

Abstract
Alzheimer's disease (AD) and type 2 diabetes mellitus (T2DM) are highly prevalent aging‐related diseases associated with significant morbidity and mortality. Some findings in human and animal models have linked T2DM to AD‐type dementia. Despite epidemiological associations between the T2DM and cognitive impairment, the interrelational mechanisms are unclear. The preponderance of evidence in longitudinal studies with autopsy confirmation have indicated that vascular mechanisms, rather than classic AD‐type pathologies, underlie the cognitive decline often seen in self‐reported T2DM. T2DM is associated with cardiovascular and cerebrovascular disease (CVD), and is associated with increased risk of infarcts and small vessel disease in the brain and other organs. Neuropathological examinations of post‐mortem brains demonstrated evidence of cerebrovascular disease and little to no correlation between T2DM and β‐amyloid deposits or neurofibrillary tangles. Nevertheless, the mechanisms upstream of early AD‐specific pathology remain obscure. In this regard, there may indeed be overlap between the pathologic mechanisms of T2DM/“metabolic syndrome,” and AD. More specifically, cerebral insulin processing, glucose metabolism, mitochondrial function, and/or lipid metabolism could be altered in patients in early AD and directly influence symptomatology and/or neuropathology.
Keywords: preclinical, pathogenesis, VCID, neuropathology, mitochondria, epidemiology
Introduction
The purpose of this article is to describe potential aspects of distinction and commonality between type 2 diabetes mellitus (T2DM) and Alzheimer's disease (AD). T2DM is a chronic metabolic disease characterized by hyperglycemia, insulin resistance (IR), and loss of pancreatic β‐cell function 137. The majority of diabetes cases worldwide are T2DM. Between 1980 and 2014, the global prevalence was reported to have risen from 108 to 422 million 108. T2DM typically presents with increased thirst, fatigue, frequent urination, and delayed wound healing 137. Major complications of T2DM include retinopathy, kidney failure, heart disease, cerebrovascular disease (CVD), neuropathy, and limb amputation 137.
AD is a neurodegenerative disease with an insidious onset and progressive course 23, 39. It is the most common form of dementia a contributing factor in approximately 70% of all dementia cases 23, 39. The neuropathologic hallmarks of AD are extracellular β‐amyloid peptide deposits, which are recognized as “amyloid plaques,” and intracellular hyperphosphorylated tau deposition which forms neurofibrillary tangles (NFT) when occurring in nerve cells 11. The typical course of AD is characterized by impairment of various cognitive domains including memory, executive function, and often comorbid psychiatric changes, ultimately culminating in death 23, 39. While speed of progression varies, the average life expectancy after diagnosis is approximately nine years 148, 185.
In this review, discussion will initially focus on existing evidence that T2DM related cognitive decline is not associated with increased AD‐type neuropathology, but is instead mediated by cerebrovascular pathology. Next, we address the emerging role of glucose, mitochondrial, and lipid metabolism abnormalities as upstream components of AD clinical and neuropathological features. The review will finish by discussing hypothesized causes of cognitive decline in T2DM patients, with or without comorbid AD.
T2DM association with clinically and pathologically defined AD
T2DM is associated with the clinical features of cognitive decline and AD‐type dementia
Cognitive dysfunction is a relatively poorly understood complication of T2DM; see Figure 1. Multiple epidemiological studies link T2DM to cognitive decline and clinically diagnosed AD 27, 117, 119, 145, 187, 198, 208, however, not all studies demonstrate this association (see below). While there may be an association between T2DM, cognitive decline, and AD‐type dementia, most of these studies lack correlation with neuropathology, ie, autopsy confirmation. A 2013 systemic review and meta‐analysis evaluated published studies to better delineate the relationship between T2DM and AD 190. The meta‐analysis identified 15 studies conducted between 1998 and 2012. Nine studies found a statistically significant correlation between T2DM and AD with risk estimates ranging from 0.83 to 2.45. Five of these studies evaluated the interaction between T2DM and Apolipoprotein E epsilon 4 allele (APOE ε4), and three of these five studies demonstrated significant association with odds ranging from 2.4 to 4.99. While there is a reported epidemiological risk association between T2DM and clinical AD, Vagelatos and Eslick noted that this association has a major confounder—cerebral infarcts 190. They found that infarcts are more common in patients with T2DM and were associated with the development of clinical AD. Based on neuropathological examination, the authors concluded: (1) cerebral infarcts are more common in T2DM than AD neuropathology is; (2) Patients with clinical dementia have both infarcts and AD‐type neuropathology on post‐mortem exam; (3) Cerebral infarcts reduce the number of AD‐type lesions needed to cause clinical dementia but do not necessarily interact synergistically with AD‐type pathology. Additionally, a recent review evaluated the association between T2DM and clinical AD diagnoses, and highlighted the complexity of the related scientific literature 94. The authors examined studies since 2015 and included a total of 10 articles. According to the authors, only 2 of 10 studies found that T2DM was independently related to cognitive decline in AD dementia.
Figure 1.
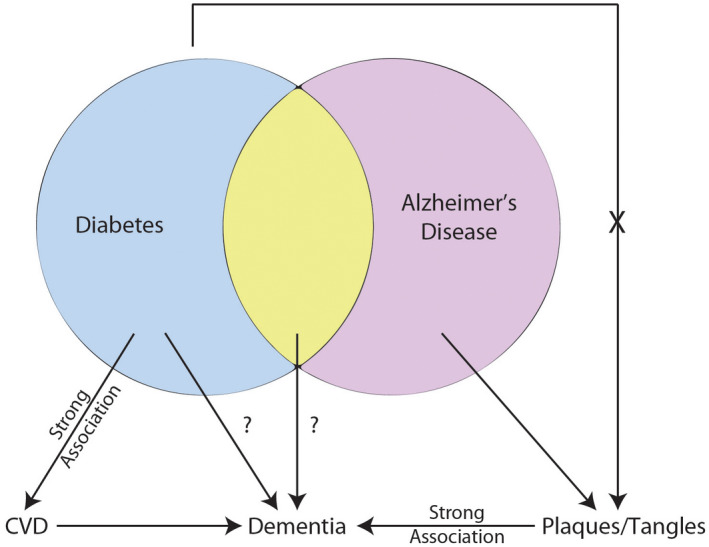
Relationship between T2DM and AD and cognitive decline. Diabetes, specifically T2DM, has a strong association with CVD that causes dementia through generation of subcortical and cortical infarcts. T2DM has been linked with dementia and AD, however, the mechanism(s) are uncertain. Amyloid plaques and neurofibrillary tangles have a strong association with cognitive status and to date, T2DM has not been associated with increased levels of plaques and tangles. T2DM, type 2 diabetes mellitus; CVD, cerebrovascular disease; AD, Alzheimer's Disease.
From a neuropathological standpoint, T2DM cognitive decline is not associated with AD lesions
The large majority of autopsy (neuropathologic) studies report no association between T2DM and amyloid plaques or NFTs 2, 10, 13, 35, 67, 76, 109, 110, 126, 140, 176. A recent multi‐center study evaluated 2365 autopsied patients with >1300 patients having available cognitive data 1. The authors concluded that T2DM status is associated with altered likelihood of being diagnosed during life with clinical “Probable AD”; yet, at autopsy, there was no association between T2DM and AD pathology. The authors utilized logistic regression modeling to evaluate the association between diabetes, CVD pathology, Braak NFT stage, and neuritic amyloid plaque score. The presence of T2DM was associated with increased odds of brain infarcts (OR = 1.57), specifically lacunae (OR = 1.71). T2DM with infarcts was associated with lower cognitive scores at end of life relative to T2DM without infarcts. Studies that have arrived at the conclusion that T2DM is associated with AD pathological hallmarks are few in number and characterized by subgrouping to determine a “positive” association. Overall, in dozens of papers, the null hypothesis—T2DM is not associated with AD‐type pathology—has been tested repeatedly, and has been strongly supported.
Cerebrovascular disease contributes to cognitive decline in T2DM
T2DM is a known risk factor for CVD 81. T2DM is associated with acute cerebral infarcts and increased stroke/brain infarction risk 44, 71. Many clinical–radiological studies report that cerebral infarcts are significantly associated with increased odds of developing dementia 38, 189, 193. This association may help account for the reported epidemiological association between T2DM and dementia 181.
Multiple mechanisms underlie CVD in T2DM. In terms of large‐vessel pathologies, vascular complications of T2DM are mediated at least partly through chronic hyperglycemia and production of reactive oxygen species (ROS) that apparently damage the vessel endothelium, and lead to atherosclerosis. Insult to vascular endothelium activates thrombotic cascades and recruits T‐cells, macrophages, and mononuclear leukocytes, impairing vascular integrity 211. From autopsy studies, T2DM is associated with cortical and subcortical atherosclerosis, and intracranial vascular stenosis is more common in those with T2DM than those without 8, 85, 111. Therefore, it is likely that T2DM association with cognitive decline is partly mediated through accelerated atherosclerosis in large blood vessels.
While microinfarcts are invisible to most radiological and gross examination techniques (by definition), they are well described in neuropathological literature and are often detected during post‐mortem microscopic examination. The location of microinfarcts (ie, cortical vs subcortical) correlates with disease subtypes. Cortical microinfarcts have been associated with cerebral amyloid angiopathy (CAA), subcortical microinfarcts with hypertensive encephalopathy, and periventricular microinfarcts with normal pressure hydrocephalus 3, 6, 37. Microinfarcts are often located in subcortical areas in diabetics; however, cortical infarcts and lacunes have also been described 1, 10, 146, 176. T2DM‐associated microinfarcts often coexist with AD neuropathological changes, however, the number of microinfarcts is not necessarily related to the severity of AD neuropathology 9, 129, 165. The typical reported microinfarct size is 0.2 mm, however, they range from 0.2 to 2.9 mm 9, 136. Several prospective cohort autopsy‐based studies evaluated the associative effect(s) of microinfarcts on cognition 9, 19, 60, 61, 120, 129, 165, 177, 180, 189, 201. According to these studies, microinfarcts are present in 18%–40% of persons, while four studies found that the prevalence of microinfarcts was higher in those with dementia than those without dementia 9, 165, 177, 180. These studies concluded that microinfarcts are independent predictors of dementia.
Classic hallmarks of AD: correlation with cognitive status and the question of “upstream” factors
Before focusing in on the possible overlapping pathogenetic mechanisms of AD and T2DM, an important concept to address is the AD‐specific lesions themselves—β‐amyloid plaques and NFTs. There has been some controversy about whether β‐amyloid and NFTs are deleterious, whether they should be considered “disease‐defining,” and/or whether these lesions are specifically associated with cognitive impairment 25, 125, 173, 175. Numerous factors have contributed to this confusion, including the strong influence of impactful “mixed” and non‐AD pathologies, some of which (eg, TDP‐43 pathology) were only relatively recently discovered 128, and others of which (eg, small vessel pathologies) have only recently been appreciated to have an association with cognitive impairment independent of other brain lesions 74, 78, 97. There also are notable biases in terms of the research volunteers that are drawn from dementia clinics 124, 164, and limitations associated with studying elderly individuals without carefully documented antemortem cognitive status. Furthermore, the dichotomous approach of “dementia yes/no” (and even the corresponding dichotomous assessment of pathologies) is prone to bias as the results are dependent on the application of imperfect and arbitrary diagnostic thresholds. Over the past several decades, new research contributions have come from large community‐based autopsy series with a new standard of cognitive assessments and longitudinal follow‐up; from biomarker (neuroimaging and body fluid) studies in clinical series; and, from genetic studies with large sample sizes and carefully assessed phenotypes. These approaches have led to an improved appreciation of, and insights into, the heterogeneity and complexity of what occurs in the aged human brain. The direct “toxicity” of β‐amyloid and NFTs still remains to be definitively proven, and autopsy evaluation is intrinsically cross sectional. However, to summarize recent studies: an evolving scientific literature has provided strong support for the hypothesis that β‐amyloid and NFTs are a part of a devastating organic disease within the complex milieu of the aged human brain, with strong adverse impact on brain function 125. For these reasons, these classic hallmarks still constitute the “gold standard” for disease instantiation and severity.
Even if one accepts the concept that β‐amyloid and NFTs “define” AD, there remain critically important questions: what occurs upstream of plaques and tangles? Can a person have the AD disease “phenotype” even before the pathologies are present? Are there common, clinically impactful features of AD that are parallel and separate from plaques and tangles? These questions get to the critical issues of causative “upstream” factors. Clearly, there is a strong genetic component to AD risk, involving APOE and other genes 163, but the exact genotype/phenotype mechanisms are still incompletely understood. To date, the association(s) between specific environmental factors and AD risk is even more controversial. Developing study designs to address those uncertainties in human studies is challenging because any influence that negatively impacts cognition increases the chance of diagnosis of AD‐type dementia, whether or not AD pathologies are the factors that underlie the symptoms. Here, we attempt to explore how the upstream biochemical pathways that contribute to AD‐type dementia may overlap with T2DM, with the important caveat that classical AD pathologic hallmarks may be irrelevant to some of these pathways.
Glucose and metabolic dysregulation in AD
While there is a wealth of information about AD pathophysiology, the initial events upstream of NFT formation and β‐amyloid deposition are still unclear. Metabolic dysregulation has been shown to be a possible contributory factor for AD neuropathology. Alterations in cerebral glucose metabolism, mitochondrial function, and lipid metabolism may be upstream triggers for NFT and β‐amyloid deposition. The next section will focus on these topics and describe the links between metabolism and AD.
AD and glucose metabolism
Studies that used fluorodeoxyglucose (FDG) positron emission tomography (PET) imaging has been interpreted to indicate likely glucose metabolism abnormalities, and/or synapse loss, in patients at risk of AD, well before symptom onset. This FDG PET neuroimaging modality has indicated that cerebral glucose metabolism may be impaired early in the adult life of persons at genetic risk for developing AD. While the exact mechanism(s) underlying this phenomenon are currently unknown, deficits in FDG PET tracer uptake begin decades before clinical onset of symptoms and—perhaps more importantly—well before the age range where abundant plaques and tangles are observed 5, 155, 160, 162.
There has been some controversy about the pathophysiologic implications of FDG PET studies. Since the radiolabeled FDG is taken up by cells but not metabolized, its uptake is largely dependent on its partitioning in the blood stream (ie, on blood flow), and hence it does not necessarily provide direct information on glucose metabolism per se, except to the extent that that affects blood flow. This neuroimaging modality also does not provide direct information on synapse loss. There are additional technical questions related to the FDG PET modality, since the observed phenomena in individuals at risk for AD may be attenuated by partial volume correction 73, 206. Knopman et al. addressed this concern by increasing the power of their study and utilizing a larger patient cohort. His group found that there was a modest age‐related reduction in cerebral glucose metabolism, and the presence of at least one APOE ε4 allele was associated with lower glucose metabolism measured in the posterior cingulate, precuneus, and/or lateral parietal regions 87. These results are similar to those found by Reiman et al. as indicated above 156.
Complementing the studies of preclinical disease, other studies have provided additional evidence that were interpreted to indicate that persons with AD‐type dementia have substantially reduced rates of cerebral glucose metabolism in posterior cingulate, parietal, temporal, and prefrontal cortices 112, 113, 114, 174. This phenomenon was demonstrated in studies conducted by Herholz et al. and Loessner et al. and was later confirmed by others 68, 130, 155, 156. Additional studies found that levels of glucose transporters in brain microvessels, frontal cortex, hippocampus, caudate nucleus, parietal, and temporal lobes were reduced in AD patients when compared with controls on autopsy studies 79, 170. A recent study using the Baltimore Longitudinal Study of Aging autopsy cohort provided further evidence supporting the hypothesis that glucose metabolism is affected early in AD 5. The authors found that higher brain tissue glucose concentrations (neural insulin resistance) and lower GLUT3 levels were associated with severity of AD neuropathology and AD clinical presentation. From these studies, the possibility emerges that glucose dysmetabolism is somehow correlated with AD‐type pathologies per se, either as a causative factor or otherwise as part of the syndrome.
Mitochondrial dysregulation in AD
Alterations of mitochondria in AD were reported as early as the 1960s 52; See Figure 2. Subsequently, studies have reported that mitochondrial structure was altered, oxygen consumption reduced, and mitochondrial‐localized enzyme activities were affected in AD 51, 53, 57, 77, 142, 202. Furthermore, mitochondrial mass, size, and copy number were shown to be reduced in AD brains, and this was linked to mitochondrial interaction with β‐amyloid peptide deposits 12, 40. Cardoso et al. demonstrated that ρ0 cells were protected from β‐amyloid peptide exposure, supporting the hypothesis that β‐amyloid peptide is detrimental to mitochondrial function 24. Others have shown that β‐amyloid peptide is capable of interacting with β‐amyloid peptide‐binding alcohol dehydrogenase (ABAD) and cyclophillin D 42, 103. Interactions of β‐amyloid peptide with ABAD deform the enzyme and prevents its interaction with nicotinamide adenine dinucleotide (NAD). When β‐amyloid peptide interacts with cyclophilin D, this causes increased mitochondrial membrane permeability. The detrimental effects of β‐amyloid peptide on the mitochondria may cause a compensatory response by increasing mitochondrial fission. The actions of mitochondrial‐shaping proteins (OPA1, MFN1, MFN2, DRP1, FIS1) play a vital role in shaping and modifying mitochondrial structure during fusion and fission 210. Specifically, mediators of mitochondrial fusion (OPA1, MNF1, and MFN2) were reduced in degenerating neurons and mediators of mitochondrial fission (FIS1, DRP1) were increased 106, 195, 196, 197. While these studies are important for elucidating the effect of β‐amyloid peptide on mitochondrial metabolism and dynamics, they do not necessarily imply a reciprocal effect—they do not prove that mitochondrial dysfunction promotes β‐amyloid peptide deposition.
Figure 2.
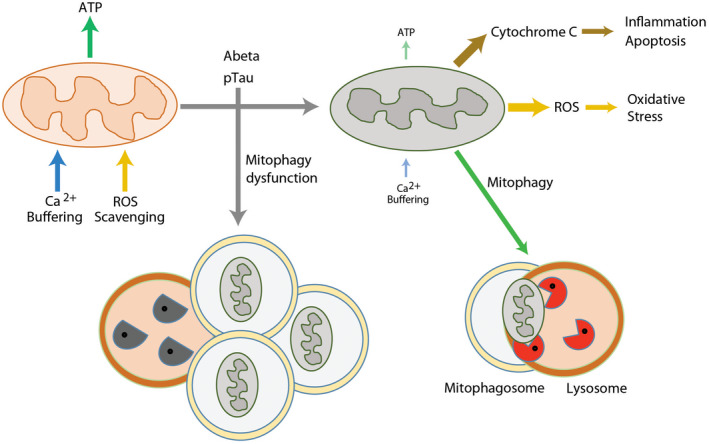
Mitochondrial autophagy in AD. Mitochondrial dysfunction could play key roles in AD pathogenesis. Damaged mitochondria not only compromise the production of cellular energy and lose the capacity for Ca2+ buffering, they also release harmful ROS and cytochrome C resulted in activation of destructive pathways. Mitophagy is a mechanism for removing aged or damaged mitochondria, however, this mechanism is impaired in AD. Functionally defective mitochondria and insufficient clearance of the damaged organelles and macromolecules may synergistically intensify the detrimental pathways of AD. ATP, adenosine triphosphosphate; ROS, reactive oxygen species.
The potential for mitochondrial metabolism to affect β‐amyloid peptide production was demonstrated when cultured SH‐SY5Y cell lines expressed AD‐ and wt‐mtDNA 83. These AD‐mtDNA “cybrid” cell preparations generated increased intracellular and extracellular levels of β‐amyloid peptide relative to controls. This result suggests that AD‐mtDNA causes functional changes which may potentiate Aβ plaque formation. Additionally, another study found that when COS cells undergo glucose deprivation, levels of α‐secretase‐derived APP product increase 56. This alteration is perhaps relevant because AD patients have reduced rates of cerebral glucose metabolism in posterior cingulate, parietal, temporal, and prefrontal cortices 112, 113, 114, 174 , as stated above. These data may indicate how reduced glucose metabolism is linked to AD phenotype though dysregulation of mitochondria, which, as indicated in the prior paragraph, may be a self‐propagating cycle with increasing β‐amyloid peptide production and worsening mitochondrial dysfunction.
Mitochondrial autophagy in AD
Both neurodegeneration and diabetes are associated with oxidative stress and inflammatory conditions in the CNS that may be mediated partly by mitochondria dysfunction. Dysfunctional mitochondria may be compromised in the production of cellular energy and lose the capacity for buffering intracellular Ca2+, and they also can release harmful ROS. Uncontrolled oxidative stress triggers the discharge of cytochrome C and activates the pro‐death apoptotic cascade 115. Increased oxidative stress also results from an imbalance in production of ROS and cells’ ROS scavenging systems from defective mitochondria. The inhibition of the clearance of damaged mitochondria, accompanied by the concurrent oxidative stress and inflammatory condition, may synergistically affect the health of neurons in AD and other conditions.
In healthy cells, mitochondria are turned over—damaged mitochondria are selectively identified, ubiquitinated, and degraded via an autophagy pathway termed “mitophagy.” This pathway is particularly important in long‐lived (post‐mitotic) cells such as neurons. Mitophagy selectively sequesters abnormal mitochondria to form autophagosomes and subsequently deliver the cargo to lysosomes for degradation. Mitophagy plays a key role in mitochondrial quality control and is an essential mechanism in tissue maintenance and cellular homeostasis, and the literature that pertains to autophagic dysregulation in AD may also be germane to mitophagy. Dysregulation of autophagy has been associated with AD pathogenesis. Nixon et al. reported the accumulation of immature autophagic vacuoles (AVs) in dystrophic neurites of AD brains 133. Later reports conflicted on how autophagy flux was affected in AD and what specific stages were dysregulated in human brains 134. Recently, Bordi et al. assessed the autophagy pathways and autophagy flux by performing microarray and immunochemical analyses of hippocampal CA1 neurons in post‐mortem tissue samples from AD subjects at different stages of disease 18. This study revealed that autophagy is upregulated and lysosomal biogenesis is increased in the early stage of AD (~10 years before clinical AD diagnosis). Additionally, autophagic flux was obstructed due to the impairment in the clearance of autophagic substrates. These studies indicate that the regulation of mitophagy plays important roles in mitochondrial homeostasis, however, how those molecular pathways interact with β‐amyloid and pTau remains mostly unknown.
Lipid metabolism dysfunction in AD
Lipid dysmetabolism is a component of the metabolic syndrome that occurs in many T2D patients, and multiple lines of evidence have implicated perturbations in lipid biochemistry in AD. The brain is one of the most lipid‐enriched organs and is partly composed of a variety of lipids such as glycerophospholipids (GPs), sphingolipids, and cholesterol 17. The involvement of lipid metabolism in the pathogenesis of AD was suspected when brains of AD patients were examined post‐mortem and found to contain “adipose inclusions” or “lipid granules” 48. Alois Alzheimer originally described this finding in his milestone study of the brain of Auguste Deter 4.
After the discovery that APOE ε4 allele is the strongest genetic risk factor for late onset AD, interest in lipid metabolism gained added momentum 15, 32. One copy of the APOE ε4 allele increases the risk of developing AD by 2–3 fold, but two copies of APOE ε4 alleles increase the risk to ~12 fold 14, 158. The APOE protein regulates cholesterol metabolism and mediates uptake of lipoprotein particles via low‐density lipoprotein (LDL) receptor related protein (LRP) 22. The APOE E4 isoform at least somewhat selectively binds β‐amyloid peptide, modulating its aggregation and clearance 22. The ε4 allele is associated with higher cholesterol levels 47, 99.
Studies demonstrated that cholesterol modulates β‐amyloid peptide levels by affecting secretase function 100. Additionally, the involvement of cholesterol has been implicated in pathogenesis of AD in epidemiological studies 22, 84. When membrane cholesterol levels are decreased, the activities of β‐secretase (BACE1) and γ‐secretase are reduced, leading to lower β‐amyloid production 46, 169, 194, 207 (Figure 3A). In addition, the inhibition of cholesterol synthesis enzymes (3‐hydroxy‐3‐methylglutaryl‐CoA‐reductase and 7‐dehydro‐cholesterol‐reductase) is able to reduce intracellular and extracellular β‐amyloid levels 46, 153, 169.
Figure 3.
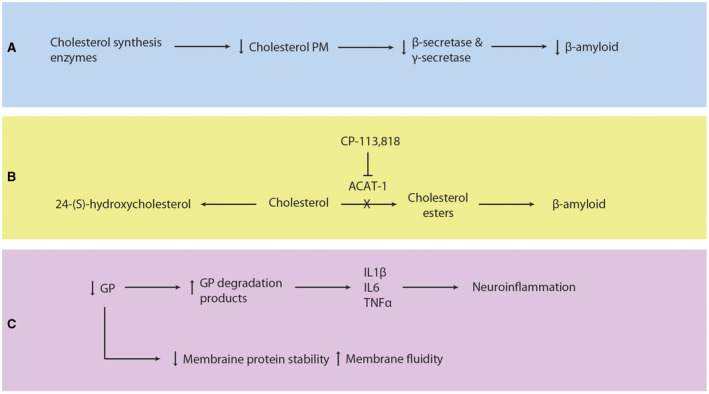
Lipid metabolism dysfunction in Alzheimer's Disease. A. Inhibition of cholesterol synthesis enzymes decreases plasma membrane cholesterol levels, β‐secretase and γ‐secretase activities, and β‐amyloid production. B. Cholesterol can be converted to 24‐(S)‐hydroxycholesterol or cholesteryl ester by CYP461A or ACAT‐1, respectively. Increased ACAT‐1 activity causes production of cholesteryl esters and increased β‐amyloid levels. Inhibition of ACAT‐1 by CP‐113,818 reduces β‐amyloid. C. Glycerophospholipids (GP) stabilize plasma membrane proteins such as ion channels and affect plasma membrane fluidity. Lower levels of GP are found in AD as evidenced by increased levels of GP degradation products, which are proinflammatory. Recruited astrocytes and microglia release IL1β, IL6, and TNFα and cause subsequent neuroinflammation. CYP461A, 24‐hydroxylase; ACAT‐1, sterol O‐acyltransferase 1; GP glycerophospholipids; AD, Alzheimer's Disease; IL1β, interleukin‐1β; IL6, interleukin‐6; TNFα tumor necrosis factor alpha.
Under normal conditions, free intracellular cholesterol is esterified to form cholesteryl‐esters by sterol O‐acyltransferase 1 (ACAT1) (Figure 3B). In cultured cells, it was demonstrated that when cholesteryl ester concentration was increased, there was a proportional rise in β‐amyloid levels 147. When ACAT1 activity was inhibited, there was a significant reduction in β‐amyloid peptide 72. Genetic deletion of ACAT1 has been shown to reduce β‐amyloid peptide and cognitive impairment in AD mouse models, supporting its role in AD pathology 21. In addition to formation of cholesteryl‐esters by ACAT1, cholesterol can be metabolized by the brain‐specific enzyme, 24‐hydroxylase (CYP46A1), into 24‐(S)‐hydroxycholesterol (cerebrosterol) that can cross the blood–brain barrier 21. When ACAT1 activity is reduced (either genetically deleted or antagonized using a small molecular inhibitor), levels of 24‐(S)‐hydroxycholesterol are increased and β‐amyloid pathology in the brain is decreased 21. Studies have found that persons with early stage AD have higher levels of 24‐(S)‐hydroxycholesterol in peripheral circulation and CSF relative to normal controls 88, 104. This increase in 24‐(S)‐hydroxycholesterol suggests that cholesterol metabolism is affected early in AD and 24‐(S)‐hydroxycholesterol is produced as a byproduct or as part of a compensatory mechanism. Furthermore, peripheral levels of 24‐(S)‐hydroxycholesterol could be used as a blood biomarker for detection of early AD 82, 104. Collectively, these studies suggest that the balance between free cholesterol and cholesteryl‐esters can alter amyloidogenesis in AD.
Lipids other than cholesterol—GPs and sphingolipids—have been implicated in AD pathogenesis. Normally, GPs interact and bind to membrane proteins and ion channels, helping them maintain correct position in the plasma membrane 139 (Figure 3C). When GPs are reduced in cell plasma membranes, the membranes become more fluid and permeable 69. Interestingly, lower levels of GPs have been reported in AD 64, 143. Further, higher levels of GP degradation products were found in the brains of AD patients 121. GP degradation products are pro‐inflammatory, and may act as signals to activate astrocytes and microglia 45. This leads to additional release of interleukin‐1β, interleukin‐6, and TNFα, producing a cascade of additional neuroinflammation 64, 143, which can result in local tissue damage and neuron cell death.
Sphingolipids make up the largest structural lipid component of CNS membranes and are highly expressed in the myelin sheath. There are different subtypes of sphinolipids, for example, ceramides are the simplest, while sphingomyelins and glycosphingolipids (eg, cerebrosides, sulfatides, and gangliosides) are more complex 101. Sulfatides play important roles in the nervous system and are abundant in the myelin sheath and in oligodendrocytes 183. In AD brains, sulfatide levels are reduced and when sulfatides are degraded, ceramide byproducts are formed 65. Sulfatides were shown to be depleted up to 93% in gray matter and up to 58% in white matter of AD brains, while other major classes of lipids were not affected 65. Also, ceramide levels were increased more than three fold in AD brains 36, 65, 66. Low levels of sulfatides are specific for AD and do not occur in patients with Parkinson's disease, Lewy body dementia, frontotemporal dementia, or multiple sclerosis 29, 101. The exact mechanism of sulfatide deficiency or how the loss of sulfatides contributes to AD neuropathology is currently unknown; however, it has been suggested that it is unlikely to be mediated directly by β‐amyloid peptide accumulation 29. A later study by the same group was inconclusive in elucidating a mechanism of sulfatide deficiency in AD 28; however, they proposed several explanations for the relationship between sulfatides and AD pathology. It was suggested that APOE mediates sulfatide depletion, sulfatides enhance β‐amyloid binding to APOE, and sulfatides enhance uptake of β‐amyloid peptides into the cell, leading to abnormal β‐amyloid accumulation in lysosomes 63. We conclude from the prior literature on lipid neurochemistry in AD that the findings are complex and, as far as we know, the various experimental “story lines” have not been reconciled together nor tied directly to T2DM. However, there appears to be compelling data in support of the hypothesis that changes in lipid biochemistry occurs early in the course of AD pathogenesis.
T2DM‐related pathways that may affect the brain
While CVD is a likely pathologic substrate of cognitive decline in T2DM, evidence exists for other mechanisms that may contribute in parallel or separately. The following section will discuss the impact of hyperglycemia, insulin resistance, inflammation, hypercorticolism, and amyloid accumulation as non‐mutually exclusive mechanisms that cause or exacerbate cognitive decline in T2DM and AD.
Hyperglycemia and cognitive decline
The relationship between blood glucose levels and degree of cognitive dysfunction in T2DM patients has been extensively evaluated. Yaffe and colleagues studied a population of 1,983 women and found that participants with HbA1c levels >7.0% had a four‐fold increase in probability of developing cognitive impairment 203. Intriguingly, this study only included “non‐diabetic” women. These findings again support the hypothesis that glucose dysregulation is associated with cognitive impairment. Other studies also found an inverse relationship between HbA1c and working memory, executive function, learning, and/or psychomotor performance in T2DM patients 123, 141, 152, 159. While impaired glucose control in the context of T2DM is associated with declining cognitive function, studies have found that impaired glucose tolerance without a formal diagnosis of diabetes (“pre‐diabetes”) is also a risk factor for cognitive dysfunction 35, 80, 90, 191. Despite the strong evidence that supports the link between impaired glucose regulation with cognitive dysfunction, it is important to note that not all studies to date demonstrate this relationship 49, 89, 98, 167.
Many of the prior studies that directly connect hyperglycemia with AD‐relevant pathways were performed in animal models. The direct relevance of these studies to the human conditions (T2DM and AD) is not firmly established. In some of these studies, hyperglycemia causes tissue damage and alters cellular function through increasing polyol pathway activation, which causes the formation of advanced glycation end products (AGEs), and protein kinase C (PKC) activation 16, 20, 86, 188. For example, streptozotocin‐treated rats were found to have increased sorbitol in cranial nerves, cerebrum, and retina. When animals were subsequently treated with tolerstat, an aldose reductase inhibitor, the accumulation was reduced 178. Another study found that when sorbinil, an aldose reductase inhibitor, was given to streptozotocin‐treated rats, it reduced brain levels of sorbitol and corrected cognitive dysfunction. However, more information is needed to definitively state whether this pathway contributes to cognitive decline in humans with T2DM.
Another potential mechanism of cognitive decline due to hyperglycemia is the formation of AGEs and receptors for AGE (RAGEs). While there may be an association between AGEs, RAGEs, and cognitive decline in T2DM, currently there is not enough evidence to support this mechanism. While animal studies demonstrated increased RAGE expression and damage to white matter and myelin, human studies on this topic produced conflicting results 188, 205. Several studies using human tissue demonstrated that patients with diabetes and AD have increased N‐carboxymethyllysine (a type of AGE) staining on post‐mortem analysis 58. However, another study failed to replicate this association 67. Little firm evidence exists for the role of PKC in relation to T2DM and cognitive decline. While some animal studies have found that PKC is highly expressed and has increased activity in diabetic animal models, other studies did not support this data 151, 168.
Some animal studies have elucidated molecular changes that occur in hippocampal neurons in response to hyperglycemia. For example, in streptozotocin‐induced diabetic rats, the NMDA currents and NMDA protein levels were reduced in the hippocampus 54. Furthermore, CA3 neurons underwent remodeling in response to hyperglycemia. This remodeling includes apical dendrite retraction and simplification. There is also an associated decrease in presynaptic vesicles 105. Another study using the streptozotocin‐induced diabetic model found evidence of apoptosis in hippocampal neurons. Cognitive deficits were associated with DNA fragmentation, positive TUNEL staining, and increased caspase‐3 levels 95.
Insulin resistance, inflammation, hypercorticolism, and amyloid accumulation
There is a growing body of evidence linking IR, a component of T2DM, to the pathogenesis of AD 116, 118, 127. Several studies have reported that the incidence of clinically diagnosed AD is 1.2–1.7‐fold greater in patients with T2DM and IR 34, 35, 90, 93, 138, 140, 203. Also, IR is reported to occur more frequently in patients with AD 76. IR has been found to impair central cholinergic activity, and diabetic animal models have reduced production and release of acetylcholine (ACh) 199, 200. Remarkably, the administration of intranasal insulin rescued memory deficits in a subset of research volunteers with clinical AD 7, 33, 135, 154.
Exactly how IR exerts its effects on cognitive function is not clear. However, several mechanisms have been proposed to help explain how IR contributes to cognitive decline in T2DM. The first mechanism is based on inflammatory markers, such as C‐reactive protein (CRP) and IL‐6 that are increased in T2DM and metabolic syndrome and are associated with reduced cognitive function 26, 102. Further, inflammatory reactants and proinflammatory cytokines have been found in CSF and β‐amyloid plaques 43, 62, 75. A study conducted by Singh‐Manoux et al. evaluated IL‐6 and CRP in 5,217 people and found that elevated IL‐6 in midlife can predict subsequent cognitive decline 171. The authors concluded that pro‐inflammatory molecules can influence cognition by inducing a prothrombic state. For example, inflammatory signals can trigger local thrombotic vascular events leading to brain infarction. Other studies have also demonstrated that persons with metabolic syndrome and elevated inflammatory markers have impaired cognitive function 107, 203.
Another hypothesized mechanism by which IR could contribute to cognitive impairment in T2DM involves dysregulation of the HPA axis, leading to higher cortisol levels. Humans and animals with T2DM have increased serum cortisol levels 92, 157, 186 and several studies found that high serum cortisol is associated with cognitive decline and dementia, an effect independent of APOE genotype. There is experimental evidence supporting the detrimental effects of cortisol on cognitive performance 131, 132, 149. For instance, healthy individuals treated with dexamethasone, corticosterone, or hydrocortisone performed worse on memory tests, and, additionally, patients with active Cushing's disease (and thus high blood cortisol levels) also demonstrate decreased performance on working memory, reasoning, and attention tests relative to controls 50, 150, 184. However, not all studies agree on the association between increased levels of cortisol and cognitive impairment 30, 91, 166.
Conclusion
T2DM is a risk factor for cognitive decline, although the exact mechanism(s) mediating this relationship are unclear. Multiple studies have found that CVD is more common in patients with T2DM than in non‐diabetics. CVD is a rather broad term, encompassing a combination of macroscopic and microscopic vascular lesions, which together contribute to cognitive impairment by impairing blood flow.
Alterations in glucose metabolism, mitochondrial metabolic dysfunction, mitochondrial autophagy, and alterations in lipid metabolism are all additional potential contributing factors to cognitive decline in T2DM. The mechanisms and processes that are occurring in aged brains still remain imperfectly characterized, and may involve the polyol pathway, formation of AGEs, PKC activation, IR, inflammation, and dysregulation of HPA axis.
Upstream events responsible for eventual β‐amyloid peptide deposition and NFT formation are also still not well understood. However, recent literature has found that metabolic dysregulation is linked with clinical and pathological AD. Abnormalities in cerebral glucose metabolism, mitochondrial function, and lipid metabolism have been reported in persons at risk for developing AD. Deeper understanding of how metabolic perturbations contribute to AD‐type pathology may help in developing new preventative and/or treatment strategies.
References
- 1. Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL et al (2016 Aug) Diabetes is associated with cerebrovascular but not Alzheimer's disease neuropathology. Alzheimers Dement 12:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B et al (2010 Sep 28) Diabetes, Alzheimer disease, and vascular dementia: a population‐based neuropathologic study. Neurology 75:1195–1202. [DOI] [PubMed] [Google Scholar]
- 3. Akai K, Uchigasaki S, Tanaka U, Komatsu A (1987 Jan) Normal pressure hydrocephalus. Neuropathological study. Acta Pathol Jpn 37:97–110. [PubMed] [Google Scholar]
- 4. Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR (1995) An English translation of Alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat 8:429–431. [DOI] [PubMed] [Google Scholar]
- 5. An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O et al (2018 Mar) Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement 14:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arboix A, Ferrer I, Marti‐Vilalta JL (1996 Jun) Clinico‐anatomopathologic analysis of 25 patients with lacunar infarction. Rev Clin Esp 196:370–374. [PubMed] [Google Scholar]
- 7. Arnold SE, Arvanitakis Z, Macauley‐Rambach SL, Koenig AM, Wang HY, Ahima RS et al (2018 Mar) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14:168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA (2017 Jan) The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol 27:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA (2011 Mar) Microinfarct pathology, dementia, and cognitive systems. Stroke 42:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z et al (2006 Dec 12) Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67:1960–1965. [DOI] [PubMed] [Google Scholar]
- 11. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011 Mar 19) Alzheimer's disease. Lancet 377:1019–1031. [DOI] [PubMed] [Google Scholar]
- 12. Baloyannis SJ (2006 Jul) Mitochondrial alterations in Alzheimer's disease. J Alzheimers Dis 9:119–126. [DOI] [PubMed] [Google Scholar]
- 13. Beeri MS, Silverman JM, Davis KL, Marin D, Grossman HZ, Schmeidler J et al (2005 Apr) Type 2 diabetes is negatively associated with Alzheimer's disease neuropathology. J Gerontol A Biol Sci Med Sci 60:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007 Jan) Systematic meta‐analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39:17–23. [DOI] [PubMed] [Google Scholar]
- 15. Bertram L, Tanzi RE (2008 Oct) Thirty years of Alzheimer's disease genetics: the implications of systematic meta‐analyses. Nat Rev Neurosci 9:768–778. [DOI] [PubMed] [Google Scholar]
- 16. Biessels GJ, van der Heide LP, Kamal A, Bleys RL, Gispen WH (2002 Apr 19) Ageing and diabetes: implications for brain function. Eur J Pharmacol 441:1–14. [DOI] [PubMed] [Google Scholar]
- 17. Bjorkhem I, Meaney S (2004 May) Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24:806–815. [DOI] [PubMed] [Google Scholar]
- 18. Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S (2016 Dec) Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12:2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brayne C, Richardson K, Matthews FE, Fleming J, Hunter S, Xuereb JH (2009) Neuropathological correlates of dementia in over‐80‐year‐old brain donors from the population‐based Cambridge city over‐75s cohort (CC75C) study. J Alzheimers Dis 18:645–658. [DOI] [PubMed] [Google Scholar]
- 20. Brownlee M (2005 Jun) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 21. Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E (2010 Feb 16) ACAT1 gene ablation increases 24(S)‐hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A 107:3081–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bu G (2009 May) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burns A, Iliffe S (2009 Feb) Alzheimer's disease. BMJ 5:b158. [DOI] [PubMed] [Google Scholar]
- 24. Cardoso SM, Santos S, Swerdlow RH, Oliveira CR (2001 Jun) Functional mitochondria are required for amyloid beta‐mediated neurotoxicity. FASEB J 15:1439–1441. [DOI] [PubMed] [Google Scholar]
- 25. Castellani RJ, Perry G (2014 Apr 15) The complexities of the pathology‐pathogenesis relationship in Alzheimer disease. Biochem Pharmacol 88:671–676. [DOI] [PubMed] [Google Scholar]
- 26. Chen J, Wildman RP, Hamm LL, Muntner P, Reynolds K, Whelton PK et al (2004 Dec) Association between inflammation and insulin resistance in U.S. nondiabetic adults: results from the Third National Health and Nutrition Examination Survey. Diabetes Care 27:2960–2965. [DOI] [PubMed] [Google Scholar]
- 27. Cheng G, Huang C, Deng H, Wang H (2012 May) Diabetes as a risk factor for dementia and mild cognitive impairment: a meta‐analysis of longitudinal studies. Intern Med J 42:484–491. [DOI] [PubMed] [Google Scholar]
- 28. Cheng H, Wang M, Li JL, Cairns NJ, Han X (2013 Dec) Specific changes of sulfatide levels in individuals with pre‐clinical Alzheimer's disease: an early event in disease pathogenesis. J Neurochem 127:733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng H, Xu J, McKeel DW, Jr, Han X (2003 Jul) Specificity and potential mechanism of sulfatide deficiency in Alzheimer's disease: an electrospray ionization mass spectrometric study. Cell Mol Biol (Noisy‐le‐grand) 49:809–818. [PubMed] [Google Scholar]
- 30. Comijs HC, Gerritsen L, Penninx BW, Bremmer MA, Deeg DJ, Geerlings MI (2010 Jan) The association between serum cortisol and cognitive decline in older persons. Am J Geriatr Psychiatry 18:42–50. [DOI] [PubMed] [Google Scholar]
- 31. Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA et al (2003 Jan) Reduced hippocampal insulin‐degrading enzyme in late‐onset Alzheimer's disease is associated with the apolipoprotein E‐epsilon4 allele. Am J Pathol 162:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW et al (1993 Aug 13) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261:921–923. [DOI] [PubMed] [Google Scholar]
- 33. Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH et al (2017)Effects of regular and long‐acting insulin on cognition and Alzheimer's disease biomarkers: a pilot clinical trial. J Alzheimers Dis 57:1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cukierman T, Gerstein HC, Williamson JD (2005 Dec) Cognitive decline and dementia in diabetes–systematic overview of prospective observational studies. Diabetologia 48:2460–2469. [DOI] [PubMed] [Google Scholar]
- 35. Curb JD, Rodriguez BL, Abbott RD, Petrovitch H, Ross GW, Masaki KH et al (1999 Mar 23) Longitudinal association of vascular and Alzheimer's dementias, diabetes, and glucose tolerance. Neurology 52:971–975. [DOI] [PubMed] [Google Scholar]
- 36. Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K et al (2004 Feb 17) Involvement of oxidative stress‐induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A 101:2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. De Reuck J, Deramecourt V, Cordonnier C, Leys D, Maurage CA, Pasquier F (2011 Jun) The impact of cerebral amyloid angiopathy on the occurrence of cerebrovascular lesions in demented patients with Alzheimer features: a neuropathological study. Eur J Neurol 18:913–918. [DOI] [PubMed] [Google Scholar]
- 38. Debette S, Markus HS (2010 Jul) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta‐analysis. BMJ 26:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dementia Fact sheet N°362 . (2015 Mar) World Health Organization. [Google Scholar]
- 40. Diana A, Simic G, Sinforiani E, Orru N, Pichiri G, Bono G (2008 Jan) Mitochondria morphology and DNA content upon sublethal exposure to beta‐amyloid(1‐42) peptide. Coll Antropol 32(Suppl 1):51–58. [PubMed] [Google Scholar]
- 41. Doens D, Fernandez PL (2014 Mar 13) Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. J Neuroinflammation 11:48‐2094‐11‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM et al (2008 Oct) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 14:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duong T, Nikolaeva M, Acton PJ (1997 Feb 21) C‐reactive protein‐like immunoreactivity in the neurofibrillary tangles of Alzheimer's disease. Brain Res 749:152–156. [DOI] [PubMed] [Google Scholar]
- 44. Ergul A, Kelly‐Cobbs A, Abdalla M, Fagan SC (2012 Jun) Cerebrovascular complications of diabetes: focus on stroke. Endocr Metab Immune Disord Drug Targets 12:148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Farooqui AA, Horrocks LA, Farooqui T (2007 May) Modulation of inflammation in brain: a matter of fat. J Neurochem 101:577–599. [DOI] [PubMed] [Google Scholar]
- 46. Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P et al (2001 May 8) Simvastatin strongly reduces levels of Alzheimer's disease beta ‐amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo . Proc Natl Acad Sci U S A 98:5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferrucci L, Guralnik JM, Pahor M, Harris T, Corti MC, Hyman BT et al (1997 Dec) Apolipoprotein E epsilon 2 allele and risk of stroke in the older population. Stroke 28:2410–2416. [DOI] [PubMed] [Google Scholar]
- 48. Foley P (2010 Aug) Lipids in Alzheimer's disease: a century‐old story. Biochim Biophys Acta 1801:750–753. [DOI] [PubMed] [Google Scholar]
- 49. Fontbonne A, Berr C, Ducimetiere P, Alperovitch A (2001 Feb) Changes in cognitive abilities over a 4‐year period are unfavorably affected in elderly diabetic subjects: results of the Epidemiology of Vascular Aging Study. Diabetes Care 24:366–370. [DOI] [PubMed] [Google Scholar]
- 50. Forget H, Lacroix A, Somma M, Cohen H (2000 Jan) Cognitive decline in patients with Cushing's syndrome. J Int Neuropsychol Soc 6:20–29. [DOI] [PubMed] [Google Scholar]
- 51. Frackowiak RS, Pozzilli C, Legg NJ, Du Boulay GH, Marshall J, Lenzi GL et al (1981 Dec) Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen‐15 and positron tomography. Brain 104(Pt 4):753–778. [DOI] [PubMed] [Google Scholar]
- 52. Friede RL (1965) Enzyme histochemical studies of senile plaques. J Neuropathol Exp Neurol 24:477–491. [DOI] [PubMed] [Google Scholar]
- 53. Fukuyama H, Ogawa M, Yamauchi H, Yamaguchi S, Kimura J, Yonekura Y et al (1994 Jan) Altered cerebral energy metabolism in Alzheimer's disease: a PET study. J Nucl Med 35:1–6. [PubMed] [Google Scholar]
- 54. Gardoni F, Kamal A, Bellone C, Biessels GJ, Ramakers GM, Cattabeni F et al (2002 Feb) Effects of streptozotocin‐diabetes on the hippocampal NMDA receptor complex in rats. J Neurochem 80:438–447. [DOI] [PubMed] [Google Scholar]
- 55. Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P et al (2001 Apr 15) Stimulation of beta‐amyloid precursor protein trafficking by insulin reduces intraneuronal beta‐amyloid and requires mitogen‐activated protein kinase signaling. J Neurosci 21:2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gasparini L, Racchi M, Benussi L, Curti D, Binetti G, Bianchetti A et al (1997 Aug 8) Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett 231:113–117. [DOI] [PubMed] [Google Scholar]
- 57. Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B et al (1988 Aug) Reduced activities of thiamine‐dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol 45:836–840. [DOI] [PubMed] [Google Scholar]
- 58. Girones X, Guimera A, Cruz‐Sanchez CZ, Ortega A, Sasaki N, Makita Z et al (2004 May 15) N epsilon‐carboxymethyllysine in brain aging, diabetes mellitus, and Alzheimer's disease. Free Radic Biol Med 36:1241–1247. [DOI] [PubMed] [Google Scholar]
- 59. Goedert M (1987 Dec 1) Neuronal localization of amyloid beta protein precursor mRNA in normal human brain and in Alzheimer's disease. EMBO J 6:3627–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gold G, Giannakopoulos P, Herrmann FR, Bouras C, Kovari E (2007 Nov) Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain 130(Pt 11):2830–2836. [DOI] [PubMed] [Google Scholar]
- 61. Gold G, Kovari E, Herrmann FR, Canuto A, Hof PR, Michel JP et al (2005 Jun) Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 36:1184–1188. [DOI] [PubMed] [Google Scholar]
- 62. Gorelick PB (2010 Oct) Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann N Y Acad Sci 1207:155–162. [DOI] [PubMed] [Google Scholar]
- 63. Han X (2010 Jun) The pathogenic implication of abnormal interaction between apolipoprotein E isoforms, amyloid‐beta peptides, and sulfatides in Alzheimer's disease. Mol Neurobiol 41:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han X, Holtzman DM, McKeel DW Jr (2001 May) Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem 77:1168–1180. [DOI] [PubMed] [Google Scholar]
- 65. Han X, M Holtzman D, McKeel DW, Jr, Kelley J, Morris JC (2002 Aug) Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J Neurochem 82:809–818. [DOI] [PubMed] [Google Scholar]
- 66. He X, Huang Y, Li B, Gong CX, Schuchman EH (2010 Mar) Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging 31:398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Heitner J, Dickson D (1997 Nov) Diabetics do not have increased Alzheimer‐type pathology compared with age‐matched control subjects. A retrospective postmortem immunocytochemical and histofluorescent study. Neurology 49:1306–1311. [DOI] [PubMed] [Google Scholar]
- 68. Herholz K, Salmon E, Perani D, Baron JC, Holthoff V, Frolich L et al (2002 Sep) Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage 17:302–316. [DOI] [PubMed] [Google Scholar]
- 69. Hishikawa D, Hashidate T, Shimizu T, Shindou H (2014 May) Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J Lipid Res 55:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z et al (2004 May) Diet‐induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. FASEB J 18:902–904. [DOI] [PubMed] [Google Scholar]
- 71. Huang J, Zhang X, Li J, Tang L, Jiao X, Lv X (2014 Jul) Impact of glucose fluctuation on acute cerebral infarction in type 2 diabetes. Can J Neurol Sci 41:486–492. [DOI] [PubMed] [Google Scholar]
- 72. Hutter‐Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A et al (2004 Oct 14) The ACAT inhibitor CP‐113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron 44:227–238. [DOI] [PubMed] [Google Scholar]
- 73. Ibanez V, Pietrini P, Furey ML, Alexander GE, Millet P, Bokde AL et al (2004 Mar 15) Resting state brain glucose metabolism is not reduced in normotensive healthy men during aging, after correction for brain atrophy. Brain Res Bull 63:147–154. [DOI] [PubMed] [Google Scholar]
- 74. Ighodaro ET, Abner EL, Fardo DW, Lin AL, Katsumata Y, Schmitt FA et al (2017 Jan) Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab 37:201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iwamoto N, Nishiyama E, Ohwada J, Arai H (1994 Aug 15) Demonstration of CRP immunoreactivity in brains of Alzheimer's disease: immunohistochemical study using formic acid pretreatment of tissue sections. Neurosci Lett 177:23–26. [DOI] [PubMed] [Google Scholar]
- 76. Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, Butler PC (2004 Feb) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53:474–481. [DOI] [PubMed] [Google Scholar]
- 77. Johnson AB, Blum NR (1970 Jul) Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer's disease: a histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol 29:463–478. [DOI] [PubMed] [Google Scholar]
- 78. Kalaria RN (2018 May 15) The pathology and pathophysiology of vascular dementia. Neuropharmacology 134(Pt B):226–239. [DOI] [PubMed] [Google Scholar]
- 79. Kalaria RN, Harik SI (1989) Abnormalities of the glucose transporter at the blood‐brain barrier and in brain in Alzheimer's disease. Prog Clin Biol Res 317:415–421. [PubMed] [Google Scholar]
- 80. Kanaya AM, Barrett‐Connor E, Gildengorin G, Yaffe K (2004 Jun 28) Change in cognitive function by glucose tolerance status in older adults: a 4‐year prospective study of the Rancho Bernardo study cohort. Arch Intern Med 164:1327–1333. [DOI] [PubMed] [Google Scholar]
- 81. Kannel WB, McGee DL (1979 May 11) Diabetes and cardiovascular disease. The Framingham study. JAMA 241:2035–2038. [DOI] [PubMed] [Google Scholar]
- 82. Khan TK, Alkon DL (2015) Peripheral biomarkers of Alzheimer's disease. J Alzheimers Dis 44:729–744. [DOI] [PubMed] [Google Scholar]
- 83. Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA et al (2000 Aug) Alzheimer's disease cybrids replicate beta‐amyloid abnormalities through cell death pathways. Ann Neurol 48:148–155. [PubMed] [Google Scholar]
- 84. Kim J, Basak JM, Holtzman DM (2009 Aug 13) The role of apolipoprotein E in Alzheimer's disease. Neuron 63:287–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Klassen AC, Loewenson RB, Resch JA (1973 Sep–Oct) Cerebral atherosclerosis in selected chronic disease states. Atherosclerosis 18:321–336. [DOI] [PubMed] [Google Scholar]
- 86. Klein JP, Waxman SG (2003 Sep) The brain in diabetes: molecular changes in neurons and their implications for end‐organ damage. Lancet Neurol 2:548–554. [DOI] [PubMed] [Google Scholar]
- 87. Knopman DS, Jack CR Jr, Wiste HJ, Lundt ES, Weigand SD, Vemuri P et al (2014 Sep) 18F‐fluorodeoxyglucose positron emission tomography, aging, and apolipoprotein E genotype in cognitively normal persons. Neurobiol Aging 35:2096–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kolsch H, Heun R, Kerksiek A, Bergmann KV, Maier W, Lutjohann D (2004 Sep 30) Altered levels of plasma 24S‐ and 27‐hydroxycholesterol in demented patients. Neurosci Lett 368:303–308. [DOI] [PubMed] [Google Scholar]
- 89. Kumari M, Marmot M (2005 Nov 22) Diabetes and cognitive function in a middle‐aged cohort: findings from the Whitehall II study. Neurology 65:1597–1603. [DOI] [PubMed] [Google Scholar]
- 90. Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T et al (1997 Oct 25) Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ 315:1045–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lara VP, Caramelli P, Teixeira AL, Barbosa MT, Carmona KC, Carvalho MG et al (2013 Aug) High cortisol levels are associated with cognitive impairment no‐dementia (CIND) and dementia. Clin Chim Acta 23:18–22. [DOI] [PubMed] [Google Scholar]
- 92. Lee ZS, Chan JC, Yeung VT, Chow CC, Lau MS, Ko GT et al (1999 Sep) Plasma insulin, growth hormone, cortisol, and central obesity among young Chinese type 2 diabetic patients. Diabetes Care 22:1450–1457. [DOI] [PubMed] [Google Scholar]
- 93. Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC et al (1997 Feb 15) Risk of dementia among persons with diabetes mellitus: a population‐based cohort study. Am J Epidemiol 145:301–308. [DOI] [PubMed] [Google Scholar]
- 94. Li J, Cesari M, Liu F, Dong B, Vellas B (2017 Feb) Effects of diabetes mellitus on cognitive decline in patients with Alzheimer disease: a systematic review. Can J Diabetes 41:114–119. [DOI] [PubMed] [Google Scholar]
- 95. Li ZG, Zhang W, Grunberger G, Sima AA (2002 Aug 16) Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res 946:221–231. [DOI] [PubMed] [Google Scholar]
- 96. Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S et al (2004 Jun) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lim AS, Yu L, Schneider JA, Bennett DA, Buchman AS (2016 Feb) Sleep fragmentation, cerebral arteriolosclerosis, and brain infarct pathology in community‐dwelling older people. Stroke 47:516–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lindeman RD, Romero LJ, LaRue A, Yau CL, Schade DS, Koehler KM et al (2001 Sep) A biethnic community survey of cognition in participants with type 2 diabetes, impaired glucose tolerance, and normal glucose tolerance: the New Mexico Elder Health Survey. Diabetes Care 24:1567–1572. [DOI] [PubMed] [Google Scholar]
- 99. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G (2013 Feb) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL et al (2007 Oct 4) Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56:66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu Q, Zhang J (2014 Apr) Lipid metabolism in Alzheimer's disease. Neurosci Bull 30:331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. de Luca C, Olefsky JM (2008 Jan 9) Inflammation and insulin resistance. FEBS Lett 582:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N et al (2004 Apr 16) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304:448–452. [DOI] [PubMed] [Google Scholar]
- 104. Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD et al (2000 Feb) Plasma 24S‐hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41:195–198. [PubMed] [Google Scholar]
- 105. Magarinos AM, McEwen BS (2000 Sep 26) Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A 97:11056–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Manczak M, Calkins MJ, Reddy PH (2011 Jul 1) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Marioni RE, Strachan MW, Reynolds RM, Lowe GD, Mitchell RJ, Fowkes FG et al (2010 Mar) Association between raised inflammatory markers and cognitive decline in elderly people with type 2 diabetes: the Edinburgh Type 2 Diabetes Study. Diabetes 59:710–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mathers CD, Loncar D (2006 Nov) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Matioli MNPDS, Suemoto CK, Rodriguez RD, Farias DS, da Silva MM, Leite REP et al (2017 Oct‐Dec) Association between diabetes and causes of dementia: evidence from a clinicopathological study. Dement Neuropsychol 11:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y et al (2010 Aug 31) Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75:764–770. [DOI] [PubMed] [Google Scholar]
- 111. Mazighi M, Labreuche J, Gongora‐Rivera F, Duyckaerts C, Hauw JJ, Amarenco P (2008 Apr) Autopsy prevalence of intracranial atherosclerosis in patients with fatal stroke. Stroke 39:1142–1147. [DOI] [PubMed] [Google Scholar]
- 112. McGeer EG, Peppard RP, McGeer PL, Tuokko H, Crockett D, Parks R et al (1990 Feb) 18Fluorodeoxyglucose positron emission tomography studies in presumed Alzheimer cases, including 13 serial scans. Can J Neurol Sci 17:1–11. [DOI] [PubMed] [Google Scholar]
- 113. Mielke R, Pietrzyk U, Jacobs A, Fink GR, Ichimiya A, Kessler J et al (1994 Oct) HMPAO SPET and FDG PET in Alzheimer's disease and vascular dementia: comparison of perfusion and metabolic pattern. Eur J Nucl Med 21:1052–1060. [DOI] [PubMed] [Google Scholar]
- 114. Minoshima S, Frey KA, Koeppe RA, Foster NL, Kuhl DE (1995 Jul) A diagnostic approach in Alzheimer's disease using three‐dimensional stereotactic surface projections of fluorine‐18‐FDG PET. J Nucl Med 36:1238–1248. [PubMed] [Google Scholar]
- 115. Mishra P, Chan DC (2014 Oct) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15:634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. de la Monte SM (2009 Aug 31) Insulin resistance and Alzheimer's disease. BMB Rep 42:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. de la Monte SM (2014 Mar) Relationships between diabetes and cognitive impairment. Endocrinol Metab Clin North Am 43:245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. de la Monte SM, Longato L, Tong M, Wands JR (2009 Oct) Insulin resistance and neurodegeneration: roles of obesity, type 2 diabetes mellitus and non‐alcoholic steatohepatitis. Curr Opin Investig Drugs 10:1049–1060. [PMC free article] [PubMed] [Google Scholar]
- 119. de la Monte SM, Tong M, Wands JR (2018) The 20‐year voyage aboard the journal of Alzheimer's disease: docking at ‘type 3 diabetes’, environmental/exposure factors, pathogenic mechanisms, and potential treatments. J Alzheimers Dis 62:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Morris JC (1993 Nov) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 121. Mulder C, Wahlund LO, Teerlink T, Blomberg M, Veerhuis R, van Kamp GJ et al (2003 Aug) Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in Alzheimer's disease. J Neural Transm (Vienna) 110:949–955. [DOI] [PubMed] [Google Scholar]
- 122. Mullins RJ, Diehl TC, Chia CW, Kapogiannis D (2017 May) Insulin resistance as a link between amyloid‐beta and tau pathologies in Alzheimer's disease. Front Aging Neurosci 3:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Munshi M, Grande L, Hayes M, Ayres D, Suhl E, Capelson R et al (2006 Aug) Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care 29:1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD et al (2010 Jan) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ et al (2012 May) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71:362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nelson PT, Braak H, Markesbery WR (2009 Jan) Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Neth BJ, Craft S (2017 Oct) Insulin resistance and Alzheimer's disease: bioenergetic linkages. Front Aging Neurosci 31:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006 Oct 6) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 129. Neuropathology Group (2001 Jan 20) Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late‐onset dementia in a multicentre, community‐based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 130. Newberg A, Alavi A, Reivich M (2002 Jan) Determination of regional cerebral function with FDG‐PET imaging in neuropsychiatric disorders. Semin Nucl Med 32:13–34. [DOI] [PubMed] [Google Scholar]
- 131. Newcomer JW, Craft S, Hershey T, Askins K, Bardgett ME (1994 Apr) Glucocorticoid‐induced impairment in declarative memory performance in adult humans. J Neurosci 14:2047–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Newcomer JW, Selke G, Melson AK, Hershey T, Craft S, Richards K et al (1999 Jun) Decreased memory performance in healthy humans induced by stress‐level cortisol treatment. Arch Gen Psychiatry 56:527–533. [DOI] [PubMed] [Google Scholar]
- 133. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A et al (2005 Feb) Extensive involvement of autophagy in Alzheimer disease: an immuno‐electron microscopy study. J Neuropathol Exp Neurol 64:113–122. [DOI] [PubMed] [Google Scholar]
- 134. Nixon RA, Yang DS (2011 Jul) Autophagy failure in Alzheimer's disease–locating the primary defect. Neurobiol Dis 43:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Novak V, Milberg W, Hao Y, Munshi M, Novak P, Galica A et al (2014) Enhancement of vasoreactivity and cognition by intranasal insulin in type 2 diabetes. Diabetes Care 37:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Okamoto Y, Ihara M, Fujita Y, Ito H, Takahashi R, Tomimoto H (2009 Jul 15) Cortical microinfarcts in Alzheimer's disease and subcortical vascular dementia. Neuroreport 20:990–996. [DOI] [PubMed] [Google Scholar]
- 137. Olokoba AB, Obateru OA, Olokoba LB (2012 Jul) Type 2 diabetes mellitus: a review of current trends. Oman Med J 27:269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999 Dec 10) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53:1937–1942. [DOI] [PubMed] [Google Scholar]
- 139. Palsdottir H, Hunte C (2004 Nov 3) Lipids in membrane protein structures. Biochim Biophys Acta 1666:2–18. [DOI] [PubMed] [Google Scholar]
- 140. Peila R, Rodriguez BL, Launer LJ (2002 Apr) Honolulu‐Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu‐Asia Aging Study. Diabetes 51:1256–1262. [DOI] [PubMed] [Google Scholar]
- 141. Perlmuter LC, Hakami MK, Hodgson‐Harrington C, Ginsberg J, Katz J, Singer DE et al (1984 Dec) Decreased cognitive function in aging non‐insulin‐dependent diabetic patients. Am J Med 77:1043–1048. [DOI] [PubMed] [Google Scholar]
- 142. Perry EK, Perry RH, Tomlinson BE, Blessed G, Gibson PH (1980 May 15) Coenzyme A‐acetylating enzymes in Alzheimer's disease: possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci Lett 18:105–110. [DOI] [PubMed] [Google Scholar]
- 143. Pettegrew JW, Panchalingam K, Hamilton RL, McClure RJ (2001 Jul) Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 26:771–782. [DOI] [PubMed] [Google Scholar]
- 144. Pivovarova O, Hohn A, Grune T, Pfeiffer AF, Rudovich N (2016 Dec) Insulin‐degrading enzyme: new therapeutic target for diabetes and Alzheimer's disease? Ann Med 48:614–624. [DOI] [PubMed] [Google Scholar]
- 145. Profenno LA, Porsteinsson AP, Faraone SV (2010 Mar 15) Meta‐analysis of Alzheimer's disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 67:505–512. [DOI] [PubMed] [Google Scholar]
- 146. Pruzin JJ, Schneider JA,, Capuano AW, Leurgans SE, Barnes LL, Ahima RS et al (2017 Jan–Mar) Diabetes, hemoglobin A1C, and regional alzheimer disease and infarct pathology. Alzheimer Dis Assoc Disord 31:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Puglielli L, Konopka G, Pack‐Chung E, Ingano LA, Berezovska O, Hyman BT et al (2001 Oct) Acyl‐coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta‐peptide. Nat Cell Biol 3:905–912. [DOI] [PubMed] [Google Scholar]
- 148. Querfurth HW, LaFerla FM (2010 Jan 28) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 149. de Quervain DJ, Roozendaal B, Nitsch RM, McGaugh JL, Hock C (2000 Apr) Acute cortisone administration impairs retrieval of long‐term declarative memory in humans. Nat Neurosci 3:313–314. [DOI] [PubMed] [Google Scholar]
- 150. Ragnarsson O, Berglund P, Eder DN, Johannsson G (2012 Sep) Long‐term cognitive impairments and attentional deficits in patients with Cushing's disease and cortisol‐producing adrenal adenoma in remission. J Clin Endocrinol Metab 97:E1640–8. [DOI] [PubMed] [Google Scholar]
- 151. Ramakrishnan R, Sheeladevi R, Suthanthirarajan N (2004 Aug 30) PKC‐alpha mediated alterations of indoleamine contents in diabetic rat brain. Brain Res Bull 64:189–194. [DOI] [PubMed] [Google Scholar]
- 152. Reaven GM, Thompson LW, Nahum D, Haskins E (1990 Jan) Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care 13:16–21. [DOI] [PubMed] [Google Scholar]
- 153. Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas‐Bryant T et al (2001 Oct) A cholesterol‐lowering drug reduces beta‐amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 8:890–899. [DOI] [PubMed] [Google Scholar]
- 154. Reger MA, Watson GS, Frey WH, 2nd, Baker LD, Cholerton B, Keeling ML et al (2006 Mar) Effects of intranasal insulin on cognition in memory‐impaired older adults: modulation by APOE genotype. Neurobiol Aging 27:451–458. [DOI] [PubMed] [Google Scholar]
- 155. Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S et al (1996 Mar 21) Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 334:752–758. [DOI] [PubMed] [Google Scholar]
- 156. Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D et al (2004 Jan 6) Functional brain abnormalities in young adults at genetic risk for late‐onset Alzheimer's dementia. Proc Natl Acad Sci U S A 101:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Reynolds RM, Strachan MW, Labad J, Lee AJ, Frier BM, Fowkes FG et al (2010 Apr) Morning cortisol levels and cognitive abilities in people with type 2 diabetes: the Edinburgh type 2 diabetes study. Diabetes Care 33:714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Roses AD (1996) Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med 47:387–400. [DOI] [PubMed] [Google Scholar]
- 159. Ryan CM, Geckle MO (2000 Oct) Circumscribed cognitive dysfunction in middle‐aged adults with type 2 diabetes. Diabetes Care 23:1486–1493. [DOI] [PubMed] [Google Scholar]
- 160. Samuraki M, Matsunari I, Chen WP, Shima K, Yanase D, Takeda N et al (2012 Oct) Glucose metabolism and gray‐matter concentration in apolipoprotein E epsilon4 positive normal subjects. Neurobiol Aging 33:2321–2323. [DOI] [PubMed] [Google Scholar]
- 161. Sasaki N, Toki S, Chowei H, Saito T, Nakano N, Hayashi Y et al (2001 Jan 12) Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer's disease. Brain Res 888:256–262. [DOI] [PubMed] [Google Scholar]
- 162. Scarmeas N, Habeck CG, Hilton J, Anderson KE, Flynn J, Park A et al (2005 Oct) APOE related alterations in cerebral activation even at college age. J Neurol Neurosurg Psychiatry 76:1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Schellenberg GD, Montine TJ (2012 Sep) The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol 124:305–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA (2009) The neuropathology of older persons with and without dementia from community vs. clinic cohorts. J Alzheimers Dis 18:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA (2007 Jul) Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Ann Neurol 62:59–66. [DOI] [PubMed] [Google Scholar]
- 166. Schrijvers EM, Direk N, Koudstaal PJ, Kirschbaum C, Hofman A, Tiemeier H et al (2011) Associations of serum cortisol with cognitive function and dementia: the Rotterdam Study. J Alzheimers Dis 25:671–677. [DOI] [PubMed] [Google Scholar]
- 167. Scott RD, Kritz‐Silverstein D, Barrett‐Connor E, Wiederholt WC (1998 Oct) The association of non‐insulin‐dependent diabetes mellitus and cognitive function in an older cohort. J Am Geriatr Soc 46:1217–1222. [DOI] [PubMed] [Google Scholar]
- 168. Sieber FE, Hurn P, Alkayed NJ, Traystman RJ (2001 Feb) Gender‐based differences in Na+ ‐K+ adenosine triphosphatase activity occur in the microcirculation of the diabetic rat brain. Anesthesiology 94:372–375. [DOI] [PubMed] [Google Scholar]
- 169. Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K (1998 May 26) Cholesterol depletion inhibits the generation of beta‐amyloid in hippocampal neurons. Proc Natl Acad Sci U S A 95:6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Simpson IA, Chundu KR, Davies‐Hill T, Honer WG, Davies P (1994 May) Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer's disease. Ann Neurol 35:546–551. [DOI] [PubMed] [Google Scholar]
- 171. Singh‐Manoux A, Dugravot A, Brunner E, Kumari M, Shipley M, Elbaz A et al (2014 Aug 5) Interleukin‐6 and C‐reactive protein as predictors of cognitive decline in late midlife. Neurology 83:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Sinha S, Lieberburg I (1999 Sep 28) Cellular mechanisms of beta‐amyloid production and secretion. Proc Natl Acad Sci U S A 96:11049–11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Skaper SD (2012) Alzheimer's disease and amyloid: culprit or coincidence? Int Rev Neurobiol 102:277–316. [DOI] [PubMed] [Google Scholar]
- 174. Smith GS, de Leon MJ, George AE, Kluger A, Volkow ND, McRae T et al (1992 Nov) Topography of cross‐sectional and longitudinal glucose metabolic deficits in Alzheimer's disease. Pathophysiologic implications. Arch Neurol 49:1142–1150. [DOI] [PubMed] [Google Scholar]
- 175. Smith MA, Drew KL, Nunomura A, Takeda A, Hirai K, Zhu X et al (2002 May) Amyloid‐beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem Int 40:527–531. [DOI] [PubMed] [Google Scholar]
- 176. Sonnen JA, Larson EB, Brickell K, Crane PK, Woltjer R, Montine TJ et al (2009 Mar) Different patterns of cerebral injury in dementia with or without diabetes. Arch Neurol 66:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD et al (2007 Oct) Pathological correlates of dementia in a longitudinal, population‐based sample of aging. Ann Neurol 62:406–413. [DOI] [PubMed] [Google Scholar]
- 178. Sredy J, Sawicki DR, Notvest RR (1991 Jan–Mar) Polyol pathway activity in nervous tissues of diabetic and galactose‐fed rats: effect of dietary galactose withdrawal or tolrestat intervention therapy. J Diabet Complications 5:42–47. [DOI] [PubMed] [Google Scholar]
- 179. Starks EJ, Patrick O'Grady J, Hoscheidt SM, Racine AM, Carlsson CM, Zetterberg H et al (2015) Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOEvarepsilon4 carriers. J Alzheimers Dis 46:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Strozyk D, Dickson DW, Lipton RB, Katz M, Derby CA, Lee S et al (2010 Oct) Contribution of vascular pathology to the clinical expression of dementia. Neurobiol Aging 31:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Sutherland GT, Lim J, Srikanth V, Bruce DG (2017) Epidemiological approaches to understanding the link between type 2 diabetes and dementia. J Alzheimers Dis 59:393–403. [DOI] [PubMed] [Google Scholar]
- 182. Svennerholm L, Bostrom K, Jungbjer B, Olsson L (1994 Nov) Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem 63:1802–1811. [DOI] [PubMed] [Google Scholar]
- 183. Takahashi T, Suzuki T (2012 Aug) Role of sulfatide in normal and pathological cells and tissues. J Lipid Res 53:1437–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Tiemensma J, Kokshoorn NE, Biermasz NR, Keijser BJ, Wassenaar MJ, Middelkoop HA et al (2010 Jun) Subtle cognitive impairments in patients with long‐term cure of Cushing's disease. J Clin Endocrinol Metab 95:2699–2714. [DOI] [PubMed] [Google Scholar]
- 185. Todd S, Barr S, Roberts M, Passmore AP (2013 Nov) Survival in dementia and predictors of mortality: a review. Int J Geriatr Psychiatry 28:1109–1124. [DOI] [PubMed] [Google Scholar]
- 186. Tojo C, Takao T, Nishioka T, Numata Y, Suemaru S, Hashimoto K (1996 Apr) Hypothalamic‐pituitary‐adrenal axis in WBN/Kob rats with non‐insulin dependent diabetes mellitus. Endocr J 43:233–239. [DOI] [PubMed] [Google Scholar]
- 187. Toro P, Schonknecht P, Schroder J (2009) Type II diabetes in mild cognitive impairment and Alzheimer's disease: results from a prospective population‐based study in Germany. J Alzheimers Dis 16:687–691. [DOI] [PubMed] [Google Scholar]
- 188. Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brussee V et al (2006 Aug) Diabetes, leukoencephalopathy and rage. Neurobiol Dis 23:445–461. [DOI] [PubMed] [Google Scholar]
- 189. Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O'Brien RJ (2008 Aug) Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol 64:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Vagelatos NT, Eslick GD (2013) Type 2 diabetes as a risk factor for Alzheimer's disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev 35:152–160. [DOI] [PubMed] [Google Scholar]
- 191. Vanhanen M, Koivisto K, Kuusisto J, Mykkanen L, Helkala EL, Hanninen T et al (1998 Mar) Cognitive function in an elderly population with persistent impaired glucose tolerance. Diabetes Care 21:398–402. [DOI] [PubMed] [Google Scholar]
- 192. Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V et al (2000 Mar 1) Neurons regulate extracellular levels of amyloid beta‐protein via proteolysis by insulin‐degrading enzyme. J Neurosci 20:1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Vermeer SE, Longstreth WT Jr, Koudstaal PJ (2007 Jul) Silent brain infarcts: a systematic review. Lancet Neurol 6:611–619. [DOI] [PubMed] [Google Scholar]
- 194. Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T et al (2002 Feb) Cholesterol‐dependent gamma‐secretase activity in buoyant cholesterol‐rich membrane microdomains. Neurobiol Dis 9:11–23. [DOI] [PubMed] [Google Scholar]
- 195. Wang X, Su B, Fujioka H, Zhu X (2008 Aug) Dynamin‐like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol 173:470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Wang X, Su B, Lee HG, Li X, Perry G, Smith MA et al (2009 Jul 15) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 29:9090–9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y et al (2008 Dec 9) Amyloid‐beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105:19318–19323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wang KC, Woung LC, Tsai MT, Liu CC, Su YH, Li CY (2012) Risk of Alzheimer's disease in relation to diabetes: a population‐based cohort study. Neuroepidemiology 38:237–244. [DOI] [PubMed] [Google Scholar]
- 199. Welsh B, Wecker L (1991 Apr) Effects of streptozotocin‐induced diabetes on acetylcholine metabolism in rat brain. Neurochem Res 16:453–460. [DOI] [PubMed] [Google Scholar]
- 200. Wenk GL (2006) Neuropathologic changes in Alzheimer's disease: potential targets for treatment. J Clin Psychiatry 67(Suppl. 3):3–7; quiz 23. [PubMed] [Google Scholar]
- 201. White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW et al (2002 Nov) Cerebrovascular pathology and dementia in autopsied Honolulu‐Asia Aging Study participants. Ann N Y Acad Sci 977:9–23. [DOI] [PubMed] [Google Scholar]
- 202. Wisniewski H, Terry RD, Hirano A (1970 Apr) Neurofibrillary pathology. J Neuropathol Exp Neurol 29:163–176. [PubMed] [Google Scholar]
- 203. Yaffe K, Blackwell T, Whitmer RA, Krueger K, Barrett Connor E (2006 Jul–Aug) Glycosylated hemoglobin level and development of mild cognitive impairment or dementia in older women. J Nutr Health Aging 10:293–295. [PubMed] [Google Scholar]
- 204. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A et al (1996 Aug 22) RAGE and amyloid‐beta peptide neurotoxicity in Alzheimer's disease. Nature 382:685–691. [DOI] [PubMed] [Google Scholar]
- 205. Yan SD, Stern D, Schmidt AM (1997 Mar)What's the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 27:179–181. [DOI] [PubMed] [Google Scholar]
- 206. Yanase D, Matsunari I, Yajima K, Chen W, Fujikawa A, Nishimura S et al (2005 Jul) Brain FDG PET study of normal aging in Japanese: effect of atrophy correction Eur J Nucl Med Mol Imaging 32:794–805. [DOI] [PubMed] [Google Scholar]
- 207. Zha Q, Ruan Y, Hartmann T, Beyreuther K, Zhang D (2004 Oct) GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol Psychiatry 9:946–952. [DOI] [PubMed] [Google Scholar]
- 208. Zhang J, Chen C, Hua S, Liao H, Wang M, Xiong Y et al (2017 Feb) An updated meta‐analysis of cohort studies: diabetes and risk of Alzheimer's disease. Diabetes Res Clin Pract 124:41–47. [DOI] [PubMed] [Google Scholar]
- 209. Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ et al (2004 Dec 8) Insulin‐degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci 24:11120–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zhou Z, Ibekwe E, Chornenkyy Y (2018 Jan 17) Metabolic alterations in cancer cells and the emerging role of oncometabolites as drivers of neoplastic change. Antioxidants (Basel) 7. 10.3390/antiox7010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Zhou H, Zhang X, Lu J (2014 Nov 9) Progress on diabetic cerebrovascular diseases. Bosn J Basic Med Sci 14:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]