

Abstract
Starting from the analysis of the hypothetical binding mode of our previous furan-based hit (I), we successfully achieved our objective to replace the nitro moiety, leading to the disclosure of a new lead exhibiting a strong activity against MbtI. Our best candidate 1 h displayed a Ki of 8.8 µM and its antimycobacterial activity (MIC99 = 250 µM) is conceivably related to mycobactin biosynthesis inhibition. These results support the hypothesis that 5-phenylfuran-2-carboxylic derivatives are a promising class of MbtI inhibitors.
Keywords: Tuberculosis, siderophores, mycobactins, drug design, antimycobacterial agent, molecular modelling
GRAPHICAL ABSTRACT

Introduction
Tuberculosis (TB) is an infectious disease caused by an obligate aerobic bacterium, known as Mycobacterium tuberculosis (Mtb). When the bacilli are inhaled, they reach the alveolar spaces of the lungs and are mainly ingested by macrophages. As a consequence, TB primarily affects the lungs, but at later stages it can also spread to other vital organs. Despite significant improvements with respect to diagnosis, treatment, and preventive measures have been successfully implemented in many healthcare systems around the world, this disease still remains the world’s biggest threat to human health causing 54 million deaths between 2000 and 2017 1 . Standard therapeutic regimens have remained substantially unchanged over the past 60 years with outdated drugs and very long therapies that are still used for the treatment of new and relapse cases. In addition to the length of the cure, other hurdles related to the management of TB infections include interactions with drugs used in comorbid conditions, especially HIV, and severe side effects. All these issues contribute to determine a poor patient compliance that, together with the improper use of TB antibiotics, has led to the insurgence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacterial strains 2–4 . As confirmed by the latest WHO report, drug resistance is becoming a real emergency; therefore, there is a growing interest in the development of novel anti-TB compounds 5–8 . A few of them reached clinical trials and two drugs, delamanid and bedaquiline, have been recently approved; however, more information on their effectiveness, safety, and tolerability are still required because severe side effects have been reported 9 , 10 . In this scenario, the research of many more TB drug candidates to sustain an effective and productive drug pipeline is pivotal. Targeting Mtb iron uptake systems is now a validated strategy for the development of antimycobacterial compounds, because iron is essential for Mtb survival in the host and its acquisition is strongly correlated with virulence 11 . Among the four different iron acquisition pathways used by Mtb, the most thoroughly characterised one is based on the production of two types of siderophores: carboxymycobactins, which acquire iron extracellularly and transport it into the cytoplasm of the bacteria, and mycobactins, which facilitate the transport of iron through the cell wall into the cytoplasm. Notably, targeting this biosynthetic process is an attractive strategy, because its impairment lowers the pathogen virulence and survival without causing toxicity issues. Indeed, as this pathway is absent in humans, the risk of off-target effects is minimal; moreover, being an unexplored biological process for the development of drugs, there is no known resistance mechanism. The first step of the biosynthesis of these siderophores is catalysed by the Mg2+-dependent enzyme salicylate synthase (MbtI), a validated pharmacological target 12–15 , whose crystal structure has been recently solved 16 .
In this context, the aim of our project is the identification of new MbtI inhibitors as potential antitubercular agents. Our previous computational studies generated a pharmacophore model, that allowed the identification of the interesting hit compound I (Table 1). Then, a structure-activity relationship study on monosubstituted derivatives underlined the importance of the nitro moiety for potency 14 . However, the nitro group is considered a structural alert for the development of a potential drug, since drugs containing nitro groups have been extensively associated with mutagenicity and genotoxicity 17 . On these bases, in the present work, we designed additional analogs (compounds 1a-p, Table 1) exploring other hitherto unconsidered pharmacophoric features and evaluating the possibility of replacing the nitro group.
Table 1.
In vitro activity of compounds 1a–p.
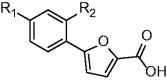 | |||||||
|---|---|---|---|---|---|---|---|
|
1a–i R1=CF3 |
1j–p R1=NO2 |
||||||
| Code | R2 | Residual activity at 100 µM (%) | MbtI IC50 (µM)* | Code | R2 | Residual activity at 100 µM (%) |
MbtI IC50 (µM)* |
| 1a | Cl | 6.1 ± 2.5 | 28.5 ± 2.6 | I** | Cl | 11.3 ± 4.2 | 17.9 ± 3.2 |
| 1b | F | 6.1 ± 1.1 | 27.6 ± 7.3 | 1j | F | 32.1 ± 2.4 | – |
| 1c | Br | 38.0 ± 3.7 | – | 1k | Br | 26.4 ± 4.8 | – |
| 1d | OH | 16.8 ± 2.7 | 35.9 ± 10.3 | 1l | OH | 13.9 ± 4.0 | 29.8 ± 4.2 |
| 1e | CH3 | 19.3 ± 2.2 | 31.5 ± 9.6 | 1m | CH3 | 13.2 ± 5.8 | 28.5 ± 1.5 |
| 1f | NH2 | 13.1 ± 2.4 | 34.6 ± 10.9 | 1n | NH2 | 13.5 ± 3.8 | 24.2 ± 5.4 |
| 1g | CN | 5.7 ± 1.5 | 18.5 ± 3.2 | 1o | CN | 5.0 ± 1.9 | 24.4 ± 5.9 |
| 1h | CF3 | 3.9 ± 1.7 | 13.1 ± 2.0 | 1p | CF3 | 25.1 ± 3.7 | 41.8 ± 5.3 |
| 1i | m-CF3 | 64.3 ± 4.5 | – | – | – | – | – |
*Only for compounds with residual activity ≤25%; **new fluorimetric assay value determined for the sake of comparison.
Materials and methods
Chemistry
Compounds 1a,b,d–i,l–n,p were synthesised by a Suzuki-Miyaura reaction 18 , followed by a base-catalysed hydrolysis of the ester function 19 . Compounds 1c,j,k,o were synthesised by a Sandmayer reaction 20 , starting from 2-furan carboxylic acid methyl ester and the diazonium salts of the appropriate amines; the ester function was then hydrolysed in basic conditions 21 . The general procedures, the synthetic pathways (Supplementary Schemes 1 and 2), the details concerning the specific synthetic steps and the analytical data are provided in the Supplementary Material.
MbtI enzymatic assays
Recombinant M. tuberculosis MbtI was produced and purified as previously reported 14 . Enzyme activity was determined measuring the production of salicylic acid by a fluorimetric assay slightly modified from Vasan et al. 12 Briefly, assays were performed at 37 °C in a final volume of 400 µL of 50 mM Hepes pH 7.5, 5 mM MgCl2, using 1–2 µM MbtI and the reactions were started by the addition of chorismic acid and monitored using a Perkin-Elmer LS3 fluorimeter (Ex. λ = 305 nm, Em. λ = 420 nm). Inhibition assays were performed in the presence of the compound at 100 µM (stock solution 20 mM in DMSO), at 50 µM chorismic acid. For significantly active compounds, IC50 and K i values were determined. To verify that the compounds were not PAINs, their ability to inhibit MbtI activity was tested in the presence of 0.1 mg/mL of bovine serum albumin (BSA) or in the presence of 0.01% (v/v) Triton X-100 to confirm that they did not act as aggregate, and in the presence of 100 mM of 1,4-dithio-DL-threitol (DTT) to exclude an enzyme inhibition due to reaction with cysteines 22 .
Minimal inhibitory concentration determinations and siderophore production assay
The MIC99 values of active compounds against M. bovis BCG were determined in low-iron Chelated Sauton’s medium 23 , using the resazurin reduction assay (REMA) 24 . Siderophore activity in the culture was tested in M. bovis BCG using the Universal CAS liquid assay. To this purpose, M. bovis was grown in 7H9 medium, subcultured in chelated Sauton’s medium and then diluted to an OD600 of 0.01 in chelated Sauton’s containing different concentrations of the test compound. After 15 days of incubation at 37 °C, cells were harvested, supernatants were used to perform CAS assay and cell pellets were used for the determination of mycobactins. For CAS assay, an aliquot of 100 µL of supernatant was mixed with 100 µL of CAS assay liquid solution in a 96-well plate, incubated for 10 min at room temperature, and the absorbance was measured at 630 nm. For mycobactin determination, cell pellets were extracted in ethanol overnight, then 0.1 M FeCl3 in ethanol was added until no color change was observed. The mixture was incubated at room temperature for 1 h, then mycobactins were extracted in chloroform, washed with water, evaporated and the residue was dissolved in methanol. The concentration of mycobactins was determined measuring the absorbance at 450 nm (1% solution of mycobactins gives an absorbance of 42.8).
Cell viability assay
MRC5 human fibroblast lung cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2, according to the supplier’s indications. Cells (104) were plated in 96-well culture plates. The day after seeding, vehicle or compounds were added at different concentrations to the medium. Compounds were added to the cell culture at a concentration ranging from 200 to 0.01 µM. Cell viability was measured after 96 h according to the supplier’s instructions (Promega, cat. n° G7571) with a Tecan M1000 PRO instrument. IC50 values were calculated from logistic dose-response curves.
Docking studies
All the compounds were docked into the minimised average structure of MbtI complexed with the lead compound I 14 . The software Gold with the four fitness functions implemented (i.e. GoldScore, ChemScore, Astex Statistical Potential, and ChemPLP) and plants were employed in this study as previously reported 14 , 25 , since they have been already used showing good results in the previous virtual screening study on MbtI inhibitors performed in our laboratory 15 . The docking site was defined as the region comprising all residues that stayed within 10 Å from the reference compound I. By applying the five docking methods, five different binding dispositions (best-scored docking pose) resulted from the docking of each ligand into the protein binding site. The RMSD of these docking poses against the remaining four was evaluated by using the rms_analysis software of the Gold suite. On this basis, for each ligand docked into the protein binding site, a 5 × 5 matrix was generated reporting the RMSD results. By using an in-house programme, these results were clustered, so that among the five results, all of the similar docking poses were grouped together 26 . We selected an RMSD clustering threshold of 2.0 Å; therefore, the so-obtained clusters contained the group of poses that were less than 2.0 Å away from all others poses belonging to the same cluster. For each compound, the binding mode belonging to the most populated cluster identified by the consensus docking evaluation was taken into consideration and subjected to MD simulations.
MD simulations
All simulations were performed using AMBER 16 27 . General amber force field (GAFF) parameters were assigned to the ligands, whereas partial charges were determined using the AM1-BCC method as implemented in the Antechamber suite of AMBER16. MD simulations were carried out employing the ff14SB force field at 300 K. Magnesium ion was inserted considering its disposition and interaction into the 2FN1 PDB code. Ligand-protein complexes were placed in a rectangular parallelepiped water-box and solvated with a 20 Å water cap by using the TIP3P explicit solvent model. Sodium ions were added as counterions in order to neutralise the system. Before MD simulations, two steps of minimisation were performed; in the first stage, a position constraint of 500 kcal/(mol⋅Å2) was applied to keep the protein fixed thus minimising only water molecules. In the second stage, the whole system was energy minimised through 5000 steps of steepest descent followed by conjugate gradient (CG), until a convergence of 0.05 kcal/(mol·Å2) and imposing a harmonic potential of 10 kcal/(mol⋅Å2) to the protein α carbon. Particle mesh Ewald (PME) electrostatics and periodic boundary conditions were used in the simulations. The time step of the simulations was 2 fs with a cutoff of 10 Å for the non-bonded interactions, while the SHAKE algorithm was applied to keep all bonds involving hydrogen atoms fixed. Constant-volume periodic boundary MD simulation was carried out for the first 0.5 ns, during which the temperature of the system was raised from 0 to 300 K. Then 19.5 ns of constant-pressure periodic boundary MD was performed at 300 K, using the Langevin thermostat in order to maintain constant the temperature of the system. A harmonic force constraint of 10 kcal/(mol·Å2) was applied to the protein α carbons during the first 3.5 ns, whereas in the last 16.5 ns no restraints were applied to the system. All the obtained MD trajectories were analyzed using the Cpptraj programme implemented in AMBER 16.
Result and discussion
In order to further develop our library of furan-based candidates, we examined the substitution pattern at the para and ortho positions of the phenyl ring. Herein, we disclose additional SAR data; firstly, we synthesised a phenyl di-substituted analog of I, characterised by the presence of the trifluoromethyl group (1a), because we knew that the more druggable p-CF3 in place of the p-NO2 group was still able to inhibit MbtI 14 . Compound 1a displayed encouraging inhibiting properties (residual enzyme activity at 100 µM, as % RA, of 6.1 ± 2.5, IC50 = 28.5 ± 2.6 µM). Therefore, we took into account this compound as a new hit and we focussed on the effects of a variety of electron donating/withdrawing or hydrophilic groups in the ortho position of the phenyl ring, keeping the CF3 group in para position (1b–i). Then, the effects of the previously analysed substituents in ortho position were evaluated in the second series of derivatives 1j–p, bearing the original p-NO2 group (Table 1).
The activity of all compounds (1a–p) was tested against the recombinant MbtI, prepared and assayed as previously reported 14 . The substitution of the chlorine atom with the fluorine in 1 b did not affect the activity, while the introduction of the bromine atom in 1c diminished the inhibitory effect of the compound (% RA 38.0 ± 3.7), due to the low capability of bromine to act as hydrogen bond acceptor together with its higher atomic radius determining a negative steric constraint. The presence of the electron donating groups in 1d, 1e, and 1f did not lead to an improvement of the biological effects. When an electron withdrawing moiety was introduced in the ortho position, as in 1 g, we detected an increased activity. This outcome was confirmed by the introduction of an o-CF3 moiety that led to the disclosure of 1 h, having an IC50 value comparable to that of the most active inhibitor (I), previously identified. Derivative 1i was then prepared and tested to assess the importance of having the electron withdrawing moiety in ortho position of the phenyl ring; interestingly, moving the CF3 to the meta position significantly decreased the inhibitory activity of the compound. A parallel trend of biological results was obtained for the p-NO2 derivatives 1j–p. These compounds were prepared and tested to compare the impact of the CF3 and NO2 groups on the activity and to better understand their role in the interaction with the target. The outcomes reflected the similar biological behaviour observed for this series (1j–p) with respect to the previous one (1a–i), confirming the ability of CF3 to act as a bioisostere of the nitro moiety, according to the bioisosterism between fluorine and carbonyl functionality 28 . Of note, the presence of two CF3 substituents in 1 h synergically contributed to enhance the activity, as shown by the comparison of the IC50 value of 1 h and the respective derivative 1p belonging to the para-nitro series (13.1 vs. 41.8 µM).
For the most active compound, 1 h, an accurate biological analysis was thus performed (Figure 1). Kinetic analysis allowed the determination of the Ki value 8.8 ± 0.7 µM, allowing the classification of this compound as competitive inhibitor of MbtI. Then, additional tests were performed to verify that 1 h was not a PAIN compound. The addition of BSA and Triton X-100 did not influence its IC50, proving that the compound did not form aggregates with the target. Similarly, the addition of DTT did not impact on 1 h activity, showing that the ligand did not interact with the cysteine residues of the protein. To evaluate the antimycobacterial activity of 1 h we used the nonpathogenic M. bovis BCG, whose siderophores closely resemble Mtb mycobactins 29 , 30 . The minimal inhibitory concentration (MIC99) value, determined in iron-limiting conditions in chelated Sauton’s medium, was similar to that of hit I (250 µM versus 500 µM). Moreover, to assess if the antitubercular activity was indeed related to iron uptake inhibition, the effects of the compound on siderophore production were evaluated by means of the Universal CAS liquid assay 30 . Firstly, the level of siderophore activity in the medium was measured and the colorimetric assay revealed that the removal of iron was inversely related to the concentration of the compound. Similarly, the quantification of mycobactins isolated from the cell pellet evidenced that the concentration of mycobactins decreased at higher 1 h concentrations. All these outcomes suggested that the inhibitory effect towards Mtb growth was due to mycobactin biosynthesis inhibition. Finally, compounds I, 1 h and 1p were selected for evaluating their antiproliferative activity against human MRC-5 fibroblasts. The biological results showed that the three compounds did not affect the growth of these normal cells (MRC-5, IC50 > 100 µM), thus indicating the potentially low level of toxicity of this class of compounds.
Figure 1.

Biological characterisation of 1h. The global reciprocal plot of data from MbtI steady-state kinetics analysis towards chorismic acid, in the presence of different concentrations of 1h (A), highlights the competitive behaviour of the inhibitor. IC50 plots of 1h in the presence of BSA (green squares), Triton X-100 (red triangles) and DTT (yellow diamonds) (B) confirm that the compound is not a PAIN. The Universal CAS assay performed on culture media of M. bovis BCG cells grown in the presence of 1h (C), together with the determination of the mycobactins in the above-mentioned cells (D), confirm that the antimycobacterial activity is related to iron uptake inhibition. All data are mean ± SD of three replicates.
With the aim of elucidating the binding mode of this class of derivatives, molecular modelling studies were performed on the most active compound of the series. Compound 1 h was subjected to a consensus docking protocol and the result was refined through 20 ns of MD simulation with explicit water molecules (see Supplementary Material for details). Figure 2 shows the minimised average structure of 1 h bound to the catalytic site of MbtI. The two aromatic rings of the ligand lay on the hydrophobic wall constituted by I207, P251, T361, and L404, thus forming lipophilic interactions with these residues. On the other side of the binding site, the ligand core moiety shows multiple interactions with K438: a hydrogen bond formed by the furan oxygen and a cation-π interaction established through the phenyl ring, which also forms additional hydrophobic contacts with the side chain of the residue. The carboxylic group of 1 h coordinates the Mg2+ ion and also forms H-bonds with the backbone nitrogen of G270 and the side chain of K205 that contribute to anchor the ligand to the MbtI binding site. According to the experimental results, the p-CF3 group of the ligand replaces the p-NO2 group of the parent compound I by forming two H-bonds with the backbone nitrogen of R405 and G419, while the o-CF3 substituent of 1 h shows an additional hydrogen bond with the side chain of H334. The same consensus docking and MD protocols were then applied on compounds 1a and 1q to gain a better interpretation of the obtained SAR. In agreement with the experimental data, the binding mode predicted for 1a confirmed the importance of the o-CF3 substituent of 1 h. In fact, the o-chlorine atom of 1a does not show any hydrogen bond with H334. Moreover, although the ligand perfectly chelates the Mg2+ ion, the presence of the chlorine atom determines a small shift in the binding orientation that hampers the formation of H-bonds with G270 and K205 predicted for 1 h (Figure S1). Nevertheless, the presence of an o-CF3 moiety was found to be detrimental when associated with the p-NO2 group originally present in I, as observed for compound 1p. Interestingly, MD simulations suggested that 1p is not able to form the H-bond and cation-π interactions with K438 (Figure S2). In order to establish the H-bond network among its p-NO2 group, the backbone nitrogen of R405 and the side chain of Y385, 1p moves toward these residues and slightly rotates toward K438, which thus moves away from the ligand to avoid steric clashes. Due to its orientation within MbtI binding site, 1p cannot even interact with H334 through the o-CF3 as predicted for 1h.
Figure 2.

Minimised average structure of 1 h within MbtI binding site.
To conclude, starting from the analysis of the hypothetical binding mode of our previous furan-based hit I, we successfully performed the bioisosteric replacement of the nitro group. Our preliminary hit optimisation study led to the disclosure of a new compound (1 h) exhibiting a strong activity against MbtI, comparable to the most potent competitive inhibitor reported so far 14 . Our best candidate 1 h, characterised by the presence of two CF3 substituents in the ortho and para positions of its phenyl ring, displayed an IC50 value of 13.1 ± 2.0 µM (Ki = 8.8 ± 0.7 µM) and the antimycobacterial activity showed by this compound (MIC99 = 250 µM) is conceivably related to mycobactin biosynthesis inhibition. Moreover, preliminary assays against noncancerous human fibroblast lung cells did not reveal cytotoxicity issues. These results support the hypothesis that 5-phenylfuran-2-carboxylic derivatives are a promising class of MbtI inhibitors and allowed us to gather new information for a further optimisation of this class of compounds.
Supplementary Material
Funding Statement
This work was funded by University of Milan (Linea B), and the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Programme (2018–2022) - Dept. of Biology and Biotechnology “L. Spallanzani”, University of Pavia.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1. Glaziou P, Floyd K, Raviglione MC. Global epidemiology of tuberculosis. Semin Respir Crit Care Med 2018;39:271–85. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y, Yew W. Mechanisms of drug resistance in Mycobacterium tuberculosis . Int J Tuberc Lung Dis 2009;13:1320–30. [PubMed] [Google Scholar]
- 3. Gandhi NR, Nunn P, Dheda K, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 2010;375:1830–43. [DOI] [PubMed] [Google Scholar]
- 4. Falzon D, Gandhi N, Migliori GB, et al. Resistance to fluoroquinolones and second-line injectable drugs: impact on multidrug-resistant TB outcomes. Eur Respir J 2013;42:156–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koul A, Arnoult E, Lounis N, et al. The challenge of new drug discovery for tuberculosis. Nature 2011;469:483–90. [DOI] [PubMed] [Google Scholar]
- 6. Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 2013;12:388–404. [DOI] [PubMed] [Google Scholar]
- 7. Fanzani L, Porta F, Meneghetti F, et al. Mycobacterium tuberculosis low molecular weight phosphatases (MPtpA and MPtpB): from biological insight to inhibitors. Curr Med Chem 2015;22:3110–32. [DOI] [PubMed] [Google Scholar]
- 8. Meneghetti F, Villa S, Gelain A, et al. Iron acquisition pathways as targets for antitubercular drugs. Curr Med Chem 2016;23:4009–26. [DOI] [PubMed] [Google Scholar]
- 9. Borisov SE, Dheda K, Enwerem M, et al. Effectiveness and safety of bedaquiline-containing regimens in the treatment of MDR- and XDR-TB: a multicentre study. Eur Respir J 2017;49:1700387. [DOI] [PubMed] [Google Scholar]
- 10. D’Ambrosio L, Centis R, Tiberi S, et al. Delamanid and bedaquiline to treat multidrug-resistant and extensively drug-resistant tuberculosis in children: a systematic review. J Thorac Dis 2017;9:2093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Voss JJ, Rutter K, Schroeder BG, et al. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc Natl Acad Sci USA 2000;97:1252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vasan M, Neres J, Williams J, et al. Inhibitors of the salicylate synthase (MbtI) from Mycobacterium tuberculosis discovered by high-throughput screening. ChemMedChem 2010;5:2079–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Manos-Turvey A, Cergol KM, Salam NK, et al. Synthesis and evaluation of M. tuberculosis salicylate synthase (MbtI) inhibitors designed to probe plasticity in the active site. Org Biomol Chem 2012;10:9223. [DOI] [PubMed] [Google Scholar]
- 14. Chiarelli LR, Mori M, Barlocco D, et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur J Med Chem 2018;155:754–63. [DOI] [PubMed] [Google Scholar]
- 15. Pini E, Poli G, Tuccinardi T, et al. New chromane-based derivatives as inhibitors of Mycobacterium tuberculosis salicylate synthase (MbtI): preliminary biological evaluation and molecular modeling studies. Molecules 2018;23:1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrison AJ, Yu M, Gardenborg T, et al. The structure of MbtI from Mycobacterium tuberculosis, the first enzyme in the biosynthesis of the Siderophore mycobactin, reveals it to be a salicylate synthase. J Bacteriol 2006;188:6081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nepali K, Lee H-Y, Liou J-P. Nitro-group-containing drugs. J Med Chem 2018. [Epub ahead of print]. doi: 10.1021/acs.jmedchem.8b00147 [DOI] [PubMed] [Google Scholar]
- 18. Porta F, Gelain A, Barlocco D, et al. A field-based disparity analysis of new 1,2,5-oxadiazole derivatives endowed with antiproliferative activity. Chem Biol Drug Des 2017;90:820–39. [DOI] [PubMed] [Google Scholar]
- 19. Masciocchi D, Gelain A, Porta F, et al. Synthesis, structure–activity relationships and stereochemical investigations of new tricyclic pyridazinone derivatives as potential STAT3 inhibitors. Medchemcomm 2013;4:1181. [Google Scholar]
- 20. Gorak YI, Obushak ND, Matiichuk VS, Lytvyn RZ. Synthesis of heterocycles from arylation products of unsaturated compounds: XVIII. 5-Arylfuran-2-carboxylic acids and their application in the synthesis of 1,2,4-thiadiazole, 1,3,4-oxadiazole, and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives. Russ J Org Chem 2009;45:541–50. [Google Scholar]
- 21. Cattò C, Grazioso G, Dell'Orto S, et al. The response of Escherichia coli biofilm to salicylic acid. Biofouling 2017;33:235–51. [DOI] [PubMed] [Google Scholar]
- 22. Dahlin JL, Nissink JWM, Strasser JM, et al. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem 2015;58:2091–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siegrist MS, Unnikrishnan M, McConnell MJ, et al. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA 2009;106:18792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Palomino J-C, Martin A, Camacho M, et al. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis . Antimicrob Agents Chemother 2002;46:2720–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tuccinardi T, Poli G, Dell'Agnello M, et al. Receptor-based virtual screening evaluation for the identification of estrogen receptor β ligands. J Enzyme Inhib Med Chem 2015;30:662–70. [DOI] [PubMed] [Google Scholar]
- 26. Poli G, Martinelli A, Tuccinardi T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J Enzyme Inhib Med Chem 2016;31:167–73. [DOI] [PubMed] [Google Scholar]
- 27. Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, et al. 2015. AMBER, Version 14. San Francisco, CA: University of California. [Google Scholar]
- 28. Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J Med Chem 2018;61:5822–80. [DOI] [PubMed] [Google Scholar]
- 29. Brosch R, Philipp WJ, Stavropoulos E, et al. Genomic analysis reveals variation between Mycobacterium tuberculosis H37Rv and the attenuated M. tuberculosis H37Ra strain. Infect Immun 1999;67:5768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwyn B, Neilands JB. Universal chemical assay for the detection and determination of siderophores. Anal Biochem 1987;160:47–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.