

We used discovery, bottom up proteomics to provide the first in-depth accessory gland proteome in D. pseudoobscura,. Computational bioinformatics identified >500 proteins in the secretory pathway of which 163 were annotated as extracellular., and therefore candidate, seminal fluid proteins. We further compared molecular rates of evolution between intra- and extra-cellular proteins, showing a hierarchy of rapid evolution with putative seminal fluid proteins evolving more rapidly than other secreted proteins, and those proteins evolving more rapidly than intra-cellular proteins.
Keywords: Protein-Protein Interactions*, Mass Spectrometry, Molecular evolution*, Protein Identification*, Networks*, experimental evolution, post-copulatory sexual selection, sexual selection
Graphical Abstract

Highlights
First deep proteomic coverage of an accessory gland proteome in Drosophila.
Discovery proteomics identified >3000 proteins of the D. pseudoobscura, accessory gland proteome.
Identified 132 putative novel seminal fluid proteins in this species.
Demonstrated the exoproteome as the most rapidly evolving subcellular component of the proteome.
Abstract
Seminal fluid proteins (SFPs), the nonsperm component of male ejaculates produced by male accessory glands, are viewed as central mediators of reproductive fitness. SFPs effect both male and female post-mating functions and show molecular signatures of rapid adaptive evolution. Although Drosophila melanogaster, is the dominant insect model for understanding SFP evolution, understanding of SFP evolutionary causes and consequences require additional comparative analyses of close and distantly related taxa. Although SFP identification was historically challenging, advances in label-free quantitative proteomics expands the scope of studying other systems to further advance the field. Focused studies of SFPs has so far overlooked the proteomes of male reproductive glands and their inherent complex protein networks for which there is little information on the overall signals of molecular evolution. Here we applied label-free quantitative proteomics to identify the accessory gland proteome and secretome in Drosophila pseudoobscura,, a close relative of D. melanogaster,, and use the dataset to identify both known and putative novel SFPs. Using this approach, we identified 163 putative SFPs, 32% of which overlapped with previously identified D. melanogaster, SFPs and show that SFPs with known extracellular annotation evolve more rapidly than other proteins produced by or contained within the accessory gland. Our results will further the understanding of the evolution of SFPs and the underlying male accessory gland proteins that mediate reproductive fitness of the sexes.
Male ejaculates typically consist of a sperm component and a nonsperm component, both of which are transferred to females during mating. The nonsperm component is seminal fluid, containing secreted peptides and proteins (SFPs)1, typically produced in the testes and specialized male exocrine glands (1, 2). SFPs have profound effects on both male and female reproductive fitness (3) and therefore significant attention has been focused on the role of SFPs in polyandrous species. Polyandry, where females mate with different males across a reproductive bout generating postcopulatory sexual selection, results in ejaculates that compete for fertilization of a limited supply of ova, and females may choose whose sperm will fertilize those limited ova (4). Polyandry also engenders sexual conflict, in which male and female reproductive interests differ, because of the disproportionate costs and benefits of mating between the sexes (5). In internally fertilizing species, postcopulatory sexual selection operates between the male ejaculate that is transferred to and stored in the female reproductive tract (6). SFPs in these species may increase female fecundity, reduce female receptivity, decrease female life span, alter female hunger, and remodel female reproductive tract morphology (2, 3, 7, 8).
SFPs were first identified by their canonical signal peptide sequence that direct proteins to the secretory pathway (2). Cross-species comparative work has found that general classes of SFPs are conserved (e.g., proteases and protease inhibitors, lectins and prohormones) suggesting that their mechanisms of action are also conserved. However, individual SFPs can rapidly evolve with signals of accelerated rates of adaptive molecular evolution found in studies of coding sequence and male-biased gene expression observed across different animal taxa (e.g., mammals (9, 10); birds (11); Drosophila (12–14)). Sex-biased genes in general show faster rates of sequence and expression divergence that is consistent with predictions from sexual selection (e.g., (15) but see (11)).
Despite these general patterns, there are limitations to understanding the evolution of SFPs and their function. For example, SFP identification and their role in influencing fitness is dominated by work in D. melanogaster,. This species is relatively highly polyandrous (16) and studies identifying SFPs in species with different mating systems is necessary to understand the evolution of reproductive proteins and their fitness consequences. The advent of high throughput proteomics using LC-MS/MS should allow identification of SFPs, even in nonmodel organisms although tests of adaptive evolution may be restricted (17).
Moreover, SFPs function in a complex network of protein-tissue interactions (1, 2)). However, the general focus on SFPs (a small subset of the accessory gland proteome) leaves open questions about the full complexity of the accessory gland proteome that supports the production of these critical reproductive proteins. Further, the larger role other accessory gland proteins play in postcopulatory sexual selection, and how the accessory gland proteome responds to such selection in toto, has been relatively ignored. For example, despite D. melanogaster, being a model system for this work, there is but a single study of its accessory gland proteome which is based on 2D gel electrophoresis (18). The recent advent of high throughput proteomics using LC-MS/MS should allow identification of not only SFPs but of the supporting proteins in the male accessory reproductive tissues. A proteomic study of the oriental fruit fly, Bactrocera dorsalis,, identified ∼3000 male accessory gland proteins by LC- MS/MS (19) but focused only on the proteins with identified signal sequences and did not further study the entire proteome. Although a recent study used LC-MS/MS to determine both the male and female accessory gland proteome of the silk worm, Bombyx mori,, no tests of molecular evolution of these proteins were performed (20). Previous studies have shown that the subcellular localization of a protein is a strong predictor of its evolutionary rate, and that extracellular proteins secreted from the cell evolve faster than intracellular proteins (21–23). Whether this pattern is observed in the male accessory gland requires testing.
Here we aimed to address these limitations by using LC-MS/MS to characterize the accessory gland proteome of Drosophila pseudoobscura,, whose mating system, although displaying lower levels of polyandry than the model species D. melanogaster,, has nonetheless proven useful for experimental evolution studies documenting rapid sex-specific responses to across-generation changes of the mating system (24, 25). Our comparative study also takes advantage the extensive genetic knowledgebase available in Drosophila melanogaster, and on SFP functional genomics and evolution. We further characterize the D. pseuddobscura, accessory gland proteome by constructing, using bioinformatics and gene ontology (GO), an accessory gland secretome (AcgS), exoproteome (exoP) and candidate SFPs. Finally, we compare rates of molecular evolution between these proteome subcomponents to test how subcellular protein localization impacts evolutionary rates in this system
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
The experimental design included D. pseudoobscura, selection lines derived from a naturally polyandrous line as previously described (25, see also Experimental Lines section below). Four replicates of each of two selection lines were used for the LFQ studies which provided the necessary replicative power for high value protein identifications and the statistical power needed for downstream GO category enrichment. We employed standard hypergeometric statistical tests as implemented in Cytoscape v3.7.1 (33, 34) and visualized using the Cluego plug-in app (35, 36).
Stock and Fly Maintenance
We used experimentally evolved sexual selection lines in which the opportunity for post- copulatory sexual selection was either eliminated or facilitated. The establishment and maintenance of the selection lines were previously described in detail (25). Briefly, an ancestral wild-caught population of D. pseudoobscura, from Tucson AZ, a naturally polyandrous species (wild caught females have been shown to be frequently inseminated by at least two males at any given time; (26)), was used to establish the selection lines. From this population, four replicate lines (replicates 1–4) of two different sexual selection treatments were established. To modify the opportunity for sexual selection, adult sex ratio in vials was manipulated by either confining one female with a single male which enforces monogamy (“monogamy” treatment, M) and eliminates postcopulatory sexual selection and sexual conflict or one female with six males promoting polyandry (“polyandry” treatment, P (NB: this treatment has also been referred to as E in other publications). Effective population sizes are equalized between the treatments as described previously (27). At each generation, offspring are collected and pooled together for each replicate line, and a random sample from this pool is used to constitute the next generation in the appropriate sex ratios, thus proportionally reflecting the differential offspring production across families. In total, eight selection lines (M1, M2, M3, M4 and P1, P2, P3, P4) are maintained, in standard vials (2.5 × 80 mm), with a generation time of 28 days. All populations are kept at 22 °C on a 12L:12D cycle, with standard food media and added live yeast.
Sample Preparation
Flies from replicates 1–4 of each of the selection lines were collected from generations 157, 156, 155, and 153 respectively. We standardized for maternal and larval environments (25), but in brief, parental flies were collected and housed in food bottles, then groups of about 30 were transferred on egg laying plates for 24 h, removed and replaced with a fresh egg plate. This second plate was removed after 24 h, then 48 h later, first instar larvae were collected in groups of 100 and housed in standard molasses/agar food vials at 22 °C. Males from these vials were collected on the day of eclosion and housed in vials of 10 individuals, until they were sexually mature (28), and then dissected when they were reproductively mature at 5 or 6 days old.
Using a Leica stereomicroscope, reproductively mature males were ether anesthetized and accessory glands dissected into a drop of PBS using fine dissection needles (supplemental Fig. S1A,, S1B,). Each accessory gland pair was moved to a fresh drop of PBS and then into a microcentrifuge tube containing 0.5 ml PBS at 4 °C until a total of 30 accessory gland pairs per replicate were acquired. Samples were then stored at −80 following a brief centrifugation. Each tube containing 30 accessory gland pairs was subsequently thawed on ice and pelleted at 20,000 rpm at 4 °C for 5 min, supernatants removed, and proteins extracted by addition of 30 μl of RIPA buffer (Sigma), containing HALT protease inhibitor mixture and phenylmethylsulfonyl fluoride as per manufacturer instructions (Thermo Fisher Scientific). Samples were then taken through three freeze/thaw cycles using dry ice then thawed at 37 °C for 30 s, vortexed then centrifuged at 20,000 rpm for 5 min at 4 °C to remove insoluble tissue
SDS-PAGE and In-gel Digestion of Proteins
Protein concentrations of the samples described above were determined using a Bradford assay followed by the addition of SDS sample buffer containing 10 mm dithiothreitol. Samples containing 50 μg/lane were then loaded onto 4–12% SDS-PAGE gels and electrophoresed as per manufacturer instructions (Invitrogen). Protein bands were visualized (supplemental Fig. S1C,) using Brilliant Blue G Colloidal Concentrate (Sigma). Each gel lane was manually cut into 33–36 pieces of approximately equivalent size and destained using 200 mm ammonium bicarbonate and 40% acetonitrile. Gel pieces were then reduced in 200 μl of a 50 mm ammonium bicarbonate buffer containing 10 mm dithiothreitol, followed by alkylation in a similar volume of a 50 mm ammonium bicarbonate containing 55 mm iodoacetamide. Gel pieces were then centrifuged at 13 Kg for 10 s and dried using a vacuum concentrator until all samples were dry (∼30 min). The dried pieces were then hydrated in a solution containing 20 μl of trypsin (New England BioLabs) and 50 μl of acetonitrile and incubated overnight at 37 °C. Peptides were extracted the following day using a standard method with a solution of 100% acetonitrile and 5% formic acid and dried down overnight in a vacuum concentrator at 30 °C. Resulting peptides were resuspended in 7.5 μl of 0.1% (v/v) formic acid, 3% (v/v) acetonitrile, sonicated in a water bath for 5 min and centrifuged at 13 × g, for 10 s, before being transferred to a sample vial and loaded into the autosampler tray of the Dionex Ultimate 3000 μHPLC system. Samples were set to run using the Xcalibur sequence system.
Liquid Chromatography-MS/MS (LC-MS/MS) Data Collection
All MS data were collected using an LTQ Orbitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific) equipped with an Easy-Spray (Thermo Fisher Scientific) ion source. Peptides were separated using an Ultimate 3000 Nano LC System (Dionex). Peptides were desalted on-line using a capillary trap column (Acclaim Pepmap100, 100 μm, 75 μm × 2 cm, C18, 5 μm; Thermo Fisher Scientific) and then separated using 60 min reverse phase gradient (3–40% acetonitrile/0.1% formic acid) on an Acclaim PepMap100 RSLC C18 analytical column (2 μm, 75 μm id × 10 cm; Thermo Fisher Scientific) with a flow rate of 0.25 μl/min. The mass spectrometer was operated in standard data dependent acquisition mode controlled by Xcalibur 2.2. The instrument was operated with a cycle of one MS (in the Orbitrap) acquired at a resolution of 60,000 at m,/z, 400, with the top 20 most abundant multiply-charged (2+ and higher) ions in a given chromatographic window further subjected to CID fragmentation in the linear ion trap. An FTMS target value of 1e6 and an ion trap MSn target value of 10,000 were used. Dynamic exclusion was enabled with a repeat duration of 30 s with an exclusion list of 500 and exclusion duration of 30 s.
Database Construction of the D. pseudoobscura Accessory Gland Proteome (AcgP)
To obtain the deepest possible coverage of the proteome, we combined the global proteomic data set of all eight replicates and searched mass spectra data files using Sequest HT within the Proteome Discover suite (Thermo Fisher Scientific, San Jose, CA; version 1.4.1.14) using Drosophila pseudoobscura pseudoobscura, fasta file (Uniprot UP000001819, 20,816 entries December, 2015 release).
Peptide matches were further analyzed and validated within Scaffold Q+ (Proteome Software; version 3.2.0) using X!Tandem. Sequest HT and X!Tandem searches were set with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 10.0 parts per million. The oxidation of methionine (15.99), carboxyamidomethyl of cysteine (57.02), and acetyl modification on peptide N terminus (42.01) were set as variable modifications. Files from Sequest HT searches within the same gel lane were merged together as Mudpit using Scaffold, which calculated False Discovery Rates (FDRs) using a reverse concatenated decoy database (FDR was set at 1.0%). Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the PeptideProphet (29) and protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet Algorithm (30). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. The data set was filtered so that every protein must be identified by at least two unique peptides in any one of the biological replicates. Although a conservative approach, this procedure ensured a robust data set devoid of potential misidentifications often caused by use of a single peptide for protein identification. To establish a working list of the AcgP, protein IDs from Scaffold were converted to D. pseudoobscura, Fly Base gene numbers (FBgns) using the Uniprot website (Uniprot.org). The resulting D. pseudoobscura, FBgns were then used to query Flybase (Flybase.org) to retrieve orthologous D. melanogaster, genes from the OrthoDB orthology tables as implemented in Flybase (flybase.org). A complete listing can be found in supplemental Table S1. Note that in all cases, except where noted, only strict 1:1 orthologs were used in this study.
Database Construction of the D. pseudoobscura Accessory Gland Secretome (AcgS) and Exoproteome (exoP)
As a secretory organ, the accessory gland is expected to contain the cellular machinery necessary for efficient and sustained secretory activity throughout the adult reproductive life cycle. To examine and focus on potential activities related to secretion, we assembled an in silico, AcgS from the 3281 FBgns of the AcgP as input into Uniprot resulting in 5624 UniProtKB IDs (which includes all predicted protein isoforms). Fasta protein sequence files from each Uniprot entry were downloaded and submitted to SignalP (31) and Phobius (32), using default settings. The protein IDs were combined and exported into Excel yielding a final list of 771 UniProt identifiers. The Uniprot IDs were mapped back to 506 unique D. pseudoobscura, FBgns (via Uniprot) which, after submission to OrthoDB (via Flybase) resulted in a final list and 506 D. melanogaster, 1:1 orthologs. Candidate SFPs were identified by first querying the list of 515 AcgS genes in Flybase for Gene Ontology (GO) terms containing “extracellular.” We also compared this list to RNAseq data of the accessory gland in this population and found that 100% of our AcgS proteins were expressed in the accessory gland (Snook, unpublished data). The resulting list of 163 proteins therefore represents the accessory gland exoproteome (exoP) and is considered to contain a representative sampling of a major fraction of SFPs.
Pathway, GO Enrichment, and Protein Interaction Network Analyses
The finalized data sets were used for downstream bioinformatic analyses and subsequent visualizations of GO enrichment. The protein coding sequences of the AcgP were downloaded from Uniprot and submitted to Blast2go for annotation and tabulation of the three major GO categories, biological process (BP), molecular function (MF) and cellular component (CC). GO enrichment and network visualization and analysis was performed with Cytoscape v3.4 (33, 34) and ClueGO plugin version 2.2.4 (35, 36). Network parameters used were specific for each dataset as detailed in figure legends and supplemental tables. Protein interaction network analysis was performed using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING), a program that calculates the degree of protein-protein network interconnectivity (37).
Molecular Evolution Parameters
We obtained genome-wide coding sequence information for two close relatives, D. lowei, and D. affinis, (http://popoolation.at/lowei_genome and http://popoolation.at/affinis_genome), (38).
These two species have the same karyotype as D. pseudoobscura, and show reasonable divergence (median pairwise dS = 0.102 versus D. lowei, and dS = 0.26 versus D. affinis,) thus avoiding substitution rate saturation. To identify orthologs of the identified AcgP D. pseudoobscura, proteins in the two other Drosophila genomes we combined two approaches. First we used gene annotation ignoring isoforms specification using only the longest isoform identified to maximize (as these are difficult to identify within a proteomic screen). We then used best BLAST hits (39) of the D. pseudoobscura, gene against each of the two other genomes but excluded gene sets for which annotation was contradictory to the D. pseudoobscura, annotation. Using a pipeline, we developed earlier (40), sequences were aligned using MUSCLE (41), uncertain sequences filtered out using ZORRO (42) and input files converted with pal2nal (43). Sequences were then analyzed using PAML v4.9 (44) to obtain dN/dS values for each gene set (one-ratio estimates). For the one-ratio estimates, median differences in dN, dS and dN/dS among groups (AcgP, AcgS, exoP) were tested using a nonparametric two-tailed test (45).
RESULTS
The D. pseudoobscura AcgP
A D. pseudoobscura, AcgP was constructed from peptide-based (“bottom-up”) shotgun MS/MS spectral data obtained from eight independent runs. For the purposes of assembling a proteome with the broadest coverage, data from all runs were pooled together resulting in a total of 3757 UniProt IDs that mapped to 3160 unique FlyBase gene names. Because a latter research goal is to quantify how sexual selection affects the production of proteins in the male accessory gland, here we present the overlap between proteins identified in the four replicates each of M and P selection lines. These data sets were highly correlated with >90% (3534/3757) overlap (Fig. 1A,). Likewise, proteins with values in all four replicates for each selection line represented most identified proteins (M-line 2103/3649; 57.6% and P-line, 2235/3642; 61.4%). A complete listing and tabulation of these results can be found in supplemental Table S1. The small number of proteins unique to each population (M-unique = 115; P-unique = 108; Fig. 1A,) most likely represent missed protein assignments because of low quantities (as measured by total spectral counts). Indeed, the average total spectral counts for the unique set of proteins (ave. 4.5; n, = 223) was 16-fold lower than the average across the entire dataset (ave. 72.8, n, = 3874). We do not consider these unique proteins further here and base the remainder of the description of male accessory gland proteins on those that are shared by each treatment.
Fig. 1.
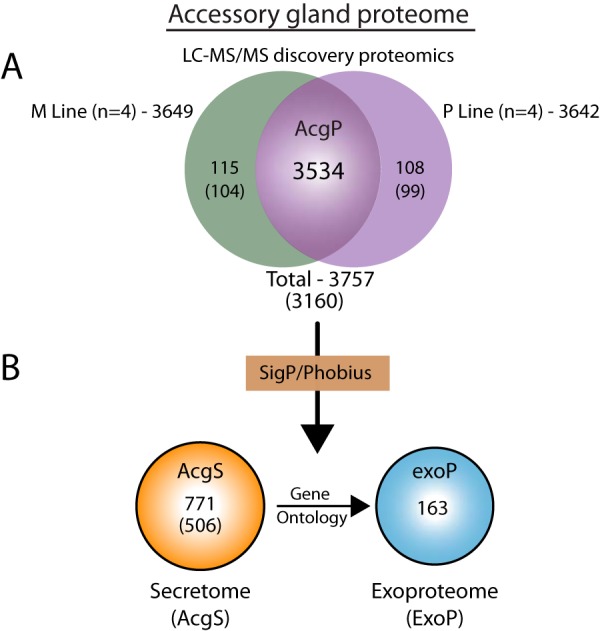
D. pseudoobscura, accessory gland summary statistics. A,, Venn diagram of combined and total Uniprot IDs from the M- and P-experimental lines used to assemble the AcgP. B,, Secretome and exoproteome from the AcgP derived in silico, (see Methods).
GO and Pathway Analysis of the AcgP
Pathway and functional analyses began by identifying orthologous D. melanogaster, genes, (3160/3281, 96.2%; supplemental Table S1), providing a useful annotated database to evaluate the overall patterns of the functional elements in the AcgP. As an overview of the major GO groupings, Blast2Go returned “signaling,” “reproduction,” and “localization” among BP- and numerous proteins annotated as extracellular involved in the CC-categories, respectively (supplemental Fig. S2). Statistical analysis for GO category enrichment (Fig. 2) identified BP terms related to intracellular transport (n, = 363, p, = 2.80E-45), translation (n, = 257, p, = 2.47E-35), establishment of protein localization (n, = 418, p, = 1.22E-44), vesicle-mediated transport (n, = 301, p, = 8.47E-42), endocytosis (n, = 106, p, = 7E-10), and secretion (n, = 140, p, = 2.71E-16). Likewise, the AcgP contains a significant number of proteins in CC categories annotated as “extracellular region” (n, = 266, p, = 3.2E-05), “endomembrane system” (n, = 583, p, = 1.04E-57), and “vesicle” (n, = 230, p, = 8.72E-25). Finally, the overall known biochemical pathways of the AcgP, analyzed using the Kyoto Encyclopedia of Genes and Genomes (KEGG), revealed a similar enrichment of 20 overview terms curated by KEGG that included ribosome biogenesis, protein export, endocytosis and the Wnt signaling pathways (Table I). We conclude the AcgP contains features expected of a tissue devoted to protein production and protein secretion. See supplemental Table S2 for a complete listing of all enriched GO categories.
Fig. 2.

AcgP GO category enrichment analysis. Distribution of major functional category groupings for Biological Process (A,) and Cellular Component (B,). Categories related to secretory processes. Categories highlighted in bold are discussed in the main text.
Table I. Enriched KEGG pathways of the AcgP.
| GOID, | GOTerm, | Nr. Genes, | p, value* |
|---|---|---|---|
| KEGG:03008 | Ribosome biogenesis | 33 | 3.5E-12 |
| KEGG:03040 | Spliceosome | 82 | 8.0E-06 |
| KEGG:04141 | Processing in endoplasmic reticulum | 82 | 5.9E-05 |
| KEGG:03018 | RNA degradation | 40 | 4.6E-04 |
| KEGG:03050 | Proteasome | 35 | 5.5E-04 |
| KEGG:04144 | Endocytosis | 74 | 5.6E-04 |
| KEGG:04310 | Wnt signaling pathway | 24 | 1.8E-03 |
| KEGG:00071 | Fatty acid degradation | 24 | 2.9E-03 |
| KEGG:04130 | SNARE vesicular transport | 17 | 2.9E-03 |
| KEGG:00330 | Arginine and proline metabolism | 34 | 4.6E-03 |
| KEGG:00230 | Purine metabolism | 40 | 1.0E-02 |
| KEGG:03060 | Protein export | 17 | 1.1E-02 |
| KEGG:00280 | Valine, leucine and isoleucine degradation | 22 | 1.3E-02 |
| KEGG:00250 | Ala, asp and glu metabolism | 20 | 1.4E-02 |
| KEGG:00020 | Citrate cycle (TCA cycle) | 27 | 2.5E-02 |
| KEGG:00030 | Pentose phosphate pathway | 16 | 2.8E-02 |
| KEGG:04068 | FoxO signaling pathway | 16 | 2.9E-02 |
| KEGG:00500 | Starch and sucrose metabolism | 20 | 4.2E-02 |
| KEGG:03010 | Ribosome | 83 | 4.3E-02 |
| KEGG:03015 | mRNA surveillance pathway | 41 | 4.4E-02 |
*Bonferroni Corrected.
The D. pseudoobscura AcgS
Signal peptides are a ubiquitous class of short (20–22 aa) N-terminal sequences, that target proteins for translocation across, and into, the endomembrane system of the cell (46, 47). Collectively proteins containing signal peptide sequences are considered part of the secretory pathway, and a subclass—those secreted into the extracellular space—are termed the secretome (also referred to as the exoproteome, exoP). Therefore, some or all components of the exoP can be considered as candidate SFPs. Given the secretory nature of the Drosophila accessory gland, we therefore queried the AcgP for proteins containing canonical signal sequences using two predictive programs, SignalP and Phobius (see Methods). SignalP (31) is a neural network-based algorithm designed to detect canonical N-terminal signal sequences and discriminate against N-terminal transmembrane regions known to reduce predictive power, and Phobius uses a combined model of both transmembrane and signal peptide predictors (32, 48). The combined output of both resulted in 771 Uniprot IDs that mapped to 535 D. pseudoobscura, FBgns (Fig. 1B,; supplemental Table S3). D. melanogaster, orthologs (OrthoDB via Flybase) subsequently returned 506 D. melanogaster, orthologs to the D. pseudoobscura, AcgS including a small percentage (8/511, 1.6%) of “1:many” matches included in the analysis to capture the greatest proteome coverage of the secretome. Thus, the AcgS represents ∼15% (535/3281; 16.3%) of the entire AcgP consistent with similar calculations for the predicted human secretome (∼15%, http://www.proteinatlas.org/humanproteome/secretome).
Gene Ontology (GO) Functional Analysis of the AcgS
The high degree of orthology between the D. pseudoobscura, AcgS genes and D. melanogaster, (506/528), supplemental Table S3) provided a putative orthologous secretome useful for GO analysis and network visualization. A significant enrichment in BP terms was observed, many related to multicellular organism reproduction (n, = 113, p, = 7E-7), reproduction (n, = 116, p, = 7E-7), behavior (n, = 53, p, = 2.1E-5), and proteolysis (n, = 66, p, = 3.5E-6). The secretome is enriched in MF terms related to oxidoreductase activity (n, = 55, p, = 7.1E-7) and hydrolase activity (n, = 138, p, = 8.5e-14; see supplemental Table S3 for a complete list of all AcgS enriched BP, CC, and MF GO categories). We also examined the subcellular localization of the AcgS using the Cerebral layout tool implemented in Cytoscape. As expected for functions related to secretion and proteins containing signal sequences targeted to the secretory pathway, the predicted subcellular localization of AcgS proteins were skewed toward extracellular and plasma membrane proteins (Fig. 3).
Fig. 3.

Functional grouping and subcellular localizaton of the AcgS mapped to subcellular regions (labeled on the right) using the Cerebral layout in Cytoscape. Shaded regions show all annotations that include the keyword “Extracellular”.
The exoP as a Proxy to Identify Putative SFPs
To identify potential candidate SFPs (essentially the exoP component of the AcgS), we queried the AcgS for the GO CC term “extracellular” and obtained a final list of 163 proteins (Fig. 1B,; supplemental Table S4). This list is a conservative estimate as 85 genes of the AcgS had no GO CC functional annotation and 28/85 were unannotated in all 3 major categories. However, inspection of the BP or MF annotation of the remaining 57 proteins revealed “extracellular” terms, suggesting several possible candidates for inclusion in the exoP including >20% (13/57) with annotations related to proteolysis and protease inhibitors (supplemental Table S4). We suggest this correlation because SFPs across diverse taxa (see below) contain several proteases, some with critical reproductive functions (13, 49–51).
In addition to identified exoP proteases, we used Cytoscape and Cluego network analysis of the annotated exoP to return enriched BP terms of major functional categories. These included insemination, sperm competition, copulation reproduction, female mating behavior, and regulation of female receptivity, and response to wounding (Table II; Fig. 4, supplemental Table S4). These are all terms in which D. melanogaster, SFPs are known to impact sex-specific fitness. We next compared our putative SFP list with a list of 212 D. melanogaster, SFPs assembled from the literature (13, 52, 53) and found an overlap of 32.1% (68/212), including 32 SFP genes that have been functionally well-studied in D. melanogaster,, such as Acp53Ea, Acp53C14d and Acp53C14c (supplemental Table S4). We also found no overlap of the 85 AcgS genes that had no CC annotation with the 212 D. melanogaster, SFPs. Likewise, comparison of our putative SFP list to that of 29 D. pseudoobscura, SFPs computationally identified from D. melanogaster, SFPs (54) found an ∼50% (14/29) overlap.
Table II. Enriched BP categories of the AcgS exoproteome (putative SFPs).
| BP Category, | Nr. Genes, | p, value* |
|---|---|---|
| Multicellular organism reproduction | 77 | 7.5E-29 |
| Insemination | 11 | 2.8E-13 |
| Drug catabolic process | 10 | 1.3E-06 |
| Response to biotic stimulus | 20 | 7.3E-06 |
| Lipid transport | 9 | 2.2E-05 |
| Response to wounding | 8 | 1.9E-03 |
| Negative regulation of catalytic activity | 9 | 2.4E-03 |
| Reproduction | 78 | 3.3E-27 |
| Multi-multicellular organism process | 11 | 2.8E-13 |
| Copulation | 11 | 2.7E-12 |
| Sperm competition | 10 | 5.6E-12 |
| Regulation of female receptivity, post-mating | 9 | 2.0E-11 |
| Regulation of female receptivity | 9 | 1.7E-09 |
| Female mating behavior | 9 | 2.0E-08 |
| Response to external biotic stimulus | 20 | 7.3E-06 |
| Response to other organism | 20 | 7.3E-06 |
| Carbohydrate derivative catabolic process | 9 | 1.1E-05 |
| Mating | 12 | 4.5E-05 |
| Reproductive behavior | 12 | 6.2E-05 |
| Mating behavior | 11 | 9.4E-05 |
| Response to bacterium | 13 | 5.5E-04 |
| Lipid localization | 9 | 9.6E-04 |
| Aminoglycan metabolic process | 8 | 2.5E-03 |
| Negative regulation of molecular function | 9 | 3.5E-03 |
*Bonferonni corrected.
Fig. 4.

GO network analysis of the exoP. The five major enriched GO BP categories visualized by Cytoscape and Cluego. Processes related to reproduction and insemination are clearly prominent in the network including as are other processes related to the adhesion and extracellular matrix assembly and maintenance. Categories of cellular functions related to wound healing and defense/immunity were also recovered.
Molecular Evolutionary Rates of Accessory Gland Protein Genes
We tested for rates of molecular evolution in male reproductive proteins by estimating omega (dN/dS substitution rates) for each set of proteins: candidate SFPs, secretome proteins (minus SFPs), and the remaining accessory gland proteome proteins using orthologs from two closely related species in the obscura group, D. lowei, and D. affinis,. We found that putative D. pseudoobscura, SFPs (Exoproteome) are evolving faster than both accessory gland proteome proteins (AcgP; median omega = 0.088 versus, 0.052, p, = E-09) and accessory gland secretome minus the Exoproteome (AcgS; median omega = 0.088 versus, 0.078, p, = 2.7E- 02; Fig. 5). AcgS proteins also evolve faster than the AcgP proteins (median omega = 0.052 versus, 0.078, p, = 7 × 10−9; Fig. 5).
Fig. 5.
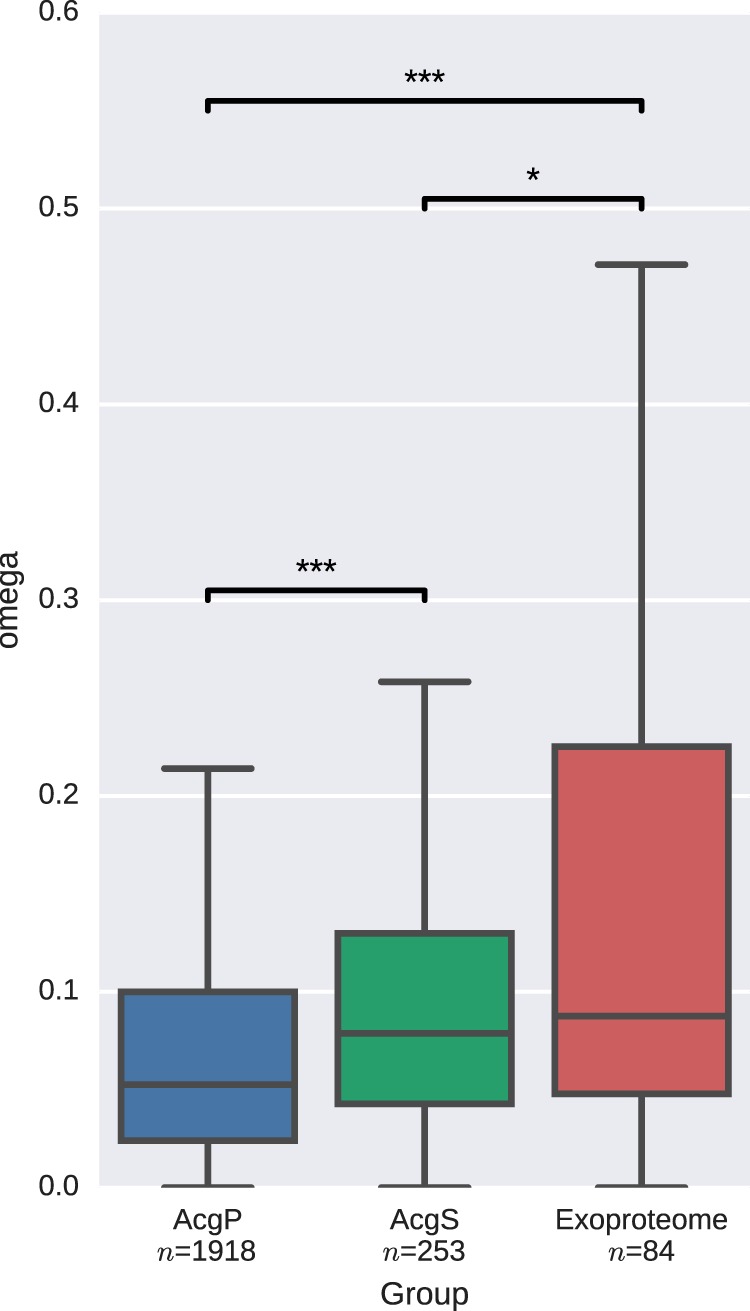
Evolutionary rate calculations of the ratio of the nonsynonymous to synonymous (dN/dS) substitution rates for (left to right) the Acg proteome (AgcP), the secretome (AcgS, excluding exoP) and the exoP proper (AcgS-Extracellular).
DISCUSSION
We used label-free quantitative proteomics to describe the accessory gland proteome and its subcomponents, including identifying candidate SFPs, in D. pseudoobscura,. This species is less polyandrous than D. melanogaster, and patterns of evolution therefore may differ, given the role SFPs play in postcopulatory sexual selection. Indeed, the microevolutionary response of sex-biased gene expression to experimental sexual selection in this species is different than that of D. melanogaster, (24, 55, 56). Here we identified 163 proteins that meet many criteria for being putative SFPs, 132 which were previously unknown for this species. GO term enrichment for biological processes for putative SFPs returned terms related to those expected to influence reproductive fitness including sperm competition. We found that only one third of the exoP overlapped with previously described D. melanogaster, SFPs. Four obvious but not mutually exclusive possibilities exist to explain the differences in the lists: (1) not all SFPs have yet been discovered in either species, (2) the different SFP discovery methods used in various studies will necessarily result in variable lists, (3) our exoP list contains false positives, and/or (4) although nearly all the proteins in the AcgS, from which we bioinformatically derived the exoP, were homologous with the D. melanogaster, genome, some of these proteins may have rapidly diversified to function as SFPs. From a discovery perspective, there is yet no AcgP and AcgS equivalent in D. melanogaster,. Such a resource would improve and extend the predictive abilities of identifying putative SFPs in related species and help understand the evolution of this tissue that generates proteins with profound fitness consequences on both sexes. Related to false positives and potential evolutionary recruitment, we computationally derived putative SFPs but reproductive proteins with extracellular function does not necessarily mean they will be transferred to females (64). Future work will require more downstream analyses of these putative SFPs that will also inform about recruitment. Such analyses include testing that these are transferred to females and functionally determining their effect on sex-specific fitness by taking advantage of the published genome of this species and the increasing use of sophisticated gene editing technology in previously nonmodel organisms (e.g., (57)). Putative SFPs that would be good targets for further investigation include the D. pseudoobscura, SFPs with protease function that were not identified in D. melanogaster,. We argue this because seminal fluid proteases in D. melanogaster, are well-known reproductive players, regulating proteolytic and post-mating reproductive processes in a variety of arthropod taxa including Drosophila (13, 20, 49, 50).
In addition to our list of potentially novel candidate SFPs, several D. melanogaster, SFPs with known impacts on postcopulatory reproductive fitness were also found. For example, we identified nearly all SFP members of the canonical Sex Peptide network (2, 53). In D. melanogaster,, Sex Peptide bound to sperm is transferred to the female seminal receptacle during copulation and is required for both long-term female resistance to remating and for sperm release from storage (58). We identified the gene duplicate pair lectins CG1652 and CG1656, aquarius (CG14061), intrepid (CG12558), antares (CG30488), seminase (CG10586), CG17575 (a cysteine-rich secretory protein), and CG9997 (a serine protease homolog) (59). CG9997 is processed in the female and males that do not produce this protein are unable to transfer the lectins, which are required to slow the rate at which CG9997 is processed in the female. All proteins identified in the D. melanogaster, SP network, except SP itself, were detected in our putative list of SFPs. Absence of detectable D. pseudoobscura, SP protein is consistent with the lack of a recognized SP ortholog in this species and raises the interesting possibility that either the D. pseudoobscura, SP ortholog has significantly diverged, or has been replaced by another gene. If indeed a bona fide D. pseudoobscura, SP gene exists, then further MS searches using algorithms to detect amino acid replacements (60) may be useful in the search for this elusive SFP.
One aim of this work was to extend the focus from solely SFPs to the functional complexity of other proteins in the accessory gland tissue. We generated a robust accessory gland proteome containing 3160 proteins, representing the first accessory gland proteome to be described in Drosophila. 96% of these proteins showed homology to D. melanogaster,. The AcgP proteins in D. pseudoobscura, were enriched for cellular components expected from a tissue whose primary function is secretory, including several cellular component GO terms related to membranes, extracellular regions, and peptidase complex. The top biological process GO terms clearly indicated a large investment in processes directly and indirectly related to protein synthesis, protein assembly, transport, and secretion. We then in silico, concatenated this list to include proteins with secretory signal sequences to identify 506 accessory gland secretome proteins, which were enriched for GO terms related to the biological processes of reproduction, behavior, and proteolysis, with these proteins heavily biased toward subcellular localizations in the plasma member or extracellular components, as predicted from proteins with secretory signals. As previously noted, describing the D. melanogaster, AcgP and AcgS, along with other related species, will provide the basis for evolutionary analyses required to understand how selection, particularly arising from postcopulatory selection pressures on males to influence female reproductive fitness, has acted on this tissue.
Documenting the AcgP then allowed testing another aim of our work—determining signals of molecular evolution in different subcomponents of accessory gland proteins. Rapid evolution at the molecular level is common for reproductive proteins including SFPs (e.g., (12, 61–63)). However, extracellular proteins in general exhibit more rapid molecular evolution than proteins restricted to functions within the cell (21–23). We therefore compared molecular evolution rates of D. pseudoobscura, with its close relatives across proteins from different subcomponents of the male reproductive accessory gland tissue that identified genes encoding SFPs with significantly faster rates of molecular evolution compared with both the AcgS (i.e., other secreted protein encoding genes that were not candidate SFPs) and to non secreted accessory gland proteome genes. Moreover, we also found that secretome genes showed higher rates of molecular evolution than nonsecreted proteins (i.e., the remainder of the AcgP). These results support not only previous work that SFP genes evolve faster than non-SFP genes, but also the more general finding that genes coding proteins which interact extracellularly evolve more rapidly than those that remain within the cytoplasm, irrespective of reproductive function (22, 23).
CONCLUSION
Increasing emphasis on understanding how SFPs impact reproductive fitness across many different organisms requires not only identifying those proteins but also understanding the protein complexity of the SFP-producing tissues. Using organisms with different mating systems and testing the extent to which signatures of rapid molecular evolution are shared across taxa, and across the different environments in which proteins function (i.e., intra- versus, extra- cellular), will generate improved understanding of the causes and consequences of SFP evolution. Here we show that the use of label-free quantitative proteomics methods can address such questions and, specifically, will serve as the basis for more detailed work in this species on the role of postcopulatory sexual selection and reproductive protein evolution.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012545.
Supplementary Material
Acknowledgments
We thank the reviewers for their careful and thoughtful comments and criticisms that helped to make this work better. We also thank the many people who have contributed to the maintenance of the Snook experimental evolution lines.
Footnotes
* Funding of the experimental evolution lines came from NSF (DEB-0093149), NERC (NE/B504065/1; NE/I014632/1), and EU (ITN-2008-213780 SPECIATION) to RRS. Funding and technical support for the proteomic work came from the University of Sheffield Biological Mass Spectrophotometry Facility (funded by Yorkshire Cancer Research (Shend01) and the Wellcome Trust). TIG was supported by a Leverhulme Early Career Fellowship (Grant ECF-2015-453) and a NERC grant (NE/N013832/1). Funding from the Kyoto Institute of Technology to TLK also supported this research.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
1 The abbreviations used are:
- SFPs
- seminal fluid proteins
- BLAST
- Basic Local Alignment Search Tool
- PCSS
- postcopulatory sexual selection
- SDS
- sodium dodecylsulfate
- SDS-PAGE
- sodium dodecylsulfate polyacrylamide gel electrophoresis
- MS
- mass spectrometry
- LC-MS/MS
- liquid chromatography-MS/MS
- AcgP
- accessory gland proteome
- FDRs
- False Discovery Rates
- AcgS
- accessory gland secretome
- exoP
- exoproteome
- LFQ
- label-free quantitation
- P
- polyandry
- M
- monandry
- GO
- gene ontology
- CC
- cellular component
- MF
- molecular function
- BP
- biological process
- STRING
- Search Tool for the Retrieval of Interacting Genes/Proteins
- DIOPT
- DRSC Integrative Ortholog Prediction Tools
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Poiani A. (2006) Complexity of seminal fluid: a review. Behav. Ecol. Sociobiol. 60, 289–310 [Google Scholar]
- 2. Avila F. W., Sirot L. K., LaFlamme B. A., Rubinstein C. D., and Wolfner M. F. (2011) Insect seminal fluid proteins: identification and function. Ann. Rev. Entomol. 56, 21–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perry J. C., Sirot L., and Wigby S. (2013) The seminal symphony: How to compose an ejaculate. Trends Ecol. Evolution 28, 414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Snook R. R. (2014) The evolution of polyandry 159–180. in The Evolution of Insect Mating Systems, Shuker D. & Simmons L., eds. Oxford University Press, Oxford, U.K. [Google Scholar]
- 5. Arnqvist G., and Rowe L. (2005) Sexual conflict, Princeton University Press, Princeton, N.J. [Google Scholar]
- 6. Chen P. S. (1984) The functional morphology and biochemistry of insect male accessory glands and their secretions. Ann. Rev. Entomol. 29, 233–255 [Google Scholar]
- 7. Mattei A. L., Riccio M. L., Avila F. W., and Wolfner M. F. (2015) Integrated 3D view of postmating responses by the Drosophila melanogaster female reproductive tract, obtained by micro-computed tomography scanning. Proc. Natl. Acad. Sci. U.S.A. 112, 8475–8480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Avila F. W., and Wolfner M. F. (2009) Acp36DE is required for uterine conformational changes in mated Drosophila females. Proc. Natl. Acad. Sci. U.S.A. 106, 15796–15800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dorus S., Evans P. D., Wyckoff G. J., Choi S. S., and Lahn B. T. (2004) Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat. Genet. 36, 1326–1329 [DOI] [PubMed] [Google Scholar]
- 10. Ramm S. A., McDonald L., Hurst J. L., Beynon R. J., and Stockley P. (2009) Comparative proteomics reveals evidence for evolutionary diversification of rodent seminal fluid and its functional significance in sperm competition. Mol. Biol. Evol. 26, 189–198 [DOI] [PubMed] [Google Scholar]
- 11. Harrison P. W., Wright A. E., Zimmer F., Dean R., Montgomery S. H., Pointer M. A., and Mank J. E. (2015) Sexual selection drives evolution and rapid turnover of male gene expression. Proc. Natl. Acad. Sci. U.S.A. 112, 4393–4398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clark N. L., Aagaard J. E., and Swanson W. J. (2006) Evolution of reproductive proteins from animals and plants. Reproduction 131, 11–22 [DOI] [PubMed] [Google Scholar]
- 13. Mueller J. L., Ravi Ram K., McGraw L. A., Bloch Qazi M. C., Siggia E. D., Clark A. G., Aquadro C. F., and Wolfner M. F. (2005) Cross-species comparison of Drosophila male accessory gland protein genes. Genetics 171, 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Panhuis T. M., Clark N. L., and Swanson W. J. (2006) Rapid evolution of reproductive proteins in abalone and Drosophila. Phil. Trans. Roy. Soc. London B 361, 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parsch J., and Ellegren H. (2013) The evolutionary causes and consequences of sex- biased gene expression. Nat. Rev. Genet. 14, 83–87 [DOI] [PubMed] [Google Scholar]
- 16. Markow T. A. (1996) Evolution of Drosophila mating systems. Evolutionary Biol. 29, 73–106 [Google Scholar]
- 17. Bayram H., Sayadi A., Immonen E., and Arnqvist G. (2019) Identification of novel ejaculate proteins in a seed beetle and division of labour across male accessory reproductive glands. Insect Biochem. Mol. Biol. 104, 50–57 [DOI] [PubMed] [Google Scholar]
- 18. Takemori N., and Yamamoto M. T. (2009) Proteome mapping of the Drosophila melanogaster male reproductive system. Proteomics 9, 2484–2493 [DOI] [PubMed] [Google Scholar]
- 19. Wei D., Li H. M., Tian C. B., Smagghe G., Jia F. X., Jiang H. B., Dou W., and Wang J. J. (2015) Proteome analysis of male accessory gland secretions in oriental fruit flies reveals juvenile hormone-binding protein, suggesting impact on female reproduction. Sci. Reports 5, 16845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong Z., Wang X., Zhang Y., Zhang L., Chen Q., Zhang X., Zhao P., and Xia Q. (2016) Proteome profiling reveals tissue-specific protein expression in male and female accessory glands of the silkworm, Bombyx mori. Amino Acids 48, 1173–1183 [DOI] [PubMed] [Google Scholar]
- 21. Sojo V., Dessimoz C., Pomiankowski A., and Lane N. (2016) Membrane proteins are dramatically less conserved than water-soluble proteins across the Tree of Life. Mol. Biol. Evol. 33, 2874–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feyertag F., Berninsone P. M., and Alvarez-Ponce D. (2017) Secreted proteins defy the expression level-evolutionary rate anticorrelation. Mol. Biol. Evol. 34, 692–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liao B. Y., Weng M. P., and Zhang J. (2010) Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol. Evol. 2, 39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crudgington H. S., Fellows S., Badcock N. S., and Snook R. R. (2009) Experimental manipulation of sexual selection promotes greater male mating capacity but does not alter sperm investment. Evolution 63, 926–938 [DOI] [PubMed] [Google Scholar]
- 25. Crudgington H. S., Beckerman A. P., Brustle L., Green K., and Snook R. R. (2005) Experimental removal and elevation of sexual selection: Does sexual selection generate manipulative males and resistant females? American Naturalist 165, S72–S87 [DOI] [PubMed] [Google Scholar]
- 26. Anderson W. W. (1974) Frequent multiple insemination in a natural population of Drosophila pseudoobscura. American Naturalist 108, 709–711 [Google Scholar]
- 27. Snook R. R., Brüstle L., and Slate J. (2009) A test and review of the role of effective population size on experimental sexual selection patterns. Evolution 63, 1923–1933 [DOI] [PubMed] [Google Scholar]
- 28. Snook R. R., and Markow T. A. (2002) Efficiency of gamete usage in nature: sperm storage, fertilization and polyspermy. Proceedings Biol. Sci. 269, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 30. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry abilities that proteins are present in a sample on the basis. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 31. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 32. Käll L., Krogh A., and Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 33. Cline M. S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., Hanspers K., Isserlin R., Kelley R., Killcoyne S., Lotia S., Maere S., Morris J., Ono K., Pavlovic V., Pico A. R., Vailaya A., Wang P.-L., Adler A., Conklin B. R., Hood L., Kuiper M., Sander C., Schmulevich I., Schwikowski B., Warner G. J., Ideker T., and Bader G. D. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat. Protocols 2, 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Demchak B., Hull T., Reich M., Liefeld T., Smoot M., Ideker T., and Mesirov J. P. (2014) Cytoscape: the network visualization tool for GenomeSpace workflows. F1000Research 3, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., and Galon J. (2009) ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bindea G., Galon J., and Mlecnik B. (2013) CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 29, 661–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Palmieri N., Kosiol C., and Schlötterer C. (2014) The life cycle of Drosophila orphan genes. Elife 3, e01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 40. Gossmann T. I., Santure A. W., Sheldon B. C., Slate J., and Zeng K. (2014) Highly variable recombinational landscape modulates efficacy of natural selection in birds. Genome Biol. Evol. 6, 2061–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu M., Chatterji S., and Eisen J. A. (2012) Accounting for alignment uncertainty in phylogenomics. PLoS ONE 7, e30288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suyama M., Torrents D., and Bork P. (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 [DOI] [PubMed] [Google Scholar]
- 45. Mann H. B., and Whitney D. R. (1947) On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Statistics 18, 50–60 [Google Scholar]
- 46. Armengaud J., Christie-Oleza J. A., Clair G., Malard V., and Duport C. (2012) Exoproteomics: exploring the world around biological systems. Expert Rev. Proteomics 9, 561–575 [DOI] [PubMed] [Google Scholar]
- 47. Tjalsma H., Bolhuis A., Jongbloed J. D., Bron S., and van Dijl J. M. (2000) Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol. Mol. Biol. Rev. 64, 515–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Käll L., Krogh A., and Sonnhammer E. L. (2007) Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–W432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sitnik J. L., Francis C., Hens K., Huybrechts R., Wolfner M. F., and Callaerts P. (2014) Neprilysins: an evolutionarily conserved family of metalloproteases that play important roles in reproduction in Drosophila. Genetics 196, 781–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu J., Baulding J., and Palli S. R. (2013) Proteomics of Tribolium castaneum seminal fluid proteins: Identification of an angiotensin-converting enzyme as a key player in regulation of reproduction. J. Proteomics 78, 83–93 [DOI] [PubMed] [Google Scholar]
- 51. LaFlamme B. A., Ram K. R., and Wolfner M. F. (2012) The Drosophila melanogaster seminal fluid protease seminase regulates proteolytic and post-mating reproductive processes. PLoS Genet. 8, e1002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Findlay G. D., Yi X., Maccoss M. J., and Swanson W. J. (2008) Proteomics reveals novel Drosophila seminal fluid proteins transferred at mating. PLos Biol. 6, e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Findlay G. D., MacCoss M. J., and Swanson W. J. (2009) Proteomic discovery of previously unannotated, rapidly evolving seminal fluid genes in Drosophila. Genome Res. 19, 886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wagstaff B. J., and Begun D. J. (2005) Comparative genomics of accessory gland protein genes in Drosophila melanogaster and D. pseudoobscura. Mol. Biol. Evol. 22, 818–832 [DOI] [PubMed] [Google Scholar]
- 55. Hollis B., Houle D., Yan Z., Kawecki T. J., and Keller L. (2014) Evolution under monogamy feminizes gene expression in Drosophila melanogaster. Nat. Commun. 5, 3482. [DOI] [PubMed] [Google Scholar]
- 56. Veltsos P., Fang Y., Cossins A. R., Snook R. R., and Ritchie M. G. (2017) Mating system manipulation and the evolution of sex-biased gene expression in Drosophila. Nat Commun 8, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gantz V. M., and Akbari O. S. (2018) Gene editing technologies and applications for insects. Curr. Opin. Insect Sci. 28, 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Avila F. W., Ravi Ram K., Bloch Qazi M. C., and Wolfner M. F. (2010) Sex peptide is required for the efficient release of stored sperm in mated Drosophila females. Genetics 186, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Findlay G. D., Sitnik J. L., Wang W., Aquadro C. F., Clark N. L., and Wolfner M. F. (2014) Evolutionary rate covariation identifies new members of a protein network required for Drosophila melanogaster female post-mating responses. PLoS Genet. 10, e1004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Starkweather R., Barnes C. S., Wyckoff G. J., and Keightley J. A. (2007) Virtual polymorphism: finding divergent peptide matches in mass spectrometry data. Anal. Chem. 79, 5030–5039 [DOI] [PubMed] [Google Scholar]
- 61. Mueller J. L., Ripoll D. R., Aquadro C. F., and Wolfner M. F. (2004) Comparative structural modeling and inference of conserved protein classes in Drosophila seminal fluid. Proc. Natl. Acad. Sci. U.S.A. 101, 13542–13547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vicens A., Borziak K., Karr T. L., Roldan E. R. S., and Dorus S. (2017) Comparative sperm proteomics in mouse species with divergent mating systems. Mol. Biol. Evol. 34, 1403–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilburn D. B., and Swanson W. J. (2016) From molecules to mating: Rapid evolution and biochemical studies of reproductive proteins. J. Proteomics 135, 12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sepil I., Hopkins B. R., Dean R., Thézénas M.-L., Charles P. D., Konietzny R., Fischer R., Kessler B. M., and Wigby S. (2019) Quantitative Proteomics Identification of Seminal Fluid Proteins in Male Drosophila Melanogaster,. Mol. Cell. Proteomics 18, S46–S58 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012545.