

Abstract
We report that alkyl-subsituted bisphosphonates have activity against Bacillus anthracis Sterne (0.40 µg/mL), Mycobacterium smegmatis (1.4 µg/mL), Bacillus subtilis (1.0 µg/mL) and Staphylococcus aureus (13 µg/mL). In many cases, there is no effect of serum binding, as well as low activity against a human embryonic kidney cell line. Targeting of isoprenoid biosynthesis is involved with74 having IC50 values of ~100 nM against heptaprenyl diphosphate synthase and 200 nM against a farnesyl diphosphate synthase. Bacillus subtilis growth inhibition was rescued by addition of farnesyl diphosphate, menaquinone-4 (MK-4) or undecaprenyl diphosphate (UP) and the combination of MK-4 plus UP resulted in a 25x increase in ED50, indicating targeting of both quinone and cell wall biosynthesis. Clostridioides difficile was inhibited by 74 and since this organism does not synthesize quinones, cell wall biosynthesis is the likely target. We also solved three X-ray structures of inhibitors bound to octaprenyl diphosphate and/or undecaprenyl diphosphate synthases.
Graphical Abstract
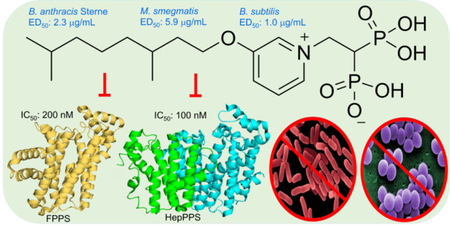
Anti-bacterial Bisphosphonates Inhibit Multiple Prenyltransferases
Introduction
There is a need for new antibiotics because of the rise in drug resistance and in our group we have been investigating enzymes involved in isoprenoid biosynthesis as potential anti-infective drug targets.1–2 These enzymes include farnesyl diphosphate synthase (FPPS) and undecaprenyl diphosphate synthase (UPPS), both of which are involved in bacterial cell wall biosynthesis, and we discovered several inhibitors,2–3 one of which was active in vivo in a mouse model of Staphylococcus aureus infection.2 We also recently discovered4 several inhibitors of S. aureus heptaprenyl diphosphate synthase (SaHepPPS), essential for the formation of menaquinone, required for electron transport (ET) and hence, ATP synthesis, in many bacteria and as with UPPS, this enzyme is not produced by humans. Here, we sought to find new compounds that might target octaprenyl diphosphate synthase (EcOPPS) or heptaprenyl diphosphate synthase (SaHepPPs), inhibiting quinone biosynthesis. A simplified version of the enzymes involved in quinone as well as cell wall biosynthesis in many bacteria is shown in Figure 1a, together with the sites of action of several antibiotics, and in Figure 1b we show the chemical structures of selected substrates/products in Staphylococcus aureus.
Figure 1.

Schematic illustration of some of the enzymes involved in cell wall and quinone biosynthesis in many bacteria, together with chemical structures of substrates and intermediates and sites of action of some inhibitors (red) and “rescue” agents (blue). (a) Enzymes, products, inhibitors and rescue agents. UP = undecaprenyl phosphate; MK-4 = menaquinone-4. (b) Chemical structures of selected enzyme substrates and products discussed in the Text. HepPPS (heptaprenyl diphosphate synthase) is a heterodimeric enzyme. Some bacteria such as E. coli use the homodimeric octaprenyl diphosphate synthase (OPPS), and also produce ubiquinones (not shown). DXP = the 1-deoxy-D-xylulose 5-phosphate pathway, found in most bacteria; MEV = the mevalonate pathway, found in e.g. S. aureus. In some bacteria, e.g. Listeria monocytogenes, both the DXP and MEV pathways are present.
Isoprenoid biosynthesis begins with the formation of two C5-isoprenoid diphosphates, isopentenyl diphosphate (1, IPP) and dimethylallyl diphosphate (2, DMAPP) which are produced by either the mevalonate (MEV) pathway, as in S. aureus, or by the so-called non-mevalonate or deoxyxylulose-5-phosphate (DXP) pathway, the latter being far more common than the MEV pathway. These two isoprenoids react to form (C10) geranyl diphosphate and then, via addition of a second IPP, farnesyl diphosphate (FPP, 3), both reactions being catalyzed by the enzyme farnesyl diphosphate synthase (FPPS). FPP is then elongated in further reactions with IPP to produce long chain (typically all-trans C30, C35, C40) diphosphates (e.g. 4) that then react with e.g. 1,4-dihydroxy-2-naphthoic acid (DHNA) to form quinone precursors, Figure 1b. In some bacteria (e.g. E. coli), both menaquinones as well as ubiquinones are formed. FPP is also converted to very long-chain cis-isoprenoids such as undecaprenyl diphosphate (UPP, 5), in reactions catalyzed by undecaprenyl diphosphate synthase (UPPS), with UPP then being converted to undecaprenyl (mono)phosphate (UP) by undecaprenyl diphosphate phosphatase (UPPP), essential for cell wall peptidoglycan biosynthesis.
OPPS is an essential gene in E. coli5 and catalyzes the formation of the C40 isoprenoid octaprenyl diphosphate (OPP) from farnesyl diphosphate (FPP) and five molecules of isopentenyl diphosphate (IPP). Its structure is known6 and is highly α-helical, resembling that of other trans-prenyltransferases, such as FPPS.7–8 Since OPPS is absent in humans, this makes it a potential anti-bacterial drug target. There are, however, no reports of bacterial OPPS inhibitors. In S. aureus, the corresponding long-chain prenyl transferase is the C35 heptaprenyl diphosphate synthase, SaHepPPS. This has a somewhat unusual heterodimeric structure4 consisting of a large, “catalytic” domain (SaHepPPS-2) and a smaller, regulatory domain (SaHepPPS-1), and both are essential for activity. Similar heterodimeric structures are found in other bacteria—such as Micrococcus luteus,9 Bacillus spp. and Enterococcus spp. and the structures of the catalytic sites in OPPS, HepPPS as well as FPPS are quite similar. In this work, we first sought to find inhibitors of OPPS and HepPPS, active in cells. Then, we extended this work to better understand inhibitor mechanisms of action, in addition to solving several structures of interest.
In order to find new, long-chain prenyl transferase inhibitors we first screened a library of previously-reported compounds including bisphosphonates, benzoic, salicylic, anthranilic and diketoacids, for OPPS inhibition, since these classes of compound were previously shown to inhibit prenyltransferases2, and some have anti-bacterial activity. We then screened a subset of compounds for bacterial cell growth inhibition (against S. aureus, B. subtilis, B. anthracis Sterne, Mycobacterium smegmatis, E. coli, Acinetobacter baumannii, Klebsiella pneumoniae, Pseudomonas aeruginosa) as well as the fungus, Candida albicans, and used the results obtained to guide the synthesis of 24 new compounds. We then investigated the mechanism of action of some of the most potent compounds in cells using enzyme inhibition assays, as well as bacterial cell growth inhibition “rescues” by putative enzyme end-products, followed by measurements of the effects of serum binding on antibacterial activity, and toxicity to a mammalian cell line. We also determined activity against Clostridioides difficile, a bacterium that does not use quinones (or hemes) in ATP biosynthesis, again to help clarify mechanisms of action. Finally, we solved several X-ray crystal structures, to determine inhibitor binding modes. Overall, the results are of general interest since we have discovered several potent multi-target antibacterials with low serum binding and low toxicity against a human cell line that act by inhibiting both quinone as well as cell wall biosynthesis.
Results and Discussion
Initial Screening Results.
We first investigated the inhibition of E. coli octaprenyl diphosphate synthase (EcOPPS) since in previous work we found that this protein expressed well and was more stable than the corresponding enzyme from S. aureus, SaHepPPS, and it has a better resolved crystal structure. We screened a diverse range of potential inhibitors (6-67): bisphosphonates, phosphonates, benzoic/anthranilic acids, and amines/diamines/diamidines, classes of compounds that in earlier work1–4, 10 we found inhibited prenyltransferases such as FPPS,3 geranylgeranyl diphosphate synthase (GGPPS),10 HepPPS,4 UPPS,2 MenA (1,4-dihydroxy-2-naphthoate polyprenyltransferase4) and dehydrosqualene synthase.1 About one third of the compounds (Supporting Information, Figure S1), had essentially no activity (IC50 >1 mM) against EcOPPS, but there were several compounds having low nM activity. All of the most potent hits were bisphosphonates, the most active species (6, Figure 2) having an IC50 value of ~6 nM. The lipophilic benzoic/anthranilic/salicylic acids were less active, the most potent species having an IC50 ~5 µM, Figure S1.
Figure 2.

Structures of some compounds that inhibit isoprenoid biosynthesis enzymes such as FPPS, GGPPS, UPPS, and UPPP that are discussed in the Text.
We then tested 6, together with 5 other compounds (33, 68–71; Figure 2 and Table S1), against S. aureus, B. subtilis, B. anthracis Sterne, M. smegmatis, E. coli, A. baumannii, K. pneumoniae, P. aeruginosa as well as C. albicans, Table S1. Three compounds were lipophilic bisphosphonates (33, 68, 69), and two were benzoic acids (70, 71), originally designed11 as phosphate/diphosphate isosteres and known to inhibit other isoprenoid biosynthesis enzymes including the cis-prenyltransferase UPPS,11–12 the trans- prenyltransferase GGPPS,12 and prenyl diphosphate phosphatase, UPPP.11 Plus, e.g. 71 has potent anti-bacterial activity.11 There was little-to-no activity (ED50 > 50 µg/mL) against the gram-negative bacteria or C. albicans for all compounds, but there was activity in the ~3–8 µg/mL range for some compounds against S. aureus, M. smegmatis, B. subtilis or B. anthracis Sterne, Table S1. The lack of activity against the gram-negative bacteria was unexpected because in previous work13 we found that lipophilic bisphosphonates such as 72 (Figure 2) had quite potent (~2 µg/mL) activity against the same gram-negative bacteria tested here, but had low activity against the gram-positive bacteria S. aureus (30 µg/mL) and B. subtilis (> 100 µg/mL). However, with 6, we see the opposite trend, and one possibility is that the presence of an aromatic group “distal” to the bisphosphonate backbone (seen also with 73, Figure 2)14 is required for transport into gram-negative bacteria.
What is also interesting about the bisphosphonate results is that the patterns of OPPS inhibition are similar to those we find with the shorter (C15) prenyl synthase FPPS, as opposed to the longer (C20) chain synthase, geranylgeranyl diphosphate synthase (GGPPS). For example, 24 (zoledronate, Figure 2) is a <1 µM inhibitor of OPPS as well as of human FPPS,15–16 but only a very weak (IC50 ~100 µM) inhibitor of human GGPPS.12 For potent FPPS inhibition, we proposed previously17–19 that there was a requirement for either a cationic or protonatable group close to the bisphosphonate backbone for activity, mimicking a reactive intermediate in FPPS catalysis. There was, however, no requirement for such a cationic feature for GGPPS inhibition.12 For example,20 6, containing a cationic charge center, has an IC50 = 100 nM for FPPS inhibition and an IC50 = 280 nM for GGPPS inhibition, while 36 (Figure 2), which lacks this feature, has an IC50 = 550 µM for FPPS inhibition but an IC50 = 590 nM for GGPPS inhibition.20 Here, we find that 36 has an IC50 = 11 µM against OPPS, while as noted above, 6 is far more potent, as discussed more below.
Synthesis and testing of novel bisphosphonates.
Based on the results described above, we reasoned that it would be of interest to synthesize a series of analogs of 6 in which we varied: 1) the nature of the aliphatic side-chain connected to the aryl group; 2) the position of the aliphatic side-chain on the ring; 3) the nature of the side-chain linker (O or CH2) between the aliphatic and aryl group and 4), the nature of the linker (NH, NHCH2, CH2) to the bisphosphonate backbone with in most cases, a protonatable/cationic feature being included. We thus synthesized the 24 compounds (74-97) shown in Figure 3 and tested each of them for B. subtilis, B. anthracis Sterne, S. aureus and M. smegmatis growth inhibition. ED50 values (in µg/mL) and computed clogP and logD7.4 values are shown in Table 1, and for convenience are shown together with their corresponding chemical structures in Table S2. There are several points of interest. First, we see that there is activity (in the ~0.6–2 µg/mL range) against B. anthracis Sterne, M. smegmatis and B. subtilis, though less so with S. aureus (~13 µg/mL). Second, with B. subtilis (where there is a large range in activity) we see (Figures S2) that there is a parabolic dependence between cell growth inhibition (log10 ED50, µg/mL) values and clogP with a Pearson correlation coefficient r = 0.81. The most active compounds have clogP ~−2 while some of the least active compounds are either much more polar (91, 93) and will have very poor cell penetration, or are far more hydrophobic (97, with a C20 side-chain) and may get “trapped” in cell membranes. What is clear, however, is that—as can be seen in Table S2—all of the most active compounds have fixed (+1) charges on the aromatic group. That is, they are either pyridiniums or imidazoliums. This is of interest since in previous work on bisphosphonate inhibition of FPPS19 we found, using solid-state NMR, that bisphosphonates with basic side-chains (such as aminopyridines) bound to FPPS with their side-chains protonated, suggesting the possibility that both FPPS as well as a very long chain trans-prenyl diphosphate transferases (such as HepPPS) might be being targeted, which would lead to inhibition of both quinone as well as cell wall biosynthesis. We thus next sought to determine which enzymes might actually be being targeted. To do this we used an expressed SaHepPPS (a hetero-dimer found in Bacilli) and for comparison, EcOPPS (a homo-dimer). Typical dose-response results for SaHepPPS are shown in Figure 4. What is clear from Figure 4 is that two of the most active compounds are 6 and 74, compounds that also have potent activity against B. anthracis Sterne, B. subtilis as well as M. smegmatis. A key feature for potent activity against HepPPS as well as these bacteria is the presence of a long-chain m-substituted pyridinium group. The amino-pyridine bisphosphonates as well as the aryl-alkyl imidazolium bisphosphonates have lower activity against HepPPS, consistent with the cell growth inhibition assay results. We also found very potent activity of some compounds against EcOPPS, typical results are shown in Figure S3. For example, 74 had a 12 nM IC50 and 6 a 20 nM IC50 value, Figure S3. These compounds thus inhibit both SaHepPPS and EcOPPS—but only have activity against the gram-positive bacteria.
Figure 3.

Structures of bisphosphonates synthesized. Compounds are rank-ordered by activity against B. subtilis from most active (74, top-left) to least active (97, bottom). Most of the active compounds contain meta-substituted pyridinium rings and a medium-size side-chain (cyan). Short or long chain substituents (red) are inactive.
Table 1.
Bacterial cell growth inhibition by bisphosphonates together with clogP and logD7.4 values.a
| Cpd# | Bs ED50 μg/mL (μM) |
Ba ED50 μg/mL (μM) |
Sa ED50 μg/mL (μM) |
Ms ED50 μg/mL (μM) |
HEK 293 μg/mL (μM) |
clogP | logD7.4 |
|---|---|---|---|---|---|---|---|
| 74 | 1.0 (2.4) | 2.3 (5.4) | 14 (33) | 5.9 (14) | 262 (620) | −2.25 | −5.82 |
| 75 | 1.7 (4.1) | 1.8 (4.4) | 26 (63) | 1.4 (3.4) | 181 (440) | −2.08 | −5.65 |
| 76 | 2.1 (4.8) | 0.6 (1.4) | 13 (30) | 6.1 (14) | 96 (220) | −0.50 | −4.07 |
| 77 | 2.2 (5.2) | 2.1 (5.0) | 13 (31) | 5.9 (14) | 194 (460) | −1.14 | −4.71 |
| 78 | 2.4 (5.7) | 0.8 (1.9) | 13 (31) | 5.5 (13) | 186 (440) | −2.50 | −6.07 |
| 79 | 2.8 (6.4) | 0.4 (0.9) | 18 (41) | 6.2 (14) | 224 (510) | −1.52 | −5.09 |
| 80 | 3.1 (7.4) | 2.2 (5.2) | 21 (50) | 7.2 (17) | 295 (700) | −1.14 | −4.71 |
| 81 | 3.6 (8.2) | 2.4 (5.5) | 51 (116) | 10 (23) | 480 (1100) | −2.46 | −6.25 |
| 82 | 4.6 (12) | 4.3 (11) | 34 (86) | 15 (38) | 170 (420) | −2.50 | −6.07 |
| 83 | 5.5 (12) | 2.2 (4.9) | 30 (67) | 7.2 (16) | 22 (48) | −0.08 | −3.65 |
| 84 | 5.7 (14) | 2.5 (5.9) | 28 (66) | 150 (350) | 420 (1000) | −2.25 | −5.82 |
| 85 | 7.0 (16) | 1.1 (2.5) | 31 (71) | 70 (160) | 440 (1000) | −1.52 | −5.08 |
| 86 | 8.3 (19) | 5.0 (12) | 23 (54) | 150 (350) | ND | −0.1 | −2.36 |
| 87 | 21 (48) | 3.2 (7.3) | 35 (80) | 75 (170) | 530 (1200) | −0.51 | −3.10 |
| 88 | 21 (49) | 7.4 (17) | 36 (84) | 150 (350) | ND | 0.48 | −2.12 |
| 89 | 26 (60) | 3.0 (6.9) | 32 (73) | 93 (210) | 480 (1100) | −0.24 | −2.00 |
| 90 | 36 (73) | 5.0 (10) | 22 (45) | 91 (180) | ND | −0.27 | −3.48 |
| 91 | 41 (120) | 8.6 (24) | 17 (48) | 65 (180) | ND | −4.24 | −7.81 |
| 92 | 75 (170) | 4.2 (9.6) | 60 (140) | 200 (460) | ND | −0.52 | −2.24 |
| 93 | 97 (340) | 2.3 (8.0) | 230 (800) | 290 (1000) | ND | −3.73 | −6.35 |
| 94 | 100 (250) | 11 (28) | 56 (140) | 96 (240) | ND | −0.36 | −2.96 |
| 95 | 110 (230) | 2.5 (5.2) | 69 (140) | 79 (160) | 630 (1300) | 1.20 | −1.63 |
| 96 | 180 (420) | 11 (26) | 52 (120) | 100 (240) | ND | 0.77 | −2.31 |
| 97 | 260 (460) | 7.7 (14) | 25 (44) | 210 (370) | ND | 1.72 | −1.85 |
Abbreviations used: Bs=B. subtilis; Ba=B. anthracis Sterne; Sa=S. aureus; Ms = Mycobacterium smegmatis; HEK293 = human embryonic kidney cell line # 293; clogP = the computed logarithm of the octanol/water partition coefficient; logD7.4 = the logarithm of the computed octanol/water partition coefficient at pH = 7.4.
Figure 4.
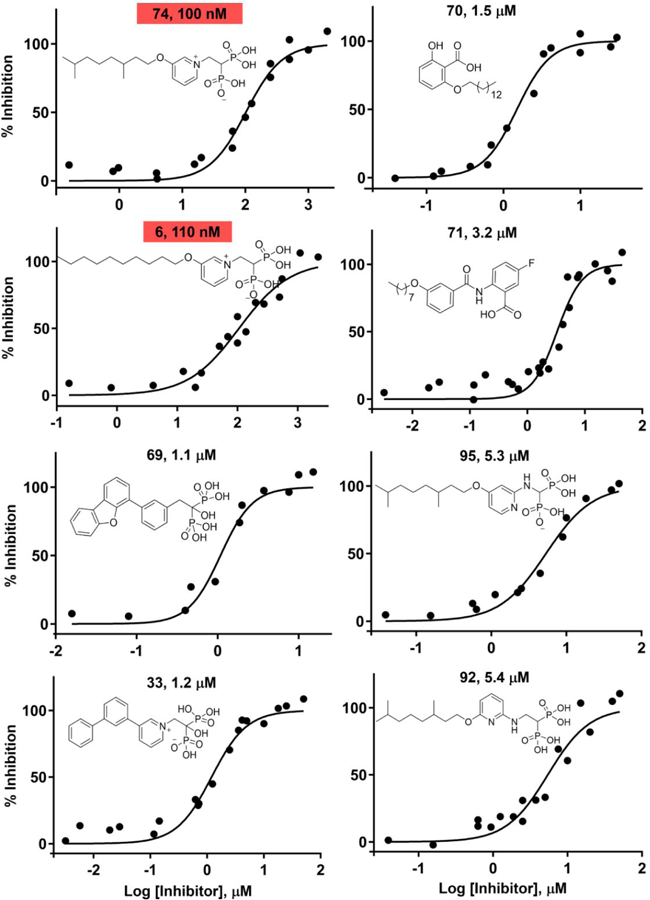
Typical dose-response curves for SaHepPPS inhibition. The most potent inhibitors also have potent activity (~10–20 nM) against EcOPPS (Figure S3), but were not active E. coli or other gram-negative bacteria. Results shown represent three pooled data sets taken on different days, fit to single dose-response curves.
The most active HepPPS, OPPS as well as cell growth inhibitors have very similar overall lengths, corresponding to ~ 13±1 contiguous “heavy” atoms appended to the bisphosphonate C1 backbone carbon. This length corresponds closely to the length of the FPP substrate of HepPPS (and OPPS), as illustrated in Figure 5, suggesting that the most potent inhibitors target a long-chain prenyltransferase, in cells. However, it also seemed possible that FPPS could be inhibited by 74. This might seem a surprising suggestion since FPP is the product of FPPS and not a substrate, and would presumably be too big to bind to the GPP substrate-binding site. However, in recent work, Park et al.21 have shown that FPP binds to the allosteric site in FPPS discovered by Jahnke et al.22, suggesting that (long-chain) FPP isosteres could also bind to this site and inhibit FPPS. We were unable to obtain active SaFPPS (from either of two commercial entities) to test this hypothesis, so we next tested for human FPPS inhibition by 74, finding an IC50=230 nM, which is remarkably similar to that found against HepPPS (100 nM), as well as the 100 nM for 6 against HsFPPS.20 This IC50 value for FPPS inhibition by 74 is also very similar to that we obtain for the potent FPPS inhibitor zoledronate (24, IC50 = 250 nM) in the same assay. In summary then, we find that 74 is a potent inhibitor of HepPPS as well as FPPS, suggesting that its activity in bacteria could be due to inhibition of both quinone as well as cell wall biosynthesis.
Figure 5.
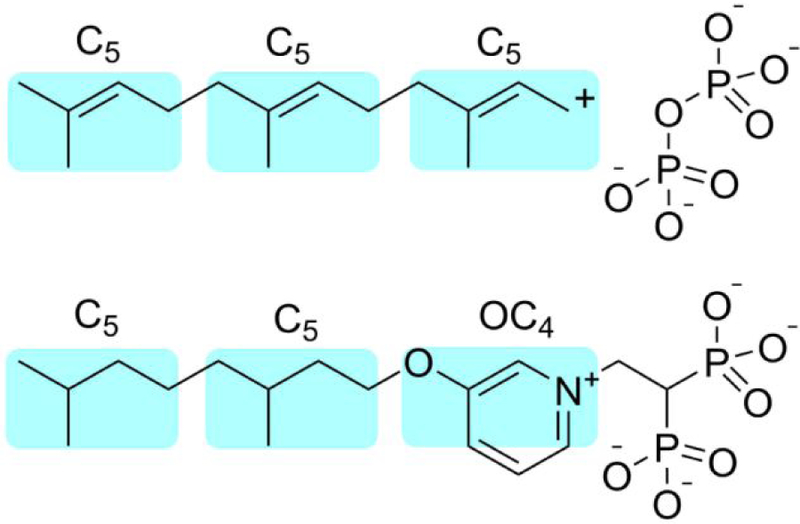
Cartoon illustration of the similarity between a putative FPP transition state/reactive intermediate (top) and a potent SaHepPPS inhibitor, 74 (bottom). Note that this mechanistic proposal would only apply to “long-chain” (~C30, C35, C40) trans prenyltransferases (which use FPP as a substrate) and not to short-chain prenyl transferases, such as FPPS, since FPP is the product and presumably would have only weak binding to FPPS. However, FPP is also known (in human FPPS) to bind to the allosteric (i.e. non-catalytic) FPPS site and acts as an FPPS inhibitor, and it is possible that FPP-analogs may also bind in this way.
Mechanisms of Action: Quinones, Cell Walls, and Clostridioides difficile.
Based on the results discussed above, we hypothesized that 74 (chosen over e.g. 75, since 74 exhibited low human cell toxicity) targets both HepPPS as well as FPPS. We tested this hypothesis in five ways. First, if HepPPS inhibition (or that of the corresponding enzymes in Bacilli spp.) is important, then it should be possible to effect a rescue (or partial rescue, if multiple targets are involved) by growing cells in the presence of a menaquinone (MK-4), an approach that we and others have used previously.23–24 Second, if FPPS inhibition is important, it should be possible to effect a growth inhibition rescue by growing cells in the presence of FPP or undecaprenyl phosphate (UP). This is because FPPS inhibition is expected to block both quinone as well as cell wall biosynthesis (Figure 1), so UP should partially rescue cell growth inhibition, as seen for example with the inhibition of UPPS by clomiphene.25 An FPP rescue should also be quite potent though more difficult to assess given its much less hydrophobic nature and hence, decreased ability to enter cells. Third, we hypothesized that a combination MK-4 plus UP rescue might be particularly effective. Fourth, we hypothesized that if FPPS inhibition is important, it should be possible to inhibit the growth of bacteria that lack quinones (and HepPPS) because cell wall biosynthesis would still be targeted. Here, pathogens such as C. difficile are ideal for testing this hypothesis since they lack all quinone biosynthesis machinery. Moreover, they also lack heme biosynthesis enzymes (which involve heme prenylation), another potential but heretofore unexplored area of prenylsynthase inhibition. Fifth, we hypothesized that an “upstream” target in the DXP pathway might be involved, so we used the IPP/DMAPP precursor 1-hydroxy-2-methyl-but-2-enyl 4-diphosphate (HMBPP) to test this hypothesis. While this would not rule out inhibition of IspH, we previously reported that IspH is not inhibited by bisphosphonates.26 Overall, there are, therefore, 10 sets of “rescue experiments” to perform: 1) HMBPP; 2) FPP; 3) UP; 4) MK-4; 5) HMBPP + FPP; 6) HMBPP + UP; 7) HMBPP + MK-4; 8) FPP + UP; 9) FPP + MK-4, and 10) UP + MK-4 and we carried each of these out using a 50 µM “rescue agent” concentration. This concentration is of course somewhat arbitrary but was chosen based on previous work with farnesol (200 μM) in gram-negative bacteria;13 MK-4 (at 10, 100 and 1000 μM) in M. tuberculosis24 and FPP (at ~ 30 μM) in Trypanosoma brucei rhodesiense.27 So 50 μM for all agents seemed reasonable and indeed, rescue effects (without toxicity) was generally observed. We also carried out additional experiments with decaprenyl phosphate (DP), but the results were the same as with UP (data not shown). We chose MK-4 (rather than MK-7) for most experiments since it is more soluble, but the results with MK-7 were the same (data not shown).
We first added menaquinone (MK-4) to B. subtilis cells and measured growth in the presence of different concentrations of 74 to determine if this quinone “rescued” cells from the growth-inhibitory effects of 74. Results are shown in Figure 6a,d and indicate that cell growth inhibition is indeed rescued by MK-4, the ED50 value for cell growth inhibition by 74 increasing by a factor of ~5. This quinone-rescue effect is consistent with 74 inhibiting quinone biosynthesis via HepPPS and/or FPPS. In a second experiment, we added undecaprenyl phosphate which is also expected to (partially) rescue cells from FPPS inhibition. There was, once again, a large (~7x) increase in the ED50 value for cell growth inhibition, consistent with FPPS as a second target. Also, addition of MK-4 plus UP resulted in a ~25x increase in IC50, Figure 6a. That is, 74 is a multi-target inhibitor that inhibits both HepPPS as well as FPPS. We also found that addition of FPP (at 50 µM) had, as expected, a significant effect on growth inhibition with a 3.5x increase in ED50, Figures 6 b-d. This increase is, perhaps, smaller than might be expected for a compound that should rescue both cell wall as well as quinone biosynthesis. However, both MK-4 and UP are for more hydrophobic than FPP and are likely to concentrate in the cell membrane which is where they actually function (in electron transfer or in peptidoglycan biosynthesis) while the more polar FPP needs to enter into cells as well as be processed (with HepPPS and UPPS), and its efficacy is evidently less than with MK-4 or UP. The combinations FPP+UP and FPP+MK-4 had larger effects (8.9x and 11x, respectively) than either agent alone, though again smaller than when using UP+MK-4 (~25x). It is also possible that there might be other targets for 74 such as UPPS or UPPP, or a target upstream of FPPS. However, we found no inhibition 0f UPPS, and UPPP inhibition was weak, ~66 μM. Upstream inhibition also seems less likely since there was only a ~2x increase in ED50 with HMBPP, Figures 6b-d. Interestingly, HMBPP potentiated the rescue effect of MK-4 (5.1x → 8.9x) and FPP (3.5x → 6.4x), probably due to a “law of mass action” effect (due to increased substrate concentration). However, with UP, the rescue effect was diminished (7.2x → 4.2x). Reasons are not known but might be related to decreased UP uptake into cells if transporters are involved.
Figure 6.
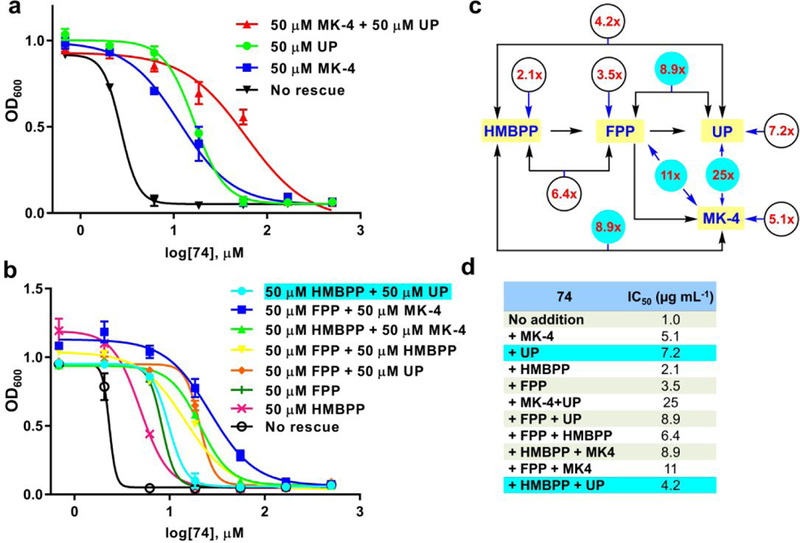
Effects of MK-4, UP, HMBPP and FPP as well as pairwise combinations on B. subtilis growth inhibition by 74. (a) Effects of MK-4, UP and MK-4 plus UP on growth inhibition by 74. (b) Effects of HMBPP, FPP with or without MK-4 or UP as well as HMBPP+FPP on 74 inhibition of B. subtilis cell growth. All rescue agents were at 50 µM. (c) Schematic illustration of the x-fold rescues by compounds or pairs of compounds on B. subtilis growth inhibition by 74. The largest effect (25x) is found with UP (50 µM) + MK-4 (50 µM). Cell growth inhibition assays were carried out in duplicate. The largest effects (~>9x) are seen with MK-4 or UP combinations and are colored cyan. d) Summary of IC50 values from data in a) and b).
Another target possibility for 74 is might be MenA, but this seems unlikely since in previous work4 on S. aureus inhibition by analogs of 75 we found a good correlation (r = 0.91) between SaHepPPS inhibition and cell growth inhibition, but only weak (~10 µM) MenA inhibition4. Nevertheless, to further test the hypothesis of FPPS+HepPPS targeting, it appeared to be of interest to investigate the extent of growth inhibition of a bacterium that does not utilize quinones in electron transfer/ATP biosynthesis, since then there would be no HepPPS (or MenA) to inhibit. One such organism is C. difficile which utilizes a complex of ferredoxins and flavoproteins to generate a Z∆ψ gradient that drives ATP synthesis via a Na+-ATP synthase.28–29 The organism lacks long-chain prenyl synthases such as HepPPS or OPPS, as well as downstream quinone biosynthesis enzymes like MenA or UbiA, and has just a single trans-prenyltransferase, FPPS, which is used in cell wall biosynthesis. Moreover, C. difficile lacks heme O synthase, the prenyltransferase28 that farnesylates heme B in many bacteria, which is then oxidized to heme A.30 We thus tested a series of known prenyl synthase inhibitors (33, 68–71, 74, 92, 95, 96, 98, 99) as well as two putative pro-drug forms (100, 101) against two strains of C. difficile (ATCC 43255 and ATCC 1870). Compound 100 is putative pro-drug form of 71, a potent anti-bacterial targeting UPPS and UPPP11, but these lipophilic benzoic acids bind to serum albumin and we reasoned that a prodrug form might have lower serum binding and perhaps, better cell penetration. Compound 101 is Piv-ester prodrug form of zoledronate (24), a very potent FPPS inhibitor, in which one phosphonate PO− group is masked and again we reasoned that such a compound might have better cell penetration than 24. Results are shown in Table S3. The compound with the most potent activity against both strains was 74 with an MIC of 6.8 µg/mL and since all the other potential targets discussed above are absent, this strongly indicates that FPPS is the major target for 74 in C. difficile. So, when all of the results described above are considered, we conclude that HepPPS and FPPS are major targets for lipophilic bisphosphonates such as 74 in the pathogens B. anthracis Sterne and S. aureus, and in B. subtilis, while only FPPS is targeted in C. difficile.
As to the bisphosphonate features that contribute to activity (or inactivity) in gram-positive and gram-negative bacteria: it appears that for activity against gram-negative bacteria, FPPS is a major target, as reflected in the ~100–200 nM activity of 72 against e.g. EcFPPS and P. aeruginosa FPPS13, and by increased EC50 values in cells that overexpress FPPS.13 Such compounds (e.g. 72, 73) have an aryl-alkyl-aryl-bisphosphonate motif, but poor activity against gram-positive bacteria.13 We suggest this is due at least in part to an uptake/efflux effect since based on structure/sequence similarities, there are no obvious differences between e.g. EcFPPS and SaFPPS X-ray structures. With the alkyl-aryl bisphosphonates such as 6 and 74, the opposite trend is seen. That is, these compounds inhibit gram-positive but not gram-negative bacteria, and there is good evidence for inhibition of HepPPS as well as FPPS. The primary difference between the two inhibitor types is, then, that the new compounds lack terminal aryl groups, which appear to be required for gram-negative activity.
Serum binding and human cell toxicity.
Next, we sought to determine to what extent the antibacterial effects of some of the more active bisphosphonates might be compromised by binding to serum proteins, as well as whether there might be potent human cell toxicity. We investigated five new compounds (74, 76, 77, 80 and 84), as well as one compound reported previously (72) that had activity in bacterial (gram-negative) cell growth.13 The effects of bovine serum on B. subtilis cell growth are shown in Table 2 and Figure S4, and effects on a human embryonic kidney cell line (HEK293) are shown in Table 2 and Figure S5. The six compounds investigated have four distinct structural motifs, illustrated for clarity (together with ED50 values) in Table 2. First, 74 and 77 have reduced geranyl (tetrahydrogeranyl) groups linked via O (74) or CH2 (77) to a meta-pyridinium bisphosphonate. Second, 84 and 80 have the same substituents but attached para to the bisphosphonate moiety. Third, 76 has an n-undecyl group (lacking any methyl substituents) located at the meta position. Fourth, 72 contains the quite different aryl-alkyl-aryl motif (Table 2) and is of interest since, as noted above, it is a potent inhibitor of gram-negative bacterial cell growth.
Table 2.
Effects of serum binding on B. subtilis growth inhibition, and toxicity to HEK293 cells.
 |
As can be seen in Table 2 and Figure S4, the addition of (4% or 10%) fetal bovine serum had little effect on bacterial cell growth with 74. Also, we see that toxicity in the HEK293 cell line is extremely low after a 24 hr incubation (620 µM) and is 76 µM at 96 hours, Figure S5. The results with the methylene analog (77) are similar (serum no effect; HEK293, 460 µM at 24 hrs, 140 µM at 96 hours). Results with the para analogs (84, 80) are different. With both compounds we find that there are quite large increases (4.9x, 34x) in ED50 at 10% serum and with both compounds there is almost no inhibition of HEK293 cell growth (IC50 values ~1 mM, Table 2). This could mean that 84, 80 simply bind tightly to serum in the B. subtilis assay, and to serum in the HEK293 cell growth inhibition assay, blocking bisphosphonate activity. Alternatively, it might be that 84/80 are simply weak inhibitors of human FPPS (the bisphosphonate target in human cells) since as noted previously, 74 is a good FPPS inhibitor (IC50 ~230 nM, in our assay—about the same strength as the commercial drug zoledronate, 24). We thus tested 84 against an expressed HsFPPS finding an IC50 = 100 µM. It thus appears that the lack of activity of 84 (and by analogy, 80) against human cells is not due to uptake/serum binding but rather, to the inefficiency of para-substituted bisphosphonates in inhibiting isoprenoid (FPP) biosynthesis.
With the meta n-alkyl substituted (n-undecyloxy) compound 76, there is good activity against B. subtilis, and serum binding has no effect on bacterial cell growth (Table 2). However, 76 is a very potent inhibitor of HEK293 cell growth (Table 2)—at least at 96 hours (the ED50 decreases from 220 µM at 24 hours to 7.3 µM at 96 hours, Table 2), making it of less interest as an antibacterial lead. We also investigated the aryl-alkyl-aryl species 72 which we previously found13 was a good inhibitor of gram-negative bacterial cell growth. In the B. subtilis assay there was only a small effect of 10% serum (a 50% increase in ED50), but we found strong toxicity (13 µM) against HEK293 cells, at least after a 96 hour incubation, Table 2. Thus, of all of the compounds investigated, 74 has, overall, the most potent activity as well as low serum binding and low toxicity.
In the future, it will be of interest to probe in more detail the molecular basis for the toxicity of compounds such as 76 since this might facilitate the design of better antibacterials. For example, is the decreased toxicity of 74 due to the presence of the two methyl groups? Are there other isoprenyl-like side-chain substituents that have better antibacterial, plasma binding and toxicity profiles? For example, are unsaturated side-chains, such as geranyl (versus saturated species) more promising? Interestingly, we find that while the tetrahydrogeranyl meta-substituted species 102 (Figure 2) is a good (1.9 µg/mL) B. subtilis growth inhibitor, the (unsaturated) geranyl analog 103 is not (ED50 ~9.2 µg/mL), indicating that side-chain flexibility is desirable for activity, facilitating perhaps, binding to multiple targets.
Crystallographic investigations.
There are currently no reported X-ray structures of very long-chain trans-prenyl diphosphate synthases such as SaHepPPS (or EcOPPS) with bound inhibitors. The only exception is, arguably, that of the S-thiolo analog of FPP (FSPP) bound to EcOPPS (PDB ID code 3WJN), since FSPP is frequently used as a non-reactive FPP substrate-analog. We thus sought to investigate how inhibitors might bind to HepPPS and OPPS, but we were unable to obtain structures with any the most potent inhibitors. We did, however, obtain structures of the bisphosphonate 69 (PDB ID code 5ZLF) as well as the salicylate 70 (PDB ID code 5ZE6), a diphosphate isostere, bound to EcOPPS, and of 70 bound to EcUPPS (PDB ID code 5ZHE), an important antibacterial target. Full data acquisition and refinement details are given in Table S4, and in the Experimental Section. Activity against EcOPPS was weak, ~330 μM for 69 and ~450 μM for 70, but the structures are still of interest since they do represent the first structures of any very long-chain bacterial prenyltransferase with bound inhibitors. Plus, both 69 and 70 are 1–2 μM HepPPS inhibitors (Figure 4).
EcOPPS has structural similarity to FPPSs (both bacterial and human) as well as GGPPS and SaHepPPS, where the Cα root mean square deviation (rmsd) between EcOPPS (PDB ID code 3WJN) and SaHepPPS (PDB ID code 5H9D) is 1.61 Å over 280 aligned residues. The Cα rmsd for EcOPPS with 69 bound (PDB ID code 5ZLF) is 2.38 Å over 244 residues versus Saccharomyces cerevisae GGPPS with 69 bound (ScGGPPS; PDB ID code 2E93). In GGPPS, there are four sites to which inhibitor/substrate/product groups can bind: the allylic substrate diphosphate/bisphosphonate inhibitor site a; the allylic substrate side-chain site b; the homoallylic diphosphate-binding site c, and a hydrophobic product (GGPP) sidechain site d. Ligands can thus bind as follows: ab; ad; bc or cd. With 69 bound to GGPPS, Figures 7a,b two molecules bind, one to site ab and one to site cd.12 We show in Figures 7a,b alignments of the new EcOPPS/69 structure (yellow; PDB ID code 5ZLF) with that of ScGGPPS containing two 69 bisphosphonate ligands (blue; PDB ID code 2E93). The single bisphosphonate inhibitor in the OPPS structure binds to the same allylic binding site ab as does 69 in ScGGPPS and has a very similar conformation, Figure 7b. As expected, the bisphosphonate binds to the two Mg2+ that are coordinated by two aspartates in the first aspartate-rich domain. The third Mg2+ seen in some GGPPS and essentially all FPPS structures is absent due, perhaps, to pH/crystallization conditions. There is no occupancy of the second 69 binding site that is found in ScGGPPS. The ab site in EcOPPS corresponds to the FPP-binding site found in the X-ray structure of EcOPPS with FSPP, the S-thiolo analog of FPP, Figure 7c, and as can be seen in Figure 7d, the FSPP (blue) and 69 (yellow) inhibitors basically overlap each other. And while we do not yet have the structure of 74 bound to either EcOPPS or SaHepPPS, the 69 ligand-bound EcOPPS structure is remarkably similar to that of the lipophilic bisphosphonate 104 (Figure 2), bound to the bifunctional Plasmodium vivax farnesyl/geranylgeranyl diphosphate synthase (PvGGPPS; PBD ID code 3RBM), Figures 8e,f, strongly suggesting that 74 will bind in a similar manner in SaHepPPS and EcOPPS.
Figure 7.
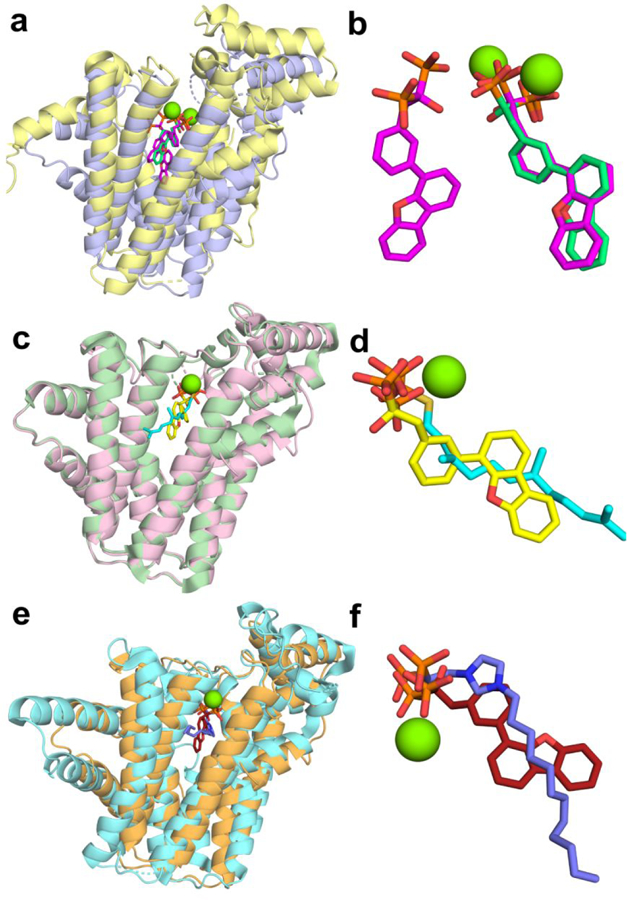
Structure of 69 bound to EcOPPS and a comparison with S. cerevisae GGPPS and P. vivax F/GGPPPS ligand-bound structures. (a) Structure superimposition EcOPPS•69 (PDB ID code 5ZLF) with ScGGPPS•69 (PDB ID code 2E93) blue = OPPS; yellow = GGPPS. (b) Superimposition of 69 ligands shown in a) pink = GGPPS; green = OPPS. (c) Superimposition of EcOPPS•69 with EcOPPS•FSPP (PDB ID code 3WJN). Pink = OPPS; cyan = FSPP. (d) Superimposition of 69 (yellow) and FSPP (cyan) ligands from (c). (e) Superimposition of EcOPPS•69 (orange) with P. vivax—F/GGPPS•105 (color; PDB ID code 3RBM). (f) Superimposition of 69 (brown) and 104 (blue) ligands from (e). 69 binds to just the allylic site (ab) in EcOPPS) and coordinates to 1 Mg2+.
Figure 8.
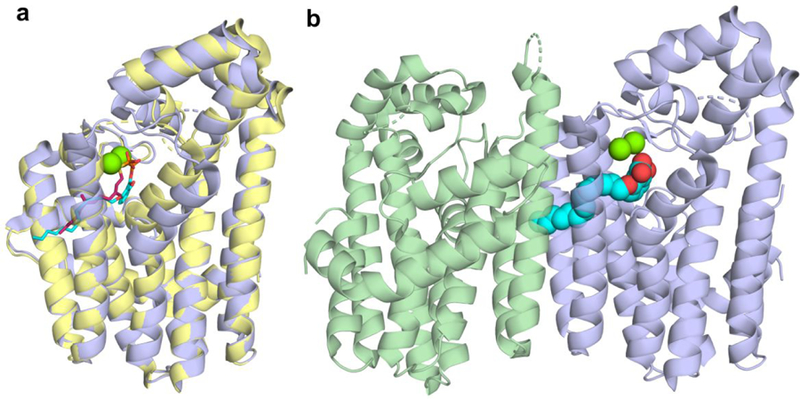
Structure of 70 and FSPP bound to EcOPPS. (a) Superimposition of EcOPPS•70 (blue; Chain B; PDB ID code 5ZE6) with EcOPPS•FSPP (yellow; PDB ID code 3WJN). (b) Illustration of 70 penetrating the dimer interface in EcOPPS•70. The ligand (cyan) is only present in Chain B and is close to the monomer surface, but is buried in the dimer interface.
The second inhibitor of interest is 70, a lipophilic salicylate. While not a bisphosphonate, the salicylates are bisphosphonate/diphosphate isosteres and inhibit prenyl diphosphate synthases with 70 having been shown to inhibit both (gram-positive) E. faecalis and E. faecium cell growth with MIC values of 4 µg/mL31 and here, we found activity against SaHepPPS with an IC50 of 1.5 μM (Figure 4), and a 19 μM IC50 for S. aureus cell growth inhibition. With this lipophilic salicylate, we again find that the inhibitor binds solely to the OPPS allylic binding site, as shown in Figure 8a (PDB ID code 5ZE6), and it closely aligns with FSPP, the allylic substrate-like analog, Figure 8a. So, the bisphosphonate and salicylate inhibitors bind to the allylic substrate-binding site, with both bisphosphonate and salicylate groups being isosteres for diphosphate. With 70, the inhibitor appears to bind with the terminal part of its side-chain in a highly solvent-exposed position—if just the monomer structure is viewed, Figure 8a. However, the side-chain is actually located in the OPPS dimer-interface and it partially penetrates the second subunit in the dimer, Figure 8b, so is protected from solvent exposure. These results in and of themselves do not explain why 70 is a good (~1.5 µM) SaHepPPS inhibitor but a poor EcOPPS inhibitor. They do, however, suggest the possibility that the difference in activity might be due to 70 binding to the regulatory subunit in SaHepPPS. This is because in earlier work using atomic force microscopy, Suzuki et al.32 discovered that FPP binds to the regulatory domain in B. subtilis HepPPS, and in our group4 we found that at high concentrations, FPP inhibits SaHepPPS. It thus appears that binding to the (essential for activity) regulatory subunit in both BsHepPPS as well as SaHepPPS by FPP-isosteres such as 70 could play a role in their inhibitory activity.
In addition to the OPPS/70 structure, we were able to obtain the structure of 70 bound to UPPS (Table 2 and PDB ID code 5ZHE). Surprisingly, unlike the two structures discussed above in which the inhibitors bind to substrate-binding sites, as can be seen in Figure 9a, 70 binds into the hydrophobic center of the protein and not to the more polar, substrate binding sites. This is (approximately) the binding pose adopted by two other inhibitors, a pyrazole33, Figure 9a,b, and clomiphene, Figure 9c,d25. This preference for a hydrophobic binding site by 70 is also seen when comparing the 70 and 69 UPPS structure (PDB 1D code 2E98) where in UPPS there are four 69 ligands. The salicylate overlaps the hydrophobic 69 side-chains in sites 1 and 4 with the long alkyl side-chain located throughout the “center” of the protein, Figures 9e,f. What is of interest about the 70/UPPS structure is that the ligand pose is very different to that we reported previously with 105 which has a 2-carbon shorter side-chain in which two 105 ligands (not one) bind to UPPS, as shown (in slate) in Figures 9g,h. Apparently, the longer (C14 alkyloxy) side-chain in 70 does not permit binding of two molecules to UPPS, while the shorter (C12) species 105 does enable such binding, due to decreased steric clashes.
Figure 9.
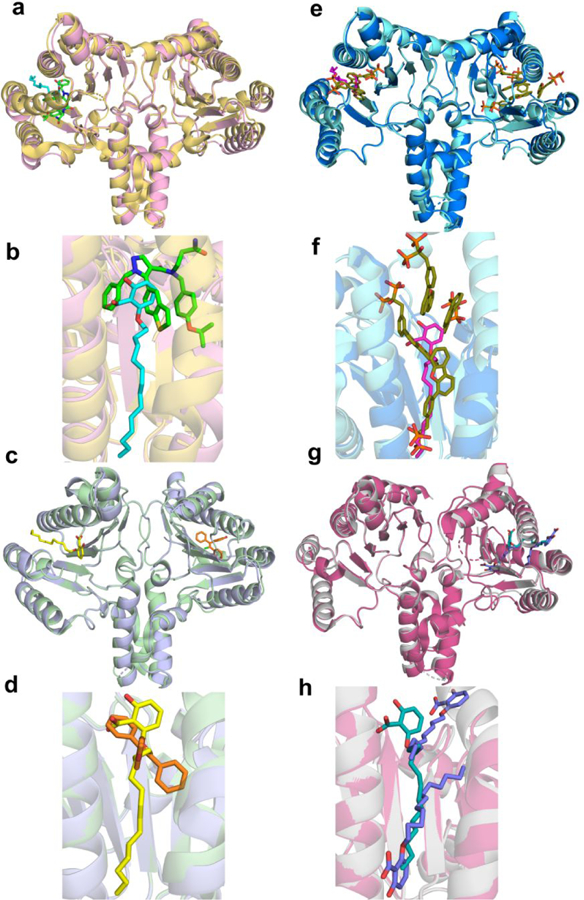
Structure of 70 bound to EcUPPS together with comparisons with pyrazole, clomiphene, 69 and 105 bound structures. (a) Structure of EcUPPS•70 (pink; PDB ID code 5ZHE) superimposed on a UPPS•pyrazole [N-(3-amino-3-oxopropyl)-5-(benzo[b]thiophen-5-yl)-1-benzyl-N-(4-isopropoxybenzyl)-1H-pyrazol e-4-carboxamide] (gold; PDB ID code 5KH5) structure (from S. pneumoniae). (b) Zoomed-in view of the ligand-binding region in (a) 70 in cyan. (c) Superimposition of EcUPPS•70 (blue) with EcUPPS•clomiphene (not all clomiphene atoms were resolved; green; PDB ID code 5CGJ). (d) Zoomed-in view of (c) 70 in yellow. (e) Superimposition of EcUPPS•70 (blue) with EcUPPS•69 (cyan; PDB ID code 2E98). (f) Zoomed-in view of (e) 70 in pink. (g) Superimposition of EcUPPS•70 (green) with EcUPPS•105 (pink; PDB ID code 3SH0). (h) Zoomed-in view of (g).
As to future structural work: The structure of the EcOPPS/69 complex is of interest since 69 is a potent (~250 nM) UPPS inhibitor that we find here has activity against S. aureus (ED50 = 19 µM), so future studies of HepPPS inhibition (IC50 = 1.1 μM, Figure 4) with additional analogs will be of interest since it may be possible to develop dual UPPS/HepPPS inhibitors. Also, of course, it will be of great interest to see whether the long chain bisphosphonates bind to the allosteric site in HsFPPS, of interest in the context of developing FPPS/GGPPS inhibitors targeting prenylation in tumor cells,34 and as vaccine adjuvants.35 Plus, whether the allosteric site is in fact present in bacterial FPPSs is worth further investigation.
Conclusions and Future Prospects.
The results we have presented here are of interest for several reasons. First, after screening a library of known prenyltransferase inhibitors for activity against the long chain prenyltransferase octaprenyl diphosphate synthase (OPPS), we tested several compounds against a variety of gram-positive and gram-negative bacteria, as well as the fungus C. albicans. We found promising activity against the gram-positive bacteria, so we next synthesized and tested 24 analogs of the most potent hit (in both enzyme and cell growth inhibition assays). We found several compounds that inhibited OPPS as well as its analog, HepPPS, that also inhibited gram-positive (but not gram-negative) bacterial cell growth in the ≈ 1–10 µg/mL range. The generally most potent hit or lead was 74, a tetrahydrogeranyl-substituted pyridinium bisphosphonate. Second, we found that the activity of 74 (and close analogs) was not inhibited by human serum, plus, there was low toxicity against the human embryonic kidney cell line, HEK293 (~0.5 mM after 24 hrs, ~70 µM at 96 hours). The potent activity against gram-positive bacteria was in sharp contrast to that we reported earlier with bisphosphonates such as 72 and 73 which only inhibited gram-negative bacterial cell growth, perhaps due to differences in bacterial influx/efflux. Third, we investigated the mechanism of action of 74. Compound 74 inhibited both EcOPPS as well as SaHepPPS with ~10–100 nM IC50 values, as well as a (human) FPPS, at ~200 nM. These results suggested the possibility that 74 (and related compounds) might act by inhibiting both short (FPPS) as well as long-chain (HepPPS) isoprenoid diphosphate synthase enzymes, in bacteria. To test this hypothesis we used a “rescue” approach in which we added downstream products FPP, menaquinone-4 (MK-4) and undecaprenyl diphosphate (UP) to cells, grown in the presence of 74. There were large increases in the ED50 for cell growth inhibition by MK-4 and UP and when added together, the ED50 for cell (B. subtilis) growth inhibition increased by a factor of 25. Addition of FPP itself also had an effect, ~3.5x, since it “rescues” FPPS inhibition. Addition of the metabolite HMBPP (E-1-hydroxy-2-methyl-but-2-enyl 4-diphosphate), which is upstream of FPP, also had a small-effect and potentiated the rescue effects of MK-4 and UP. One obvious explanation for these observations is that addition of the very hydrophobic species MK-4 and UP is extremely effective in rescuing cells from inhibition of their biosynthesis simply because being so hydrophobic (MK-4, clogP = 8.48; UP, clogP = 17.32), they rapidly bind to bacterial cell membranes—which is where they normally function. On the other hand, while addition of FPP might be expected to help rescue both quinone as well as cell wall biosynthesis, it is not particularly effective because not only does it have to be transported into the cytoplasm, it also has to be processed by UPPS and HepPPS, and HepPPS is actually inhibited by e.g. 74. Fourth, we solved the structures of three prenyl-transferase inhibitor complexes. The structure of the bisphosphonate 69 bound to OPPS showed that it bound to the (allylic) substrate-binding site much better, as did the salicylate inhibitor 70. However, 70 was a surprisingly bad (several hundred-fold) HepPPS inhibitor than an OPPS inhibitor and we propose that the salicylate (70), a diphosphate isostere, binds to the regulatory subunit because in previous work we found that FPP inhibits HepPP product formation at high concentrations (Kd ~35 µM) in SaHepPPS, and other workers have shown that FPP binds to the regulatory subunit, in BsHepPPS. That is, FPP can regulate (i.e. inhibit) isoprenoid biosynthesis, in both FPPS20 and in HepPPS, and some inhibitors (such as 70 with HepPPS and 74 with FPPS) may act in a similar manner. Indeed, it is now well known that other isoprenoid biosynthesis enzymes such as mevalonate kinase36 as well as UPPS37 are subject to feedback inhibition, and are also targets for (antibacterial) allosteric site inhibitors. Overall then, the results we have presented above are of general interest since we report the discovery of a series of compounds, lipophilic bisphosphonates, that target bacterial cell growth via inhibition of both bacterial cell wall, as well as quinone, biosynthesis.
Experimental Section.
Chemical Synthesis and Characterization
We synthesized 24 compounds using primarily the general methods (A-D) described below. Full synthesis details including the production of intermediates (designated as IM-1, IM-2, etc) and schemes are given in the Supporting Information. All chemicals used were of reagent grade. Product formation was monitored by 1H and where applicable, 31P NMR, 13C NMR, and 19F NMR on Varian or Bruker spectrometers at 400 or 500 MHz for 1H. Chemical shifts are reported in parts per million from TMS (1H, 13C), 85% H3PO4 (31P) or CFCl3 (19F). Purity was determined by 1H qNMR (500 MHz Bruker Cryoprobe) using Mnova software, and by HRMS or LCMS. All compounds were ≥ 95% pure except for 101 (92.7%).
General Synthetic Methods
Procedure A (alkylation of a hydroxypyridine).
To a solution of a hydroxypyridine (1.0 equivalent) and alkyl bromide (1.0 equivalent) in anhydrous DMF was added K2CO3 (2.0 equivalents) portion-wise under a N2 atmosphere.38 The mixture was stirred at 80 °C until TLC analysis (silica gel; 30% EA/PE) showed complete consumption of hydroxypyridine. The mixture was cooled to RT, and DMF evaporated under reduced pressure to give a residue. To the crude product, 10 mL of water was added, and the product extracted with 2 × 20 mL ethyl acetate. The combined organic layers were washed with brine, the product dried over Na2SO4 then evaporated to give the crude product as a dark brown oil. The crude product was purified by flash column chromatography on silica (10% to 50 % EA/PE) to give the alkylated product.
Procedure B (synthesis of 2-(pyridinium-1-yl)ethylidene-1,1-bisphosphonic acid).
A mixture of a substituted pyridine (1.0 equivalent) and vinylidene-1,1-bisphosphonic acid (0.95 equivalents) in water was refluxed for 2 h. The reaction mixture was cooled to RT, and water was evaporated in vacuo to give a semi-solid.39 The residue was triturated with absolute ethanol, and the resulting white suspension filtered and then washed with ethanol to afford the corresponding bisphosphonic acid as a white powder.
Procedure C (reaction of a heteroaryl amine with tetraethyl/methyl vinylidene bisphosphonate).
A solution of a heteroaryl amine (1.0 equivalent) and tetraethyl or tetramethylvinylidene bisphosphonate (0.55 equivalents) in CHCl3 was stirred at RT for 20 hrs. The progress of the reaction was monitored by TLC (50% EA/PE).40 To the reaction mixture, silica was added, and all volatiles were removed in vacuo. The crude product was purified by flash column chromatography (40% EA/PE to 2% MeOH/EA) to afford a pure compound as a viscous, colourless oil.
Procedure D (TMSBr-mediated deprotection of a bisphosphonate ester).
A bisphosphonate ester (1.0 equivalent) was dissolved in anhydrous DCM and cooled to 0 ºC. A solution of TMSBr (14.0 equivalents) in DCM was added dropwise over a period of 5 min at 0 ºC. The consumption of ester was monitored by TLC (10% MeOH/EA). After 24 h, anhydrous MeOH was added dropwise,41 and the mixture stirred for another 30 min. Removal of volatiles in vacuo yielded a semi-solid. The crude product was triturated with ethanol, and the resulting white suspension filtered and washed with ethanol, affording a pure (pyridinium-1-yl)ethylidene-1,1-bisphosphonic acid as a white powder.
Hydrogen(2-(3-((3,7-dimethyloctyl)oxy)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (74).
According to Procedure B, 74 was prepared as a white powder (69%). 1H NMR (D2O, 500 MHz): δ 8.71 (s, 1H), 8.58 (d, J = 6.0 Hz, 1H), 7.99 (dd, J = 9.0 Hz, 1.5 Hz, 1H), 7.85 (dd, J = 6.5, 6.0 Hz, 1H), 4.93 (td, J = 12.8, 6.8 Hz, 2H), 4.34–4.30 (m, 2H), 2.40 (tt, J = 20.8, 13.3 Hz, 1H), 1.93–1.89 (m, 1H), 1.72–1.67 (m, 2H), 1.58–1.53 (m, 1H), 1.39–1.18 (m, 6H), 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.5 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 14.06; ESI HRMS: m/z [M+H]+ calculated for C17H32NO7P2+, 424.1649; found, 424.1639 ; purity = 99.9 % (qNMR).
Hydrogen(2-(1-(4,8-dimethylnonyl)-1H-imidazol-3-ium-3-yl)-1- phosphonoethyl) phosphonate (75).
According to Procedure B, 75 was prepared as a white powder (32%). 1H NMR (D2O, 500 MHz): δ 8.82 (s, 1H), 7.58 (s, 1H), 7.46 (s, 1H), 4.63 (td, J = 13.8, 7.0 Hz, 2H), 4.17 (t, J = 7.0 Hz, 2H), 2.68 (tt, J = 21.5, 7.0 Hz, 1H), 1.93–1.81 (m, 2H), 1.52–1.44 (m, 2H), 1.31–1.12 (m, 8H); 0.85 (d, J = 6.5 Hz, 3H), 0.84 (d, J = 6.5 Hz, 6H); 31P (202 MHz, D2O): δ 14.29; ESI HRMS: m/z [M+H]+ calculated for C16H33N2O6P2+, 411.1808; found, 411.1801; purity = 96.6 % (qNMR).
Hydrogen (1-phosphono-2-(3-undecylpyridin-1-ium-1-yl)ethyl)phosphonate (76).
According to Procedure B, 76 was prepared as a white powder (66%). 1H NMR (D2O, 500 MHz): δ 8.82 (s, 1H), 8.41 (d, J = 6.0 Hz, 1H), 8.28 (d, J = 8.0 Hz, 1H), 7.85 (dd, J = 8.0 Hz, 1H), 4.92 (td, J = 13.0, 6.3 Hz, 2H), 2.86 (t, J = 7.8 Hz), 2.34 (tt, J = 21.0, 6.5, 1H), 1.74–1.71 (m, 2H), 1.36–1.30, (m, 16H), 0.88 (t, J = 7.5 Hz, 3H); 31P NMR (D2O, 202 MHz): δ 14.38. ESI HRMS: m/z [M+H]+ calculated for C18H34NO6P2+, 422.1856; found, 422.1837; purity = 99.1 % (qNMR).
Hydrogen(2-(3-(4,8-dimethylnonyl)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (77).
According to Procedure B, 77 was prepared as a white powder (73%). 1H NMR (D2O, 500 MHz): δ 8.77 (s, 1H), 8.72 (d, J = 7.5 Hz, 1H), 8.26 (d, J = 10.0 Hz, 1H), 7.82 (dd, J = 10.0, 7.8 Hz, 1H), 4.87 (td, J = 16.0, 8.8 Hz, 2H), 2.84–2.77 (m, 2H), 2.36 (tt, J = 26.0, 17.3 Hz, 1H), 1.70–1.66 (m, 2H), 1.50–1.06 (m, 10H), 0.81 (d, J = 8.0 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 13.41; ESI HRMS: m/z [M+H]+ calculated for C18H34NO6P2+, 422.1856; found, 422.1853; purity = 98.4 % (qNMR).
Hydrogen(2-(3-((3,7-dimethyloctyl)amino)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (78).
According to Procedure B, 78 was prepared as a pale brown solid (26%). 1H NMR (D2O, 500 MHz): δ 8.21 (s, 1H), 8.13 (d, J = 6.0 Hz, 1H), 7.59 (dd, J = 8.8, 5.8 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 4.83–4.80 (m, 2H), 3.29–3.19 (m, 2H), 2.32 (tt, J = 21.0, 6.6, 1H), 1.72–1.47 (m, 2H), 1.37–1.17, (m, 6H), 0.95 (d, J = 6.5 Hz, 3H), 0.87 ( J = 7.0 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 14.72; ESI HRMS: m/z [M+H]+ calculated for C17H33N2O6P2+, 423.1790; found, 423.1795; purity = 96.8 % (qNMR).
Hydrogen(2-(3-((3,7-dimethyloctyl)thio)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (79).
According to Procedure B, 79 was prepared as a white powder (71.4%). 1H NMR (D2O, 500 MHz): δ 8.88 (s, 1H), 8.73 (d, J = 6.0 Hz, 1H), 8.31 (d, J = 8.5 Hz, 1H), 7.83 (dd, J = 8.5, 8.0 Hz, 1H), 4.92 (td, J = 12.5, 6.5 Hz, 2H), 3.27–3.17 (m, 2H), 2.35 (tt, J = 21.0, 6.5, 1H), 1.76–1.52 (m, 2H), 1.35–1.16, (m, 6H), 0.94 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.5 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 14.11; ESI HRMS: m/z [M+H]+ calculated for C17H32NO6P2S+, 440.1420; found, 440.1412; purity = 97.5 % (qNMR).
Hydrogen(2-(4-(4,8-dimethylnonyl)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (80).
According to Procedure B, 80 was prepared as a white powder (78%). 1H NMR (D2O, 500 MHz): δ 8.71 (d, J = 6.5 Hz, 2H), 7.75 (d, J = 6.0 Hz, 2H), 4.86–4.80 (m, 2 H), 2.90–2.81 (m, 2H), 2.31 (tt, J = 20.5, 6.8 Hz, 1H), 1.78–1.65 (m, 2H), 1.52–1.43 (m, 2H), 1.36–1.05 (m, 8H), 0.81 (d, J = 6.5 Hz, 9H); 31P (202 MHz, D2O): δ 14.10 ESI HRMS: m/z [M+H]+ calculated for C18H34NO6P2+, 422.1856; found, 422.1862; purity = 95.4 % (qNMR).
Hydrogen(2-(3-((3,7-dimethyloctyl)oxy)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (81).
Bromoacetic acid (0.27 g, 1.95 mmol) was added to a solution of the 3-((3,7-dimethyloctyl)oxy)pyridine (0.46 g, 1.95 mmol) in ethyl acetate (3 mL), and the mixture was stirred at RT for 2 days yielding substituted 1-carboxymethylpyridinium bromide as a white precipitate, basically as described previously.18 The product was then filtered, washed with ethyl acetate (2 × 3 mL), and dried in vacuo. The resulting white powder was added to a mixture of H3PO3 (0.49 mL, 9.77 mmol) and toluene (6 mL) and heated to 80 °C, while stirring. After all solids melted, POCl3 (0.91 mL, 9.77 mmol) was added dropwise, and the mixture vigorously stirred at 80 °C for 5 h. Upon cooling, the supernatant was decanted, and 6 N HCl (3 mL) added to the residue. The resulting solution was refluxed for 1 h, then most solvent was removed in vacuo. 2-Propanol (25 mL) was added to precipitate the title compound as a white powder. The powder was filtered, washed with 2-propanol (5 × 5 mL), then dried and further purified by recrystallization from H2O/2-PrOH to yield hydrogen(2-(3-((3,7-dimethyloctyl)oxy)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (0.51, 1.21 mmol, 62%) as a white powder. 1H NMR (D2O, 500 MHz): δ 8.61 (s, 1H), 8.54 (d, J = 6.0 Hz, 1H), 8.02 (dd, J = 8.5 Hz, 1H), 7.83 (dd, J = 9.0, 6.5 Hz, 1H), 4.95 (t, J = 9.0 Hz, 2H), 4.36–4.28 (m, 2H), 1.93–1.88 (m, 1H), 1.74–1.67 (m, 3H), 1.39–1.19 (m, 6H) 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.5 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 13.28; ESI HRMS: m/z [M+H]+ calculated for C17H32NO8P2+, 440.1598; found,440.1586; purity = 95.0 % (qNMR).
Hydrogen(2-(1-(3,7-dimethyloctyl)-1H-imidazol-3-ium-3-yl)-1-phosphonoethyl) phosphonate (82).
According to Procedure B, 82 was prepared as a white powder (28%). 1H NMR (D2O, 500 MHz): δ 8.81 (s, 1H), 7.57 (s, 1H), 7.42 (s, 1H), 4.61–4.54 (m, 2H), 4.22–4.19 (m, 2H), 2.43–2.35 (m, 1H), 1.95–1.88 (m, 1H), 1.73–1.67 (m, 1H), 1.52–1.15 (m, 6H), 0.94 (d, J = 6.5 Hz, 3H), 0.85 (d, J = 6.5 Hz, 6H); 31P (D2O, 202 MHz): δ 14.37; ESI HRMS: m/z [M+H]+ calculated for C15H31N2O6P2+, 397.1652; found, 397.1651; purity = 96.1 % (qNMR).
Hydrogen (1-phosphono-2-(3-tridecylpyridin-1-ium-1-yl)ethyl)phosphonate (83)
According to Procedure B, 83 was prepared as a white powder (75%). 1H NMR (D2O, 500 MHz): δ 8.77 (s, 1H), 8.72 (d, J = 6.0 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.80 (dd, J = 7.0 Hz, 1H), 4.87 (td, J = 13.3, 6.7 Hz, 2H), 2.80 (t, 2H), 2.30 (tt, J = 20.8, 6.5 Hz, 1H), 1.68 (quintet, J = 6.9 Hz, 2H), 1.31–1.24 (m, 20H), 0.82 (t, J = 6.8 Hz, 3H); 31P (202 MHz, D2O): δ 14.09; ESI HRMS: m/z [M+H]+ calculated for C20H33NO6P2+,450.2169; found, 450.2158; purity = 95.9 % (qNMR).
Hydrogen (2-(4-((3,7-dimethyloctyl)oxy)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (84).
According to Procedure B, 84 was prepared as a white powder (76.2%). 1H NMR (D2O, 500 MHz): δ 8.61 (d, J = 7.5 Hz, 2H), 7.31 (d, J = 7.5 Hz, 2H), 4.72 (td, J = 13.0, 7.0 Hz, 2H), 4.37–4.35 (m, 2H), 2.64–2.55 (m, 2H), 2.31 (tt, J = 20.8, 6.7 Hz, 1H), 1.90–1.84 (m, 12H), 1.66–1.63 (m, 2H); 1.50 (quintet, J = 6.7 Hz, 1H), 1.36–1.13 (m, 6H), 0.91 (d, J = 6.0 Hz, 3H), 0.82 (d, J = 7.0 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 14.09; ESI HRMS: m/z [M+H]+ calculated for C17H32NO7P2+, 424.1649; found, 424.1634; purity = 96.5 % (qNMR).
Hydrogen(2-(4-((3,7-dimethyloctyl)thio)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (85).
According to Procedure B, 85 was prepared as a white powder (76%).. 1H NMR (D2O, 500 MHz): δ 8.55 (d, J = 7.5 Hz, 2H), 7.69 (d, J = 7.5 Hz, 2H), 4.84–4.75 (m, 2H), 3.32–3.20 (m, 2H), 2.33 (tt, J = 20.8, 6.6 Hz, 1H), 1.82–1.51 (m, 4H), 1.40–1.17 (m, 6H), 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.5 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 13.53; ESI HRMS: m/z [M+H]+ calculated for C17H32NO6P2S+, 440.1420; found, 440.1409; purity = 96.9 % (qNMR).
(2-((4-(3,7-Dimethyloctyl)thiazol-2-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (86).
According to Procedure D, 86 was prepared as a white powder (71%). 1H NMR (D2O, 500 MHz): δ 6.28 (s, 1H), 3.58–3.51 (m, 2H), 3.68 (td, J = 13.5, 6.8 Hz, 2H), 5.58–2.50 (m, 2H), 2.27 (tt, J = 20.5, 6.7 Hz, 1H), 1.64–1.15 (m, 10H), 0.91 (d, J = 5.0 Hz, 3H), 0.86 (d, J = 6.5 Hz, 6H); 31P (202 MHz, D2O): δ 16.47; ESI HRMS: m/z [M+H]+ calculated for C15H31N2O6P2S+, 429.1373; found, 429.1365; purity = 96.5 % (qNMR).
(2-((2-((3,7-Dimethyloctyl)oxy)pyridin-4-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (87).
According to Procedure D, 87 was prepared as a white powder (71%). 1H NMR (D2O, 500 MHz): δ 7.70 (d, J = 6.0 Hz, 1H), 6.43 (d, J = 6.0 Hz, 1H), 6.07 (s, J = 2.0 Hz, 1H), 4.26–4.20 (m, 2 H), 3.54 (td, J = 14.0, 7.5 Hz, 2H), 2.05 (tt, J = 21.0, 6.8 Hz, 1H), 1.83–1.53 (m, 4H), 1.37–1.17 (m, 6 H), 0.95 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.5 Hz, 6H); 31P (D2O, 202 MHz): δ 16.95; ESI HRMS: m/z [M+H]+ calculated for C17H33N2O7P2+, 439.1758; found, 439.1760; purity = 96.4 % (qNMR).
(2-((5-(3,7-Dimethyloctyl)thiazol-2-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (88).
According to Procedure D, 88 was prepared as a white powder (67%). 1H NMR (500 MHz, D2O): δ 6.78 (s, 1H), 3.62 (td, J = 20.8, 6.8 Hz, 2H), 2.76–2.64 (m, 2H), 2.11 (tt, J = 20.5, 7.0 Hz, 1H), 1.65–1.16 (m, 10H), 0.91 (d, J = 1.5 Hz, 3H), 0.88 (d, J = 1.5 Hz, 6H); 31P (202 MHz, D2O): δ 16.62; ESI HRMS: m/z [M+H]+ calculated for C15H31N2O6P2S+, 429.1373; found, 429.1368; purity = 97.2 % (qNMR).
(((6-(4,8-Dimethylnonyl)pyridin-2-yl)amino)methylene)bis(phosphonic acid) (89).
According to Procedure D, 89 was prepared as a white powder (69%). 1H NMR (D2O, 500 MHz): δ 7.58 (dd, J = 7.8 Hz, 1H), 6.65 (d, J = 7.0 Hz, 1H), 6.56 (d, J = 8.0 Hz, 1H), 3.63 (td, J = 14.0, 7.0 Hz, 2H), 2.64–2.55 (m, 2H), 2.13 (tt, J = 20.8, 7.0 Hz, 1H), 1.73–1.10 (m, 12H), 0.87 (d, J = 6.5 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 17.20; ESI HRMS: m/z [M+H]+ calculated for C18H35N2O6P2+, 437.1965; found, 437.1961; purity = 98.1 % (qNMR).
Hydrogen(1-phosphono-2-(3-((3,7,11-trimethyldodecyl)oxy)pyridin-1-ium-1-yl)ethyl) phosphonate (90).
According to Procedure B, 90 was prepared as a white powder (75.2%). 1H NMR (500 MHz, D2O): δ 8.75 (s, 1H), 8.61 (d, J = 5.5 Hz, 1H), 7.95–7.88 (m, 2H), 4.94 (td, J = 12.5, 7.5 Hz, 2H), 4.34–4.26 (m, 2H), 2.50 (tt, J = 20.0, 7.3 Hz, 1H), 1.94–1.90 (m, 1H), 1.72–1.12 (m, 16H), 0.97 (d, J = 6.0 Hz, 3H), 0.90 (d, J = 7.0 Hz, 9H); 31P (202 MHz, D2O): δ 13.26; ESI HRMS: m/z [M–H]− calculated for C22H41NO7P2−, 492.2282; found, 492.2280; purity = 96.5 % (qNMR).
Hydrogen (2-(3-(isopentyloxy)pyridin-1-ium-1-yl)-1-phosphonoethyl)phosphonate (91).
According to Procedure B, 91 was prepared as a white powder (73.8%). 1H NMR (D2O, 500 MHz): δ 8.71 (s, 1H), 8.57 (d, J = 5.5 Hz, 1H), 7.99 (dd, J = 8.8, 2.3 Hz, 1H), 7.85–7.82 (m, 2H), 4.93 (td, J = 13.0, 5.0 Hz, 2H), 4.31 (t, J = 6.8 Hz, 1H), 2.46 (tt, J = 20.8, 6.5 Hz, 1H), 1.85 (septet, J = 6.7 Hz, 1H), 1.78–1.74 (q, J = 5 Hz, 1H) 0.98 (d, J = 6.5 Hz, 6H); 31P (202 MHz, D2O): δ 13.33; ESI HRMS: m/z [M+H]+ calculated for C12H22NO7P2+, 354.0866; found, 354.0866; purity = 98.3 % (qNMR).
(2-((6-((3,7-Dimethyloctyl)oxy)pyridin-2-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (92).
According to Procedure D, 92 was prepared as a white powder (65%). 1H NMR (D2O, 500 MHz): δ 7.51 (dd, J = 8.0 Hz, 1H), 6.26 (d, J = 6.5 Hz, 1H), 6.08 (d, J = 8.0 Hz, 1H), 4.14–4.09 (m, 2H), 3.53 (td, J = 18.5, 6.0 Hz, 2H), 1.93 (tt, J = 21.0, 6.5 Hz, 1H), 1.77–1.48 (m, 4H), 1.32–1.13 (m, 6H), 0.90 (d, J = 6.5 Hz, 3H), 0.81 (d, J = 6.5 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 17.62; ESI HRMS: m/z [M–H]− calculated for C17H31N2O7P2−, 437.1612; found, 437.1611; purity = 96.1 % (qNMR).
(2-((5-(6-Methylheptan-2-yl)thiazol-2-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (94).
According to Procedure D, 94 was prepared as a white powder (70%). 1H NMR (500 MHz, D2O): δ 6.80 (s, 1H), 3.65 (td, J = 13.8, 7.0 Hz, 2H), 2.96–2.90 (m, 1H), 2.18 (tt, J = 20.5, 7.3 Hz, 1H), 1.57–1.50 (m, 3H), 1.31–1.17 (m, 7H), 0.86–0.84 (m, 6H); 31P (D2O, 202 MHz): δ 16.41; ESI HRMS: m/z [M+H]+ calculated for C13H2N2O6P2S+, 401.1060; found, 401.1071; purity = 98.0 % (qNMR).
(((4-((3,7-Dimethyloctyl)oxy)pyridin-2-yl)amino)methylene)bisphosphonic acid (95).
In a 5 mL round-bottom flask, (((4-((3,7-dimethyloctyl)oxy)pyridin-2-yl)amino) methylene) bis phosphonate (0.020 g, 0.037 mmol) and 6 N HCl (0.30 mL) were refluxed for 4 h.42 The progress of the reaction was monitored by TLC (10% MeOH/EA). The volatile components were removed in vacuo to yield pure (((4-((3,7-dimethyloctyl)oxy)pyridin-2-yl)amino)methylene)bisphosphonic acid (0.013 g, 0.030 mmol, 80%) as a white solid. 1H NMR (D2O, 500 MHz): δ 7.68 (d, J = 6.0 Hz, 1H), 6.16 (d, J = 6.0 Hz, 1H), 6.01 (s, 1H), 4.13–4.12 (m, 2H), 1.77–1.73 (m, 1H), 1.62–1.49 (m, 3H), 1.31–1.13 (m, 6H), 0.89 (d, J = 6.5 Hz), 0.83 (d, J = 6.5 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 14.90; ESI HRMS: m/z [M+H]+ calculated for C16H31N2O7P2+, 425.1601; found, 425.1610; purity = 96.6 % (qNMR).
(2-((4-((3,7-Dimethyloctyl)oxy)pyridin-2-yl)amino)ethane-1,1-diyl)bis(phosphonic acid) (96).
According to Procedure D, 96 was prepared as a white powder (65%). 1H NMR (D2O, 500 MHz): δ 7.76 (d, J = 6.0 Hz, 1H), 6.30 (d, J = 6.0 Hz, 1H), 6.17 (s, 1H), 5.58 (td, J = 13.8, 7.2, 2H), 4.13–4.12 (m, 2H), 2.14–2.03 (m, 1H), 1.78–1.47 (m, 4H), 1.30–1.13 (m, 6H), 0.89 (d, J = 6.5 Hz), 0.82 (d, J = 6.5 Hz, 6H); 31P NMR (D2O, 202 MHz): δ 18.04; ESI HRMS: m/z [M+H]+ calculated for C17H33N2O7P2+, 439.1758; found, 439.1761; purity = 98.1% (LCMS).
Hydrogen(1-phosphono-2-(3-((3,7,11,15-tetramethylhexadecyl)oxy)pyridin-1-ium-1-yl)ethyl)phosphonate (97).
According to Procedure B, 97 was prepared as a white powder (72.8%). 1H NMR (500 MHz, D2O): δ 8.77 (s, 1H), 8.63 (d, J = 4.5 Hz, 1H), 7.87–7.82 (m, 2H), 4.93–4.90 (m, 2H), 4.28–4.26 (m, 2H), 2.42 (tt, J = 20.0, 6.8 Hz, 1H), 1.95–1.16 (m, 24H), 0.97 (d, J = 5.5 Hz, 3H), 0.93 (d, J = 6.0 Hz, 12H); 31P (202 MHz, D2O): δ 13.45; ESI HRMS: m/z [M–H]− calculated for C27H51NO7P2−, 562.3063; found, 562.3069; purity = 96.4 % (qNMR).
6-Fluoro-1-[3-(nonyloxy)benzoyl]-2H-3,1-benzoxazin-4-one (100).
To a solution of 5-fluoro-2-(3-(octyloxy)benzamido)benzoic acid (0.015 gm, 0.039 mmol) and chloromethyl isopropyl carbonate (5.9 μL, 0.043 mmol) in 0.5 mL of DMF at RT, K2CO3 (5.8 mg, 0.043 mmol) was added, and the mixture stirred at 50 °C for 18 h. The mixture was then cooled to room temperature, 3 mL of water added, and the product extracted into 2 × 5 mL of ethyl acetate. The combined organic layers were dried over Na2SO4. The crude compound was purified by flash column chromatography over silica (10% EA/PE) to give 101 (9.0 mg, 0.022 mmol, 58%) as a pale yellow solid. 1H NMR (CDCl3, 500 MHz): δ 7.78 (dd, J = 8.0, 3.0 Hz, 1H), 7.42 (broad s, 1H), 7.35–7.24 (m, 2H), 7.07–7.04 (m, 3H), 5.71 (s, 2H), 3.96 (t, J = 6.5 Hz, 2H), 1.78 (quintet, J = 7.1 Hz, 2H), 1.47–1.25 (m, 10H), 0.87 (t, J = 6.8 Hz, 3H); 19F NMR(CDCl3, 376 MHz): δ −113.77 (d, J = 2.6 Hz); 13C NMR (CDCl3, 125 MHz): δ 169.4, 161.8 (d, J = 2.4 Hz), 159.9 (d, J = 247.0 Hz), 159.5, 137.5 (d, J = 2.8 Hz), 133.9, 129.94, 125.4 (d, J = 7.6), 121.9 (d, J = 23.3 Hz), 120.2 (d, J = 8.0 Hz), 120.1, 118.5, 116.3, 114.1, 76.5, 68.4, 31.8, 29.3, 29.2, 29.1, 25.9, 22.7, 14.1. ESI HRMS: m/z [M+H]+ calculated for C24H29FNO4+, 414.2075; found, 414.2062; purity = 96.1 % (qNMR).
(1-Hydroxy-1-(hydroxy((pivaloyloxy)methoxy)phosphoryl)-2-(1H-imidazol-1-yl)ethyl)phosphonic acid (101).
A solution of the tris-tetrabutyl ammonium salt of zoledronic acid (0.08 gm, 0.08 mmol) and iodomethyl pivalate (0.010 gm, 0.04 mmol, 0.5 equivalent) in 1.5 mL ACN was stirred at room temperature for 24 h. Then, another 0.5 equivalent of iodomethyl pivalate was added and mixture stirred for 24 h. The organic solvent was removed, and the crude product converted to the NH4+ form by treating with 1.5 mL of DOWEX NH4+ resin. The product was then purified by methanol washing (2 × 1 mL), to give 102 (6.8 mg, 0.016 mmol, 19 %) as a white crystalline solid. 1H NMR (D2O, 500 MHz): δ 8.63 (s, 1H), 7.50 (d, J = 1.5 Hz, 1H), 7.35 (d, J = 1.5 Hz, 1H), 5.51–5.43 (m, 2H), 4.72–4.57 (m, 2H), 1.20 (s, 9H); 31P NMR (D2O, 205 MHz): δ 15.07 (d, J = 18.2 Hz, 1P), 15.63 (d, J = 18.2 Hz, 1P). ESI HRMS: m/z [M+H]+ calculated for C11H21N2O9P2+, 387.0722; found, 387.0715; Purity = 92.7 % (qNMR).
Hydrogen (2-(3-(3,7-dimethyloctyl)pyridin-1-ium-1-yl)-1-phosphonoethyl) phosphonate (102).
According to Procedure B, 103 was prepared as a white powder (39 %). 1H NMR (D2O, 500 MHz): δ 8.83 (s, 1H), 8.78 (d, J = 6.0 Hz, 1H), 8.31 (d, J = 8.0 Hz, 1H), 7.86 (dd, J = 7.5, 6.5 Hz, 1H), 4.93 (td, J = 13.0, 6.5 Hz, 2H), 2.94–2.82 (m, 2H), 2.39 (tt, J = 20.8, 6.5 Hz, 1H), 1.77–1.71 (m, 1H), 1.60–1.51 (m, 3H), 1.39–1.17 (m, 6H), 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.5 Hz, 9H); 31P NMR (D2O, 202 MHz): δ 13.38; ESI HRMS: m/z [M+H]+ calculated for C17H32NO6P2+, 408.1705; found, 408.1704; purity = 95.4 % (qNMR).
3-[(2E)-3,7-dimethylocta-2,6-dien-1-yl]-1-(2-hydrogen phosphonato-2-phosphonoethyl)pyridin-1-ium (103).
According to Procedure B, 104 was prepared as a white powder (29 %). 1H NMR (D2O, 500 MHz): δ 8.81 (s, 1H), 8.79 (d, J = 6.5 Hz, 1H), 8.31 (d, J = 6.5 Hz, 1H), 7.92 (dd, J = 6.0, 6.0 Hz, 1H), 5.45–5.42 (m, 1H), 5.22–5.20 (m, 1H), 4.93 (td, J = 12.8, 7.5 Hz, 2H), 3.63 (d, J = 7.5 Hz, 2H), 2.39 (tt, J = 19.8, 7.5 Hz, 1H), 2.20–2.16 (m, 4H), 1.76 (s, 3H), 1.70 (s, 3H), 1.64 (s, 3H); 31P NMR (D2O, 202 MHz): δ 13.47; ESI HRMS: m/z [M+H]+ calculated for C17H28NO6P2+, 404.1386; found, 404.1389; purity = 95.4 % (qNMR).
Biological Assays
Cells.
Bacteria were kindly provided by Professor Douglas A. Mitchell and were as follows: B. subtilis from subsp. subtilis (Ehrenberg) Cohn ATCC 6051; E. coli (K12, ATCC® 29425™); S. aureus (Newman strain); Mycobacterium smegmatis (MC2 155, ATCC 700084); B. anthracis (str. Sterne); A. baumannii (Bouvet and Grimont, ATCC 19606); K. pneumoniae (subsp. pneumoniae Schroeter Trevisan ATCC 27736); P. aeruginosa PA01. C. albicans (CAI-4) was from Peter Orlean. HEK293 cells were from ATCC, ATCC CRL-1573.
Chemicals.
Decaprenyl monophosphate (DP) and undecaprenyl monophosphate (UP) were from Larodan AB, Sweden (product # 62–1050-2 and 62–1055-2). Menaquinone-4 (MK-4) was from Sigma-Aldrich (product # 47774). DP and UP were dissolved in 1:1 MeOH:DMSO, while MK-4 was prepared in ethanol.
B. subtilis growth inhibition assay:
An overnight starter culture (in LB both) of B. subtilis was diluted 1000-fold (in fresh LB media) and grown to an OD600 of ~0.3 (approx. 3.5 hours at 37 °C). This log-phase culture was then diluted 500-fold into fresh LB broth to generate the working solution. 180 µL of this working solution was then transferred into every well in a flat-bottom 96-well plate, except for the first column. Inhibitors were added at specific starting concentrations (100 µM – 1 mM) with a total volume of 360 µL (diluted with working solution) into the first column. The inhibitors were then sequentially diluted two-fold across 12 wells. Plates were incubated at 37 °C, shaking at 200 rpm, for 12 hours. The OD600 values were then measured to determine ED50 values, using GraphPad Prism software [Version 7.04]. Experiments were carried out in duplicate or triplicate.
B. subtilis cell growth inhibition rescue assays:
An overnight starter culture (in LB both) of B. subtilis was diluted 1000-fold (in fresh LB media) and grown to an OD600 value of ~0.3 (approx. 3.5 hours at 37 °C). The working solution was prepared by 500-fold dilution of this log phase-culture into fresh LB broth. Then, 50 µM UP MK-4, FPP or HMBPP as well as the 6 pairwise combinations were prepared using the 500-fold diluted working solution; 200 µL of working solution was then transferred into every well in a flat-bottom 96-well plate, except for the first column. Inhibitors were added at specific starting concentrations (100 µM–1 mM) with a total volume of 300 µL (diluted with working solution) into the first column. The inhibitors were then sequentially diluted threefold across 12 wells. Plates were incubated at 37 °C, shaking at 200 rpm for 12 hours. The OD600 values were then measured to determine bacterial growth inhibition rescue effects, basically as described above for B. subtilis alone.
S. aureus growth inhibition assay:
An overnight starter culture of S. aureus was diluted 1000-fold (in fresh Tryptic soy media) and grown to an OD600 value of ~0.3 (approx. 3.5 hours at 37 °C). This log-phase culture was diluted 500-fold into fresh Tryptic soy broth to generate the working solution. 180 µL of this working solution was then transferred into every well in a flat-bottom 96-well plate, except for the first column. Inhibitors were added at specific starting concentrations (100 µM – 1 mM) with a total volume of 360 µL (diluted with working solution) into the first column. The inhibitors were then sequentially diluted twofold across 12 wells. Plates were incubated at 37 °C, shaking at 200 rpm for 12 hours. The OD600 values were then measured to determine bacterial growth inhibition.
B. anthracis Sterne growth inhibition assay:
An overnight starter culture of B. anthracis Sterne was diluted 1000-fold (in fresh Mueller-Hinton Broth 2 media) and grown to an OD600 value of ~0.3 (approx. 3.5 hours at 37 °C). This log-phase culture was diluted 500-fold into fresh MHB broth to generate the working solution. 180 µL of this working solution was then transferred into every well in a flat-bottom 96-well plate, except for the first column. Inhibitors were added at specific starting concentrations (100 µM–1 mM) with a total volume of 360 µL (diluted with working solution) into the first column. The inhibitors were then sequentially diluted twofold across 12 wells. Plates were incubated at 37 °C, shaking at 200 rpm for 12 hours. The OD600 value was then measured to determine bacterial growth inhibition.
M. smegmatis growth inhibition assay:
M. smegmatis (grown for 36–48 hours) was diluted 1000-fold in fresh Middlebrook 7H9 (plus 10% ADC supplement, Sigma: M0553–1VL; 0.5% glycerol; 0.05% Tween 80) media to generate a working solution. 200 µL of this working solution was then transferred into every well in a flat-bottom 96-well plate except for the second column and peripheral wells. Inhibitors were added at specific starting concentrations (100 µM–1 mM) with a total volume of 300 µL (diluted with working solution) into the second column. The inhibitors were then sequentially diluted threefold across 10 wells; 200 µL of water was added to each peripheral well to prevent water evaporation from the plate. Plates were incubated at 37 °C, shaking at 200 rpm for 48 hours. The OD600 values were then measured to determine bacterial growth inhibition, as described above.
C. difficile growth inhibition assay.
Clostridioides difficile were grown anaerobically on brain heart infusion supplemented agar plates (Brain heart infusion medium, BD, supplemented with yeast extract, L-cysteine, vitamin K1 and hemin) at 37˚C for 48 hours. Then, a bacterial solution equivalent to a 0.5 McFarland standard was prepared and diluted in brain heart infusion-supplemented broth to achieve a bacterial concentration of ~5 × 105 CFU/mL and cells were then seeded into 96-well well plates. Inhibitors were added at a concentration of 64 µM in the first row of the plates and diluted along the plates to achieve a range of 64–0.5 µM. Plates were then incubated anaerobically at 37˚C for 48 hours. MICs values reported are the minimum concentrations of the inhibitor that visually inhibited cell growth.
C. albicans growth inhibition assay:
C. albicans growth inhibition was carried out according to a reported protocol43 except that YPD media was used instead of RPMI 1640.
B. subtilis serum binding assay:
An overnight starter culture (in LB both) of B. subtilis was diluted 1000-fold (in fresh LB media) and grown to an OD600 value of ~0.3 (approx. 3.5 hours at 37 °C). A working solution was then prepared by 500-fold dilution of this log phase-culture into fresh LB broth. Then, 0%, 4%, and 10% of serum working solutions were prepared using the 500-fold diluted working solution; 200 µL of this working solution was then transferred into every well in a flat-bottom 96-well plate, except for the first column. Inhibitors were added at specific starting concentrations (100 µM–1 mM) with a total volume of 300 µL (diluted with working solution) into the first column. The inhibitors were then sequentially diluted threefold across 12 wells. Plates were incubated at 37 °C, shaking at 200 rpm for 12 hours. The OD600 values were then measured to determine bacterial growth inhibition.
E. coli growth inhibition assay:
An overnight starter culture of E. coli (K-12), was diluted 1000-fold and grown to an OD600 value of ~0.3. These log-phase cultures were then diluted 500-fold into fresh LB broth to generate a working solution; 200 μL of working solution was transferred into each well of a 96-well culture plate (Corning 3370). Inhibitors were then added at 1 mM and sequentially diluted threefold to 46 nM, keeping volume and culture broth composition constant. Plates were incubated for 12 h at 37 °C, shaking at 200 rpm. The OD600 value was then measured to determine bacterial growth inhibition.
Gram-negative bacterial cell growth inhibition assays:
As with the E. coli inhibition assays, overnight cultures (in cation-adjusted Mueller-Hinton broth, CAMHB) of A. baumannii; K. pneumoniae; and P. aeruginosa were diluted 1000-fold (in fresh CAMHB) to create a working solution. Working solutions were then transferred into flat-bottom 96-well plates and inhibitors added at 1 mM and sequentially diluted 3x to 46 nM. Plates were incubated at 37°C, shaking at 200 RPM, overnight. OD600 values were then measured to determine bacterial growth inhibition.
HEK293 cell growth inhibition assay:
A frozen stock of human embryonic kidney cells (HEK293, ATCC CRL-1573) was used to grow a first generation of cells in DMEM (4.5 g/L glucose with L-glutamine) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (10,000 U mL−1). This generation was harvested in 0.25% trypsin/2.1 µM EDTA, and cells were counted under a light microscope. A working solution was then generated containing 105 cells/mL, which was then transferred into every well of a flat-bottom 96-well plate, except for the second column and peripheral wells (the outermost row that encircles all of the inner wells). Then, 15–45 µL of 10 mM inhibitor solutions were added into the second column, diluted to a total volume of 150 µL using working solution. The inhibitors were then sequentially diluted threefold across 10 wells; 200 µL of PBS was added to each peripheral well to prevent evaporation. Plates were incubated at 37 °C for 24 or 96 hours. MTT solution (10 µL, 5 mg/mL in PBS) was then added to each well and the plate incubated for 4 hours. HCl in isopropanol (100 µL of a 100 mM solution) was added to each well and the absorbance at 570 nm measured to determine ED50 values. Experiments were carried out in duplicate.
Enzyme inhibition assays: General methods.
Enzyme inhibition was determined with a continuous spectrophotometric assay for inorganic phosphate release using the substrate 2-amino-6-mercapto-7-methylpurine (MESG) and the enzyme purine nucleoside phosphorylase (PNP).44 For the prenyltransferase assays, inorganic pyrophosphatase (Ppase) was added to the reaction mixture to convert each pyrophosphate to two inorganic phosphates. The inorganic phosphate then reacts with MESG in a PNP catalyzed reaction. The 2-amino-6-mercapto-7-methylpurine product has an absorption peak at 360 nm and coupled reactions catalyzed by Ppase and PNP are very rapid, so the kinetics of FPPS, OPPS, HepPPS, UPPS and UPPP can be directly monitored at 360 nm. The total volume of each reaction was 200 μL, with 100 μL reaction mix being added to 100 μL of protein mix. For the UPP phosphatase assay, there was no need to add Ppase (since Pi is a product). Reactions were continuously monitored on a spectrophotometer at room temperature until there was no longer any significant increase in absorbance at 360 nm, indicating reaction completion. 20 mM IPP, FPP, and GPP substrate stock solutions were prepared in water. Basically the same procedure was followed for inhibition assays except that prior to the addition of reaction mix to the protein mix, the protein was incubated with serial dilutions of the compound for 30 minutes, at room temperature. 10 mM compound stock solutions in H2O were used. All assays were performed at least 3 times on different days and with different inhibitor concentrations. All data sets (typically ~15 points) were then pooled, and doseresponse curves calculated in GraphPad Prism using a nonlinear regression function.
FPPS assay.
Expression, purification, and storage of HsFPPS was conducted as previously described.16 Assays were performed at 25˚C using a mixture of 50 μM IPP and 50 μM GPP, 0.3 units of PNP (Sigma-Aldrich, 10 units/mg), 0.6 unit of Ppase (Sigma-Aldrich, 500 units/mg), 0.1 mg/mL MESG, and 0.15 μM HsFPPS in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl and 500 μM MgCl2.
OPPS assay.
Expression, purification, and storage of EcOPPS were carried out as described previously.45 Assays were performed at 30°C in a reaction mixture of 50 μM IPP and 5 μM FPP, 125 nM EcOPPS, 0.3 unit of PNP, 0.6 unit of Ppase, 0.1 mg/mL MESG in activity buffer containing 100 mM HEPES-KOH (pH 7.5), 0.1% Triton X-100, 50 mM KCl and 0.5 mM MgCl2.
HepPPS assay.
Expression, purification, and storage of HepPPS was as described previously.4 Assays were conducted at 30°C in a reaction mixture of 40 μM IPP, 10 μM FPP, 300 nM SaHepPPS, 0.3 unit of PNP, 0.6 unit of Ppase, 0.1 mg/mL MESG in activity buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM MgCl2.
UPPS assay.
Expression, purification, and storage of EcUPPS was as in a previous paper.3 Assays were conducted at 25°C in a reaction mixture of 50 μM IPP, 6 μM FPP, 63 nM EcUPPS, 0.3 unit of PNP, 0.6 unit of Ppase, 0.1 mg/mL MESG in activity buffer containing 100 mM HEPES (pH 7.5), 50 mM KCl, 0.5 mM MgCl2, and 0.1% Triton X-100 (v/v).
UPPP assay.
Expression, purification, and storage of EcUPPP were as reported previously.46 Assays were performed at 25°C using a mixture of 25 μM FPP, 125 nM EcUPPP, 0.3 unit of PNP, 0.1 mg/mL MESG in activity buffer containing 50 mM HEPES (pH 7.0), 150 mM NaCl, 1 mM CaCl2 and 0.016% DDM (w/v).
Protein expression and purification for X-ray crystallography.
EcOPPS was expressed and purified as described previously.6 Briefly, a pET46Ek/LIC-EcOPPS plasmid was transformed into E. coli BL21trxB (DE3) for expression. EcOPPS was induced with 0.8 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 37 ºC for 4 h and the target protein purified by using a Ni-NTA column, then a DEAE Sepharose Fast Flow column (GE Healthcare Life Sciences). The eluted EcOPPS was then dialyzed twice against 5 L buffer (25 mM Tris•HCl, pH 7.5, 150 mM NaCl) and concentrated to 3 mg/mL for crystallization screening. The pET46Ek/LIC-EcUPPS plasmid was transformed into E. coli BL21(DE3) for expression. EcUPPS was induced with 1.0 mM IPTG at 37 ºC for 4 h, and the target protein purified by using a Ni-NTA column, then subjected to FXa digestion to remove the His tag. The mixture was then passed through a Ni-NTA column for purification.
Crystallization, data collection, structure determination and refinement.
All crystallization experiments were conducted at 22 ºC using the sitting-drop vapor-diffusion method. In general, 2 μL of EcOPPS or EcUPPS-containing solution (25 mM Tris•HCl, 150 mM NaCl, pH 7.5; 3 mg/mL) was mixed with 2 μL of reservoir solution in 24-well Cryschem Plates (Hampton Research) and then equilibrated against 300 μL of reservoir solution. The optimized crystallization condition for EcOPPS was 0.3 M MgCl2, 0.1 M Tris•HCl, pH 8.5, and 24% w/v PEG 3350. Within 3 to 4 days, the crystals reached dimensions suitable for X-ray diffraction. EcOPPS crystals in complex with 69 and 70 were obtained by soaking the apo-crystals with cro-protectant solution (0.3M MgCl2, 0.1M Tris•HCl, pH 8.5, 28 % w/v polyethylene glycol 3350, and 4 % v/v glycerol) containing 5 mM 69 or 5 mM 70, for 3 h. The optimized crystallization condition for EcUPPS was 20 % ethylene glycol and 5 % PEG 35000, and crystals were soaked with cryoprotectant solution containing 5 mM 70, 30 % ethylene glycol, and 5% PEG35000, for 3 h.
X-ray diffraction data-sets for EcOPPS in complex with 69 and 70 and EcUPPs in complex with 70 were collected at beam line BL13B1 of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). Crystals were mounted in a cryoloop and soaked with their cryoprotectant solution prior to data collection at 100 K. The diffraction images were processed by using HKL2000.47 Prior to structure refinement, 5 % randomly selected reflections were set aside for calculating Rfree48 as a monitor of model quality. The complex structures of EcOPPS•69 and EcOPPS•70 were determined by using the molecular replacement method with Phaser49 from the CCP4i program suite50 using the refined EcOPPS structure (PDB code 3WJK6) as a search model. The model and map were further improved by refinement using Refmac551 and Coot.52 The complex structure of EcUPPS•70 was determined by using the same method as for the EcOPPS structures, with the previously reported EcUPPS structure (PDB ID code 1X0653) as the search model. Data collection and refinement statistics are summarized in Table S4. All figures were prepared by using the PyMOL program (http://pymol.sourceforge.net/).
Supplementary Material
ACKNOWLEDGEMENTS.
This work was supported in part by the United States Public Health Service (National Institutes of Health grants GM065307 and CA158191); the National Natural Science Foundation of China (grants 31870790, 31470240, 31570130 and 31700057) and the Chinese Academy of Sciences (grant KFZD-SW-215–01). We thank Professor Douglas A. Mitchell for providing bacteria, and Xinxin Feng for carrying out initial cell growth inhibition assays.
ABBREVIATIONS USED
- FPPS
farnesyl diphosphate synthase
- UPPS
undecaprenyl diphosphate synthase
- SaHepPPS
S. aureus heptaprenyl diphosphate synthase
- EcOPPS
E. coli octaprenyl diphosphate synthase
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- HMBPP
1-hydroxy-2-methyl-but-2-enyl 4-diphosphate
- MEV
mevalonate
- DXP
deoxyxylulose-5-phosphate
- FPP
farnesyl diphosphate
- DHNA
1,4-dihydroxy-2-naphthoic acid
- UPP
undecaprenyl diphosphate
- MK
menaquinone
- DP
decaprenyl phosphate
- UP
undecaprenyl phosphate
- MenA
1,4-dihydroxy-2-naphthoate polyprenyl transferase
- GGPPS
geranylgeranyl diphosphate synthase
- IC50
half maximal inhibitory concentration
- ED50
effective dose for 50% inhibition of the population
- MIC
minimum inhibitory concentration required to inhibit the growth of 90% of organisms
- Bs
B. subtilis
- Ba
B. anthracis Sterne
- Sa
S. aureus
- Ms
M. smegmatis
- Sc
Saccharomyces cerevisae
- Pv
Plasmodium vivax
- HEK293
human embryonic kidney cell line # 293
- MESG
2-amino-6-mercapto-7-methylpurine
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
Footnotes
ASSOCIATED CONTENT
Supporting Information
Supporting Information consisting of Figures S1-S5, Tables S1-S5, synthesis schemes and procedures, compound characterization (1H, 31P, 19F, and 13C NMR, high-resolution mass spectrometry), purity by 1H qNMR and molecular formula strings (CSV) is available free of charge at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
PDB ID Codes
Authors will release the atomic coordinates and experimental data upon article publication. PDB IDs are provided in the Figure legends.
REFERENCES
- 1.Liu CI; Liu GY; Song Y; Yin F; Hensler ME; Jeng WY; Nizet V; Wang AH; Oldfield E, A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu W; Zhang Y; Sinko W; Hensler ME; Olson J; Molohon KJ; Lindert S; Cao R; Li K; Wang K; Wang Y; Liu YL; Sankovsky A; de Oliveira CA; Mitchell DA; Nizet V; McCammon JA; Oldfield E, Antibacterial drug leads targeting isoprenoid biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu W; Wang Y; Li K; Gao J; Huang CH; Chen CC; Ko TP; Zhang Y; Guo RT; Oldfield E, Antibacterial drug leads: DNA and enzyme multitargeting. J. Med. Chem 2015, 58, 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai J; Liu YL; Wei H; Liu W; Ko TP; Guo RT; Oldfield E, Structure, function, and inhibition of Staphylococcus aureus heptaprenyl diphosphate synthase. ChemMedChem 2016, 11, 1915–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okada K; Minehira M; Zhu X; Suzuki K; Nakagawa T; Matsuda H; Kawamukai M, The ispB gene encoding octaprenyl diphosphate synthase is essential for growth of Escherichia coli. J. Bacteriol 1997, 179, 3058–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han X; Chen CC; Kuo CJ; Huang CH; Zheng Y; Ko TP; Zhu Z; Feng X; Wang K; Oldfield E; Wang AH; Liang PH; Guo RT; Ma Y, Crystal structures of ligand-bound octaprenyl pyrophosphate synthase from Escherichia coli reveal the catalytic and chain-length determining mechanisms. Proteins 2015, 83, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tarshis LC; Yan M; Poulter CD; Sacchettini JC, Crystal structure of recombinant farnesyl diphosphate synthase at 2.6-Å resolution. Biochemistry 1994, 33, 10871–10877. [DOI] [PubMed] [Google Scholar]
- 8.Hosfield DJ; Zhang Y; Dougan DR; Broun A; Tari LW; Swanson RV; Finn J, Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J. Biol. Chem 2004, 279, 8526–8529. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki D; Fujihashi M; Okuyama N; Kobayashi Y; Noike M; Koyama T; Miki K, Crystal structure of heterodimeric hexaprenyl diphosphate synthase from Micrococcus luteus B-P 26 reveals that the small subunit is directly involved in the product chain length regulation. J. Biol. Chem 2011, 286, 3729–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CK-M; Hudock MP; Zhang Y; Guo RT; Cao R; No JH; Liang PH; Ko TP; Chang TH; Chang SC; Song Y; Axelson J; Kumar A; Wang AH; Oldfield E, Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: a crystallographic and computational investigation. J. Med. Chem 2008, 51, 5594–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y; Desai J; Zhang Y; Malwal SR; Shin CJ; Feng X; Sun H; Liu G; Guo RT; Oldfield E, Bacterial Cell Growth Inhibitors Targeting Undecaprenyl Diphosphate Synthase and Undecaprenyl Diphosphate Phosphatase. ChemMedChem 2016, 11, 2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo RT; Cao R; Liang PH; Ko TP; Chang TH; Hudock MP; Jeng WY; Chen CK; Zhang Y; Song Y; Kuo CJ; Yin F; Oldfield E; Wang AH, Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai J; Wang Y; Wang K; Malwal SR; Oldfield E, Isoprenoid biosynthesis inhibitors targeting bacterial cell growth. ChemMedChem 2016, 11, 2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leon A; Liu L; Yang Y; Hudock MP; Hall P; Yin F; Studer D; Puan KJ; Morita CT; Oldfield E, Isoprenoid biosynthesis as a drug target: bisphosphonate inhibition of Escherichia coli K12 growth and synergistic effects of fosmidomycin. J. Med. Chem 2006, 49, 7331–7341. [DOI] [PubMed] [Google Scholar]
- 15.Rondeau JM; Bitsch F; Bourgier E; Geiser M; Hemmig R; Kroemer M; Lehmann S; Ramage P; Rieffel S; Strauss A; Green JR; Jahnke W, Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem 2006, 1, 267–273. [DOI] [PubMed] [Google Scholar]
- 16.Kavanagh KL; Guo K; Dunford JE; Wu X; Knapp S; Ebetino FH; Rogers MJ; Russell RG; Oppermann U, The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 7829–7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin MB; Arnold W; Heath HT 3rd; Urbina JA; Oldfield E, Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem. Biophys. Res. Commun 1999, 263, 754–758. [DOI] [PubMed] [Google Scholar]
- 18.Sanders JM; Song Y; Chan JM; Zhang Y; Jennings S; Kosztowski T; Odeh S; Flessner R; Schwerdtfeger C; Kotsikorou E; Meints GA; Gomez AO; Gonzalez-Pacanowska D; Raker AM; Wang H; van Beek ER; Papapoulos SE; Morita CT; Oldfield E, Pyridinium-1-yl bisphosphonates are potent inhibitors of farnesyl diphosphate synthase and bone resorption. J. Med. Chem 2005, 48, 2957–2963. [DOI] [PubMed] [Google Scholar]
- 19.Mao J; Mukherjee S; Zhang Y; Cao R; Sanders JM; Song Y; Zhang Y; Meints GA; Gao YG; Mukkamala D; Hudock MP; Oldfield E, Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase-bisphosphonate complexes. J. Am. Chem. Soc 2006, 128, 14485–14497. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y; Cao R; Yin F; Lin FY; Wang H; Krysiak K; No JH; Mukkamala D; Houlihan K; Li J; Morita CT; Oldfield E, Lipophilic pyridinium bisphosphonates: potent γδ T cell stimulators. Angew. Chem. Int. Ed. Engl 2010, 49, 1136–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J; Zielinski M; Magder A; Tsantrizos YS; Berghuis AM, Human farnesyl pyrophosphate synthase is allosterically inhibited by its own product. Nat. Commun 2017, 8, 14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jahnke W; Rondeau JM; Cotesta S; Marzinzik A; Pelle X; Geiser M; Strauss A; Gotte M; Bitsch F; Hemmig R; Henry C; Lehmann S; Glickman JF; Roddy TP; Stout SJ; Green JR, Allosteric non-bisphosphonate FPPS inhibitors identified by fragment-based discovery. Nat. Chem. Biol 2010, 6, 660–666. [DOI] [PubMed] [Google Scholar]
- 23.Dhiman RK; Mahapatra S; Slayden RA; Boyne ME; Lenaerts A; Hinshaw JC; Angala SK; Chatterjee D; Biswas K; Narayanasamy P; Kurosu M; Crick DC, Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol 2009, 72, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li K; Schurig-Briccio LA; Feng X; Upadhyay A; Pujari V; Lechartier B; Fontes FL; Yang H; Rao G; Zhu W; Gulati A; No JH; Cintra G; Bogue S; Liu YL; Molohon K; Orlean P; Mitchell DA; Freitas-Junior L; Ren F; Sun H; Jiang T; Li Y; Guo RT; Cole ST; Gennis RB; Crick DC; Oldfield E, Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem 2014, 57, 3126–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farha MA; Czarny TL; Myers CL; Worrall LJ; French S; Conrady DG; Wang Y; Oldfield E; Strynadka NC; Brown ED, Antagonism screen for inhibitors of bacterial cell wall biogenesis uncovers an inhibitor of undecaprenyl diphosphate synthase. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11048–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K; Wang W; No JH; Zhang Y; Zhang Y; Oldfield E, Inhibition of the Fe(4)S(4)-cluster-containing protein IspH (LytB): electron paramagnetic resonance, metallacycles, and mechanisms. J. Am. Chem. Soc 2010, 132, 6719–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin MB; Sanders JM; Kendrick H; de Luca-Fradley K; Lewis JC; Grimley JS; Van Brussel EM; Olsen JR; Meints GA; Burzynska A; Kafarski P; Croft SL; Oldfield E, Activity of bisphosphonates against Trypanosoma brucei rhodesiense. J. Med. Chem 2002, 45, 2904–2914. [DOI] [PubMed] [Google Scholar]
- 28.Biegel E; Schmidt S; Gonzalez JM; Muller V, Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci 2011, 68, 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chowdhury NP; Klomann K; Seubert A; Buckel W, Reduction of flavodoxin by electron bifurcation and sodium ion-dependent reoxidation by NAD+ catalyzed by ferredoxin-NAD+ reductase (Rnf). J. Biol. Chem 2016, 291, 11993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown BM; Wang Z; Brown KR; Cricco JA; Hegg EL, Heme O synthase and heme A synthase from Bacillus subtilis and Rhodobacter sphaeroides interact in Escherichia coli. Biochemistry 2004, 43, 13541–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruble JC; Wakefield BD; Kamilar GM; Marotti KR; Melchior E; Sweeney MT; Zurenko GE; Romero DL, Structure-activity relationships of bioisosteres of a carboxylic acid in a novel class of bacterial translation inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 4040–4043. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki T; Zhang YW; Koyama T; Sasaki DY; Kurihara K, Direct observation of substrate-enzyme complexation by surface forces measurement. J. Am. Chem. Soc 2006, 128, 15209–15214. [DOI] [PubMed] [Google Scholar]
- 33.Concha N; Huang J; Bai X; Benowitz A; Brady P; Grady LC; Kryn LH; Holmes D; Ingraham K; Jin Q; Pothier Kaushansky L; McCloskey L; Messer JA; O’Keefe H; Patel A; Satz AL; Sinnamon RH; Schneck J; Skinner SR; Summerfield J; Taylor A; Taylor JD; Evindar G; Stavenger RA, Discovery and characterization of a class of pyrazole inhibitors of bacterial undecaprenyl pyrophosphate synthase. J. Med. Chem 2016, 59, 7299–7304. [DOI] [PubMed] [Google Scholar]
- 34.Xia Y; Liu YL; Xie Y; Zhu W; Guerra F; Shen S; Yeddula N; Fischer W; Low W; Zhou X; Zhang Y; Oldfield E; Verma IM, A combination therapy for KRAS-driven lung adenocarcinomas using lipophilic bisphosphonates and rapamycin. Sci. Transl. Med 2014, 6, 263ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia Y; Xie Y; Yu Z; Xiao H; Jiang G; Zhou X; Yang Y; Li X; Zhao M; Li L; Zheng M; Han S; Zong Z; Meng X; Deng H; Ye H; Fa Y; Wu H; Oldfield E; Hu X; Liu W; Shi Y; Zhang Y, The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell 2018, 175, 1059–1073. [DOI] [PubMed] [Google Scholar]
- 36.Kudoh T; Park CS; Lefurgy ST; Sun M; Michels T; Leyh TS; Silverman RB, Mevalonate analogues as substrates of enzymes in the isoprenoid biosynthetic pathway of Streptococcus pneumoniae. Bioorg. Med. Chem 2010, 18, 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danley DE; Baima ET; Mansour M; Fennell KF; Chrunyk BA; Mueller JP; Liu S; Qiu X, Discovery and structural characterization of an allosteric inhibitor of bacterial cis-prenyltransferase. Protein Science 2015, 24, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murthi KK; Kostler R; Smith C; Brandstetter T; Kluge AF, Derivatives of squaric acid with anti-proliferative activity. US 20080200523 A1, September 21, 2007.
- 39.Zhang Y; Leon A; Song Y; Studer D; Haase C; Koscielski LA; Oldfield E, Activity of nitrogen-containing and non-nitrogen-containing bisphosphonates on tumor cell lines. J. Med. Chem 2006, 49, 5804–5814. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto K; Hayashi K; Murata-Hirai K; Iwasaki M; Okamura H; Minato N; Morita CT; Tanaka Y, Targeting cancer cells with a bisphosphonate prodrug. ChemMedChem 2016, 11, 2656–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Recher M; Barboza AP; Li ZH; Galizzi M; Ferrer-Casal M; Szajnman SH; Docampo R; Moreno SN; Rodriguez JB, Design, synthesis and biological evaluation of sulfur-containing 1,1-bisphosphonic acids as antiparasitic agents. Eur. J. Med. Chem 2013, 60, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin MB; Grimley JS; Lewis JC; Heath HT 3rd; Bailey BN; Kendrick H; Yardley V; Caldera A; Lira R; Urbina JA; Moreno SN; Docampo R; Croft SL; Oldfield E, Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J. Med. Chem 2001, 44, 909–916. [DOI] [PubMed] [Google Scholar]
- 43.CLSI. Reference Method for Broth dilution Antifungal Susceptibility Testing of Yeasts. In Approved Standard-Third ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2008. [Google Scholar]
- 44.Webb MR, A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 4884–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X; Han X; Ko TP; Chen CC; Zhu Z; Hua E; Guo RT; Huang CH, Preliminary X-ray diffraction analysis of octaprenyl pyrophosphate synthase from Escherichia coli. Acta. Crystallogr 2013, F69, 328–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Touze T; Blanot D; Mengin-Lecreulx D, Substrate specificity and membrane topology of Escherichia coli PgpB, an undecaprenyl pyrophosphate phosphatase. J. Biol. Chem 2008, 283, 16573–16583. [DOI] [PubMed] [Google Scholar]
- 47.Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr 1993, D49, 24–36. [DOI] [PubMed]
- 49.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Potterton E; Briggs P; Turkenburg M; Dodson E, A graphical user interface to the CCP4 program suite. Acta Crystallogr 2003, D59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- 51.Murshudov GN; Skubak P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA, REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr 2011, D67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of coot. Acta Crystallogr 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo RT; Ko TP; Chen AP; Kuo CJ; Wang AH; Liang PH, Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate: roles of the metal ion and conserved residues in catalysis. J. Biol. Chem 2005, 280, 20762–20774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.