0.08 | 0.0818 | 1.11 ± 0.07 | 0.98 ± 0.11 | 0.0244 |
| IVSs (mm) | 1.87 ± 0.09 | 1.76 ± 0.12 | 0.0442 | 1.83 ± 0.09 | 1.73 ± 0.16 | 0.1610 | 1.89 ± 0.09 | 1.74 ± 0.15 | 0.0328 |
| LVIDs (mm) | 1.05 ± 0.09 | 1.61 ± 0.26 | 0.0071 | 1.07 ± 0.13 | 1.82 ± 0.32 | 0.010 | 1.21 ± 0.13 | 1.8 ± 0.19 | 0.0001 |
| LVPWs (mm) | 1.84 ± 0.11 | 1.56 ± 0.31 | 0.0199 | 1.81 ± 0.31 | 1.53 ± 0.07 | 0.0222 | 1.82 ± 0.11 | 1.58 ± 0.13 | 0.0017 |
| FS (%) | 64.25 ± 1.40 | 51.21 ± 3.98 | 0.0001 | 65.3 ± 3.09 | 50.9 ± 4.36 | 0.0001 | 62.6 ± 2.78 | 51.05 ± 3.01 | 0.0003 |
| Weight (g) | 22.35 ± 1.19 | 22.43 ± 1.61 | 0.9104 | 26.7 ± 1.32 | 25.8 ± 1.7 | 0.5937 | 29.6 ± 1.87 | 28.0 ± 2.37 | 0.1341 |
| Heart rate (bpm) | 550.9 ± 29.05 | 543.8 ± 43.21 | 0.6802 | 533.65 ± 37.77 | 530.3 ± 37.44 | 0.8553 | 531 ± 54.61 | 525.3 ± 33.99 | 0.7984 |
FS, fractional shortening; IVSd/s, intraventricular septum thickness in diastole/systole; LVIDd/s, left ventricular end-diastolic/systolic diameter; LVPWd/s, LV posterior wall thickness in diastole/systole.
One mouse unexpectedly died in the 19 weeks group. Two-tailed P-values are reported for each group. The echocardiography data were analysed with repeated measures ANOVA test. Data reported are mean ± SD.
Furthermore, Masson’s histological analysis showed deposition of interstitial collagen vs. perivascular collagen, and reduced myofibrils and disorganized cardiomyocytes in mice lacking Dlk1 (Figure 5C; Supplementary material online, Figures S3 and S4). This pattern was paralleled by an enhancement in mRNA expression of the pro-fibrotic markers/ECM components such as procollagen-1, CTGF, LOX, MMP-9, and TIMP-1 in the Dlk1-null mice (Figure 5D). These data indicate that the absence of Dlk1 triggers myofibroblasts accumulation, resulting in excess ECM deposition leading to pathological fibrosis and cardiac dysfunction.
Delta-like homologue 1 expression is down-regulated in ischaemic human hearts and within the scar of infarcted pigs’ myocardium
To obtain translational potential, we determined the expression of Dlk1 in ischaemic cardiac tissues from human patients and from pigs subjected to MI for 1 month. Delta-like homologue 1 mRNA (Figure 6A; upper panel) and protein (Figure 6A; lower panel) expression was down-regulated in ischaemic tissues from human patients compared to healthy individuals. Similarly, Dlk1 mRNA (Figure 6B; upper panel) and protein (Figure 6B; lower panel) levels were decreased in the border and scar zones of infarcted pigs’ hearts compared to the remote area or sham hearts (Figure 6B). The mRNA expression of pro-fibrotic molecules, collagen1a, LOX and αSMA was significantly up-regulated in the scar area (Figure 6C), confirming the phenotype seen in porcine tissues. Interestingly, membrane-bound Dlk1 isoforms were the predominant variants in both, healthy humans (Figure 6A) and sham pigs (Figure 6B), since primers designed against the protease site barely detected any soluble isoforms.
Figure 6.

Delta-like homologue 1 is down-regulated in human ischaemic/fibrotic hearts and within the scar of infarcted pigs’ myocardium. (A) qRT-PCR of total delta-like homologue 1 and soluble/cleaved-encoding delta-like homologue 1 mRNA variants from healthy and ischaemic human cardiac (upper panel), and representative western blot of DLK1 in ischaemic patients (lower panel). (B) qRT-PCR of total delta-like homologue 1 and soluble/cleaved-encoding delta-like homologue 1 mRNA variants from sham pigs and from the remote, border and scar tissues of infarcted pigs’ hearts (upper panel) and western blot of delta-like homologue 1 in pigs’ scar lysates (lower panel). Commassie-blue staining is a loading control in A and B. (C) qRT-PCR mRNA quantification of collagen-1a, lysyl oxidase and α-smooth muscle actin.
Delta-like homologue 1 regulates TGFβ signalling and cardiac fibrotic response through activation of miR-370
We next attempted to determine the potential molecular mechanism(s) underlying Dlk1’s effects on TGFβ signalling and myocardial fibrosis. Considering that we have previously demonstrated a direct interaction between Dlk1 and Notch1 and that Dlk 1 inhibits Notch signalling in different cell systems,6,16,17 we examined the potential significance of Notch activity in this case. Cardiac Notch signalling didn’t seem to be affected by the absence of Dlk1. As shown in Supplementary material online, Figure S5A, mRNA expression levels of Notch receptors (1–4) and their downstream transcription factors, Hes1 and hey2, were unchanged in cardiac tissue from Dlk1-null mice compared to wild-type hearts. Furthermore, using antibodies against the active intracellular form of Notch [Notch intracellular domain (NICD)], we demonstrated that protein expression of NICD1 and NICD2 is not significantly different between Dlk1-null and wild-type hearts (Supplementary material online, Figure S5B), suggesting a Notch-independent action of Dlk1.
TGFβ receptor (TGFβ-R) 1 and 2 protein expression levels were significantly elevated in Dlk1−/− mice (Figure 7A), suggesting a transcriptional regulation. Our bioinformatics target prediction interrogation of Dlk1-Dio3 cluster-containing microRNAs that regulate TGFβ-R signalling identified miR-370 as a strong candidate that targets TGFβ-R2. The mRNA sequence of Tgfb-R2 is predicted to contain a conserved ‘seed’ sequence complimentary to miR-370 in the 3′-untranslated region, which is conserved between rodents, human and pig (Figure 7B). Evaluation of miR-370 expression in cardiac myocytes from Dlk1−/− mice shows a significant down-regulation of miR-370 compared to wild types (Figure 7C). Isolated cardio-fibroblasts also expressed miR-370 but to a much lesser extent compared to cardiomyocytes (Figure 7C). Interestingly, miR-370 was also decreased in human ischaemic/fibrotic hearts and within the scar of infarcted pig myocardium (Figure 7C), paralleling the expression of Dlk1 in these tissues (Figure 6A, B). We next examined if Dlk1 regulates the expression of miR-370. Transfection of cardiomyocytes from Dlk1−/− or wild-type hearts with Ad.Dlk1 virus showed increased expression of miR-370 compared to control transfection (Figure 7D), suggesting that Dlk1 mediates the expression of miR-370 in cardiomyocytes.
Figure 7.

Delta-like homologue 1 activates miR-370 and inhibits TGF-β1/Smad3 signaling. (A) Representative western blot analysis and densitometry quantification of TGFβ-R1 and 2 in wild-type and delta-like homologue 1-null hearts; Tubulin is a loading control. (B) Bioinformatics analysis identified TGFβ-R2 as putative target gene of miR-370. (C) qRT-PCR quantification of miR-370 in cardiac fibroblasts (CF) and cardiomyocytes (CM) isolated from wild-type and delta-like homologue 1-null mice, in healthy human and ischaemic patients, and in scar area in pigs vs. sham hearts. (D) qRT-PCR quantification of miR-370 following Ad.Dlk1 infection of cardiomyocytes (MOI100) from wild-type and delta-like homologue 1-null mice. (E) qRT-PCR quantification of TGFβ-R2, α-smooth muscle actin, connective tissue growth factor, and lysyl oxidase in fibroblasts isolated from Dlk1−/− or wild-type hearts treated with control miR (miR-Ctrl), miR-370 mimics (miR-370), control AntimiR (AntimiR-Ctrl), or miR-370 inhibitor/antimiR (AntimiR-370). 10% FBS is a positive control. Multiple comparisons were performed by analysis of variance (ANOVA) with post hoc Bonferroni’s test. (F) Western blot analysis and quantification of Smad3 phosphorylation (normalized to total Smad3) in wild-type and delta-like homologue 1-null fibroblasts ± miR-370 mimics.
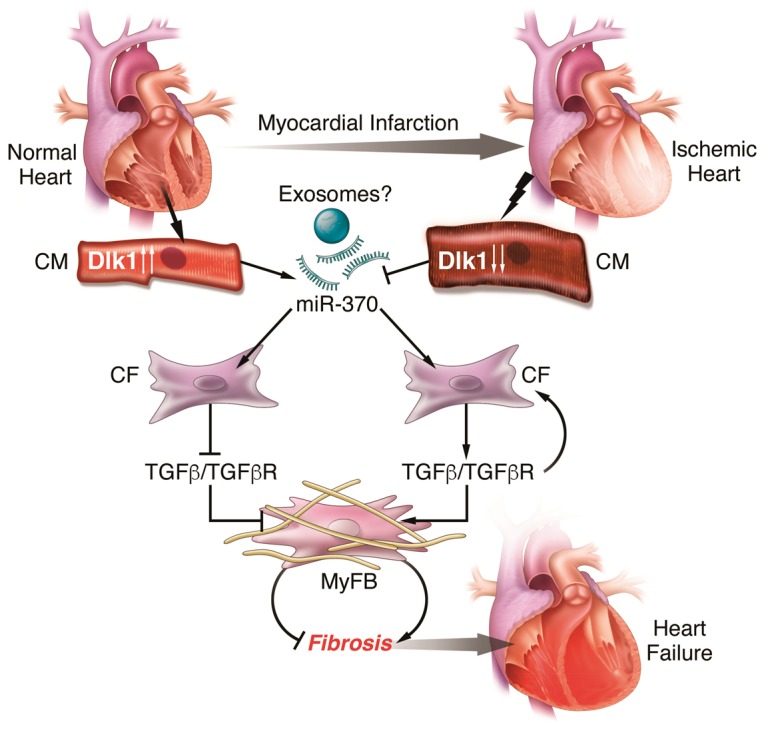
Take home figure.
In normal heart, Dlk1 expression is high in cardiomyocytes (CM) leading to increased secretion of miR-370 likely via exosomes. miR-370 acts on cardiac fibroblasts (CF) to block TGFβ signalling and transdifferentiation of myofibroblasts (MyFB) and fibrosis. However, under stress condition (i.e. myocardial infarction), Dlk1 expression is suppressed leading to downregulation of miR-370, stimulation of TGFβ signalling, and activation of myFB and induction of fibrosis and heart failure.
To further characterize the function of miR-370 in TGFβ signalling, we examined the effects of miR-370 on myofibroblast differentiation molecular program. The expression levels of TGFβ-R2, αSMA, CTGF, and LOX were all significantly reduced in fibroblasts isolated from Dlk1−/− or wild-type hearts treated with miR-370 mimics compared to fibroblasts treated with either control miR (miR-Cont) or miR-370 inhibitor (AntimiR-370) (Figure 7E). Furthermore, miR-370 mimics also attenuated the increase in Smad3 phosphorylation induced by the absence of Dlk1 in cardio-fibroblasts (Figure 7F). Interestingly, inhibition of miR-370 with AntimiR-370 in cardiac fibroblasts stimulated with 10% serum or TGF-β1 produced a myofibroblast phenotype very similar to Dlk1−/− fibroblasts (Supplementary material online, Figure S6). Altogether, these data provide strong evidence that Dlk1 controls the fibrotic response through the regulation of the miR-370/TGFβ-R2 axis, highlighting new opportunities to target cardiac fibrosis.
Discussion
Increasing evidence has demonstrated a role for Dlk1 in inhibiting differentiation of many cells, including adipocyte,9 osteoblasts,18 chondrocytes,19 haematopoietic,20 and neuronal cells.11 In line with this rationale, we asked whether Dlk1 may also be involved in cardiac fibroblast-to-myofibroblast differentiation, a process initiated after MI and is critical for the remodelling of the injured heart.2,4 Herein, we report for the first time that cardiac myocytes and fibroblasts express different Dlk1 isoforms and its absence in fibroblasts accelerates the process of differentiation into myofibroblasts. We demonstrate that Dlk1-null mice myocardium exhibited activation of the pro-fibrotic TGF-β1/Smad3 pathway and induction of the ECM EDA fibronectin variant, resulting in infiltration/accumulation of myofibroblasts and the formation of fibrotic areas. This is interesting as TGF-β1 expression upsurges in myocardium in experimental and human heart diseases.21 Specifically, activation of TGF-β1/Smad3 signalling in the infarct border is critical in the pathogenesis of fibrotic cardiac remodelling, contributing to cardiac dysfunction.4 We also show that mouse cardiomyocytes lacking Dlk1 display abnormalities in the number and arrangement of myofibrils, and overall reduction in cardiac performance and function. Additionally, our data demonstrate that Dlk1 expression in cardiac tissue from human ischaemic patients was significantly down-regulated compared to healthy donors, and in tissues from the border and scar areas in a porcine model of MI. Again, these observations reinforce our hypothesis that Dlk1 may be required to prevent cardiac tissue demise and cardiac performance decline.
Ischaemic stress activates cardiac fibroblasts, normally quiescent in healthy hearts,15 and induces their migration (to the injured area), proliferation, and differentiation into myofibroblasts,22 resulting in profound alterations in the composition of the ECM. Myofibroblasts, morphologically and functionally different from fibroblasts, are characterized by the presence of a contractile apparatus that contains αSMA,15 and are the main source of ECM proteins in the infarcted and remodelling heart; their appearance is a hallmark of the cardiac fibrotic response.2,15 The most important factors that mediate fibroblast differentiation are: (i) TGF-β1, which induces αSMA expression in fibroblasts through activation of Smad35 and (ii) alterations in the mechanical properties and composition of ECM, such as de novo expression of EDA-FN.13,23 Our results highlight a new candidate, Dlk1, as a primary regulator of fibroblast-to-myofibroblast conversion. We found that isolated, therefore activated, cardiac fibroblasts express mostly soluble Dlk1, but its expression was significantly down-regulated when they differentiate to myofibroblasts. Furthermore, isolated fibroblasts from Dlk1-null mice displayed a marked acceleration in the process of fibroblasts conversion. These data are consistent with our finding of accumulated myofibroblasts in the myocardium of mice lacking Dlk1. The fact that myofibroblasts are absent from healthy heart prompted us to examine the TGF-β/Smad3 pathway. Our data revealed higher Smad3 phosphorylation not only in fibroblasts/myofibroblasts, but also in cardiomyocytes from mice lacking Dlk1. These findings suggest that Dlk1 acts as an inhibitor of TGF-β1 signalling; indeed, isolated Dlk1-null fibroblasts exhibited an enhanced expression of TGF-β1-induced pro-fibrotic genes in both basal condition and following TGF-β1 treatment. Furthermore, TGF-β1 stimulation of Smad3 phosphorylation was further increased in fibroblasts lacking Dlk1. Our results showing that gene transfer of Dlk1 prevents fibroblast-to-myofibroblast differentiation support the hypothesis that Dlk1 is a negative mediator of this process. However, fibroblasts lacking Dlk1 were somewhat less responsive to Dlk1 overexpression. This discrepancy may be due to a remarkable acceleration in reaching a steady-state of fully differentiated myofibroblasts that makes them insensitive to changes to external factors.12 Additionally, mice lacking Dlk1 had high deposition of EDA-FN surrounding myocytes, and indeed, myofibroblasts were found embedded in this ECM. It is known that TGF-β1 increases total fibronectin by promoting accumulation of the EDA variant,24 and the presence of EDA allows TGF-β1 to induce fibroblast differentiation.13 These reports clearly support our hypothesis that Dlk1 interferes with the TGF-β1 pathway. This is an important finding because EDA fibronectin-deficient mice exhibited less fibrosis and better heart function after MI.23 Therefore, our findings strongly argue for a critical role for Dlk1 in maintaining an adequate ‘under-control’ TGF-β1 signalling in the heart, since its absence results in a phenotype clearly characterized by a hyperactive TGF-β1, with accumulation of myofibroblasts, excessive EDA variant deposition, dilated LV dimensions, and reduced myofibril integrity and organization, collectively leading to cardiac dysfunction.
To gain molecular mechanistic insight regarding Dlk1’s effects on TGFβ signalling,25 we examined the activation of the Notch pathway since recent findings show that augmented Notch activity enhances TGFβ signalling. In addition, we have previously demonstrated a direct interaction between Dlk1 and Notch1 and that Dlk1 inhibits Notch signalling in different cell systems.6,16,17 However, to our surprise, cardiac Notch signalling does not seem to be affected by the absence of Dlk1. This may partially be explained by the fact that Notch signalling is tightly regulated and is extremely age- and cell-context dependent, and/or the time window at which we examined Notch activity is characterized by a limited interaction between Dlk1 and Notch complexes. It is also possible that, unlike other cells types, in the adult heart Notch signalling is essential for normal cardiac function and response to injury. Notch inhibition accelerates fibroblast-to-myofibroblast transformation26 and leads to cardiac hypertrophy, apoptosis and development of fibrosis27; therefore, Dlk1’s inhibition of Notch in this case may not be beneficial for the maintenance of functional and structural integrity of the myocardium. This observation and the finding that enhanced TGFβ signalling stimulated by increased TGFβ receptor (TGFβ-R) 1 and 2 expression levels in Dlk1-knockout mice prompted us to explore potential transcriptional regulation mediated by microRNAs, given the key roles these molecules play in cardiac function and fibrosis.28 We focused our search on microRNAs that target TGFβ receptors and are at the same time expressed within the mammalian Dlk1-Dio3 genomic imprinted region. We reasoned that the Dlk1 cluster and the microRNAs it hosts may share a common regulatory program. miR-370 was identified as a strong candidate since (i) its expression pattern dovetails with that of Dlk1, (ii) Dlk1 appears to modulate the expression of miR-370, (iii) miR-370 directly targets TGFβ-R2, and (iv) miR-370 attenuated the myofibroblast differentiation molecular signature while its inhibition accelerated it, providing evidence that Dlk1 controls the fibrotic response through the regulation of the miR-370/TGFβ-R2 axis. How miR-370 expression varies depending on the fibroblast differentiation stages, whether miR-370 is primarily secreted by cardiomyocytes via exosomes and acts on fibroblasts in a paracrine manner, and what is the role of miR-370 in the maintenance of cardiac function and structure integrity are focus questions of other ongoing investigations.
In summary, we have identified a novel role for Dlk1 in regulating the process of cardiac fibroblast-to-myofibroblast differentiation. Delta-like homologue 1 exerts these effects via activation of miR-370 leading to inhibition of TGF-β1/Smad3 signalling pathway. Given the deleterious effects of continuous activation of this pathway, we propose Dlk1 as a new potential candidate for therapy in cases where aberrant TGF-β1 signalling leads to chronic fibrosis.
Funding
National Institutes of Health (Grant R01HL097357 to D.L.); Ministerio de Economía y Competitividad (MINECO) SAF 2012-31338, CSD 2007-00020, Comunidad de Madrid ‘Fibroteam’ S2010/BMD-2321 to S.L.
Conflict of interest: none declared.
Supplementary Material
References
- 1. Kong P, Christia P, Frangogiannis NG.. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC.. Cardiac fibrosis: the fibroblast awakens. Circ Res 2016;118:1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J.. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 2010;7:30–37. [DOI] [PubMed] [Google Scholar]
- 4. Bujak M, Frangogiannis NG.. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 2007;74:184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG.. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 2010;107:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baladrón V, Ruiz-Hidalgo MJ, Nueda ML, Díaz-Guerra MJM, García-Ramírez JJ, Bonvini E, Gubina E, Laborda J.. Dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp Cell Res 2005;303:343–359. [DOI] [PubMed] [Google Scholar]
- 7. Smas CM, Green D, Sul HS.. Structural characterization and alternate splicing of the gene encoding the preadipocyte EGF-like protein pref-1. Biochemistry 1994;33:9257–9265. [DOI] [PubMed] [Google Scholar]
- 8. Mei B, Zhao L, Chen L, Sul HS.. Only the large soluble form of preadipocyte factor-1 (Pref-1), but not the small soluble and membrane forms, inhibits adipocyte differentiation: role of alternative splicing. Biochem J 2002;364:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laborda J. The role of the epidermal growth factor-like protein dlk in cell differentiation. Histol Histopathol 2000;15:119–129. [DOI] [PubMed] [Google Scholar]
- 10. Smas CM, Sul HS.. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell 1993;73:725–734. [DOI] [PubMed] [Google Scholar]
- 11. Ferrón SR, Charalambous M, Radford E, McEwen K, Wildner H, Hind E, Morante-Redolat JM, Laborda J, Guillemot F, Bauer SR, Fariñas I, Ferguson-Smith AC.. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 2011;475:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV.. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res 2014;101:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G.. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 1998;142:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, Roman J, Muro AF.. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp Cell Res 2010;316:2644–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA.. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349–363. [DOI] [PubMed] [Google Scholar]
- 16. Rodríguez P, Higueras MA, González-Rajal A, Alfranca A, Fierro-Fernández M, García-Fernández RA, Ruiz-Hidalgo MJ, Monsalve M, Rodríguez-Pascual F, Redondo JM, de la Pompa JL, Laborda J, Lamas S.. The non-canonical NOTCH ligand DLK1 exhibits a novel vascular role as a strong inhibitor of angiogenesis. Cardiovasc Res 2012;93:232–241. [DOI] [PubMed] [Google Scholar]
- 17. Nueda ML, Baladron V, Sanchez-Solana B, Ballesteros MA, Laborda J.. The EGF-like protein dlk1 inhibits notch signaling and potentiates adipogenesis of mesenchymal cells. J Mol Biol 2007;367:1281–1293. [DOI] [PubMed] [Google Scholar]
- 18. Abdallah BM, Jensen CH, Gutierrez G, Leslie RG, Jensen TG, Kassem M.. Regulation of human skeletal stem cells differentiation by Dlk1/Pref-1. J Bone Miner Res 2004;19:841–852. [DOI] [PubMed] [Google Scholar]
- 19. Chen L, Qanie D, Jafari A, Taipaleenmaki H, Jensen CH, Saamanen AM, Sanz ML, Laborda J, Abdallah BM, Kassem M.. Delta-like 1/fetal antigen-1 (Dlk1/FA1) is a novel regulator of chondrogenic cell differentiation via inhibition of the Akt kinase-dependent pathway. J Biol Chem 2011;286:32140–32149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakajiri S, O'Kelly J, Yin D, Miller CW, Hofmann WK, Oshimi K, Shih LY, Kim KH, Sul HS, Jensen CH, Teisner B, Kawamata N, Koeffler HP.. Dlk1 in normal and abnormal hematopoiesis. Leukemia 2005;19:1404–1410. [DOI] [PubMed] [Google Scholar]
- 21. Li RK, Li G, Mickle DA, Weisel RD, Merante F, Luss H, Rao V, Christakis GT, Williams WG.. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997;96:874–881. [DOI] [PubMed] [Google Scholar]
- 22. Virag JI, Murry CE.. Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol 2003;163:2433–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP.. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res 2011;108:582–592. [DOI] [PubMed] [Google Scholar]
- 24. Kocher O, Kennedy SP, Madri JA.. Alternative splicing of endothelial cell fibronectin mRNA in the IIICS region. Functional significance. Am J Pathol 1990;137:1509–1524. [PMC free article] [PubMed] [Google Scholar]
- 25. Ohnuki H, Jiang K, Wang D, Salvucci O, Kwak H, Sanchez-Martin D, Maric D, Tosato G.. Tumor-infiltrating myeloid cells activate Dll4/Notch/TGF-beta signaling to drive malignant progression. Cancer Res 2014;74:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fan YH, Dong H, Pan Q, Cao YJ, Li H, Wang HC.. Notch signaling may negatively regulate neonatal rat cardiac fibroblast-myofibroblast transformation. Physiol Res 2011;60:739–748. [DOI] [PubMed] [Google Scholar]
- 27. High FA, Epstein JA.. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet 2008;9:49–61. [DOI] [PubMed] [Google Scholar]
- 28. Gurha P. MicroRNAs in cardiovascular disease. Curr Opin Cardiol 2016;31:249–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.