

Conspectus

The biotechnological revolution has made it possible to create enzymes for many reactions by directed evolution. However, because of the immense number of possibilities, the availability of enzymes that possess a basal level of the desired catalytic activity is a prerequisite for success. For new-to-nature reactions, artificial metalloenzymes (ARMs), which are rationally designed hybrids of proteins and catalytically active transition-metal complexes, can be such a starting point.
This Account details our efforts toward the creation of ARMs for the catalysis of new-to-nature reactions. Key to our approach is the notion that the binding of substrates, that is, effective molarity, is a key component to achieving large accelerations in catalysis. For this reason, our designs are based on the multidrug resistance regulator LmrR, a dimeric transcription factor with a large, hydrophobic binding pocket at its dimer interface. In this pocket, there are two tryptophan moieties, which are important for promiscuous binding of planar hydrophobic conjugated compounds by π-stacking. The catalytic machinery is introduced either by the covalent linkage of a catalytically active metal complex or via the ligand or supramolecular assembly, taking advantage of the two central tryptophan moieties for noncovalent binding of transition-metal complexes.
Designs based on the chemical modification of LmrR were successful in catalysis, but this approach proved too laborious to be practical. Therefore, expanded genetic code methodologies were used to introduce metal binding unnatural amino acids during LmrR biosynthesis in vivo. These ARMs have been successfully applied in Cu(II) catalyzed Friedel–Crafts alkylation of indoles. The extension to MDRs from the TetR family resulted in ARMs capable of providing the opposite enantiomer of the Friedel–Crafts product. We have employed a computationally assisted redesign of these ARMs to create a more active and selective artificial hydratase, introducing a glutamate as a general base at a judicious position so it can activate and direct the incoming water nucleophile.
A supramolecularly assembled ARM from LmrR and copper(II)–phenanthroline was successful in Friedel–Crafts alkylation reactions, giving rise to up to 94% ee. Also, hemin was bound, resulting in an artificial heme enzyme for enantioselective cyclopropanation reactions. The importance of structural dynamics of LmrR was suggested by computational studies, which showed that the pore can open up to allow access of substrates to the catalytic iron center, which, according to the crystal structure, is deeply buried inside the protein.
Finally, the assembly approaches were combined to introduce both a catalytic and a regulatory domain, resulting in an ARM that was specifically activated in the presence of Fe(II) salts but not Zn(II) salts.
Our work demonstrates that LmrR is a privileged scaffold for ARM design: It allows for multiple assembly methods and even combinations of these, it can be applied in a variety of different catalytic reactions, and it shows significant structural dynamics that contribute to achieving the desired catalytic activity. Moreover, both the creation via expanded genetic code methods as well as the supramolecular assembly make LmrR-based ARMs highly suitable for achieving the ultimate goal of the integration of ARMs in biosynthetic pathways in vivo to create a hybrid metabolism.
1. Introduction
The creation of enzymes for the catalysis of reactions that are new to nature will be key to achieving a more sustainable approach to chemical synthesis. Metalloenzymes are particularly of interest because metal cofactors can significantly expand the catalytic repertoire of enzymes beyond what is achievable using canonical amino acids only. Yet even the synthetic repertoire of natural metalloenzymes is limited compared with the vast number of metal-catalyzed reactions at the disposal of the synthetic chemist. Hence metalloenzymes using abiotic metal cofactors, also known as artificial metalloenzymes (ARMs),1 present an attractive approach toward achieving the enzymatic catalysis of “new-to-nature reactions”, that is, reactions that do not occur in nature.2 Because these ARMs are man-made and do not have the benefit of billions of years of evolution, they are a major test case for our understanding of enzyme design and our ability to create novel “designer” enzymes for a given reaction.
The two general approaches toward novel designer enzymes are either rational, structure-based and/or computationally assisted design, where mechanistic knowledge of the reaction of interest is translated in a protein structure that provides the desired interactions to stabilize the transition states involved,3,4 or the nonrational approach involving iterative random mutagenesis and subsequent screening or selection for the activity of interest, also known as directed evolution.5,6 Whereas these approaches are often contrasted, they are actually complementary. Rational design approaches have produced some impressive demonstrations of novel enzymes;1,7,8 however, in general, their activities are low, at least compared with natural enzymes. This is because whereas we can construct a rudimentary catalytic site, the fine-tuning of the interactions to achieve highly accelerated catalysis is still beyond our understanding. In contrast, directed evolution does allow for exploring sequence space to achieve optimal stabilizing interactions, but this approach itself is limited by the required availability of a suitable, evolvable starting point that already possesses a basal level of the desired catalytic activity.5
Hence these approaches are complementary: Rational design can give rise to a rudimentary enzyme that can then can be subjected to the power of biotechnology to give highly active and selective enzymes for new-to-nature reactions. This is where ARMs enter the picture: Rational design, that is, judicious choice of the protein scaffold and a catalytically active abiological metal cofactor and mode and position of attachment, can produce a rudimentary artificial metalloenzyme that can then be evolved toward high activity and selectivity in the catalytic reaction of interest.9
This Account details the approach used in my research group toward the creation of ARMs for new-to-nature reactions, with the ultimate goal of integrating them in metabolic pathways in vivo and, in this way, augmenting biological synthesis with “unnatural” chemical reactions.
2. Design Considerations
The starting point for our ARM design approach is the notion that the binding of substrates, giving rise to high effective molarities of substrates, is a very important contributor to the enzymatic rate enhancement of bimolecular reactions by removing the entropic cost from the rate-limiting step. Hence the design starts by selecting a protein scaffold that provides a suitable binding pocket (Figure 1a).
Figure 1.

(a) Schematic representation of the artificial metalloenzyme (ARM) design concept. (b) Space-filling representation. (c) Ribbon representation of the structure of LmrR (PDB: 3F8C).10
However, in the context of enzyme design, it is not advisable to start with a highly specific binding pocket because these are very difficult to optimize. On the contrary, such systems will usually require initial “reverse” engineering. In our experience with DNA-based and micellar catalysis, a pocket that provides more generic binding interactions such as hydrophobic interactions is a good starting point to achieve the moderate binding of substrates and, as a result, significant rate accelerations.11−13
The next step is installing the catalytic machinery that is needed for the reaction of interest. Whereas in nature this often is done by combining residues that by themselves show no appreciable activity in the reaction of interest, in our approach, we introduce an unnatural catalytic transition-metal complex that already has some basal activity in the reaction of interest and that is compatible with the protein scaffold, both structurally and in terms of reactivity. The fact that the design starts from binding and then focuses on introducing the catalytic site has implications. First, the protein binding pocket has to be sufficiently large to accommodate both substrates and the metal cofactor. Additionally, the position of the metal cofactor in the protein scaffold is an important design variable because it should be placed judiciously to allow for effective interactions between the substrates and the catalytic metal. Hence multiple positions for incorporation need to be evaluated, and the one that gives rise to the best results in catalysis is developed further. This is in contrast with most other approaches, which usually start from a predetermined position of the metal cofactor.1
Combined, this results in a rudimentary ARM, which can then be optimized and specialized for the reaction of interest by fine-tuning the second coordination sphere, that is, the interactions provided by the protein scaffold to bind the substrates and to stabilize transition states with respect to ground states, by rational redesign of the newly created active or directed evolution, as described above.
With this in mind, we have selected multidrug resistance regulators (MDRs) as the basis for our designs. MDRs are regulatory proteins that are involved in the recognition of foreign agents, that is, antibiotics, and the regulation of the subsequent cellular response, which usually involves transcription/translation of efflux pumps.14 Many MDRs are inherently promiscuous in the recognition and binding of exogenous agents, thus providing a “broad spectrum” defense against antibiotics. A significant number of MDRs contain a large hydrophobic binding pocket for the binding of hydrophobic drugs. These are attractive because organic substrates, which often are hydrophobic, will like to bind here, without this binding being too specific.
We have been particularly interested in the lactococcal multidrug resistance regulator (LmrR), which is a member of the PadR family of MDRs.15 LmrR is homodimeric and possesses a characteristic typical β-winged helix-turn-helix domain with an additional C-terminal helix involved in dimerization (Figure 1b,c).10 It shows a unique large hydrophobic pore at the dimer interface where planar hydrophobic drug molecules can bind, sandwiched between the indole rings of two tryptophan residues, one from each monomer, that is, W96 and W96′. Crystal and nuclear magnetic resonance (NMR) studies, both with and without drug molecules bound, show that the structure is highly flexible and readily adapts to the bound guest molecule.10,16 The size of the pore varies somewhat depending on the guest molecule bound, but a typical volume is ∼1400 Å3 (in the case of PDB: 6FUU).17,18
Other interesting MDRs for ARM design are those from the TetR family, which includes proteins like QacR,19 CgmR,20 and RamR.21 These are also homodimeric, but in this case, each monomer has a separate hydrophobic binding site that is capable of promiscuous binding of hydrophobic drugs.
Finally, there are a number of practical considerations for the ARM design. One is ease of assembly: For directed evolution and application in vivo, it is essential that the ARM is readily assembled without chemical modification and subsequent purification steps. This effectively rules out covalent anchoring approaches. The other is the flexibility of the design. There is only a limited number of proteins that meet the requirements mentioned above. Hence it is desirable to have a general design of a rudimentary enzyme that can be readily adapted for a new chemical reaction. This means that the initially created ARM preferably has several promiscuous catalytic activities that can be evolved toward the desired activity.5
3. Artificial Metalloenzymes with Covalently Attached Metal Cofactors
Our initial efforts were directed toward the covalent anchoring of transition-metal complexes to LmrR, that is, anchoring the metal complexes by conjugating the ligand to a reactive site in the protein. Whereas this is ultimately not the desired approach, as detailed above, it was surmised that this would, in the initial stages of the project, allow for the greatest control over the positioning of the transition-metal complex within the hydrophobic pore of LmrR. On the basis of our previous experience in DNA-based catalysis, we opted for the incorporation of Lewis acidic Cu(II) complexes of 2,2′-bipyridine and phenanthroline ligands.22 For this purpose, cysteine residues were introduced into LmrR through mutagenesis at positions M89 and N19. Because the protein is a homodimer, all changes to the protein occur twice, which also means that two ligands will be present in the hydrophobic pore (Scheme 1). The selected residues are at the far end of the hydrophobic pore, thus reducing the possibility of the formation of catalytically inactive ligand/Cu(II) 2:1 complexes. The ligands were attached by cysteine alkylation using bromoacetamide-substituted bipyridine and phenanthroline ligands. The catalytic potential of these ARMs was demonstrated in the Cu(II)-catalyzed Diels–Alder reaction of azachalcone with cyclopentadiene (Scheme 1b). The results, including the enantiomeric preference of the reaction, proved to be highly dependent on the nature of the ligand and the site of attachment. The best results were obtained when the Cu(II) complex was anchored at position 89, with 66% ee of the (−) enantiomer of the endo product in the case of an attached Cu(II)–bpy complex (LmrR_M89C_bpy), whereas LmrR_M89C_phen gave rise to the (+) enantiomer of the DA product in the highest yield and 97% ee. In both cases, the reaction was significantly protein-accelerated; that is, much higher yields were obtained in the same time with the ARM compared with the copper complex alone.
Scheme 1.

(a) Schematic representation of the preparation of ARMs with covalently attached transition-metal complexes by chemical conjugation of bromoacetamide-functionalized phenanthroline and bipyridine ligands to genetically introduced cysteine residues. For clarity, the modification of only one of the monomers is shown. (b) Diels–Alder reaction of azachalcone with cyclopentadiene. (c) Enantioselective conjugate addition of water.
This ARM was evaluated in a more challenging reaction: the enantioselective conjugate addition of water to enones, giving rise to the formation of β-hydroxyketone products (Scheme 1c).23 This is a reaction that still has no equivalent in “conventional” transition-metal catalysis. The challenges are the small size of water, the limited reactivity of water at neutral pH, and the reversibility of the reaction. Using LmrR_M89C_phen-Cu(II) as catalyst resulted in 80% yield of the β-hydroxyketone product with 84% ee, which is the highest ee reported for this reaction to date.24 Again, the reaction proved to be significantly protein-accelerated. A limited mutagenesis study revealed that residues F93 and D100, which are close to the central tryptophan pair, are of crucial importance. It was proposed that F93 is important because of the steric bulk it provides. D100 was suggested to act as a general base in the reaction, activating and directing the water molecule to one prochiral face of the enone (see below).
This study highlighted both the potential of mutagenesis as well as the limitations of covalent anchoring by chemical modification: Because of the additional handling steps associated with covalent modification, the study of site-directed mutants is tedious, let alone that a practical directed evolution protocol could be envisaged. Therefore, this approach was abandoned.25
As an alternative, we decided to explore methods that allow for the incorporation of a metal-binding ligand during protein biosynthesis using expanded genetic code methods. This can be achieved using the powerful stop codon suppression methodology introduced by Schultz et al.,26 which allows for the robust and reliable incorporation of an unnatural amino acid (uAA) in response to the amber stop codon (UAG). Since its introduction, the method has been continuously improved, and the robust and reliable incorporation of >200 amino acids can be achieved.27,28 This includes a number of uAAs that contain metal-binding moieties as a side chain,29,30 such as (2,2′-bipyridin-5yl)alanine (BpyA).31
Using an evolved tRNA/aminoacyl-tRNA synthetase (aaRS) pair from Methanococcus jannaschii, BpyA was incorporated at various positions in the hydrophobic pore of LmrR (Scheme 2).32 Upon binding of Cu(II), the resulting ARMs were evaluated in the enantioselective Friedel–Crafts alkylation of indoles with α,β-unsaturated 2-acylimidazoles, which showed that incorporation at position 89 (LmrR_M89BpyA) gave rise to the best results in catalysis. The Friedel–Crafts product was obtained in 52–80% ee, which could be slightly improved by introducing an F93W mutation.
Scheme 2. Schematic Representation of the Creation of Artificial Metalloenzymes by Using the Stop Codon Suppression Methodology for the in Vivo Incorporation of BpyAla and the Catalyzed Friedel–Crafts Alkylation Reaction.

*Sign of the optical rotation of the major enantiomer was erroneously assigned in the original report. Adapted with permission from ref (32). Copyright 2015 The Royal Society of Chemistry.
This approach was extended to MDRs of the TetR family, notably QacR, CgmR, and RamR.33 In each of these proteins, at various positions, the BpyA was introduced. The best results were obtained with QacR_Y123BpyA, which gave rise to up to 94% ee of the Friedel–Crafts alkylation product. Interestingly, compared with LmrR, in this case, the opposite enantiomer of the product was obtained in excess, showing that these various MDR-based ARMs are complementary.
Having established a proof of principle, we returned to the enantioselective hydration reaction. LmrR_M89BpyA-Cu(II) was indeed proven to be active in this reaction, giving rise to a moderate yield and ee of the β-hydroxyketone product. Instead of immediately embarking on a directed evolution campaign, we decided to first optimize the catalytic machinery using a combination of chemical knowledge and computational methods.
The water addition is known to benefit from base catalysis.34 On the basis of this, combined with the proposed role of the D100 residue in the covalently anchored system described above, we surmised that judicious positioning of a glutamate residue that could function as a general base in the newly created active site would make it possible to activate and direct the water nucleophile toward the addition from one preferred prochiral face of the enone, giving rise to a faster and more enantioselective reaction.
Using a multiscale computational approach comprising a combination of quantum mechanical (QM) calculations of the substrate-bound Cu(II) complexes, subsequent protein ligand dockings, and MD simulations, (Figure 2), we proposed three mutants that would have a glutamate within the required distance of the β position of the enone to form a prereactive conformation with a water nucleophile during significant amounts of time.35
Figure 2.

Molecular dynamics simulations of (A) LmrR_M89X, (B) LmrR_M89X_D100E, (C) LmrR_M89X_W96E, and (D–F) LmrR_M89X_V15E, where X = BpyA, showing prereactive conformations, which are identified by the distance of a water molecule to the electrophilic carbon of the enone substrate and the hydrogen bonding to an aspartate or glutamate. Reproduced with permission from ref (35). Copyright 2017 The Royal Society of Chemistry.
The mutants D100E, V15E, and W96E were prepared, and the designs were experimentally validated (Scheme 3). Gratifyingly, all three mutants gave rise to an increased conversion of the enone in the same time compared with the LmrR_M89BpyA, as predicted from the simulations. The D100E mutant gave rise to a somewhat lower ee, of the same enantiomer as the wild-type LmrR.
Scheme 3. Hydration Reaction Catalyzed by Designed LmrR-Based ARMs.

The W96E mutant was predicted to give rise to the formation of the opposite enantiomer of the product compared with LmrR_M89BpyA. Experimentally, near-racemic product was obtained, which does imply that the artificial enzyme has an increased preference for the formation of the opposite enantiomer.
The mutant V15E gave rise to both an increase in yield and enantioselectivity of the β-hydroxyketone product. The Michaelis–Menten kinetics showed a three-fold increase in kcat/KM for the LmrR_M89BpyA_V15E mutant compared with LmrR_M89BpyA itself. Notably, the corresponding glutamine mutant, that is, V15Q, gave rise to significantly decreased yields and ee, further supporting the proposal that the placement of a general base at a judicious position with respect to the substrate is indeed a good approach to the design of metallohydratases.
The approach of creating artificial metalloproteins via the incorporation of metal-binding uAAs using the expanded genetic code methodology is not limited to the formation of copper enzymes. It was shown that other divalent first-row transition-metal ions, such asNi(II), Co(II), and Zn(II), are also efficiently bound to BpyA.36 The corresponding iron protein could also be created, albeit this required the introduction of additional carboxylate moieties in the vicinity of the metal-BpyA site, most likely to act as additional ligands to the iron center.
These artificial metalloproteins were investigated for their potential catechol dioxygenase activity. Surprisingly, upon the addition of di-tert-butylcatechol (DTB-C), the formation of a stable radical species was observed that showed characteristic absorptions in the UV–vis spectrum and exhibited an electron paramagnetic resonance (EPR) signal at g = 2.003, which suggested it to be a bound semiquinone radical (DTB-SQ, Scheme 4a).37 DTB-SQ is the intermediate product in the oxidation of DTB-C to the o-quinone product (DTB-Q) but can also be generated independently by comproportionation of DTB-C and DTB-Q. The formation of DTB-SQ was observed, to a variable extent, with all of the different metal ions investigated. Yet the EPR signal of DTB-SQ was the same regardless of the metal ion used, suggesting that there was no direct interaction (Scheme 4c). This implies that the metal complex acts as a counterion for the DTB-SQ, which is a radical anion. Remarkably, this species proved to be stable for at least 4 weeks. Presumably, DTB-SQ is bound between the two tryptophan moieties in the hydrophobic pocket, where it is shielded from bulk water, which results in its stabilization. This could be an important step toward harnessing the chemistry of unstable radical species, with potential for novel catalytic chemistry.
Scheme 4. Formation of the DTB-SQ Radical Anion and EPR Spectra of ARM-Bound DTB-SQ.

(a) As one electron-oxidized intermediate product in the oxidation of DTB-C to DTB-Q. (b) By comproportionation of DTB-C and DTB-Q. (c) EPR spectra of LmrR_M89BpyA with various divalent metal ions and bound DTB-SQ. Reproduced with permission from ref (36). Copyright 2017 American Chemical Society.
4. Supramolecular Assembly
Not only is the promiscuous binding pocket of LmrR suitable for substrate binding but also, in particular, the two central tryptophan residues, W96 and W96′, could be used for the binding of catalytically active planar coordination complexes of aromatic ligands. This approach obviates the need for the covalent attachment of the transition-metal complex and would result in a more dynamic, self-adjustable system. A similar approach has proven beneficial in our previous work on DNA-based asymmetric catalysis.38
Our first studies focused on Cu(II)–phen, which was shown to exhibit low micromolar binding affinity for LmrR.39 Combined tryptophan fluorescence and fluorescence lifetime measurements supported the binding of the Cu(II)–phen between the two tryptophan moieties. This was further supported by the fact that the binding affinity was found to decrease with one order of magnitude for the LmrR_W96A mutant.
The supramolecularly assembled ARM, LmrR⊂Cu(II)–phen, showed excellent selectivity in the Cu(II) catalyzed Friedel–Crafts alkylation of indoles with α,β-unsaturated 2-acylimidazoles with up to 94% ee achieved (Scheme 5). The LmrR_W96A⊂Cu(II)–phen system gave rise to a significantly slower reaction and <5% ee of the product in the case of the 5-OMe indole. This further supports the proposed binding of the Cu(II)–phen complex in between the indole moieties of the central tryptophan moieties.
Scheme 5. Schematic Representation of Supramolecular Assembly of an ARM from LmrR and Cu(II)–phen and an Example of the Catalyzed Friedel–Crafts Alkylation of Indoles.

Adapted with permission from ref (39). Copyright 2015 American Chemical Society.
The generality of this assembly principle was demonstrated by the creation of an artificial heme enzyme from LmrR and a catalytically active hemin cofactor.17 Recently, heme enzymes have attracted considerable attention because of their ability to catalyze abiological carbene transfer reactions, including cyclopropanations, olefinations, and X–H insertion reactions.40,41
Using tryptophan fluorescence titration, the hemin was found to bind LmrR with nanomolar affinity, that is, KD = 38 nM. In contrast, no heme binding was observed in the case of the W96A mutant, showing the importance of the central tryptophans. The LmrR/hemin complex formation was further supported by the crystal structure, which showed electron density attributed to the bound heme between the tryptophan indole moieties (Figure 3). Whereas the protein structure was well resolved, the heme, without the axial chloride ligand, could be modeled in four different orientations. This already suggested significant structural dynamics in the heme binding, which proved to be important for catalysis.
Figure 3.
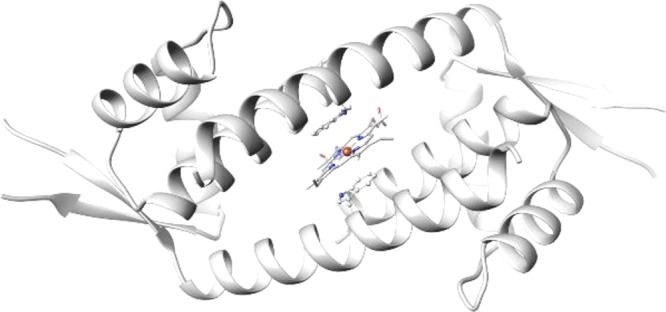
Crystal structure of LmrR⊂heme (PDB: 6FUU).17 The heme is stacked in between the indole side chains of W96/W96′. Four binding modes of the heme were modeled, which differed by rotation around the central heme axis and a flip of the heme resulting from the crystallographic two-fold symmetry. For clarity, only one of the heme binding orientations is shown.
The LmrR⊂heme assembly was evaluated as a catalyst in the cyclopropanation of styrenes with diazoacetate esters as carbene precursors (Scheme 6). Good activities, that is, several hundreds of turnovers of styrene and a moderate ee of the 1R,2R enantiomer of the trans-cyclopropane product, were obtained. This could further be increased by introducing an M8A mutation, which increased the ee up to 51%. The W96A mutant gave rise to similar activity but significantly decreased ee.
Scheme 6. Schematic Representation of an LmrR-Based Artificial Heme Enzyme Created by Supramolecular Assembly and an Example of the Catalyzed Enantioselective Cyclopropanation Reaction.

TTN = total turnover number. Adapted with permission from ref (17). Copyright 2018 John Wiley & Sons.
On the basis of the crystal structure, it is not obvious why the LmrR⊂heme ARM is active because the catalytic iron atom is fully buried within the protein and hence is not accessible. Therefore, computational studies were performed to understand the conformational changes required to make the catalytic iron site available for substrates and allowing for the reaction to take place.
On the basis of the crystal structure, docking studies of heme and the heme-bound carbene intermediate were carried out. For the latter, several low-energy solutions were found with the W96′ rotated away toward the solvent, resulting in space becoming available for the heme-bound carbene. These docked structures were used as a starting point for MD studies (Figure 4), which showed that most clusters of the MD simulation of LmrR⊂heme were in agreement with the crystal structure. However, in the case of the docking of the heme–carbene intermediate, some clusters showed structural changes in the orientation of the α4 helix at the front entrance, accompanied by a flip of W96′ outward, thus creating space. This open space is indeed sufficient to accommodate the heme–carbene complex in the pocket, which was found to be directed toward the solvent, making it accessible for the styrene substrate. Finally, the calculated transition-state structure that leads to the formation of the major enantiomer was studied. The majority of the clusters showed a broader dimer interface, and W96 and W96′ separated further, making the accommodation of the catalytic complex possible. The M8A mutant was subjected to the same analysis, which showed the effect of this mutation to be predominantly steric: It frees up space where the benzene ring of the styrene can be accommodated.
Figure 4.

Representative structures resulting from 400 ns MD simulations of (a,b) the LmrR heme system, (c,d) the LmrR–heme–carbene system, and (e,f) the transition state of the cyclopropanation reaction in the case of (e) LmrR and (f) LmrR_M8A (100 ns MD simulation). Reproduced with permission from ref (17). Copyright 2018 John Wiley & Sons.
These results suggest the importance of structural dynamics in ARMs. This further adds to the attractiveness of MDRs as a scaffold for ARM design because their biological role already requires them to be structurally flexible and dynamic.
5. Combining Covalent Attachment and Supramolecular Assembly
As described above, LmrR allows for the covalent attachment of the transition-metal complex as well as supramolecular assembly. Hence we envisioned that it would be possible to combine these different approaches to create ARMs that contain both a catalytic and a regulatory domain. The regulation of catalytic activity is very important in nature, and hence the regulation of ARMs can be expected to become important in hybrid metabolic pathways.42
The design involved the covalent attachment of a bipyridine ligand at position 104 by alkylation of a genetically introduced cysteine (LmrR_E104C) with the bromoacetamide-substituted 2,2′-bipyridine ligand used in our initial ARM studies (vide supra).22 When combined with Cu(II)–phen, instead of binding between W96/W96′, the copper complex binds to the bipyridines via its open coordination sites (Scheme 7).43 The result is that the Cu(II)–phen can longer interact with the enone substrate, and hence no catalysis can occur. The addition of a Fe(II) salt causes the dissociation of the Cu(II)–phen from the bipyridine ligands because the binding of Fe(II) by the two bipyridines, one from each monomer, is thermodynamically preferred. Thus the Cu(II)–phen complex binds between W96/96′, and the catalysis of the Friedel–Crafts alkylation reaction of indoles is “turned on”. The activation is metal-ion-selective: Zn(II) salts do not displace the Cu(II)–phen and therefore do not activate the ARM.
Scheme 7. Schematic Representation of the Concept of a Metal-Ion-Regulated LmrR-Based Artificial Metalloenzyme That Is Selectively Activated by Fe2+ but Not Zn2+ Ions.

6. Conclusions and Outlook
The work described here shows that LmrR is one of the privileged protein scaffolds for ARMs. The unique promiscuous hydrophobic pore can bind many organic substrates and thus is an excellent starting point for the creation of a novel active site following our design strategy. LmrR is without a doubt one of the most versatile protein scaffolds because it allows for the creation of ARMs via the covalent attachment of transition-metal complexes, either via chemical modification or biosynthetic incorporation using expanded genetic code methods as well as supramolecular assembly.
This versatility is due to its unique structure: The large hydrophobic pore, with its characteristic two tryptophan residues, offers a highly versatile promiscuous binding pocket where, in addition to a catalytic transition-metal complex, a multitude of different organic substrates can bind. The result is a rudimentary metalloenzyme that exhibits moderate levels of catalytic activity for various reactions. Importantly, the binding is not very specific, which means that the rudimentary ARMs can be further optimized, that is, specialized, for the desired reaction.
We propose that in addition to the structural layout of the binding pocket, the structural flexibility and dynamics of LmrR are a key factor in its success. This makes the protein readily adapt its structure to the substrates and the catalyzed reaction. This point is illustrated by the large structural changes of the protein during the cyclopropanation reaction catalyzed by the LmrR-based artificial heme enzyme, as suggested by computation.
The LmrR-based ARMs have proven to be remarkably tolerant to mutagenesis, with both canonical amino acids as well as uAAs. Moreover, LmrR is compatible with a range of different metal complexes, reaction types, substrates, and reaction conditions.
The fact that multiple assembly approaches are readily combined suggests the LmrR structure can be equipped with multiple catalytic functions that can work in concert. For example, synergistic catalysis can be envisioned, in which one reagent is activated by one catalytic moiety and the other is activated by another. This will give access to a completely new classes of reactions.
Finally, the fact that active metalloenzymes can be created by expanded genetic code methods or a supramolecular assembly creates excellent prospects for applications in vivo. This will allow for practical directed evolution approaches to create optimized ARMs for new-to-nature reactions but also suggests the feasibility of integrating the MDR-based ARMs into biosynthetic pathways in vivo to create a hybrid metabolism in which biological chemistry is augmented with new-to-nature ARM-catalyzed reactions.
Acknowledgments
I wish to thank all of the former and current members of my research group and the many collaborators that have contributed to the work described here. This work was generously supported by grants from The Netherlands Research School Combination on Catalysis (NRSC-C), European Research Council (starting grant 280010), The Netherlands Organization for Scientific Research (NWO, vici grant 724.013.003), and The Netherlands Ministry of Education Culture and Science (Gravitation programme no. 024.001.035)
Biography
Gerard Roelfes obtained his Ph.D. from the University of Groningen, The Netherlands, under the supervision of Prof. Ben L. Feringa. His Ph.D. research was on synthetic models for nonheme iron oxygenases, which was a joint project with Unilever Research and Prof. Lawrence Que, Jr. (University of Minnesota), in whose lab he carried out part of the work. After his Ph.D., he went for a postdoctoral stay with Prof. Donald Hilvert at the ETH-Zürich (Switzerland), where he worked on semisynthetic strategies towards selenoproteins. In 2003, he returned to the University of Groningen, where is now a Professor of Biomolecular Chemistry & Catalysis. He is interested in catalysis at the interface of chemistry and biology and is currently focusing on bioorthogonal catalysis, enzyme design, and in vivo catalysis.
The author declares no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Artificial Metalloenzymes and Abiological Catalysis of Metalloenzymes”.
References
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Köhler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Arnold F. H. Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes To Create New Catalysts. J. Am. Chem. Soc. 2015, 137, 13992–14006. 10.1021/jacs.5b09348. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- Huang P.; Boyken S. E.; Baker D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Renata H.; Wang Z. J.; Arnold F. H. Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem., Int. Ed. 2015, 54, 3351–3367. 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeymer C.; Hilvert D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. 10.1146/annurev-biochem-062917-012034. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Röthlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas C. F. III; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St. Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science 2010, 329, 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markel U.; Sauer D. F.; Schiffels J.; Okuda J.; Schwaneberg U. Towards evolution of artificial metalloenzymes – A protein engineer’s perspective. Angew. Chem., Int. Ed. 2019, 10.1002/anie.201811042. [DOI] [PubMed] [Google Scholar]
- Madoori P. K.; Agustiandari H.; Driessen A. J. M.; Thunnissen A. W. H. Structure of the transcriptional regulator LmrR and its mechanism of multidrug recognition. EMBO J. 2009, 28, 156–166. 10.1038/emboj.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati F.; Oelerich J.; Roelfes G. Dramatic micellar rate enhancement of the Cu2+ catalyzed vinologous Friedel-Crafts alkylation in water. Chem. Commun. 2010, 46, 7804–7806. 10.1039/c0cc02584d. [DOI] [PubMed] [Google Scholar]
- García-Fernández A.; Megens R. P.; Villarino L.; Roelfes G. DNA-Accelerated Copper Catalysis of Friedel-Crafts Conjugate Addition/Enantioselective Protonation Reactions in Water. J. Am. Chem. Soc. 2016, 138, 16308–16314. 10.1021/jacs.6b08295. [DOI] [PubMed] [Google Scholar]
- Otto S.; Engberts J.; Kwak J. Million-fold acceleration of a Diels-Alder reaction due to combined Lewis acid and micellar catalysis in water. J. Am. Chem. Soc. 1998, 120, 9517–9525. 10.1021/ja9816537. [DOI] [Google Scholar]
- Grkovic S.; Brown M.; Skurray R. Regulation of bacterial drug export systems. Microbiol. Mol. Biol. Rev. 2002, 66, 671–701. 10.1128/MMBR.66.4.671-701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agustiandari H.; Lubelski J.; van den Berg van Saparoea H. B.; Kuipers O. P.; Driessen A. J. M. LmrR is a transcriptional repressor of expression of the multidrug ABC transporter lmrCD in Lactococcus lactis. J. Bacteriol. 2008, 190, 759–763. 10.1128/JB.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K.; Tokunaga Y.; Imai M.; Takahashi H.; Shimada I. Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci. Rep. 2015, 4, 6922. 10.1038/srep06922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino L.; Splan K. E.; Reddem E.; Alonso-Cotchico L.; Gutiérrez de Souza C.; Lledós A.; Maréchal J.; Thunnissen A. W. H.; Roelfes G. An Artificial Heme Enzyme for Cyclopropanation Reactions. Angew. Chem., Int. Ed. 2018, 57, 7785–7789. 10.1002/anie.201802946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.; Chen C.; Lei X.; Zhao J.; Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363. 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher M.; Miller M.; Grkovic S.; Brown M.; Skurray R.; Brennan R. Structural mechanisms of QacR induction and multidrug recognition. Science 2001, 294, 2158–2163. 10.1126/science.1066020. [DOI] [PubMed] [Google Scholar]
- Itou H.; Okada U.; Suzuki H.; Yao M.; Wachi M.; Watanabe N.; Tanaka I. The CGL2612 protein from Corynebacterium glutamicum is a drug resistance-related transcriptional repressor - Structural and functional analysis of a newly identified transcription factor from genomic DNA analysis. J. Biol. Chem. 2005, 280, 38711–38719. 10.1074/jbc.M505999200. [DOI] [PubMed] [Google Scholar]
- Yamasaki S.; Nikaido E.; Nakashima R.; Sakurai K.; Fujiwara D.; Fujii I.; Nishino K. The crystal structure of multidrug-resistance regulator RamR with multiple drugs. Nat. Commun. 2013, 4, 2078. 10.1038/ncomms3078. [DOI] [PubMed] [Google Scholar]
- Bos J.; Fusetti F.; Driessen A. J. M.; Roelfes G. Enantioselective Artificial Metalloenzymes by Creation of a Novel Active Site at the Protein Dimer Interface. Angew. Chem., Int. Ed. 2012, 51, 7472–7475. 10.1002/anie.201202070. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Coquière D.; Geerdink D.; Rosati F.; Feringa B. L.; Roelfes G. Catalytic enantioselective syn hydration of enones in water using a DNA-based catalyst. Nat. Chem. 2010, 2, 991–995. 10.1038/nchem.819. [DOI] [PubMed] [Google Scholar]
- Bos J.; García-Herraiz A.; Roelfes G. An enantioselective artificial metallo-hydratase. Chem. Sci. 2013, 4, 3578–3582. 10.1039/c3sc51449h. [DOI] [Google Scholar]
- Laureanti J. A.; Buchko G. W.; Katipamula S.; Su Q.; Linehan J. C.; Zadvornyy O. A.; Peters J. W.; O’Hagan M. Protein scaffold activates catalytic CO2 hydrogenation by a rhodium bis(diphosphine) complex. ACS Catal. 2019, 9, 620–625. 10.1021/acscatal.8b02615. [DOI] [Google Scholar]
- Wang L.; Brock A.; Herberich B.; Schultz P. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- Young T. S.; Ahmad I.; Yin J. A.; Schultz P. G. An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361–374. 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- Dumas A.; Lercher L.; Spicer C. D.; Davis B. G. Designing logical codon reassignment - Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. 10.1039/C4SC01534G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Hilvert D.; Green A. P. Engineered Metalloenzymes with Non-Canonical Coordination Environments. Chem. - Eur. J. 2018, 24, 11821–11830. 10.1002/chem.201800975. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Hu C.; Xia L.; Wang J. Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851–1863. 10.1021/acscatal.7b03754. [DOI] [Google Scholar]
- Xie J.; Liu W.; Schultz P. G. A genetically encoded bidentate, metal-binding amino acid. Angew. Chem., Int. Ed. 2007, 46, 9239–9242. 10.1002/anie.200703397. [DOI] [PubMed] [Google Scholar]
- Drienovská I.; Rioz-Martínez A.; Draksharapu A.; Roelfes G. Novel artificial metalloenzymes by in vivo incorporation of metal-binding unnatural amino acids. Chem. Sci. 2015, 6, 770–776. 10.1039/C4SC01525H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersellini M.; Roelfes G. Multidrug resistance regulators (MDRs) as scaffolds for the design of artificial metalloenzymes. Org. Biomol. Chem. 2017, 15, 3069–3073. 10.1039/C7OB00390K. [DOI] [PubMed] [Google Scholar]
- Jin J.; Hanefeld U. The selective addition of water to C = C bonds; enzymes are the best chemists. Chem. Commun. 2011, 47, 2502–2510. 10.1039/c0cc04153j. [DOI] [PubMed] [Google Scholar]
- Drienovská I.; Alonso-Cotchico L.; Vidossich P.; Lledós A.; Maréchal J.; Roelfes G. Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem. Sci. 2017, 8, 7228–7235. 10.1039/C7SC03477F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségaud N.; Drienovská I.; Chen J.; Browne W. R.; Roelfes G. Artificial Metalloproteins for Binding and Stabilization of a Semiquinone Radical. Inorg. Chem. 2017, 56, 13293–13299. 10.1021/acs.inorgchem.7b02073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulas G.; Lemmin T.; Wu Y.; Gassner G. T.; DeGrado W. F. Designed metalloprotein stabilizes a semiquinone radical. Nat. Chem. 2016, 8, 354–359. 10.1038/nchem.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma A. J.; Megens R. P.; Feringa B. L.; Roelfes G. DNA-based asymmetric catalysis. Chem. Soc. Rev. 2010, 39, 2083–2092. 10.1039/b811349c. [DOI] [PubMed] [Google Scholar]
- Bos J.; Browne W. R.; Driessen A. J. M.; Roelfes G. Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold. J. Am. Chem. Soc. 2015, 137, 9796–9799. 10.1021/jacs.5b05790. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Brandenberg O. F.; Fasan R.; Arnold F. H. Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr. Opin. Biotechnol. 2017, 47, 102–111. 10.1016/j.copbio.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Lebrun V.; Kitanosono T.; Mallin H.; Köhler V.; Haussinger D.; Hilvert D.; Kobayashi S.; Ward T. R. Upregulation of an Artificial Zymogen by Proteolysis. Angew. Chem., Int. Ed. 2016, 55, 11587–11590. 10.1002/anie.201605010. [DOI] [PubMed] [Google Scholar]
- Bersellini M.; Roelfes G. A metal ion regulated artificial metalloenzyme. Dalton Trans. 2017, 46, 4325–4330. 10.1039/C7DT00533D. [DOI] [PubMed] [Google Scholar]