

Abstract
When using liquid chromatography/mass spectrometry (LC/MS) to perform untargeted metabolomics, it is now routine to detect tens of thousands of features from biological samples. Poor understanding of the data, however, has complicated interpretation and masked the number of unique metabolites actually being measured in an experiment. Here we place an upper bound on the number of unique metabolites detected in Escherichia coli samples analyzed with one untargeted metabolomic method. We first group multiple features arising from the same analyte, which we call “degenerate features”, using a context-driven annotation approach. Surprisingly, this analysis revealed thousands of previously unreported degeneracies that reduced the number of unique analytes to ~2,961. We then applied an orthogonal approach to remove non-biological features from the data using the 13C-based credentialing technology. This further reduced the number of unique analytes to less than 1,000. Our 90% reduction in data is five fold greater than any previously published studies. On the basis of the results, we propose an alternative approach to untargeted metabolomics that relies on thoroughly annotated reference data sets. To this end, we introduce the creDBle database (http://creDBle.wustl.edu), which contains accurate mass, retention time, and MS/MS fragmentation data as well as annotations of all credentialed features.
INTRODUCTION
It has become increasingly popular to perform untargeted metabolomics by using liquid chromatography/mass spectrometry (LC/MS). This is at least in part due to the large number of signals or features that are typically detected from most biological samples.1–3 While it is often assumed that these tens of thousands of detected signals provide “global” coverage of the metabolome, the number of metabolites being measured in an experiment has not been rigorously assessed. The major barrier preventing this type of analysis has been the challenge of identifying metabolites.4 To date, the overwhelming majority of the detected signals in any one untargeted metabolomics experiment have not been named. Even comprehensive efforts to identify as many metabolites as possible in a data set by using the most advanced informatic resources currently available have resulted in small percentages of the total number of signals being identified.5–7 Thus, the basic question of how many unique metabolites are being profiled in an untargeted metabolomics experiment has remained outstanding.
Uncertainty in the number of unique metabolites being profiled (i.e., experimental coverage) has not prevented the widespread application of the untargeted metabolomics technology. Improvements in instrumentation and software have made acquiring untargeted metabolomics data with LC/MS relatively routine.8 The number of research cores offering LC/MS untargeted metabolomics services therefore has increased dramatically over the last decade.9 The untargeted workflows used by many research facilities, however, do not directly address the issue of experimental coverage.10 Their experimental output is a long list of signals or features, often with minimal annotation. The data sets are either mined in a targeted fashion for specific metabolites with known retention times and fragmentation patterns, or only the small subset of signals that have a statistically significant difference between sample classes are further investigated.11 Most dysregulated signals remain unidentified because feature relationships are under-annotated, and their accurate mass and fragmentation patterns do not match data in metabolomic databases.12 Although it is common to refer to these unmatched signals as “unknown metabolites”, rarely is such a designation justified because signals associated with contaminants, artifacts, and many adducts also do not return matches from metabolomic databases. These possibilities and others must be ruled out before gaining confidence that a signal is a bona fide, unique metabolite with an unknown structure.
The total number of features in a LC/MS-based metabolomic data set that result from the sum of contaminants, artifacts, and degeneracies (such as complex adduct formation) has not been comprehensively evaluated. Previous assessments of data set composition have only focused on specific subsets of feature inflation.13 CAMERA analysis, for example, has been effectively used to search for common relationships between pairs of peaks.14 Intensity correlations across samples and peak profiles have also been applied to estimate signal degeneracy in the case of RAMclust and others.1,15 Analysis of peak-pairs has been applied to estimate the frequency of some common analyte transformations.16 Additionally, isotopic approaches such as IROA have also shed light on the complexity of metabolomic data sets. Here, analyte signals from organisms cultured in 13C-enriched media exhibit isotopic signatures that provide a mechanism to reduce features.17–21 Such isotopic techniques address another specific subset of feature inflation related to contaminants and artifacts. Despite the availability of these types of approaches, limitations have prevented their simultaneous application to a single metabolomic data set at the comprehensive level. Moreover, recent targeted analyses from our laboratory have suggested that additional sources of degeneracy within a metabolomic data set are still unannotated, even with the aforementioned resources.22 Thus, the number of unique metabolites being measured in a metabolomics experiment has remained elusive.
The goal of the current study was to accurately enumerate contaminants, artifacts, and degeneracies at the systems level within the same data set to get an upper estimate of the number of unique metabolites detected in a representative LC/MS-based metabolomics experiment. For the purposes of this work, contaminant refers to a detected signal that does not originate from the biological sample being measured (e.g., solvent impurities and plastic leechables). Artifacts refer to features detected due to informatic error. As an example, artifacts can be caused by baseline fluctuations and poorly resolved components.23,24 Finally, degeneracy refers to multiple signals arising from a single analyte. There are many causes of degeneracy including: fragmentation, analyte adduction with various charge carriers (e.g., a proton, sodium, potassium, etc.), and the detection of naturally occurring isotopes (e.g., 13C,15N, etc.).16,22,25 Fragmentation and adduction are dependent on the ionization conditions employed. Annotation of these features in datasets will enable their formation to be evaluated and minimized in future work. A final, largely under-annotated source of degeneracy is the adduction of an analyte with other species present, including other analytes or the chemical background.
Although some degenerate relationships are well known and commonly annotated with the approaches described above, the prevalence of many degenerate relationships has not been previously estimated.14 Here we introduce and apply an approach that recovers relationships implied by the experimental data, rather than relying on a hypothetical predetermined list as is typically done in metabolomics.1 The approach allows for more comprehensive annotation, especially in the case of under-annotated adducts that may be specific to a single laboratory or experiment. Here, we use the term “annotation” to refer to feature relationships rather than structural identifications.
In this work, we have focused on Escherichia coli cells that were extracted and analyzed with a representative untargeted metabolomics method. In positive-ion mode, we detected 25,230 high-quality metabolomic signals or features. Strikingly, we found that more than 90% of these detected signals were due to contaminants, artifacts, and degeneracy. This order-of- magnitude reduction in data is significantly greater than that which has been estimated by previous studies examining only subsets of feature inflation. On the basis of our results, we introduce a new approach for performing untargeted metabolomic studies that relies on thoroughly annotated reference data sets in a resource such as creDBle, described here. Our work has important implications for the experimental coverage, design, quantitation, and interpretation of discovery profiling experiments.
METHODS
Generating credentialed samples
Credentialed samples were either generated in house or obtained from Cambridge Isotopes Laboratories (MSK-CRED-KIT). For in-house generation, E. coli (K12 MG1655) was grown in both uniformly enriched 13C-medium and natural-abundance medium to generate cellular material suitable for credentialing analysis. Cell pellets weighing 2.5 mg (dry) were extracted with a 2:2:1 methanol:acetonitrile:water mixture and reconstituted in 100 μL of 1:1 acetonitrile water. Detailed methods can be found in Supplementary Methods.
Data set generation
Each sample was analyzed five times via LC/MS. A reversed-phase UPLC separation (Waters Cortecs T3 column) was coupled to a Q-Exactive Plus mass spectrometer for analysis. Chromatographic features were detected by using a model-based peak detection and the skew normal distribution for fitting. The process resulted in a set of features detected in each replicate run. Subtle variations from run to run cause many features to be integrated differently and sometimes not integrated in each file. The Warpgroup algorithm (https://github.com/nathaniel-mahieu/warpgroup) was applied to resolve these inconsistencies and generate a consensus data set from the individual replicate runs.26 This resulted in 25,230 “high-quality” features in our representative data set.
Mz.unity based annotation
Mz.unity was applied to the data set to detect mass and charge ([m, z]) relationships between eluting signals derived from a single analyte.22 We use [m, z] to denote the mass and charge of a species, where both are specified as opposed to m/z where the two are convolved. These searches find sets of features that have [m,z]s differing by a specific amount. Differences are specific to relationships, for example, loss of 12C and gain of 13C ([+1.003355, 0]), or loss of water ([−18.01057, 0]).
Searches were first performed for the following relationships: isotopes, common charge carriers, common neutral losses, and common adducts. We then searched for dimers between coeluting features. The dimer search posits each eluting [m, z] as a possible adduct former. The charge state was specified based on observed isotopes, or assumed to be a charge of 1. As dimers are normally formed with a charge from only one constituent, we also assumed the loss of a proton [1.00783, +1] for each pair.
Mz.unity is available at https://github.com/nathaniel-mahieu/mz.unity.
Frequent intrinsic relationships
Groups of features eluting within 1 second of each other were taken, and their pairwise [m, z] differences were calculated after assuming a charge state of 1. A Gaussian kernel density estimation was performed on the mass differences with a bandwidth of 0.00001 Da (our observed scan-to-scan mass error). Local maxima of the density estimate were detected along with the estimated density at those locations. The heights of the local maxima represent the frequency and mass dispersion of each mass difference. Mass differences that are more frequent and more similar in mass will have larger density estimates.We took enriched mass differences larger than 15 Da and occurring more than 50 or 200 times throughout the data set into the mz.unity search.
Situational adducts
Background ions that lack a chromatographic peak shape are an ever-present set of species that often form adducts with eluting analytes. These situational adducts are then detected as features having a chromatographic peak shape. A low mass background spectrum was collected, containing detected ions above 50 Da. This spectrum was deisotoped and background species appearing at higher than 200,000 intensity were used to seed possible adduct relationships. The [m, z]s of each background peak were included in the dimer search, as above after specifying the charge state based on observed isotopes or assuming a charge of 1.
Credentialing
A high confidence set of features were recovered from the 12+13C data set by applying version 3.0 of the credentialing algorithm, which is available at https://github.com/pattilab/credential. Credentialing searches for pairs of peaks that have precise isotopic spacing expected from U-12C and U-13C analytes.27 This provides a filter against many forms of noise, contaminants, and artifactual features. Credentialing was run with the parameters: ppmwid, 8; rtwid, 1.2; cd, 1.00335; mpc, c(12, 120); ratio, 1; ratio.lim, 0.1; maxnmer, 4. Credentialed features from the 12+13C data set were then matched to the 12C data set by applying retention time and mass correction as above before grouping.
Credentialed feature characterization
The set of credentialed features were further characterized for deposition in the creDBle database. Targeted MS/MS was performed on the credentialed features with a 0.4 Da window width and a stepped collision energy of 10, 30, and 90 V. Annotations and feature groupings of the credentialed features were taken from the previously performed mz.unity annotations.
The creDBle database
All credentialed features (and associated annotations) from the data set were deposited in the creDBle database. The data are freely available at http://credble.wustl.edu/ and easily downloadable in JSON format via the REST API. This includes m/z, retention time, annotation grouping, MS/MS spectra, credentialed isotopes, extracted ion chromatograms, and identifications (when available).
RESULTS AND DISCUSSION
Generating a representative untargeted metabolomic data set
In untargeted metabolomics, putative metabolite signals are often referred to as features. A feature is a detected ion with a peak shape, unique m/z, and retention time. To estimate the number of unique analytes detected in a representative untargeted metabolomic data set, we set out to annotate three types of features: (i) degenerate features, (ii) contaminant features, and (iii) artifactual features. This work represents the first attempt to accurately enumerate each of these types of features simultaneously for the same data set. We annotated degenerate features by using mz.unity and a new contextual approach to find degeneracies implied by the data. We annotated contaminant features and artifactual features by using the credentialing approach.27 Uniform 13C-labeling is a requirement of the credentialing appraoch. Given that there are convenient and well-established methods to culture E. coli on a uniformly labeled carbon source, we chose to focus our work on E. coli. We anticipate that our results from E. coli will be generally consistent with other sample types and change mostly as a function of feature intensities. Artifacts depend primarily on the amount of raw signal detected, for example, and artifact number will therefore increase linearly with signal amount. Dimer formation, on the other hand, is combinatorial. As sample complexity and analyte coelution increase, the number of dimers will increase superlinearly.
Metabolites from E. coli cells were extracted and analyzed with an LC/MS-based untargeted metabolomics platform, as detailed in Methods. These and similar experimental methods are commonly applied in untargeted metabolomics.28–30 To process the resulting LC/MS data, we employed an iterative, two-phase peak detection process (Figure 1). A model- based feature detection algorithm was run on each of five individual replicates. To refine the features detected in the five replicates, we utilized the Warpgroup algorithm.26 Warpgroup considers all files in concert to identify “consensus features”, a set of feature integrations supported by all replicates. The result is a near one-to-one matching of features between samples (Figure S1A-B) and decreased variation introduced by informatic processing (Figure S1C-D). The Warpgroup refined feature detection is highly sensitive, allowing the recovery of features that, when processed in isolation, would be challenging to detect (Figure S1E). This resulted in 25,230 high-quality features in our representative data set. It is worth mentioning that analysis of our data set with the standard XCMS software resulted in the detection of more features compared to Warpgroup (see Table S1). These data show that our informatic methods are not contributing to atypically high feature counts.
Figure 1.
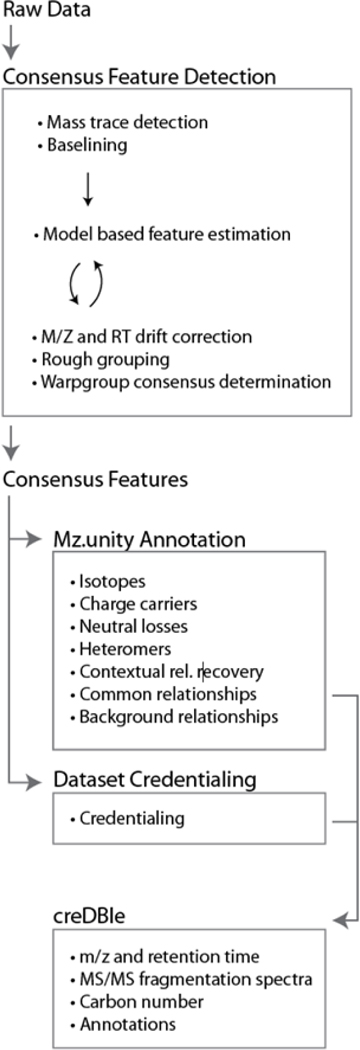
Our informatic workflow. Raw data were processed with in-house algorithms to first identify high-quality, consensus features (i.e., recurring features between replicates) and discriminate against processing artifacts. This consensus data set was further characterized by mz.unity (to estimate signal degeneracy) and credentialing (to estimate contaminants and artifacts). The resulting annotated data set was catalogued in the creDBle database.
We note that there is no universally accepted experimental platform for untargeted metabolomics at this time. The extraction techniques, chromatography, mass spectrometers, and peak detection algorithms used vary between laboratories and are often multiplexed.31,32 However, it is routine to detect tens of thousands of signals from a biological sample in most LC/MS experiments.33,34 Our detection of 25,230 consensus features from five replicates resulted in a data set with complexity that is typical of an untargeted metabolomic experiment.
Simple annotations
As a first step, we filtered features present in the blank that were not at least two-fold higher than the signal detected in extraction blanks. These features represent contaminants or artifacts that are introduced during the sample extraction or data-processing steps. This reduced our list of 25,230 features to 12,797 (Figure 2A).
Figure 2.
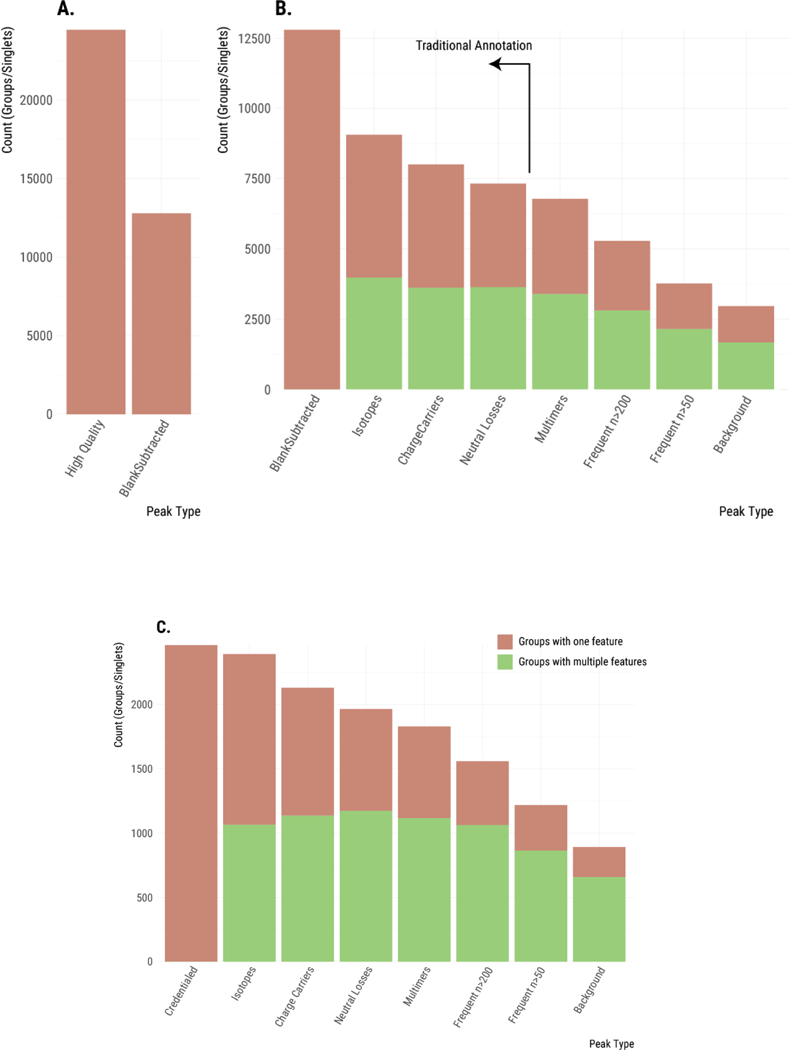
Plotting the maximum number of unique analytes detected throughout the steps of our annotation process. (A) Removal of features occurring in the blank. (B) Features are grouped as additional relationships are annotated. This reduces the maximum number of unique analytes. When a feature group contains multiple features, it is shown in green. When a feature group contains only a single feature (i.e., is a singlet), then it is shown in pink. Relationships from left to right: no relationships; isotopes; charge carriers; neutral losses; complex dimers (single and multi-analyte dimers); frequent intrinsic relationships; situational adducts (background). (C) Similar annotation of features that were credentialed.
Next, we set out to annotate degenerate features (i.e., those features arising from the same analyte). We started our analysis by identifying simple relationships that are already commonly annotated in untargeted metabolomics.14,35–37 This included degeneracy due to carbon and other isotopes as well as common adducts and neutral losses. Annotations were made by using mz.unity, and degenerate features were grouped together.22 Because features within the same group arise from the same analyte, the number of “feature groups” provides a much better estimate of the maximum number of unique analytes detected in an experiment than the number of total features (Table S2 and Figure 2B-C). In our subsequent descriptions, we will therefore transition from counting features to counting feature groups. A feature for which no degeneracy has been identified constitutes its own feature group, which we refer to as a singlet. Figure 2B shows the progressive decrease in the number of feature groups as isotopes, common charge carriers, and common neutral losses are annotated.
When isotopes, common charge carriers, and neutral losses are annotated, the number of feature groups decreases from 12,797 to 7,318. We note that currently employed annotation approaches end here with the identification of simple relationships (see vertical line in Figure 2B). These results might suggest that there are as many as 7,318 unique analytes detected in the sample, but two observations suggested that much degeneracy still remained unannotated in our E. coli data set. First, about 50% of our feature groups still contain only a single feature (i.e., singlets with no detected relationships). Although in some cases singlets result from low- abundance analytes with no natural isotopes detected above noise level, the prevalence of singlets suggested that additional relationships remained unannotated. Second, we also know that the set of relationships annotated thus far are only a small subset of the possible degeneracies. A recent targeted study of glutamate demonstrated that many additional, complex sources of degeneracy can exist in LC/MS-based metabolomics that are not currently annotated with existing informatic resources.22 Glutamate was found to produce over 100 spectral peaks and exhibited complex adduct formation. Our objective was to comprehensively characterize these additional sources of degeneracy within a data set (E. coli) for the first time.
Single-analyte and multi-analyte multimers
We then expanded our search for degenerate relationships to complex adducts (i.e., two or more species non-covalently bound to one another, such as dimers, trimers, etc.). Our search included analytes adducted with themselves, as well as analytes adducted with different analytes. We considered all coeluting features as potential multimer partners evaluating all [m, z] values as possible adduct formers. As an example, a complex, multi-analyte-relationship between three detected features will satisfy: [m1, z1] + [m2, z2] - [1.00783, 1] = [m3, z3]. Grouping these detected complex adducts reduced the number of feature groups in our data set to 3,400 (see “multimers” bar in Figure 2B-C).
Frequent intrinsic relationships show previously unannotated degeneracy
Current annotation approaches in untargeted metabolomics face the major challenge of determining the set of relationships to search for. While some relationships are well known and occur ubiquitously (such as the commonly annotated sodium or potassium adducts), constraining annotation to only these is significantly limiting. Other degenerate relationships are specific to experimental methodologies or the materials and reagents used during the analysis. Since there is no way to determine these relationships a priori, they have gone unannotated to date. Here we introduce an informatic approach to find data set wide, experimentally unique relationships that are implied by their context in the data. We then estimate their prevalence within our E. coli data set.
Common adducts and fragments will always coelute with the original analyte and will occur multiple times throughout the run.22 We leverage this fact and recover “frequent intrinsic relationships” by performing a frequency analysis of mass differences between all pairs of features eluting within one second of each other.16 Unrelated but coeluting analytes will exhibit mass spacing that is random and, as such, will not be enriched in the frequency distribution. Thus, frequently occurring mass differences represent probable degenerate relationships. Mass differences were calculated assuming a charge state of 1, a simplification that limits the analysis to relationships that do not include a charge-state conversion. In our experience, adducts and fragments occuring only with a charge-state conversion are rare and this simplification is not limiting. A Gaussian kernel density estimation was performed on the observed mass differences with a bandwidth of 0.00001 Da (our observed scan-to-scan mass error) (Figure 3A). The heights of the local maxima represent the frequency and mass dispersion of each mass difference. Mass differences that are frequent and similar in mass will have large density estimates. The 24 most frequently observed mass differences are listed in Table 1.
Figure 3.
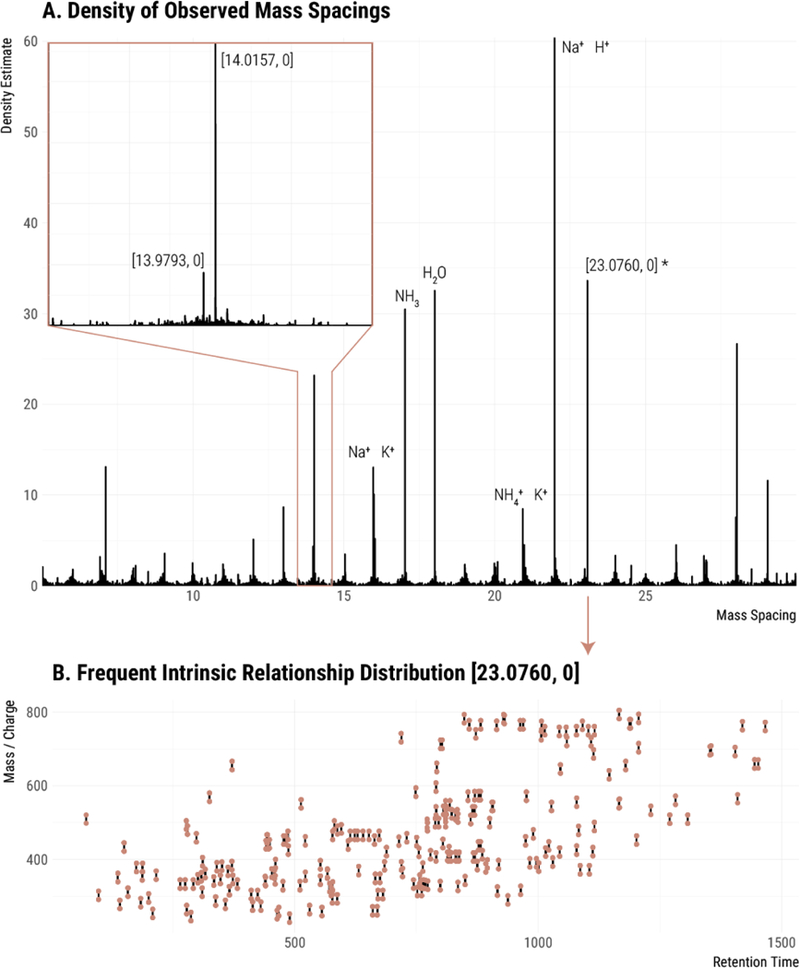
Detection of frequent intrinsic relationships. (A) The Gaussian kernel density of all pairwise peak relationships in the data set. Inset is a zoomed-in section around 14 Da. Known relationships are labeled with a formula. Unknown relationships are labeled with mass and charge transitions [m, z]. (B) Peak pairs of the recovered frequent intrinsic relationship [23.0760, 0] plotted in mass/charge and retention time (points). Line segments connect pairs with the specified spacing.
Table 1.
Recovered frequent intrinsic relationships. Not all recovered relationships shown were used in the annotation. The local maxima of the density are ordered by the number of occurrences. These frequently occurring differences are good candidates for peak relationships. Several well-known relationships are present, including alternative charge carriers at the top of the list.
| Δ Mass | Δ Charge | Density | Known Species |
|---|---|---|---|
| 21.9820 | 0 | 60.4 | H+ ↔ Na+ |
| 4.9554 | 0 | 55.2 | NH4+ ↔ Na+ |
| 23.0760 | 0 | 33.6 | |
| 18.0107 | 0 | 32.5 | H2O |
| 17.0266 | 0 | 30.5 | NH3 |
| 28.0314 | 0 | 26.7 | C2H4 |
| 45.0580 | 0 | 23.4 | C2H7N |
| 14.0157 | 0 | 23.2 | CH2 |
| 65.1230 | 0 | 19.6 | |
| 87.1046 | 0 | 18.2 | C5H13N |
| 42.0470 | 0 | 16.6 | C3H6 |
| 44.0262 | 0 | 15.3 | C2H4O |
| 39.9926 | 0 | 13.3 | C2O |
| 7.1020 | 0 | 13.1 | |
| 15.9740 | 0 | 13.0 | K+ ↔ Na+ |
| 70.0783 | 0 | 12.5 | |
| 29.0518 | 0 | 11.6 | |
| 36.0713 | 0 | 11.3 | |
| 15.9949 | 0 | 10.1 | |
| 1.9967 | 0 | 9.3 | 41K ↔ 39K |
| 56.0627 | 0 | 9.3 | |
| 12.9952 | 0 | 8.7 | |
| 35.0373 | 0 | 8.7 | |
| 20.9292 | 0 | 8.5 | NH4+ ↔ K+ |
The effectiveness of the approach was confirmed by the recovery of two commonly known relationships as the most frequent relationships in our data set: the exchange of H+ and Na+ and the exchange of Na+ and NH4+. This result indicated that the analysis of frequent intrinsic relationships offers novel insight into the nature of features detected in metabolomic data sets. Notably, the approach returned a multitude of relationships that had not been included in our prior searches. These commonly occurring relationships are likely adducts or fragments, and may be specific to our sample or experimental equipment/materials. Figure 3B shows the peak pairs observed with mass difference [23.0760, 0] throughout the data set.
We recognize that the recovery of frequent intrinsic relationships can also return relationships between commonly coeluting, non-degenerate analyte pairs. Fully saturated and partially unsaturated lipids, for example, commonly coelute and have a mass difference of [2.0156, 0] (H2).38 We observed 176 occurrences of such a mass difference in our experiment. To minimize the risk of grouping unrelated features, we did not annotate relationships with mass differences smaller than 15 Da and we applied two frequency cutoffs to illustrate the possible range of degeneracy. The conservative cutoff annotated and grouped frequent intrinsic relationships occurring more than 200 times (see bar labeled “commons n>200” in Figure 2B-C), while the aggressive cutoff annotated and grouped frequent intrinsic relationships occurring more than 50 times (see bar labeled “commons n>50” in Figure 2B-C). The inclusion of frequent intrinsic relationships in our data set annotation reduced the number of feature groups to 5,281 or 3,769, depending on the cutoff.
Situational adducts due to background ions contribute significantly to degeneracy
To further expand the scope of our annotation, we considered a source of adduct ions that are present throughout the run: the chemical background. These ions lack a chromatographic peak shape, but they are detected throughout the experiment due to the ionization of solvents, their additives, or any contaminants present. Because the background ions coelute with every feature, it is reasonable to expect that they will produce many adducts. We refer to adducts between analytes and other simultaneously observed species (such as background ions) as “situational adducts”. Spectra of a situational adduct and its constituents are shown in Figures S2 and S3.
A low-mass spectrum was collected, deisotoped, and background ions appearing at intensities higher than 200,000 were used as potential participants in situational adduct formation (Figure 4). Annotation of the identified situational adducts reduced our number of feature groups to approximately 3,000 (see bar labeled “background” in Figure 2B-C). This significant reduction in feature groups indicates that background ions are indeed a major source of feature inflation in our experiment. We also note that annotation of situational adducts reduced the number of feature groups containing only a single feature (i.e., singlets) to 1,288.
Figure 4.
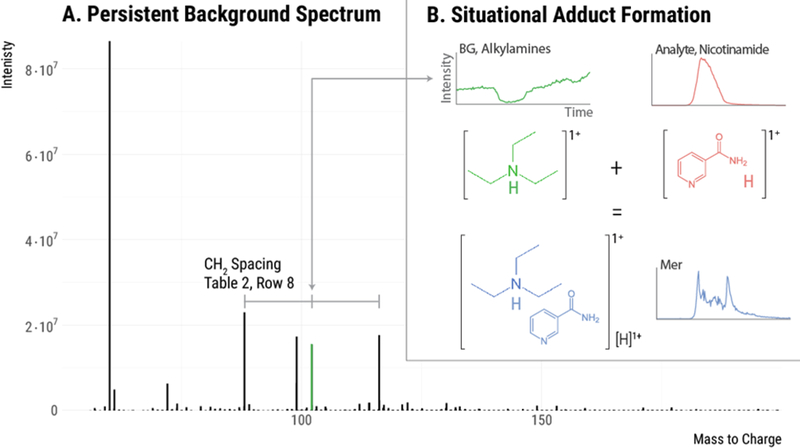
Situational adducts. (A) The persistent background spectrum observed in this experiment. The three indicated background peaks have mass spacings that correspond to a methylene group. These are likely an alkyl amine series with carbon numbers 5, 6, and 7. When these background species adduct with an analyte, situational adducts are formed. (B) An example of a situational adduct formed between background ion 102.1280 (a six carbon alkyl amine) and an eluting analyte. This process likely occurs with all three alkyl amine species throughout the run, giving rise to the frequent intrinsic relationships of mass 14.0157 (see Table 1, Row 8).
Background ions give rise to some frequent intrinsic relationships
Some frequent intrinsic relationships that we detected are indicative of novel adduction or fragmentation phenomena in our untargeted metabolomic data set, and we were interested in the origin of these unknown relationships. We speculated that some of the frequent intrinsic relationships that we discovered were the result of analyte adduction with the chemical background described above. In the simplest of cases, we found that some frequently occurring mass-to-charge differences between features corresponded to the mass-to-charge values of background ions. However, we also noted that mass differences in the background ions were found in features. This indicated that a single analyte formed adducts with multiple background ions (Figure 4, Figure 5, and Supplemental Figure S4) and therefore multiple situational adducts were detected for the same analyte. As the spacings between the background ions determine the spacings in the situational adduct features, we expect these repeatedly occurring spacings to be returned as frequent intrinsic relationships. Inspecting the returned frequent intrinsic relationships, we found several mass differences that also appear in the chemical background. This result is an additional confirmation of the effectiveness of frequent intrinsic relationship discovery and suggests that chemical background is a large source of feature inflation.
Figure 5.
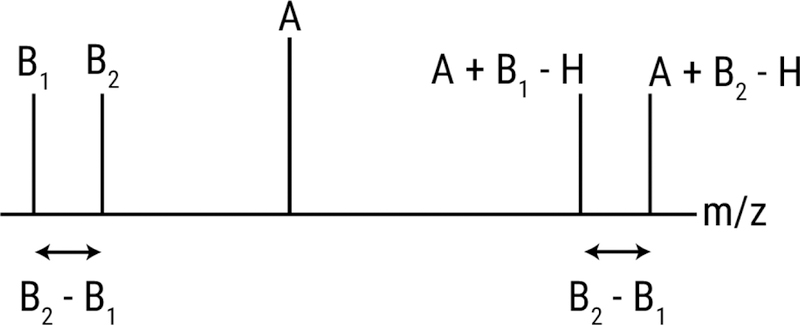
Schematic showing how background ions give rise to frequent intrinsic relationships. Analyte A is detected as an adduct of each background ion (B1 and B2). The spacing between the adducts (A+B1-H and A+B2-H) is equal to the spacing between the background ions.
We also performed formula decomposition on the frequent intrinsic relationships to further elucidate their origins. Interestingly, chemical formula CH2, C2H4, and C3H6 were found in the frequent intrinsic relationships exhibited by the chemical background. Additional analysis of the background ions indicated that they were an alkyl amine series. These alkyl amine species are known to form strong adducts and are commonly found as contaminants in alcohol solvents.39 We note that our laboratory does not perform ion-pairing experiments and the source of these reagents was solvent impurity as indicated by the series rather than sole presence of triethylamine. In developing our methods, we attempted to find solvents with the lowest possible levels of chemical background (Burdick & Jackson brand purchased from Honeywell). Unfortunately, alkyl amines seem to be ubiquitous in methanol and isopropanol LC/MS solvents.
Removing artifacts and contaminants by credentialing
The degenerate relationships that we annotated above led to a striking reduction in the number of feature groups, indicating that fewer than 15% of the total 25,230 features that were detected in E. coli correspond to unique analytes. Even after this extensive annotation process, however, two sources of feature inflation remained in artifacts and contaminants. We applied an alternative experimental approach called credentialing to filter these features associated with artifacts and contaminants. The credentialing process introduces an isotopic signature into biological analytes during E. coli growth.27 Features in our data set displaying this isotopic signature are deemed “credentialed”, as they are known to be of E. coli origin. In contrast, features that do not display this isotopic signature are annotated as artifacts or contaminants. Credentialing does not rely on any of the relationship annotation approaches that we described above, and is thus an orthogonal and highly complementary approach to data analysis.
We first filtered non-credentialed features from the raw data set on the basis of isotopic signatures. The resulting set of features is free of artifacts, noise, and contaminants. This process returned 2,462 high-quality, credentialed features. We then took these credentialed features through the same annotation process as the full data set to remove degeneracy. Annotation of degeneracy reduced the estimated number of unique E. coli analytes being measured to 892 (Figure 2C). Thus, out of the 25,230 total features we detected, only ~3% passed our filters for being a unique and bona fide metabolite.
creDBle: a database for thoroughly annotated reference data sets
An alternative approach to each investigator having to identify the relatively small number of features corresponding to unique, bona fide metabolites from every experiment is to create thoroughly annotated reference data sets. Reference data sets have been shown to be effective in other profiling sciences, such as genomics (for example, during the EST collection era of gene identification in the 1990’s).40,41 The idea is for one laboratory to first identify all of the unique metabolites that can be detected from a given sample with a given experimental methodology. Then, other laboratories performing the same experiment benefit by having to target only these reference analytes in their subsequent experiments. Of course, the major challenge of this strategy is that there are a multitude of experimental methods currently being used in untargeted metabolomics, each of which will have to be annotated for different sample types.31
There may also be other benefits to having a repository of thoroughly annotated data sets. Knowing the comprehensive list of unique metabolites that can be detected with specific experimental protocols, for example, will be invaluable to designing LC/MS-based metabolomic experiments. Although the number of detected features is often used as an indicator of experimental coverage, our work suggests that this is an unreliable metric.42,43 Instead, it would be preferred if researchers based their experimental design on the numbers of unique metabolites known to be detected. Additionally, even if the sample of interest has not been annotated, researchers might be able to use annotated data from other sample types (e.g., E. coli) as a touchstone to evaluate data from their experiments and to compare it to others.
As a first step in establishing a repository for thoroughly annotated reference data sets, we have created the creDBle database. All credentialed features for the reference E. coli data set described here have been deposited in creDBle. Degeneracy annotations as well as accurate mass, retention times, and fragmentation patterns are included. creDBle is freely available on the Web at http://creDBle.wustl.edu/ and provides a convenient companion resource for credentialed E. coli standards (Figure S5). All data within creDBle (including fragmentation patterns for identified metabolites) can be freely downloaded.
The addition of more analyses to creDBle will greatly expand its applicability. Our first goal is to repeat the annotation processes above for credentialed E. coli samples analyzed with different methods (e.g., different extraction protocols, chromatography, mass spectrometers, etc.). Notably, identification of metabolites from these annotated experiments will provide a readily available set of complex standards. As the number of credentialed E. coli experiments within creDBle increases, we hope that it will eventually provide a common reference point with enough observations in each experiment to model and normalize some of the variation that has historically prevented cross-laboratory data comparisons. This, in turn, will make data sets present in repositories, when run with a credentialed standard extract, more amenable to reprocessing and meta-analysis.
CONCLUSION
Detecting tens of thousands of LC/MS features from biological samples is typical in untargeted metabolomics, however, to date it has been unclear how many unique metabolites are actually being profiled. Our work here evaluated one representative untargeted metabolomic data set from E. coli to set an upper bound on the number of unique metabolites being measured. By using a new context-driven approach to identify degenerate features arising from the same metabolite, we determined that the ~25,000 features detected in our experiment corresponded to fewer than 2,961 unique analytes. An orthogonal and complementary approach using credentialing isotope signatures to identify artifacts and contaminants similarly reduced the number of unique analytes detected. Out of the total ~25,000 features detected, only 892 passed both our degeneracy and credentialing filters. This major reduction in data is more than five fold greater than any previously reported in studies examining only a subset of feature inflation and suggests that curating reference data sets in a resource such as creDBle might enable more efficient approaches to performing untargeted metabolomics. Accurate masses, retention times, fragmentation patterns, and degeneracy annotations for these 892 features have been deposited in the creDBle database.
We wish to emphasize that our work is only indirectly related to the size of the E. coli metabolome and should not be interpreted as an indication of the total number of intracellular metabolites present. There are certainly more than 892 E. coli metabolites.44 The purpose of our work is only to assess how many unique metabolites are being measured in a representative untargeted metabolomic experiment. Additionally, we note that our context-driven analysis of degeneracy is not exhaustive. Relationships that are uncommon and not indicated by background ions remain unannotated and may further reduce the number of unique analytes detected. Notwithstanding, our results suggest that there are an order of magnitude more features than unique metabolites in untargeted metabolomic experiments. This has important implications for designing untargeted metabolomics experiments and influences strategies for interpreting the data produced before establishing metabolite identifications.
Supplementary Material
Acknowledgements
This work was supported by funding from NIH Grants R01 ES022181 (G.J.P.) and R21 CA191097 (G.J.P.) as well as grants from the the Camille & Henry Dreyfus Foundation (G.J.P.) and the Pew Scholars Program in the Biomedical Sciences (G.J.P.).
Footnotes
Notes
G.J.P. is a scientific advisory board member for Cambridge Isotope Laboratories. No other authors have competing financial interests.
Supporting Information Available
Additional material as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Broeckling CD; Afsar FA; Neumann S; Ben-Hur A; Prenni JE Anal. Chem. 2014, 86 (14), 6812–6817. [DOI] [PubMed] [Google Scholar]
- (2).Uppal K; Walker DI; Liu K; Li S; Go Y-M; Jones DP Chem. Res. Toxicol. 2016, 29 (12), 1956–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Xia J; Sinelnikov IV; Han B; Wishart DS Nucleic Acids Res. 2015, 43, W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dunn WB; Erban A; Weber RJM; Creek DJ; Brown M; Breitling R; Hankemeier T; Goodacre R; Neumann S; Kopka J; Viant MR Metabolomics 2013, 9, 44–66. [Google Scholar]
- (5).Benton HP; Ivanisevic J; Mahieu NG; Kurczy ME; Johnson CH; Franco L; Rinehart D; Valentine E; Gowda H; Ubhi BK; others; Tautenhahn R.; Gieschen A.; Fields MW; Patti GJ.; Siuzdak G. Anal. Chem. 2014, 87 (2), 884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stanstrup J; Gerlich M; Dragsted LO; Neumann S Anal. Bioanal. Chem. 2013, 405 (15), 5037–5048. [DOI] [PubMed] [Google Scholar]
- (7).Tautenhahn R; Cho K; Uritboonthai W; Zhu Z; Patti GJ; Siuzdak G Nat. Biotechnol. 2012, 30 (9), 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mahieu NG; Genenbacher JL; Patti GJ Curr. Opin. Chem. Biol. 2016, 30, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS; Sumner S; Subramaniam S Nucleic Acids Res. 2016, 44, D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cho K; Mahieu NG; Johnson SL; Patti GJ Curr. Opin. Biotechnol. 2014, 28, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Patti GJ; Yanes O; Siuzdak G Nat. Rev. Mol. Cell Biol. 2012, 13 (4), 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nikolskiy I; Mahieu NG; Chen Y-J; Tautenhahn R; Patti GJ Anal. Chem. 2013,(16), 7713–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee TS; Ho YS; Yeo HC; Lin JPY; Lee D-Y Metabolomics 2013, 9 (6), 1301–1310. [Google Scholar]
- (14).Kuhl C; Tautenhahn R; Böttcher C; Larson TR; Neumann S Anal. Chem. 2012, 84 (1), 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Scheltema R; Decuypere S; Dujardin J; Watson D; Jansen R; Breitling R Bioanalysis 2009, 1 (9), 1551–1557. [DOI] [PubMed] [Google Scholar]
- (16).Brown M; Dunn WB; Dobson P; Patel Y; Winder CL; Francis-McIntyre S; Begley P; Carroll K; Broadhurst D; Tseng A; Swainston N; Spasic I; Goodacre R; Kell DB Analyst 2009, 134 (7), 1322–1332. [DOI] [PubMed] [Google Scholar]
- (17).Chokkathukalam A; Jankevics A; Creek DJ; Achcar F; Barrett MP; Breitling R Bioinformatics 2013, 29 (2), 281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhou R; Tseng C-L; Huan T; Li L Anal. Chem. 2014, 86 (10), 4675–4679. [DOI] [PubMed] [Google Scholar]
- (19).Stupp GS; Clendinen CS; Ajredini R; Szewc MA; Garrett T; Menger RF; Yost RA; Beecher C; Edison AS Anal. Chem. 2013, 85 (24), 11858–11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).de Jong FA; Beecher C Bioanalysis 2012, 4 (18), 2303–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bueschl C; Kluger B; Lemmens M; Adam G; Wiesenberger G; Maschietto V; Marocco A; Strauss J; Bödi S; Thallinger GG; Krska R; Schuhmacher R Metabolomics 2014, 10 (4), 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mahieu NG; Spalding JL; Gelman SJ; Patti GJ Anal. Chem. 2016, 88 (18), 9037–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tong H; Bell D; Tabei K; Siegel MM J. Am. Soc. Mass Spectrom. 1999, 10 (11), 1174–1187. [Google Scholar]
- (24).Zhu J; Cole RB J. Am. Soc. Mass Spectrom. 2000, 11 (11), 932–941. [DOI] [PubMed] [Google Scholar]
- (25).Brown M; Wedge DC; Goodacre R; Kell DB; Baker PN; Kenny LC; Mamas MA; Neyses L; Dunn WB Bioinformatics 2011, 27 (8), 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mahieu NG; Spalding JL; Patti GJ Bioinformatics 2016, 32 (2), 268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mahieu NG; Huang X; Chen Y-J; Patti GJ Anal. Chem. 2014, 86 (19), 9583–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cajka T; Fiehn O Anal. Chem. 2016, 88 (1), 524–545. [DOI] [PubMed] [Google Scholar]
- (29).Contrepois K; Jiang L; Snyder M Mol. Cell. Proteomics 2015, 14 (6), 1684–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ivanisevic J; Zhu Z-J; Plate L; Tautenhahn R; Chen S; O’Brien PJ; Johnson CH; Marietta MA; Patti GJ; Siuzdak G Anal. Chem. 2013, 85 (14), 6876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Vinayavekhin N; Saghatelian A In Current Protocols in Molecular Biology; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2010; Vol. Chapter 30, p Unit 30.1.1–24. [DOI] [PubMed] [Google Scholar]
- (32).Wishart DS Nat. Rev. Drug Discov. 2016, 15 (7), 473–484. [DOI] [PubMed] [Google Scholar]
- (33).Melamud E; Vastag L; Rabinowitz JD Anal. Chem. 2010, 82 (23), 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Milne SB; Mathews TP; Myers DS; Ivanova PT; Brown HA Biochemistry 2013, 52 (22), 3829–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Daly R; Rogers S; Wandy J; Jankevics A; Burgess KEV; Breitling R Bioinformatics 2014, 30 (19), 2764–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kessler N; Walter F; Persicke M; Albaum SP; Kalinowski J; Goesmann A; Niehaus K; Nattkemper TW PLoS One 2014, 9 (11), e113909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zeng Z; Liu X; Dai W; Yin P; Zhou L; Huang Q; Lin X; Xu G Anal. Chem. 2014, 86 (8), 3793–3800. [DOI] [PubMed] [Google Scholar]
- (38).Han X; Yang K; Gross RW Mass Spectrom. Rev. 2012, 31 (1), 134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Keller BO; Sui J; Young AB; Whittal RM Anal. Chim. Acta 2008, 627 (1), 71–81. [DOI] [PubMed] [Google Scholar]
- (40).Barbazuk WB; Korf I; Kadavi C; Heyen J; Tate S; Wun E; Bedell JA; McPherson JD; Johnson SL Genome Res. 2000, 10 (9), 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hillier LD; Lennon G; Becker M; Bonaldo MF; Chiapelli B; Chissoe S; Dietrich N; DuBuque T; Favello A; Gish W; Hawkins M; Hultman M; Kucaba T; Lacy M; Le M; Le N; Mardis E; Moore B; Morris M; Parsons J; Prange C; Rifkin L; Rohlfing T; Schellenberg K; Bento Soares M; Tan F; Thierry-Meg J; Trevaskis E; Underwood K; Wohldman P; Waterston R; Wilson R; Marra M Genome Res. 1996, 6 (9), 807–828. [DOI] [PubMed] [Google Scholar]
- (42).Masson P; Alves AC; Ebbels TMD; Nicholson JK; Want EJ Anal. Chem. 2010, 82 (18), 7779–7786. [DOI] [PubMed] [Google Scholar]
- (43).Yanes O; Tautenhahn R; Patti GJ; Siuzdak G Anal. Chem. 2011, 83 (6), 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sajed T; Marcu A; Ramirez M; Pon A; Guo AC; Knox C; Wilson M; Grant JR; Djoumbou Y; Wishart DS Nucleic Acids Res. 2016, 44, D495–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.