

Abstract
The P2Y14 receptor (P2Y14R) mediates inflammatory activity by activating neutrophil motility, but few classes of antagonists are known. We have explored the structure activity relationship of a 3-(4-phenyl- 1H-1,2,3-triazol-1-yl)-5-(aryl)benzoic acid antagonist scaffold, assisted by docking and molecular dynamics (MD) simulation at a P2Y14R homology model. A computational pipeline using the High Throughput MD Python environment guided the analogue design. Selection of candidates was based upon ligand-protein shape and complementarity and the persistence of ligand-protein interactions over time. Predictions of a favorable substitution of a 5-phenyl group with thiophene and an insertion of a three-methylene spacer between the 5-aromatic and alkyl amino moieties were largely consistent with empirical results. The substitution of a key carboxylate group on the core phenyl ring with tetrazole or truncation of the 5-aryl group reduced affinity. The most potent antagonists, using a fluorescent assay, were a primary 3-aminopropyl congener 20 (MRS4458) and phenyl p-carboxamide 30 (MRS4478).
Keywords: G protein-coupled receptor, nucleotides, molecular dynamics, inflammation, P2Y receptor
Graphical abstract
Introduction
Extracellular nucleotides released by tissue and organs during stress or injury activate a class of cell-surface receptors (P2Rs) to boost the innate and adaptive immune responses.1–3 This mechanism acts as a time-dependent component of the signaling purinome, along with the anti-inflammatory adenosine receptors (ARs, also termed P1 receptors), to protect the organism in various challenged circumstances. The P2Y14 receptor (P2Y14R) responds to endogenous agonists uridine-5'-diphosphoglucose and uridine-5'-diphosphate to mediate inflammatory activity, in part by activating human neutrophil motility.4–6 Mouse neutrophils do not express the P2Y14R,8 and the recruitment of neutrophils in the mouse lung induced by LPS is mediated by a platelet P2Y14R, which has no effect on hemostasis.45 Structurally, the P2Y14R belongs to the δ-branch of rhodoposin-like G protein-coupled receptors (GPCRs) in a subfamily of three P2YRs that are preferentially coupled to inhibition of adenylate cyclase through guanine nucleotide inhibitory (Gi) protein. P2Y14R and other P2YR subtypes are also associated with proinflammatory effects, and their antagonists are desired for their anti-inflammatory activity.7–14 Selective P2Y14R antagonists are sought as potential agents for treating asthma, sterile inflammation of the kidney and possibly diabetes and neurodegeneration.7–12 However, only a few classes of antagonists are known, so there is a clear need for more diverse competitive P2Y14R antagonists.26 Selective P2Y14R antagonists endowed with good bioavailability properties are not available.
As the three-dimensional structure of the P2Y14R has not yet been elucidated, X-ray crystallographic structures of the closely related P2Y12R have been used to predict the binding mode of various competitive agonist and antagonist ligands.15,16 The sequence homology between human (h) P2Y12R and P2Y14R is ~48%, and key amino acids for coordinating the bound nucleotides are common to both receptors. Thus, homology modeling has predicted detailed interactions of P2Y14R agonists and antagonists in a manner consistent with their structure activity relationship (SAR).17,18 Antagonists of the P2Y14R were first reported by Black and colleagues,19 and of the two classes reported, naphthoic acids and pyrido[4,3-d]pyrimidines, only the former appeared to be competitive antagonists. We have explored both the pharmacology of a reported high affinity antagonist in that series, 4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid (PPTN, 1, Chart 1) and the SAR of related heterocyclic scaffolds.17,20 Compound 1 was shown to be highly specific for interaction with the P2Y14R with high affinity (Ki 0.4 nM in a functional assay).6 However, it was poorly bioavailable, and its amphiphilic and hydrophobic character leads to low solubility in either aqueous or apolar solvents. A prodrug of 1 containing a dimethylaminocarboxymethyl ester improved the utility of this antagonist in vivo.27 Recently, we reported that a 3-(4-phenyl-1H-1,2,3-triazol-1-yl)-5-(phenyl)benzoic acid scaffold was suitable for the design of P2Y14R antagonists. The combination of a core benzene ring and triazole (shown in blue in Chart 1) was predicted by modeling to be a bioisosteric replacement for the naphthalene moiety. Here, we also used a structure-based approach that included docking and molecular dynamics (MD) simulation at a hP2Y14R homology model. This study extends the SAR reported by Junker et al.20 with replacement of specific features attached to the core benzene ring of the scaffold: the 5-aryl group and the carboxylate group.
Chart 1.
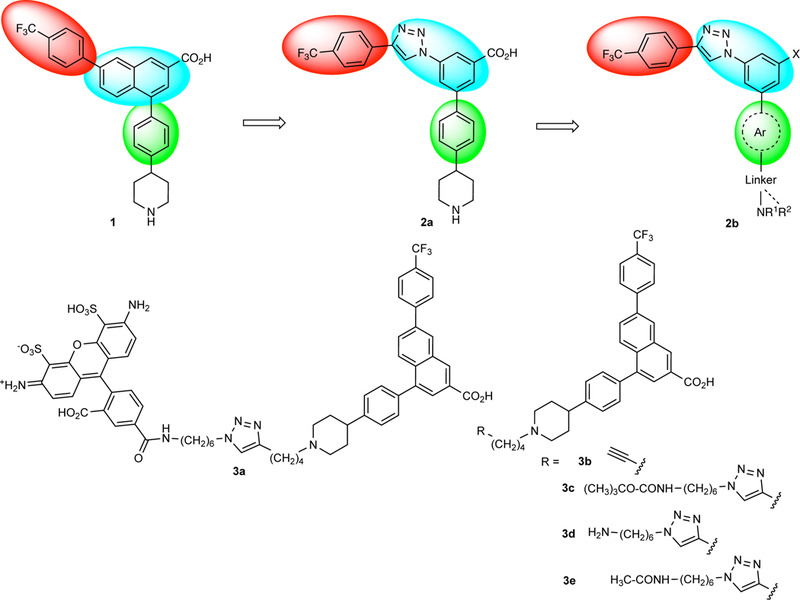
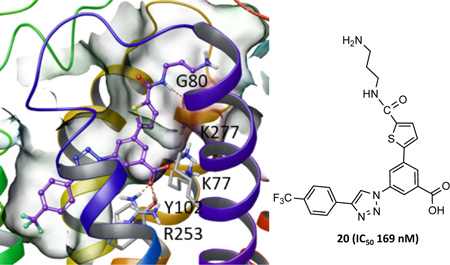
Progression of the structural modifications of naphthoic acid derivative 1 to the present set of derivatives 2b. The major focus was the introduction of diverse heteroaromatic rings in place of the bridging phenyl ring shown in green. The p-trifluoromethylphenyl moiety shown in red and the core benzene ring and adjacent triazole shown in blue were not modified in this study. Compound 3a is the fluorescent antagonist containing structure 1 as a pharmacophore that was used in the flow cytometric assay. Group X is a carboxylic acid or a bioisosteric replacement.
Results
Molecular modeling and compound design
We based our computational approach to the design of 1-substituted 3-(4-phenyl-1H-1,2,3-triazol-1-yl)-5-(aryl)benzene derivatives as new P2Y14R antagonists on the assumption that this chemical series binds at the orthosteric site of the receptor, located at the upper portion of the transmembrane (TM) domain facing the extracellular environment. Confidence in this assumption has accrued from previously published studies in which the predicted detailed interactions of P2Y14R agonists and antagonists were largely consistent with their SAR.17,18 We used a homology model of the human (h) P2Y14R based on the high resolution X-ray structure of an agonist-bound hP2Y12R (PDB ID: 4PXZ),16 which was previously refined using MD simulation,20 and set up a computational pipeline to guide the design of the new scaffold and prioritize the synthesis of the newly designed analogues. Similar to our previous approach, the pipeline consisted of docking of potential candidates at the hP2Y14R homology model followed by molecular dynamics (MD) refinement (30 ns of MD simulation run in triplicate for each structure) of a few selected hits in the membrane-embedded solvated environment (Figure 1). At variance with the previously reported strategy,20 in this study we accelerated the pipeline by docking the compounds at the rigid receptor and automating the MD refinement phase by exploiting the recently developed High Throughput MD (HTMD) module,31 a programmable environment based on Python that enables performing an automatic system setup, equilibration and production by running a single script. The design strategy was to retain the optimal, hydrophobic 4-(4-trifluoromethyl)-phenyl moiety (red in Charts 1 and 2), that was present in the original lead compound 1 and modify other parts of the scaffold. This trifluoromethyl-phenyl moiety was found to be optimal among 64 differently substituted 4-phenyl derivatives20 and predicted to be deeply buried in a narrow, hydrophobic region of the orthosteric hP2Y14R binding site. As a result, the 4-(4-trifluoromethyl)-phenyl analogue 2a from Junker et al.20 was only 6-fold less potent than 1 in a fluorescence binding assay using flow cytometry in hP2Y14R-expressing Chinese hamster ovary (CHO) cells.17,20 The adjacent triazole moiety, introduced with the previous set of P2Y14R antagonists, was also retained in the current set (Chart 2 and Table 1), due to its ability to be synthesized from readily available intermediates through a click ([2+3]cycloaddition of azide and alkyne) reaction. On another region of this molecular series, a primary or secondary alkylamine was included, similar to the piperidine moiety in 1 and 2a. This moiety is predicted to interact with the P2Y14R extracellular loop (EL) region, which is more flexible than the deeper binding region surrounding the minimal pharmacophore. Although our SAR exploration of 1 predicted that a charged amino group is not essential,17 we preserved the amino function in various analogues for interaction with polar groups present in this region of the receptor. The expectation from molecular modeling that the amino moiety could be substituted with various extended or tethered bulky groups enabled the introduction of the high affinity fluorescent antagonist ligand 3a used in the flow cytometry assay.17 We, therefore, devoted an effort to more clearly define possible interactions of this ligand series with the ELs using molecular modeling. An intermediate structure of the fluorescent probe 3a that terminated in a primary amino group, containing the triazole ring, but not the amide-linked fluorophore, i.e. 3d, was built in place starting from the docking pose of 1 in the hP2Y14R homology model. Then, the region around the ligand (10 Å) was subjected to Monte Carlo sampling (see Supplementary Information). In the resulting refined complex (Figure S1), the primary amino group of 3d extended toward the extracellular side and established additional H-bond and salt-bridge interactions with the backbone and sidechain of Glu166 (EL2).
Figure 1.
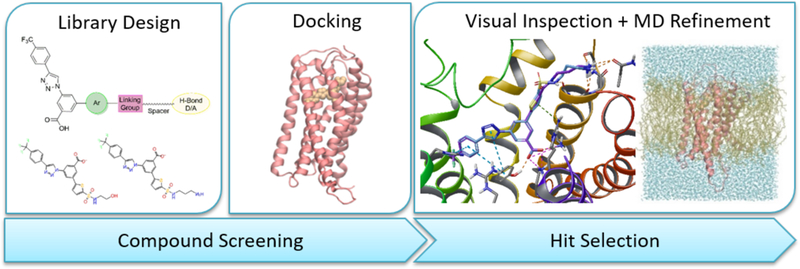
Computational pipeline applied for the design of new derivatives as hP2Y14R antagonists. A library of compounds was designed according to the general scheme reported in Chart 2. The compounds were docked at a hP2Y14R homology model (Glide, SP scoring function). Hit selection was performed by visual inspection followed, for a few selected candidates, by membrane molecular dynamics (MD) refinement (30 ns, run in triplicate, CHARMM36/CGenFF, POPC lipid model). The MD refinement phase was sped up using High Throughput MD (HTMD).
Chart 2.
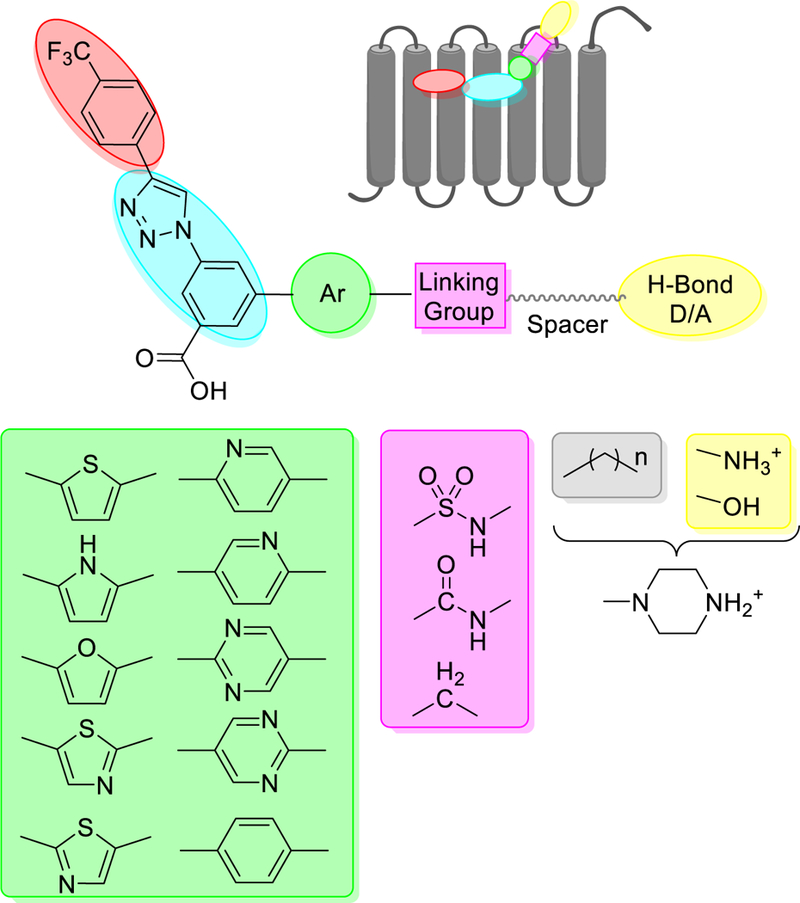
Detailed general approach for the design of new derivatives. The expected orientation of the scaffold with respect to the hP2Y14R is shown in a symbolic representation. Diverse 5- and 6- membered heteroaromatic rings linking the core (cyan) to the group facing the EC side of the receptor (yellow) were considered, although not all of these analogues were synthesized. Various linking groups or linkers (magenta), including amide, sulfonamide and methylene, were inserted to provide conformational diversity in the terminal alkylamino moiety. The terminal group on the spacer moiety was an H-bond donor/acceptor group, an amino, protected amino or a hydroxyl group. The spacer group consisted of either a straight chain alkyl moiety or a six-membered ring containing the secondary amine.
Table 1.
Structures, affinity and cLogP values of hP2Y14R antagonists.a
 | ||||
|---|---|---|---|---|
| No. | R1 = | R2 = | IC50, μM |
cLogPf |
| 1 | |
CO2H | 0.00796 ± 0.0035 | 6.18 |
| 2a | > |
CO2H | 0.0317 ± 0.0080e | 4.64 |
| 3bb | - | - | 0.763 ± 0.024 | 7.54 |
| 3cb | - | - | 0.133 ± 0.013 | 7.40 |
| 3db | - | - | 0.0254 ± 0.0049 | 4.04 |
| 3eb | - | - | 0.0835 ± 0.0064 | 7.40 |
| 4 | 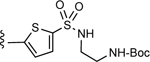
|
CO2H | 5.92 ± 0.42 | 3.65 |
| 5 | 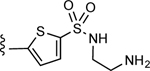
|
CO2H | 2.26 ± 0.21 | 0.40 |
| 6 | 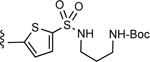
|
CO2H | 9.33 ± 0.24 | 3.77 |
| 7 | 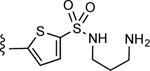
|
CO2H | 0.756 ± 0.092 | 0.50 |
| 8 | 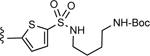
|
CO2H | 13.6 ± 0.8 | 3.89 |
| 9 | 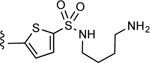
|
CO2H | 2.18 ± 0.56 | 0.66 |
| 10 | 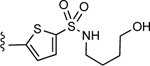
|
CO2H | 8.42 ± 0.87 | 3.11 |
| 11 | 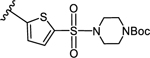
|
CO2H | 54.7 ± 14.6 | 3.72 |
| 12 | 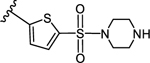
|
CO2H | 1.51 ± 0.45 | 2.86 |
| 13c | 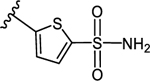
|
CO2H | 6.44 ± 1.54 | 3.47 |
| l4 | 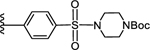
|
CO2H | >50 | 2.79 |
| l5 | 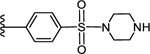
|
CO2H | 1.64 ± 0.23 | 1.50 |
| l6c | 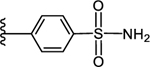
|
CO2H | 0.608 ± 0.080 | 3.57 |
| 17 | |
CO2H | 67.2 ± 7.2 | 4.68 |
| 18 | |
CO2H | 1.96 ± 0.20 | 3.46 |
| l9 | 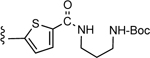
|
CO2H | 2.28 ± 0.26 | 4.66 |
| 20 | 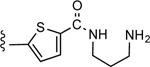
|
CO2H | 0.169 ± 0.042 | 0.84 |
| 21 | 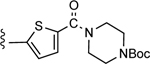
|
CO2H | 53.0 ± 10.8 | 4.31 |
| 22 | 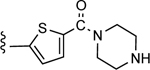
|
CO2H | 12.6 ± 0.6 | 3.08 |
| 23 | 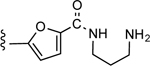
|
CO2H | 10.9 ± 1.5 | 0.73 |
| 24c | 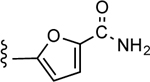
|
CO2H | 3.11 ± 0.84 | 3.03 |
| 25 | 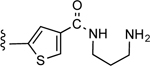
|
CO2H | 0.735 ± 0.280 | 0.87 |
| 26 | 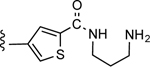
|
CO2H | 1.75 ± 0.25 | 0.85 |
| 27d | 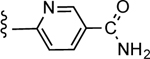
|
CO2H | 0.516 ± 0.243 | 3.07 |
| 28d | 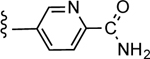
|
CO2H | 1.41 ± 0.31 | 3.13 |
| 29d | 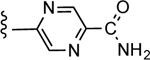
|
CO2H | 0.566 ± 0.200 | 2.32 |
| 30c | 
|
CO2H | 0.269 ± 0.121 | 3.63 |
| 31d | 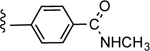
|
CO2H | 0.563 ± 0.198 | 3.95 |
| 32d | |
CO2H | 0.899 ± 0.417 | 4.25 |
| 33d | |
CO2H | 1.49 ± 0.83 | 4.60 |
| 34d | |
CO2H | 7.53 ± 0.52 | 4.90 |
| 35 | |
 |
0.958 ± 0.357 | 4.15 |
| 36a | 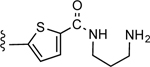
|
 |
1.19 ± 0.26 | 2.97 |
| 36b | 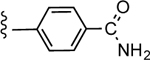
|
 |
2.43 ± 0.88 | 3.25 |
| 36c | |
 |
20.7 ± 7.0 | 4.40 |
| 37a | −OH | CO2H | 16.4 ± 4.8 | 3.11 |
| 37b | −Br | CO2H | 26.8 ± 7.8 | 3.85 |
| 37c | −Br | 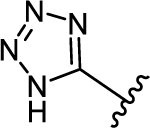 |
33.1 ± 7.3 | 3.45 |
| 37d | −OH | CO2H | 3.20 ± 0.64 | 5.22 |
IC50 in inhibition of hP2Y14R antagonist binding, determined using flow cytometry of whole hP2Y14R-CHO cells in the presence of a fixed concentration (20 nM) of 3a (mean ± SEM, n = 3–6).
structure shown in Chart 1.
tested as the triethylammonium salt.
tested as the sodium salt.
data from Junker et al.20
cLogP calculated using ALOGPS 2.1 program (www.vcclab.org/lab/alogps/).46
The major focus in the present set of P2Y14R antagonist analogues is, therefore, the aryl ring (green, in Chart 1 and 2) bridging the polar amino chain to the rest of the molecule. In compounds 1 and 2a, this aryl group consists of a p-substituted phenyl ring attached at the 5 position of the core benzene ring. We therefore designed compounds bearing heterocyclic 5-and 6-membered rings in place of the p-substituted phenyl ring that could be linked via a sulfonyl chloride intermediate to the amino function as shown in the general scaffold (Chart 2, linker = SO2NH). The selection of promising candidates was performed mainly by visual inspection according to ligand-protein shape and complementarity and, for a few selected derivatives, the persistency of ligand-protein interactions over time. As part of this inspection, we selected docking poses that maintained the expected interaction pattern for the hydrophobic 4-(4-trifluoromethyl)-phenyl (common to chemical series 1 and 2a) and the carboxylic acid moieties rather than relying on the ranking by docking score (listed in Table S1). In particular, the key polar interactions of the carboxylate group identified previously are: electrostatic pairing with the side chains of Lys772.60 (Ballesteros-Weinstein numbering22) and Lys2777.35, and H-bonding with the phenol of Tyr1023.33. Furthermore, the terminal secondary amino group in 1 and 2a interacted with the hP2Y14R EL region through H-bonding to the helical backbone carbonyl group of Gly802.63. Notably, as discussed below, the computational predictions, based on this interaction pattern in qualitative agreement with the observed binding data, were able to identify specific geometric and electrostatic requirements for receptor binding.
Selection of the aromatic ring.
Among the computationally tested analogues containing 5-membered heteroaromatic rings such as thiophene, pyrrole, furan, and thiazole (green box in Chart 2), the thiophene ring was predicted to better fit a narrow pocket connecting the orthosteric binding site to the EL environment. As depicted in Figure 2A, along with the abovementioned interactions, the thiophene ring of 13 enabled an additional π-cation interaction with Arg2747.32. Therefore, 13 was selected as the first target heterocycle to synthesize in the new series. Among the computationally tested analogues containing 6-membered aromatic rings such as benzene and different isomers of pyridine and pyrimidine (see green box in Chart 2), the compounds were ranked as follows: benzene and pyridine were predicted to better fit the receptor than pyrimidine. Among the sulfonamide derivatives, the highest ranked 6-membered rings were predicted to establish the additional π-cation interaction with Arg2747.35 (e.g. benzene-containing compound 16 in Figure 2B). However, as the scaffolds of the analogues bearing a thiophene and a benzene ring superimposed well in the binding site, it was challenging to predict which of the two rings would lead to a more potent compound.
Figure 2.
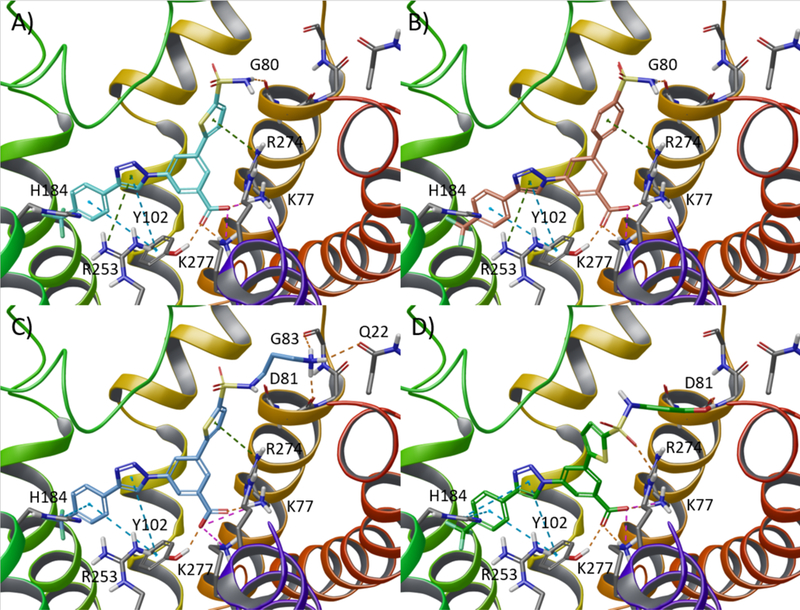
Upper panels: Selection of the aromatic ring. Docking pose of (A) thiophene derivative 13 (cyan carbon atoms) and (B) benzene derivative 16 (salmon carbon atoms) at the hP2Y14R. Both the thiophene and the benzene rings establish an additional π-cation interaction with Arg2747.32 (dark green dashed lines). Lower panel: Selection of the optimal spacer between the aromatic and amine functions. Docking poses of derivatives bearing a spacer of three methylene groups and a (C) terminal primary amine (compound 7, light blue carbon atoms) or (D) a hydroxyl group (dark green carbon atoms) at the hP2Y14R. Side chains of residues important for ligand recognition are reported as sticks (grey carbon atoms). H-bonds, salt bridges, and π-π stacking interactions are pictured as orange, magenta, and cyan dashed lines, respectively. Nonpolar hydrogen atoms are omitted.
Selection of the optimal spacer between the aromatic and amine functions.
Proceeding with the selected 5-membered heteroaromatic ring, i.e. thiophene, we evaluated the length of the spacer connecting the aromatic moiety to the amine function via a sulfonamide linker. In this stage, we also tested the possibility of replacing the amino group (positively charged at physiological pH and therefore giving rise to zwitterionic compounds) with another hydrogen bond donor/acceptor moiety, such as the hydroxyl group (Table S1). As shown in Figure 2C, a spacer consisting of three methylene groups was predicted to yield a derivative, compound 7, amenable to establish additional H-bond interactions with residues residing at the extracellular tip of TM2. Moreover, the docking pose predicted for the charged amino group resulted in a better geometry and a more favorable pattern of H-bond interactions with respect to the derivative bearing a hydroxyl moiety (Figure 2D).
Selection of the optimal spacer geometry.
An alternative binding pose of 7 (Figure S2A) suggested the possibility of rigidifying the flexible alkyl spacer by enclosing it in a 6-membered ring. We therefore docked several piperazine analogues bearing linkers of different geometries such as sulfonamide (12), amide (22), and methylene (18). The docking poses suggested that both the amide (compound 22, Figure S2C) and the sulfonamide (compound 12, Figure S2B) would allow the optimal orientation of the functional group, thus, retaining the H-bond pattern with residues located in the extracellular tip of TM2. On the other hand, a methylene spacer (compound 18, Figure S2D) was predicted to place the piperazine group in an unfavorable orientation with respect to the abovementioned H-bond network. However, the bioassay result (see below) contradicted this prediction, suggesting that some degree of flexibility either in the linker or in the spacer is required. We then evaluated the differences between the sulfonamide and amide linkers in the acyclic series by comparing the two corresponding derivatives bearing three methylene groups, previously predicted as the optimal spacer (Figure 3, upper panel). The superimposition of the docking poses of the compounds bearing the sulfonamide group (7) and the amide (20) did not predict significant changes in the H-bond pattern around the primary amino function (Figure 3A). We therefore subjected the two selected hits to 30 ns of MD simulation (run in triplicate, replicate analyses in Table S2, Supporting Information) and observed the stability of the ligand-protein interactions over time. As shown by the visualization of the trajectories (Video S1), both ligands were anchored in the binding site by a stable interaction pattern around the carboxylate moiety consisting of a H-bond with the Tyr1023.33 sidechain and two salt bridges with Lys772.60 and Lys2777.35 for the 7-hP2Y14R complex and with Lys772.60 and Arg2536.55 for the 20-hP2Y14R complex. The ligands were further stabilized by constant π–π stacking interactions connecting the 4-(4-trifluoromethyl)-phenyl moiety and the triazole ring to the His1845.36 and Tyr1023.33 sidechains, respectively. With respect to the H-bond pattern anchoring the primary amine to the extracellular (EC) tip of TM2, only the H-bond with the Gly802.63 sidechain was preserved during the MD simulations. Analogously, the π-cation interaction with Arg2747.35 was not stable over time, as the residue engaged in either a salt-bridge with the Asp812.64 sidechain or a H-bond with the Gln16 (N-Term) sidechain (Video S1, Supporting Information). No striking differences were noticed in the simulations of the two complexes, except a higher rigidity of the amine moiety in the amide compared with that in the sulfonamide-linked compound. This rigidity anchored the ligand in a slightly deeper position by allowing the carboxylate moiety to establish an additional salt bridge interaction with Arg2536.55 though requiring higher conformational rearrangement by the protein (Video S1 and Figure 3D). Indeed, as reported in Table S2, the 20-hP2Y14R complex returned higher average protein alpha-carbon atom (Cα) RMSD values with respect to the 7-hP2Y14R complex. A comparison between the ligand-protein complexes characterized by the lowest ligand-protein interaction energy (IEmin) extracted from the selected trajectories (Figure 3C and D), highlighted the presence of an additional salt bridge for the amide derivative (20). Although it was challenging to unambiguosly identify which of the two compounds would lead to a more potent derivative, knowledge gained from a previous SAR study20 and the MD results described above provided a hypothesis. Specifically, moieties displaying a higher conformational rigidity, a planar geometry, and an aromatic character would fit better within the narrow pocket connecting the orthosteric binding site with the extracellular (EC) environment delimited by the conserved disulfide bridge between Cys943.25 and Cys172 (EL2), on one side, and TM2, on the other side (Figure 3B).
Figure 3.
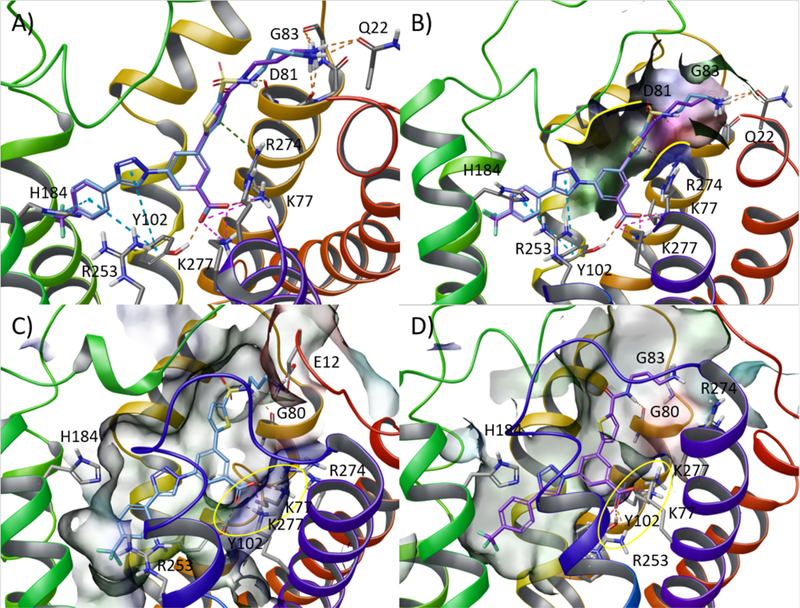
Selection of the optimal linker geometry: alkyl sulfonamide vs amide. Upper panel: docking analysis. (A) Superimposition of docking poses of sulfonamide derivative 7 (light blue carbon atoms) and amide derivative 20 (purple carbon atom) representing the starting geometry for subsequent MD simulations. (B) Detail of the shape (highlighted with a solid yellow line) of the narrow pocket surrounding the thiophene ring delimited by the conserved disulfide bridge and the EC tip of TM2 of the hP2Y14R. Lower panel: lowest interaction energy (IE) structure extracted from the selected trajectory of 30 ns of MD simulation of the (C) 7-hP2Y14R and (D) 20-hP2Y14R complexes. Side chains of residues important for ligand recognition are reported as sticks (grey carbon atoms). H-bonds and salt bridges are pictured as orange and magenta dashed lines, respectively. Nonpolar hydrogen atoms and π-π stacking interactions are omitted. The binding site surface is color-coded by residue property (blue: positively charged, red: negatively charged, green: hydrophobic, cyan: polar). The interaction pattern around the carboxylate moiety is highlighted with a yellow circle.
Replacement of the carboxylate group.
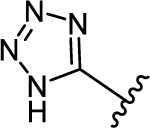
The 3-carboxylate group, in combination with an amino group on one side of the molecule and a hydrophobic portion on the other, contributed to limited aqueous solubility of some of the analogues. Therefore, we sought to substitute the carboxylate with known bioisosteres,24 beginning with a tetrazole moiety, which might also improve the bioavailability. Docking predictions suggested that the replacement of the carboxylate group with a bulkier tetrazole ring would require the scaffold to turn to fit in the narrow binding site while maintaining at least two out of the three key interactions with charged residues in TM7 and TM2 (data not shown).
Retrospective analysis.
Modeling analysis (Supplementary Information, Video S2 and Figure S3) of a subsequent potent analogue (primary carboxamide 30, see below) suggested that the scaffold might adopt a different orientation in the hP2Y14R binding site stabilized by a specific H-bond interaction between the ligand’s amide moiety and the sidechain of the non-conserved Asn903.21 allowing the ligand to interact with residues located in the EC tip of TM3 and EL1. As this EC region considerably differs among P2YRs receptors, it seems that derivative 30 might be a good lead compound for the design of more selective antagonists.
In an effort to explain the difference in the affinity of the new derivatives with respect to the parent compounds, we performed a MD analysis of the 2a-hP2Y14R complex. The initial structure was extracted from a previously published MD simulation20 and subjected to the same HTMD protocol described above (see replica analysis in Table S1). The interaction pattern that emerged from the trajectory visualizations (see selected run in Video S3) was consistent with the previously published MD analysis: the ligand was anchored in the binding site by its carboxylate moiety establishing salt-bridges with of Lys772.60 and Lys27 7 7.35 sidechains and H-bonds with Tyr1023.33 sidechain. The 4-(4-trifluoromethyl)-phenyl moiety and the triazole ring established stable π–π stacking interactions with Tyr102 3.33 and His1845.39, respectively. The ligand was positioned slightly higher in the binding site with respect to the initial structure, with the piperidine ring pointing toward TM3 and anchored to the Asn903.21 sidechain thorough a H-bond. The only noticeable difference between the MD analyses of the 2a-hP2Y14R complex and the complexes with new derivatives is a higher conformational plasticity around the bond connecting the p-substituted phenyl (30) or the thiophene ring (7 and 20) to the core benzene ring. It is likely that the H-bond anchoring the piperidine ring of 2a to TM3 restricts the conformation to ensure an optimal fit of the p-substituted phenyl in the narrow pocket connecting the orthosteric binding site with the EC environment. Consistent with this hypothesis, the amide derivative (20) displayed a higher rigidity in the linker, i.e. the region in the immediate proximity of the core benzene ring, to contribute to increased potency relative to the sulfonamide (7) derivative.
Chemical synthesis
The synthesis of the 3-(4-phenyl-1H-1,2,3-triazol-1-yl)-5-(aryl)benzene derivatives was performed as shown in Schemes 1–4. Scheme 1 shows the preparation by click chemistry, involving reaction of azide 40 and 4-(trifluoromethyl)phenylethylene, of a common intermediate 41 containing a methyl ester as a protected carboxylate group. A dioxaborolane group served as the preactivated attachment point for an additional aryl ring through a Suzuki reaction. The carboxylic group of 38 was first protected as the methyl ester 39, followed by conversion of the amine functionality into an azide 40, which was reacted with 4-(trifluoromethyl)phenylethylene in presence of copper sulfate to afford triazole 41.
Scheme 1:
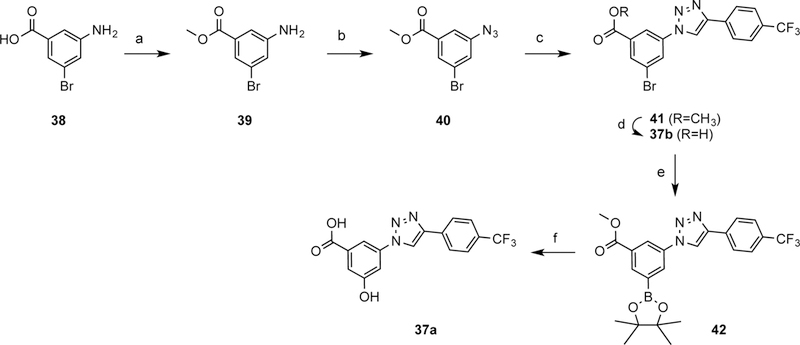
a) SOCl2, CH3OH, rt, 15 h, 98%; b) PTSA, NaNO2 then NaN3, H2O, CH3CN, rt, 15 h, 63%; c) 4-ethynyltrifluorotoluene, sodium ascorbate, CuSO4, THF, H2O, rt, 1 h, 84%; d) LiOH, MeOH, 60 °C, 2 h, 76%; e) B2pin2, KOAc, PdCl2(dppf), dioxane, 70 °C, 15 h, 87%; f) i) 10% NaOH, H2O2, THF, 0 °C, 0.5 h; ii) KOH, MeOH, water, 50 °C, 5 h, 86%.
Scheme 4:
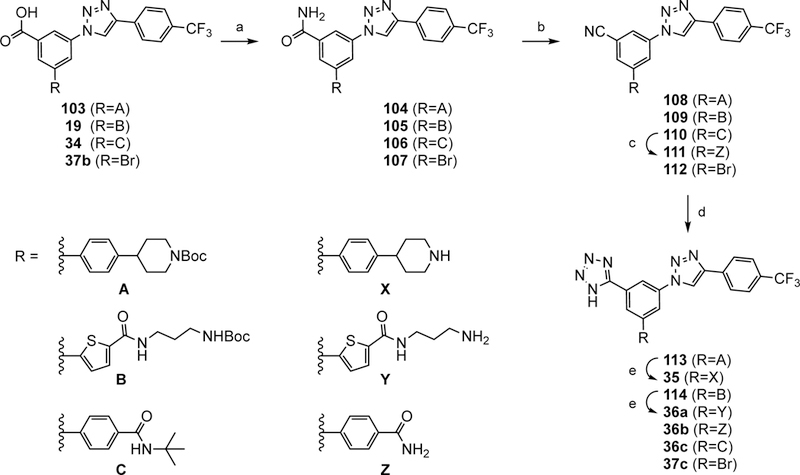
a) NH4Cl, HATU, DIPEA, DMF, rt, 1 h, 80–99%; b) (CF3CO)2O, Et3N, DCM, 0 °C to rt, 1 h, 64–76%; c) TMSOTf, anisole, μW, 140 °C, 1 h, 80%; d) CuSO45H2O, NaN3, DMSO, 140 °C, 6 h, 69–99%; e) TFA, rt, 10 min; 44–74%.
Dioxaborolane 42, which was the substrate for the Suzuki reaction, was synthesized by coupling aryl bromide 41 with bis(pinacolato)diboron (B2pin2) under a basic condition. Two of these synthetic intermediates, i.e. 41 and 42, were deprotected to yield truncated analogues 37b and 37a, respectively for biological evaluation. Thus, the dioxoborolane group of 42 was converted to a hydroxyl group under basic conditions to obtain 37a.
Scheme 2 shows the preparation of a series of aryl bromides 55–78 having either a carboxamide or sufonamide as the terminal group or as a linker to a an extended aminoalkyl (or hydroxyalkyl) chain. The amide and sulfonamide groups were intended for increasing aqueous solubility. For the synthesis of lead compound 2a, tert-butyl 4-(4-bromophenyl)piperidine-1-carboxylate (55) was prepared from compound 43, which was synthesized according to a previously reported procedure.17 To achieve the synthesis of various aryl bromides 56–78, three different strategies were employed. Thiophene and benzene sulfonyl chlorides were converted to sulfonamides (56–63) in the presence of triethylamine. Compound 64 was obtained by reductive amination of 5-bromothiophene-2-carbaldehyde (46).43 The carboxamides 65–78 were obtained with two different methods; compounds 65–73 were synthesized from the carboxylic acids 47–53 under HATU coupling conditions, while 4-bromobenzoyl chloride (54) was reacted with an appropriate amine to afford compounds 74–78. Scheme 3 shows the preparation of the condensed compounds through a Suzuki reaction with dioxaborolane 42 and aryl bromides 55–78 to afford compounds 79–102, which were then deprotected by one of three methods to yield compounds 2a and 4–34. In most cases, the hydrolysis of the methyl ester was accomplished using potassium hydroxide, followed by removal of a N-Boc group, when present on R, with trifluoroacetic acid.
Scheme 2:
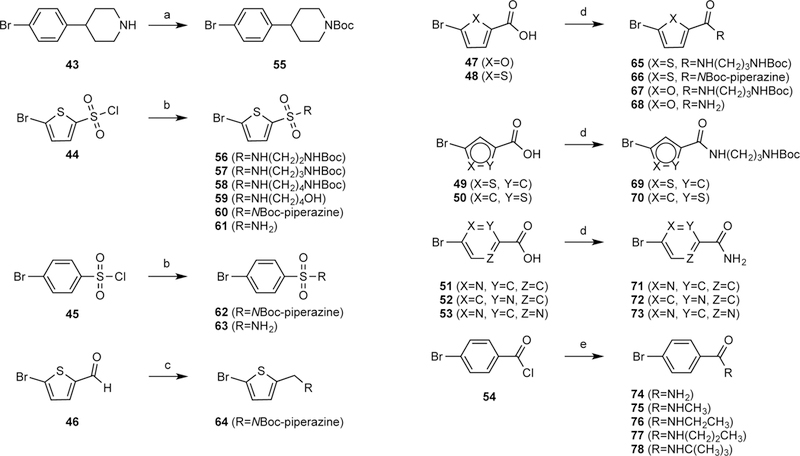
a) Boc2O, DCM, 0 °C, 0.5 h, 70%; b) amine, Et3N, DCM, rt, 78 – 99%; c) N-Boc piperazine, Et3N, NaBH(OAc)3, DCE, rt, 15 h, 90%; d) amine, HATU, DIPEA, DMF, rt, 14 – 88%; e) amine, Et3N, CH3CN, 0 °C, 73–99%.
Scheme 3:
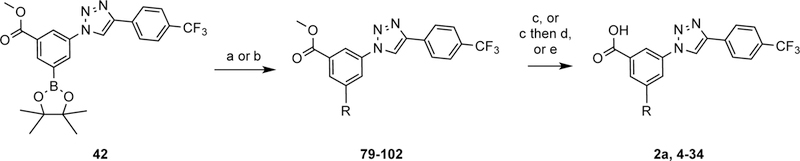
a) PdCl2(dppf), 2M Na2CO3, DME, 50 °C, 25 – 73%; b) Pd(PPh3)4, Na2CO3 (KOAc for 79), DMF, H2O, 11 – 43%; c) KOH, MeOH, H2O, 50 – 70 °C, 30 – 99%; d) TFA, rt, 34 – 92%; e) AlCl3, Me2S, rt, 15 h, 29 – 61%. The methyl esters of 79 - 102 were hydrolyzed using KOH to give compound 4, 6, 8, 10, 11, 13, 14, 16, 17, 19, 21 and 27 - 34. The N-Boc groups of compounds 103,4,6, 8, 11, 14, 17, 19 and 21 were removed with TFA to give compound 2a, 5, 7, 9, 12, 15, 18, 20 and 22, respectively. Compound 23 and 25 - 26 were obtained using AlCL3 and Me2S.
However, compound 25 and 26 were obtained by deprotection using AlCl3 and Me2S, because these two compounds were unstable in basic environments.
Scheme 4 shows the preparation of tetrazole analogues 35,-36a–c and 37c of compounds 2a, 20, 30, 34 and 37b through benzonitrile precursors 108–112. The carboxylic acid was converted into a carboxamide group and then into a nitrile group with trifluoroacetic anhydride. The conversion of the nitriles 108–112 to tetrazoles 113, 114, 36c, 36b and 37c, respectively, was accomplished using NaN3 in the presence of catalytic CuSO45H2O.44 Removing the N-Boc protecting group of 113–114 gave tetrazole derivatives 35 and 36a.
As previously described, the fluorescent antagonist 3a was prepared from the primary amino intermediate 3d. As a model compound to examine the effect of distal changes on the affinity in this chain-extended series, we prepared the terminal N-acetyl derivative 3e as described in Scheme 5. Dioxaborolane 119 was synthesized according to literature procedures reported.17 Suzuki reaction of 119 with compound 55 yielded 120, followed by removal of the ethyl ester and N-Boc protecting groups to provide 1. This route to prototypical P2Y14R antagonist 1 was improved over the previous route17 by avoiding catalytic reduction as the final step. The triazole linker leading to the fluorescent analogue was obtained following the previously reported procedure.20 The ethyl ester of 123 was hydrolyzed under basic condition to afford N-Boc derivative 3c, and then the free amine derivative 3d was obtained by acid treatment. The free amine 3d was masked with an acetyl group to give the acetyl derivative 3e. The truncated derivative 37d for biological evaluation was obtained by deprotection of synthetic intermediate 118. The synthetic procedures for truncated derivatives 37 are described in the Supporting Information.
Scheme 5:
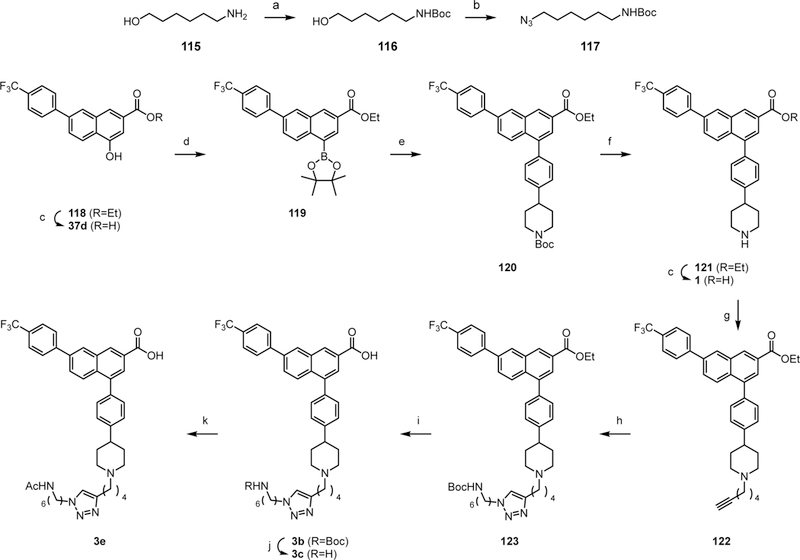
a) Boc2O, Et3N, DCM, rt, 3 h, 80%; b) i) MsCl, Et3N, DCM, 0 °C, 5 min; ii) NaN3, NBu4Br, H2O, 100 °C, 15 h, 59%; c) KOH, MeOH, water, 50 °C, 10 h; d) i) Tf2O, pyr, -78 °C to rt, 1 h; ii) B2pin2, PdCl2(dppf), KOAc, dioxane, 85 °C, 4 h, 61%; e) tert-butyl 4-(4-bromophenyl)piperidine-1- carboxylate 55, Pd(PPh3)4, K2CO3, DMF, 80 °C, 5 h, 59%; f) TFA:THF=1:1, rt, 1 h, 93%; g) 6- bromohexyne-1, K2CO3, DMF, rt, 15 h, 70%; h) 117, CuSO45H2O, sodium ascorbate, DCM:tBuOH:H2O=1:1:1, rt, 15 h, 58%; i) LiOH, MeOH, 80 °C, 15 h, 76%; j) TFA, rt, 30 min, 78%; k) Ac2O, pyr, rt, 1 h, 81%.
Biological characterization
The flow cytometric assay for measuring affinity at the hP2Y14R expressed in CHO cells was based on inhibition of cell surface binding of fluorescent antagonist 3a.20 The procedure was further adapted here to a more efficient 96-well format to increase the throughput. The IC50 values determined for prototypical P2Y14R antagonist 1 using the two method variations were nearly identical (8.0±0.4 nM using the 96-well format compared to 6.0±0.1 nM reported previously).20 Representative fluorescent binding inhibition curves are shown in Figure 4.
Figure 4.
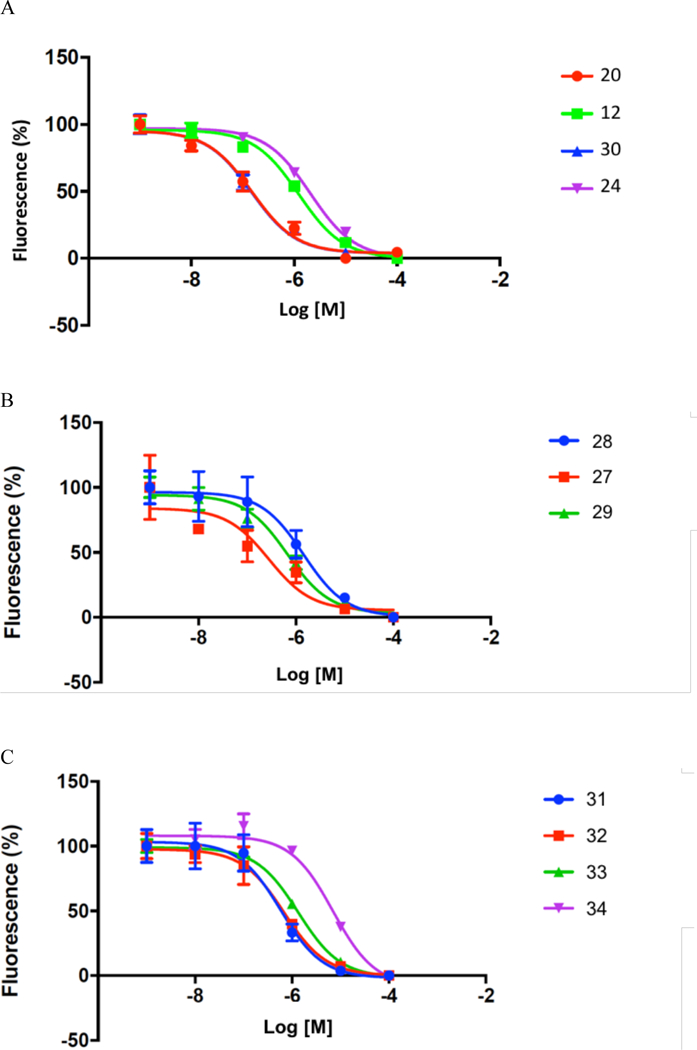
Fluorescent assay of specific hP2Y14R binding of selected 3-(4-trifluorophenyl-1H-1,2,3- triazol-1-yl)-5-(aryl) derivatives. AlexaFluor488-labeled tracer 3a binding was determined by flow cytometry using P2Y14R-expressing CHO cells. (A) 5-Thienyl 12 and 20, 5-furyl 24 and primary 4- carboxamidophenyl 30 derivatives. (B) Primary 4-substituted carboxamides with 5-pyridyl 27 and 28 and 5-pyrimidyl 29 aromatic groups. (C) N-alkyl elaboration of a potent 4-carboxamidophenyl congener 30: Me, 31; Et, 32; n-Pr, 33; tert-Bu, 34. The IC50 values are given in Table 1.
The most potent free amino derivative in the 2,5-disubstituted thiophene series was 20 (IC50 169 nM, (Figure 4A), which had a three-methylene spacer adjacent to a terminal primary amine and was anchored on the thiophene moiety through an amide linker. In the series of linear amino derivatives, carboxamides 19 and 20 were 4-fold more potent than the corresponding sulfonamides 6 and 7. However, in the series of cyclic amines, thienyl amide 22 was 2-fold less potent than thienyl sulfonamide 12. The effect of linear alkyl chain length and the terminal functionality was probed in the sulfonamide series. The optimal chain length among the primary amines was found to be three methylenes in 7 (IC50 756 nM), with the primary amino analogues containing two and four methylenes, 5 and 9, respectively, being 3-fold less potent in P2Y14R binding. When isolated, the Boc intermediates used for preparation of the free amines were tested for P2Y14R affinity. In each case of sulfonamide and amide derivatives with linear amine moieties, the free amine derivative, i.e. 5, 7, 9 and 20, was more potent than the corresponding Boc-protected amine. For example, the 3-aminopropyl congener 7 was 12-fold more potent than its Boc derivative 6 (IC50 9.33 μM), but the difference was less pronounced for amines 5 and 9. Similarly, for cyclic amines, the Boc group reduced potency, e.g. piperazine sulfonamide 15 (IC50 1.64 μM) compared to its inactive Boc derivative 14 (IC50 >50 μM). However, none of the piperazine free amine derivatives surpassed μM IC50 values. Methylpiperazine 18 was the most potent in the group consisting of piperazine sulfonamide 12, methylpiperazine 18 and piperazine carboxamide 22, while its Boc derivative 17 was 34-fold weaker. A primary alcohol derivative 10 was 4-fold less potent than the corresponding primary aminobutyl congener 9.
In a comparison of aryl groups, primary phenyl p-sulfonamide 16 was 10-fold more potent than the corresponding 2,5-disubstituted thienyl sulfonamide 13. However, p-phenyl sulfonamido-piperazine derivative 15 was similar in potency to the corresponding thienyl derivative 12. In the series of linear amino derivatives, 2,5-disubstituted thiophene carboxamides, e.g. Boc-protected 19 and free amine 20 were more potent than the corresponding thiophene sulfonamides 6 and 7, respectively. The phenyl primary sulfonamide 16 and phenyl primary carboxamide 30 displayed a sub-μM affinity (IC50 608 nM and 269 nM, respectively) and could serve as leads for additional analogues. Unlike most analogues in the initial set, the compounds are not Zwitterionic. It is recognized that pharmaceutical drugs include fewer Zwitterionic compounds than acidic, basic or neutral compounds, because of generally low oral bioavailability.23
The affinities of isomers of the most potent thiophene derivative 20 were compared. The alternate substitution patterns of thiophene, e.g. primary 3-aminopropyl derivatives 25 and 26, were less potent than the 2,5-disubstituted lead compound. Similarly, sulfur replacement with oxygen in furan 23 reduced affinity 39-fold. The furan primary carboxamide 24 was 10-fold less potent than the corresponding p-phenyl substituted carboxamide 30. The corresponding thiophene 5-carboxamide related to 20 was not prepared.
The introduction of nitrogen atoms in the p-substituted phenyl ring of 30 reduced the potency by 2 to 5-fold in 27–29 (Figure 4B). Compounds 31–34 are homologated N-alkyl amides based on potent antagonist 30. The N-methyl 31 and ethyl 32 analogues were roughly equipotent to the parent carboxamide, but the N-propyl analogue 33 was less potent (Figure 4C). The N-tert-Bu analogue 34 was 15-fold less potent than the primary carboxamide 30.
Carboxylate bioisosteres were considered,24 although this group was deemed essential in previous studies of P2Y14R antagonists. The substitution of the carboxylate of 2a20 with a tetrazole moiety in 35 reduced the affinity by 30-fold. However, the difference between carboxylate and tetrazole isostere was less in comparing 20 and 36a (7-fold). In comparing potent carboxamide analogues 30 and 34 and their tetrazole derivatives 36b and 36c, the affinity was either reduced 9-fold or 3-fold, respectively. The truncated derivatives 37 were greatly reduced in potency in the triazole series, but less so in the naphthalene series (37d) with an IC50 value of 3.2 μM. Thus, carboxylic acid 37d is a relatively simple, novel molecule with moderate affinity at the P2Y14R.
The aryl ring (green in Chart 1) contribution to the overall affinity was explored with compound 37a, in which that ring is replaced with a hydroxyl group. Compared to phenylcarboxamide 30, compound 37a was 60-fold weaker in its interaction with the P2Y14R. Thus, this aryl ring provides a major contribution to the P2Y14R binding.
To further explore the interaction of long chains, including amino functionalized chains with the outer P2Y14R regions, compounds 3c (alkyne), 3c (Boc-protected), 3d (primary amine) and 3e (acetamido derivative), all related to the naphthalene-containing fluorescent probe 3a, were prepared and tested. The order of affinity in the fluorescent P2Y14R assay was 3d > 3e > 3c > 3b, and all three congeners were more potent than the most potent triazole derivatives prepared in the present study, i.e. 20 and 30.
Off-target and ADME-tox testing
Off-target interactions of selected antagonists (1, 2a, 3d, 3e, 16, 20, 30 and 37d) were determined at 45 receptors, transporters and ion channels by the Psychoactive Drug Screening Program (PDSP).21 The previously reported20 Ki values (μM) of 1 were 6.79 (D3 dopamine receptor) and 2.75 (δ-opioid receptor); of 2a: α2c-adrenergic, 1.32; H1-histamine, 0.17. Compounds 16, 20 and 30 also bound to the δ-opioid receptor with Ki values (μM) of 3.26±0.64, 0.941 and 1.47, respectively. Carboxamide 30 did not bind appreciably to any other sites in the comprehensive PDSP screen, and truncated naphthalene derivative 37d bound only to NET (5.8 μM). Therefore, this compound series is generally not promiscuous, i.e. PAINS compounds.25
Curiously, the long-chain primary amino derivative 3d displayed extensive off-target interactions with biogenic amine receptors. The Ki values (μM) of 3d that were below 10 μM were: 5HT1A, 5.65; 5HT2A, 8.4; α1A, 1.77; α1B, 1.11±0.37; α1D, 1.27; α2A, 0.028; α2B, 0.984; D1, 7.60; D2, 8.21; D3, 1.60; D5, 1.07; H2, 0.365. Also, interactions of 3d with the dopamine transporter (DAT, 1.07 μM), norepinephrine transporter (NET, 1.87 μM), and sigma2 receptor (1.2 μM) were observed. We hypothesize that the terminal amino group interacts with the common amine-binding residue in biogenic amine receptors (D2.50), because the off-target activities at α-adrenergic and dopamine receptors were greatly reduced in the terminal N-acetyl derivative 3e. Thus, the nine interactions below 10 μM of 3c were (Ki, μM): 5HT2B, 2.78; 5HT6, 7.9;; α2A, 5.28; β3, 2.36±0.48; H2, 0.829; H3, 2.77; KOR, 4.06; TSPO, 5.60; SERT, 2.08. However, compound 20 was also a primary amine, but it did not display extensive off-target interactions with biogenic amine receptors.
cLogP (calculated logarithm of partition coefficient between n-octanol and water), a good predictor of solubility, is shown for each analogue in Table 1.46 Based on the range of cLogP values, the carboxylate analogues 4–34 are expected to have more favorable (lower) hydrophobicity (0.40–4.90) than compound 1 (6.18) and its N-alkylated congeners 3b–3e (4.04–7.54). cLogS and cLogD at pH 7.4 of 30 were calculated to be-3.24 and 1.51, respectively, indicating predicted moderate aqueous solubility when ionized at physiological pH (Figure S4, Supporting Information). However, 20 as a Zwitterion has increased solubility predicted at pH extremes compared to physiological pH, with cLogS and cLogD at pH 7.4 of-6.5 and 1.9.
The tetrazole derivatives 35, 36 and 37c displayed intermediate cLogP values (2.97–4.40). The effect, based on cLogD and cLogS (Figure S4, Supporting Information), of replacing a carboxylate group with tetrazole was generally to reduce predicted solubility at pH 7.4 (ΔcLogD): for 2a and 35 (-0.3); for 20 and 36a (-0.3); for 30 and 36b (-1.3).
Predicted pharmacokinetic parameters for some of the more potent compounds 7, 16, 20, 25, 26, 29–32 and 35 (Table S3, Supporting Information)47 indicated satisfactory intestinal absorption (63–100%) and unbound fraction (18–27%), with no AMES toxicity, renal OGT substrate, or PGP I inhibition properties. However, for all compounds, PGP substrate properties were predicted. Compounds 7, 16, 20, 25, 26, 32 and 35 were predicted as CYP 3A4 substrates.
The pharmacokinetics of a representative non-Zwitterionic analogue 30 was studied in the male Sprague Dawley rat following oral administration (1–10 mg/kg) indicated a lack of bioavailability. Consistent with this finding, 30 lacked CACO-2 cell permeability (Papp, apical to basal, was 0.0 X 10−6 cm/sec). Its plasma half-life when administered i.v. (0.5 mg/kg) was short (0.20 ± 0.14 h, Table S4, Supporting Information). Other preclinical assays were performed as reported.42 30 did not inhibit CYP450 enzymes (IC50 ≥ 30 μM at 1A2, 2C9, 2C19, 2D6 and 3A4 isoforms) and was stable in simulated gastric and intestinal fluids (100% remaining after 240 min) and in plasma of three species (94 – 100% remaining at 120 min; human, rat and mouse). 30 was not cytotoxic to Hep-G2 cells (CC50 > 30 μM). Its aqueous solubility was 138±4 μg/mL, and the IC50 at hERG was 0.166 μM.
Discussion and Conclusions
We have expanded the SAR of heterocyclic P2Y14R antagonists, guided by computational methods. Although oral bioavailability was lacking in the one compound tested (30), it displayed high aqueous solubility and stability in simulated body fluids. We now know that 30 has a hERG liability, but this can likely be overcome with additional structural modification.
In the previous modeling analysis,20 the required aryl carboxylate of this chemical series interacted with a pair of Lys residues in TMs 2 and 7 that are specific to the Gi-coupled subfamily of P2YRs. Furthermore, a Tyr residue which forms a putative π interaction with the naphthalene ring of 1 was predicted to shift to the vicinity of the triazole ring in its bioisosteric equivalent in 2a, to form a stabilizing π interaction.20 This interaction was not observed for a synthesized bioisosteric alkyne derivative, which was suggested as an alternative scaffold, consistent with the greater affinity of the triazole-containing scaffold compared to the alkyne. We have, therefore, conserved the CF3-phenyl ring of 2a and the triazole moiety in the present set of analogues and concentrated on modification of the terminal phenyl-piperidine rings of 1 and 2a. In addition, we tried to find a bioisosteric replacement for the carboxylate group on the central ring.
Thus, a modeling pipeline based on a hP2Y14R homology model guided analogue design. Many but not all predictions based on this approach were confirmed experimentally. In particular, we were able to identify (i) the bioisostere replacement of the p-substituted phenyl ring with other 5- and 6-membered rings, (ii) suitable linker and spacer connecting the aromatic moiety to the amine function, and, to some degree, (ii) the optimal spacer length and geometry between the linker and the amine moiety. With respect to the latter aspect, the experimental data suggested that some degree of flexibility either in the linker or in the spacer is required. In particular, if the amine was enclosed in a rigidified spacer, e.g. a piperazine ring, more flexible linkers yielded more active compounds (12 and 18 more active than 22). Conversely, when the amine was attached to a flexible alkyl spacer, the more rigidified the linker the more potent was the corresponding compound (e. g. 7 vs 20).
In conclusion, we have used a computational pipeline to predict the P2Y14R interactions of 5-aryl ring substituents and functional groups, which were largely consistent with empirical results. The substitution of a key carboxylate group on the core phenyl ring with tetrazole or truncation of the 5-aryl group reduced affinity. The most potent antagonists, using a fluorescent assay, were a thienyl-linked primary 3-aminopropyl congener 20 and a phenyl p-carboxamide 30.
The activity of a P2Y14R antagonist in reducing neutrophil motility and inflammation, a predictor of utility of selective P2Y14R antagonists in inflammatory diseases, has been shown. Future studies will focus on the use of these leads to discovery of more drug-like molecules for application to animal models.
Experimental Section
Molecular Modeling
Protein preparation
In this study, we used a previously published20 hP2Y14R homology model based on the high resolution X-ray structure of an agonist-bound hP2Y12R (PDB ID: 4PXZ)16 that was further refined using molecular dynamics (MD) simulation. The structure extracted from the MD trajectory was prepared for docking using the Protein Preparation Wizard tool implemented in the Schrödinger suite28 by removing water molecules and minimizing protein sidechains with OPLS3 (convergence criterion: heavy atoms RMSD < 0.30 Å). The protonation state of histidine residues was determined according to H-bond patterns analysis. In particular, His184/217/280 were protonated on the Nδ and His264 was considered doubly protonated.
Docking
Ligand structures were built using the Builder tool implemented in the Schrödinger suite29 and minimized using the OPLS3 force field. Molecular docking was performed with Glide30, by selecting the barycenter of PPTN in the MD-refined hP2Y14 homology model as the grid center (inner box: 10 × 10 × 10 Å; outer box extended by 30 Å in each direction from the inner box). Docking was performed considering the protein binding site residues rigid by using the standard precision (SP) scoring function and generating 5 poses for each ligand. Selection of hits were performed by visual inspection of optimal ligand-protein interactions rather than on the basis of docking scores. In particular, for each ligand we selected the docking poses establishing H-bond and salt bridge interactions with the Tyr1023.33, Lys772.60 and Lys27 77.35 sidechains.
Molecular Dynamics
MD system setup, equilibration, and production were performed with the HTMD31 module (Acellera, Barcelona Spain, version 1.5.4). The ligand-protein complexes were embedded into an 80 x 80 Å 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane leaflet generated through the VMD Membrane Plugin tool32. Overlapping lipids (within 0.6 Å) were removed upon protein insertion and the systems were solvated with TIP3P33 water and neutralized by Na+/Cl− counter-ions (final concentration 0.154 M). MD simulations were carried out with the ACEMD engine (Acellera, version 2016.10.27)34 using the CHARMM3635,36/CGenFF(3.0.1)37,38 force fields for lipid and protein, and ligand atoms, respectively and periodic boundaries conditions. Ligand parameters were retrieved from the ParamChem service (https://cgenff.paramchem.org, accessed 07/2017, version 1.0.0) with no further optimization. The systems were equilibrated through a 5000-step minimization followed by 40 ns of MD simulation in the NPT ensemble by applying initial constraints (0.8 for the ligand atoms, 0.85 for alpha carbon atoms, and 0.4 for the other protein atoms) that were linearly reduced after 20 ns. During the equilibration procedure, the temperature was maintained at 310 K using a Langevin thermostat (damping constant = 1 ps−1) and the pressure was maintained at 1 atm using a Berendensen barostat. Bond lengths involving hydrogen atoms were constrained using the M-SHAKE39 algorithm. The equilibrated systems were subjected to 30 ns of unrestrained MD simulations run in triplicate for each ligand-protein complex (NVT ensemble, timestep = 2 fs, damping constant = 0.1 ps−1). Long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)40 with grid size rounded to the approximate integer value of cell wall dimensions. A non-bonded cutoff distance of 9 Å with a switching distance of 7.5 Å was used. All simulations were run on three NVIDIA GeForce GTX (two 980Ti, and one 1080).
MD Trajectory Analysis
MD trajectory analysis was performed with an in-house script exploiting the NAMD 2.1041 mdenergy function and the RMSD trajectory tool (RSMDTT) implemented in VMD32. All simulations were run in triplicate and selection of representative trajectories and of lowest interaction energy (IE) ligand-protein complexes were based upon the total ligand–protein interaction energy (IEtot) expressed as the sum of van der Waals (IEvdw) and electrostatic (IEele) contribution as previously described.42
Chemical synthesis
Reagents and instrumentation
All reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO), Ark Pharm, Inc. (Libertyville, IL; 6-bromonicotinic acid, 5-bromopicolinic acid and 5- bromopyrazine-2-carboxylic acid) and Enamine LLC (Cincinnati, OH; 5-bromopyrazine-2-carboxlic acid). 1H NMR spectra were obtained with a Bruker 400 spectrometer using CDCl3, CD3OD3, and DMSO-d6 as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. NMR spectra were collected with a Bruker AV spectrometer equipped with a z-gradient [1H,13C,15N]-cryoprobe. TLC analysis was carried out on glass sheets precoated with silica gel F254 (0.2 mm) from Sigma-Aldrich. The purity of final compounds was checked using a Hewlett-Packard 1100 HPLC equipped with a Zorbax SB-Aq 5 μm analytical column (50 × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 5 mM tetrabutylammonium dihydrogen phosphate–CH3CN from 100:0 to 0:100 in 15 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed >95% purity by HPLC analysis (detection at 254 nm). Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (MicromassWaters) using external calibration with polyalanine, unless noted. Observed mass accuracies are those expected based on known instrument performance as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time dependent drift in mass accuracy. tPSA (total polar surface area) was calculated using ChemDraw Professional (PerkinElmer, Boston, MA, v. 16.0). cLogP was calculated as reported.46 Simulated pharmacokinetic parameters were calculated as reported.47 Compound 3b was prepared as reported.17
4-(4-(3-Carboxy-6-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)phenyl)piperidin-1-ium chloride (1)
The mixture of compound 121 (55 mg, 0.109 mmol) in methanol (2 mL) and water (1 mL) was added potassium hydroxide (30 mg, 0.545 mmol), and then this reaction mixture was stirred at 70 °C for 15 h. After being neutralized with 1M HCl, a white precipitate was collected by filtration, washed with water and dried under vacuum to afford compound 1 as a white solid (40 mg, 77%).
4-(4-(1-(4-(1-(6-((tert-Butoxycarbonyl)amino)hexyl)-1H-1,2,3-triazol-4-yl)butyl)piperidm-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic acid (3c)
A mixture of compound 123 (30 mg, 36.3 |imol) and lithium hydroxide (9 mg, 0.363 mmol) in methanol (5 mL) was stirred at 80 °C for 15 h. After cooling at room temperature, this mixture was neutralized with 1N HCl until pH was 5–6. The slightly acidic mixture was evaporated under reduced pressure and purified by silica gel column chromatography (dichloromethane:methanol=5:1) to afford compound 3b (22 mg, 76%) as a white solid; HPLC purity 95% (Rt = 13.99 min); 1H NMR (400 MHz, CD3OD) δ 8.62 (s, 1H), 8.37 (s, 1H), 8.04 (s, 1H), 7.95 (d, J = 8.04 Hz, 2H), 7.90 (d, J = 8.80 Hz, 1H), 7.82 (s, 1H), 7.78–7.74 (m, 3H), 7.49 (d, J = 7.44 Hz, 2H), 7.35 (d, J = 7.72 Hz, 2H), 4.36 (t, J = 7.08 Hz, 2H), 3.65 (d, J = 11.6 Hz, 2H), 3.19 (t, J = 7.68 Hz, 2H), 3.08–2.97 (m, 4H), 2.94–2.91 (m, 1H), 2.83 (t, J = 7.06 Hz, 2H), 2.09 (s, 4H), 1.90–1.80 (m, 6H), 1.43 (s, 9H), 1.33–1.31 (m, 6H); MS (ESI, M/Z) 798.4 [M+1]+; ESI-HRMS calcd. m/z for C46H55N5O4F3 798.4206 found 798.4217 [M+1]+.
4-(4-(1-(4-(1-(6-Ammohexyl)-1H-1,2,3-triazol-4-yl)butyl)piperidm-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic acid (3d)
A mixture of compound 3c (22 mg, 27.6 μmol) in trifluoroacetic acid (0.2 mL) was stirred at room temperature for 30 min. The solvent was evaporated with methanol under reduced pressure. The residue was purified by semipreparative HPLC (0.1% TFA-water:acetonitrile=20:80 to 80:20 in 40 min) to afford compound 3d (15 mg, 78%) as a white solid; Rt = 32.7 min; HPLC purity 96% (Rt = 11.84 min); 1H NMR (400 MHz, CD3OD) 8.78 (s, 1H), 8.46 (s, 1H), 8.04–7.99 (m, 4H), 7.96–7.93 (m, 1H), 7.84–7.78 (m, 3H), 7.55 (d, J = 8.24 Hz, 2H), 7.50 (d, J = 8.24 Hz, 2H), 4.42 (t, J = 7.04 Hz, 2H), 3.76 (d, J = 11.96 Hz, 2H), 3.29–3.23 (m, 4H), 3.21–3.14 (m, 2H), 3.11–3.05 (m, 1H), 2.93 (t, J = 7.56 Hz, 2H), 2.83 (t, J = 7.24 Hz, 2H), 2.28 (d, J = 14.96 Hz, 2H), 2.15–2.05 (m, 1H), 2.00–1.82 (m, 5H), 1.71–1.64 (m, 2H), 1.50–1.37 (m, 4H); MS (ESI, M/Z) 698.4 [M+1]+; ESI-HRMS calcd. m/z for C41H47N5O2F3 698.3682, found 698.3689 [M+1]+.
4-(4-(1-(4-(1-(6-Acetamidohexyl)-1H-1,2,3-triazol-4-yl)butyl)piperidm-4-yl)phenyl)-7-(4- (trifluoromethyl)phenyl)-2-naphthoic acid (3e)
To a solution of compound 3e (2.24 mg, 3.21 μmol) in pyridine (0.3 mL) was added acetic anhydride (10 μl), and then this reaction mixture was stirred at room temperature for 1 h. After all volatiles were evaporated under reduced pressure, the residue purified by semipreparative HPLC (10 mM triethylammonium acetate buffer:acetonitrile=20:80 to 80:20 in 40 min) to afford compound 3d (1.9 mg, 81%) as a white solid; Rt = 34.2 min; HPLC purity 97% (Rt = 13.07 min); 1H NMR (400 MHz, CD3OD) 8.59 (s, 1H), 8.37 (s, 1H), 8.04 (s, 1H), 8.01–7.98 (m, 3H), 7.84–7.80 (m, 4H), 7.50 (d, J = 7.96 Hz, 2H),7.43 (d, J = 7.96 Hz, 2H), 4.39 (t, J = 7.06 Hz, 2H), 3.50–3.43 (m, 2H), 3.15 (t, J = 7.02 Hz, 2H), 2.88–2.81 (m, 4H), 2.74–2.68 (m, 1H), 2.13–1.99 (m, 2H), 1.96–1.89 (m, 7H), 1.78 (m, 4H), 1.54–1.47 (m, 2H), 1.43–1.27 (m, 6H); MS (ESI, M/Z) 740.4 [M+1]+; ESI-HRMS calcd. m/z for C43H49N5O3F3 740.3788, found 740.3779 [M+1]+.
General procedure for carboxylic acid derivative deprotection reactions
Method A:
A mixture of the condensed compound (41, 79–90, 92, 95–102, 118, 121, 123; 1 eq, 5–10 mg) and potassium hydroxide (5 eq) in a methanol (1 mL):water (0.5 mL) mixture was stirred at 50 °C for 15 h. This mixture was neutralized with 1N HCl until pH 5–6. The slightly acidic mixture was evaporated under reduced pressure, and the product was purified by silica gel column chromatography (dichloromethane:methanol:acetic acid=95:5:0.1) or semipreparative HPLC (10 mM triethylammonium acetate buffer:acetonitrile=20:80 to 80:20 in 40 min) to afford the product as a white solid. The salt form obtained was determined using NMR.
Method B:
A solution of the carboxylic derivative (4, 6, 8, 11, 14, 17, 19, 21, 113, 114; 1 eq, 5 mM) in trifluoroacetic acid was stirred at room temperature for 10 min. The solvent, with methanol added, was evaporated under reduced pressure. The residue was purified by semipreparative HPLC (10 mM triethylammonium acetate buffer:acetonitrile=20:80 to 80:20 in 40 min) to afford the compound as a white solid.
Method C:
For compounds that decomposed using Method A or B, the condensed compound (91, 93, 94; 1 eq, 5 mM) was dissolved in dimethyl sulfide and treated with aluminum chloride (15 eq), and the reaction mixture was stirred at room temperature for 15 h. The resulting suspension was filtered through a bed of silica gel, which was washed with dichloromethane:methanol:acetic acid (100:20:1) to separate residual product from the aluminum salts. The combined solution was evaporated to dryness under reduced pressure. The residue was purified by semipreparative HPLC (triethylammonium acetate buffer and acetonitrile=20:80 to 80:20 in 40 min) to afford the product as a white solid.
3-(5-(N-(2-((tert-Butoxycarbonyl)amino)ethyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (4)
Method A:
Yield 72%; HPLC purity 99% (Rt = 13.46 min); 1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 8.54 (s, 1H), 8.52 (s, 1H), 8.29 (s, 1H), 8.21 (d, J = 8.4 Hz, 2H), 8.06 (broad s, 1H), 7.91 (d, J =8.4 Hz, 2H), 7.88 (d, J = 4.0 Hz, 1H), 7.67 (d, J = 4.0 Hz, 1H), 6.84 (broad s, 1H), 3.04–3.02 (m, 2H), 2.95–2.93 (m, 2H), 1.35 (s, 9H); MS (ESI, M/Z) 538.1 [M+1-Boc]+, 582.0 [M+1-isopropyl]+; ESI- HRMS calcd. m/z for C22H19N5O4F3S2 538.0831, found 538.0841 [M+1-Boc]+.
3-(5-(N-(2-Aminoethyl)sulfamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (5)
Method B:
Yield 34%; Semipreparative HPLC: Rt = 27.6 min; HPLC purity 95% (Rt = 10.40 min); 1H NMR (400 MHz, DMSO-d 6) δ 9.56 (s, 1H), 8.42 (s, 1H), 8.19 (s, 1H), 8.16 (s, 1H), 8.10 (d, J = 8.0 Hz, 2H), 7.81 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 4.0 Hz, 1H), 7.69 (d, J = 4.0 Hz, 1H), 6.59 (broad s, 2H; NH2), 2.97 (t, J = 6.4 Hz, 4H); MS (ESI, M/Z) 538.1 [M+1]+; ESI-HRMS calcd. m/z for C22H19N5O4F3S2 538.0831, found 538.0837 [M+1]+.
3-(5-(N-(3-((tert-Butoxycarbonyl)amino)propyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (6)
Method A:
Yield 45%; HPLC purity 98% (Rt = 14.00 min); 1H NMR (400 MHz, CD3OD) δ 9.26 (s, 1H), 8.51 (s, 1H), 8.42–8.41 (m, 2H), 8.19 (d, J = 8.12 Hz, 2H), 7.81 (d, J = 8.28 Hz, 2H), 7.68 (d, J = Hz, 1H), 7.65 (d, J = 3.96 Hz, 1H), 3.10 (t, J = 6.62 Hz, 2H), 3.05 (t, J = 7.08 Hz, 2H), 1.70 (t, J = 6.90 Hz, 2H), 1.41 (s, 9H); MS (ESI, M/Z) 552.1 [M+1-Boc]+; ESI-HRMS calcd. m/z for C23H21N5O4F3S2 552.0987, found 552.0979 [M+1-Boc]+.
3-(5-(N-(3-Ammopropyl)sulfamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3- triazol-1-yl)benzoic acid (7)
Method B:
Yield 73%; Semipreparative HPLC: Rt = 30.9 min; HPLC purity 98% (Rt = 10.47 min); 1H NMR (400 MHz, DMSO-d 6) δ 9.49 (s, 1H), 8.37 (s, 1H), 8.06–8.04 (m, 3H), 7.78 (s, 1H), 7.75 (d, J =8.4 Hz, 2H), 7.60 (s, 2H), 3.12 (t, J = 6.8 Hz, 2H), 2.87 (t, J = 6.8 Hz, 2H), 1.80–1.76 (m, 2H); MS (ESI, M/Z) 552.0 [M+1]+; ESI-HRMS calcd. m/z for C23H21N5O4F3S2 552.0987, found 552.0991 [M+1]+.
3-(5-(N-(4-((tert-Butoxycarbonyl)amino)butyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (8)
Method A:
Yield 71%; HPLC purity 98% (Rt = 14.07 min); 1H NMR (400 MHz, CD3OD) δ 9.23 (s, 1H), 8.54 (s, 1H), 8.41 (s, 1H), 8.38 (s, 1H), 8.15 (d, J = 7.2 Hz, 2H), 7.78 (d, J = 7.6 Hz, 2H), 7.63 (s, 2H; thiophene), 3.05–3.02 (m, 4H), 1.54 (m, 4H), 1.41 (s, 9H); MS (ESI, M/Z) 664.1 [M-1]−, 778.1 [M- H+TFA]−; ESI-HRMS calcd. m/z for C29H29N5O6F3S2 664.1511, found 664.1507 [M-H]−.
3-(5-(N-(4-Ammobutyl)sulfamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (9)
Method B:
Yield 40%; Semipreparative HPLC: Rt = 28.5 min; HPLC purity 95% (Rt = 10.88 min); 1H NMR (400 MHz, DMSO-d 6) δ 9.62 (s, 1H), 8.40 (s, 1H), 8.23 (s, 1H), 8.22 (s, 1H), 8.20 (d, J = 8.4 Hz, 2H), 7.88 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 4.0 Hz, 1H), 7.63 (d, J = 4.0 Hz, 1H), 2.92 (s, 2H), 2.67 (s, 2H), 1.49 (s, 4H); MS (ESI, M/Z) 566.1 [M+1]+; ESI-HRMS calcd. m/z for C24H23N5O4F3 S2 566.1144, found 566.1145 [M+1]+.
3-(5-(N-(4-Hydroxybutyl)sulfamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3- triazol-1-yl)benzoic acid (10)
Method A:
Yield 30%; Semipreparative HPLC: Rt = 25.1 min; HPLC purity 97% (Rt = 12.13 min); 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.49 (s, 1H), 8.46 (s, 1H), 8.27 (s, 1H), 8.20 (d, J = 8.4 Hz, 2H), 7.95 (t, J = 5.8 Hz, 1H; NH), 7.91 (d, J = 8.0 Hz, 2H), 7.83 (d, J = 4.0 Hz, 1H), 7.65 (d, J = 4.0 Hz, 1H), 4.42 (broad s, 1H; NH), 3.36 (merged with water peak), 2.92 (q, J = 6.6 Hz, 2H), 1.49–1.42 (m, 4H); MS (ESI, M/Z) 567.1 [M+1]+; ESI-HRMS calcd. m/z for C24H22N4O5F3S2 567.0984, found 567.0981 [M+1]+.
3-(5-((4-(tert-Butoxycarbonyl)piperazin-1-yl)sulfbnyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (11)
Method A:
Yield 93%; HPLC purity 98% (Rt = 15.22 min); 1H NMR (400 MHz, CD3OD) δ 9.21 (s, 1H), 8.51 (s, 1H), 8.39 (s, 1H), 8.33 (s, 1H), 8.11 (d, J = 7.2 Hz, 2H), 7.76 (d, J = 8.0 Hz, 2H), 7.70 (s, 1H), 7.63 (s, 1H), 3.58 (s, 4H), 3.11 (s, 4H), 1.42 (s, 9H); MS (ESI, m/z) 564.1 [M+1-Boc]+, 608.1 [M+1-isobutyl]+, 664.2 [M+1]+; ESI-HRMS calcd. m/z for C29H29N5O6F3S 664.1511, found 664.1523 [M+1]+.
3-(5-(Piperazin-1-ylsulfbnyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (12)
Method B:
Yield 54%; Semipreparative HPLC: Rt = 29.3 min; HPLC purity 96% (Rt = 12.22 min); 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.34 (s, 1H), 8.03 (s, 1H), 7.97 (d, J = 8.0 Hz, 2H), 7.92 (s, 1H), 7.74 (d, J = 4.0 Hz, 1H), 7.72 (d, J = 4.0 Hz, 1H), 7.68 (d, J = 8.0 Hz, 2H), 3.18 (s, 4H), 3.11 (s, 4H); MS (ESI, M/Z) 564.1 [M+1]+; ESI-HRMS calcd. m/z for C24H21N5O4F3S 564.0987, found 564.0980 [M+1]+.
3-(5-Sulfamoylthiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (13)
Method A:
Yield 64%; Semipreparative HPLC: Rt = 29.8 min; HPLC purity 99% (Rt = 12.71 min); 1H NMR (400 MHz, DMSO-d6) δ 9.62 (s, 1H), 8.42 (s, 1H), 8.30 (s, 1H), 8.24 (s, 1H), 8.20 (d, J = 8.4 Hz, 2H), 7.89 (d, J = 8.0 Hz, 2H), 7.82 (s, 2H; NH2), 7.70 (d, J = 4.0 Hz, 1H), 7.62 (d, J = 4.0 Hz, 1H), 2.99–2.97 (m, 6H; Et3N salt), 1.14 (t, J = 7.4 Hz, 9H; Et3N salt); MS (ESI, M/Z) 495.0 [M+1]+; ESI-HRMS calcd. m/z for C20H14N4O4F3S2 495.0409, found 495.0402 [M+1]+.
4'-((4-(tert-Butoxycarbonyl)piperazm-1-yl)sulfonyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3-carboxylic acid (14)
Method A:
Yield 82%; HPLC purity 99% (Rt = 14.54 min); 1H NMR (400 MHz, CD3OD) δ 9.26 (s, 1H), 8.60 (s, 1H), 8.47–8.46 (m, 2H), 8.18 (d, J = 8.12 Hz, 2H), 8.06 (d, J = 8.44 Hz, 2H), 7.95 (d, J = 8.44 Hz, 2H), 7.81 (d, J = 8.24 Hz, 2H), 3.56–3.53 (m, 4H), 3.05 (t, J = 4.88 Hz, 4H), 1.42 (s, 9H); MS (ESI, M/Z) 558.2 [M+1-Boc]+, 602.1 [M+1-isobutyl]+, 658.3 [M+1]+; ESI-HRMS calcd. m/z for C27H23N5O6F3S 602.1321, found 602.1326 [M+1-isobutyl]+.
4'-(Piperazm-1-ylsulfonyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3-carboxylic acid (15)
Method B:
Yield 69%; Semipreparative HPLC: Rt = 28.8 min; HPLC purity 96% (Rt = 11.79 min); 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.50 (s, 1H), 8.31 (s, 1H), 8.25 (s, 1H), 8.12 (d, J = 8.28 Hz, 2H), 8.07 (d, J = 8.20 Hz, 2H), 7.91 (d, J = 8.40 Hz, 2H), 7.81 (d, J = 8.20 Hz, 2H), 3.16 (s, 8H); MS (ESI, M/Z) 558.1 [M+1]+; ESI-HRMS calcd. m/z for C26H23N5O4F3S 558.1423, found 558.1422 [M+1]+.
4'-Sulfamoyl-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3-carboxylic acid (16)
Method A:
Yield 99%; Semipreparative HPLC: Rt = 28.4 min; HPLC purity 99% (Rt = 12.77 min); 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.51 (s, 1H), 8.40 (s, 1H), 8.34 (s, 1H), 8.21 (d, J = 8.08 Hz, 2H), 8.05 (d, J = 8.48 Hz, 2H), 7.98 (d, J = 8.48 Hz, 2H), 7.91 (d, J = 8.28 Hz, 2H), 7.47 (s, 2H; NH2), 2.78 (broad s, 6H; Et3N salt), 1.07 (t, J = 6.94 Hz, 9H; Et3N salt); MS (ESI, M/Z) 489.1 [M+1]+; ESI-HRMS calcd. m/z for C22H16N4O4F3S 489.0844, found 489.0842 [M+1]+.
3-(5-((4-(tert-Butoxycarbonyl)piperazin-1-yl)methyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (17)
Method A:
Yield 45%; HPLC purity 95% (Rt = 15.25 min); 1H NMR (400 MHz, CD3OD) δ 9.26 (s, 1H), 8.44 (s, 1H), 8.39 (t, J = 1.78 Hz, 1H), 8.35 (s, 1H), 8.18 (d, J = 8.16 Hz, 2H), 7.81 (d, J = 8.24 Hz, 2H), 7.54 (d, J = 3.64 Hz, 1H), 7.09 (d, J = 3.64 Hz, 1H), 3.89 (s, 2H), 3.51–3.50 (m, 4H), 2.60 (t, J = 4.76 Hz, 4H), 1.47 (s, 9H); MS (ESI, M/Z) 514.2 [M+1-Boc]+, 558.1 [M+1-isobutyl]+, 614.2 [M+1]+; ESI-HRMS calcd. m/z for C30H31N5O4F3S 614.2049, found 614.2042 [M+1]+.
3-(5-(Piperazin-1-ylmethyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (18)
Method B:
Yield 25%; Semipreparative HPLC: Rt = 27.2 min; HPLC purity 96% (Rt = 11.70 min); 1H NMR (400 MHz, D2O+DMSO-d6) δ 8.23 (s, 1H), 8.19–8.17 (m, 4H), 8.09 (s, 1H), 7.86 (d, J = 8.28 Hz, 2H), 7.49 (d, J = 3.52 Hz, 1H), 7.00 (d, J = 3.44 Hz, 1H), 3.66 (s, 2H; merged with H2O), 2.70 (s, 4H), 2.38 (s, merged with DMSO); MS (ESI, M/Z) 514.1 [M+1]+; ESI-HRMS calcd. m/z for C25H23N5O4F3 514.1525, found 514.1697 [M+1]+.
3-(5-((3-((tert-Butoxycarbonyl)amino)propyl)carbamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (19)
Method A:
Yield 93%; HPLC purity 95% (Rt = 13.69 min); 1H NMR (400 MHz, CD3OD) δ 9.27 (s, 1H), 8.53 (s, 1H), 8.44 (s, 2H), 8.19 (d, J = 8.0 Hz, 2H), 7.82 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 4.0 Hz, 1H), 7.67 (d, J = 4.0 Hz, 1H), 3.45–3.42 (m, 2H), 3.18–3.15 (m, 2H), 1.81–1.78 (m, 2H), 1.49 (s, 9H); MS (ESI, M/Z) 516.2 [M+1-Boc]+, 638.2 [M+Na]+; ESI-HRMS calcd. m/z for C29H28N5O5F3SN 638.1661, found 638.1653 [M+Na]+.
3-(5-((3-Aminopropyl)carbamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (20)
Method B:
Yield 92%; Semipreparative HPLC: Rt = 26.7 min; HPLC purity 95% (Rt = 10.31 min); 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.88 (broad s, 0.5H, C(O)-NH), 8.42 (s, 1H), 8.31 (s, 1H), 8.28 (s, 1H), 8.19 (d, J = 8.0 Hz, 2H), 7.95 (d, J = 4.0 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 3.6 Hz, 1H), 3.47 (merged with water peak), 2.93 (t, J = 7.4 Hz, 2H), 1.88–1.86 (m, 2H); 13C NMR (150 MHz, DMSO-d6) δ 161.4, 161.2, 146.6, 146.4, 140.3, 137.1, 134.8, 134.2, 129.5, 128.9, 128.6, 126.2, 126.5, 126.3, 125.6, 123.8, 121.6, 120.6, 117.2, 40.9, 40.5, 37.2; MS (ESI, M/Z) 516.1 [M+1]+; ESI- HRMS calcd. m/z for C24H21N5O3F3S 516.1317, found 516.1324 [M+1]+.
3-(5-(4-(tert-Butoxycarbonyl)piperazine-1-carbonyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (21)
Method A:
Yield 72%; HPLC purity 98% (Rt = 14.47 min); 1H NMR (400 MHz, CD3OD) δ 9.21 (s, 1H), 8.43 (s, 1H), 8.42 (s, 1H), 8.33 (s, 1H), 8.19 (d, J = 8.08 Hz, 2H), 7.81 (d, J = 8.24 Hz, 2H), 7.64 (d, J = 3.88 Hz, 1H), 7.50 (d, J = 3.88 Hz, 1H), 3.82 (s, 4H), 3.57 (s, 4H), 1.50 (s, 9H); MS (ESI, M/Z)572.1 [M+1-isobutyl]+, 628.2 [M+1]+; ESI-HRMS calcd. m/z for C26H21N5O5F3S 572.1216, found 572.1213 [M+1-isobutyl]+.
3-(5-(Piperazine-1-carbonyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (22)
Method B:
Yield 71%; Semipreparative HPLC: Rt = 25.9 min; HPLC purity 97% (Rt = 10.72 min); 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.44 (s, 1H), 8.41 (s, 1H), 8.25 (s, 1H), 8.21 (d, J = 8.12 Hz, 2H), 7.90 (d, J = 8.28 Hz, 2H), 7.75 (d, J = 3.84 Hz, 1H), 7.51 (d, J = 3.88 Hz, 1H), 3.69 (s, 4H), 2.88 (s, 4H); MS (ESI, M/Z) 528.1 [M+1]+; ESI-HRMS calcd. m/z for C25H21N5O3F3S 528.1317, found 528.1322 [M+1]+.
3-(5-((3-Aminopropyl)carbamoyl)furan-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (23)
Method C:
Yield 35%; Semipreparative HPLC: Rt = 26.7 min; HPLC purity 95% (Rt = 10.19 min); 1H NMR (400 MHz, DMSO-d6) δ 9.60 (s, 1H), 8.76 (broad s, 1H), 8.42 (s, 1H), 8.36 (s, 1H), 8.32 (s, 1H), 8.23 (d, J = 8.12 Hz, 2H), 7.88 (d, J = 8.36 Hz, 2H), 7.23 (d, J = 3.52 Hz, 1H), 7.21 (d, J = 3.56 Hz, 1H), 3.36 (merged with water peak), 2.71–2.67 (m, 2H), 1.70–1.65 (m, 2H); MS (ESI, M/Z) 500.1 [M+1]+; ESI-HRMS calcd. m/z for C24H21N5O4F3 500.1546, found 500.1544 [M+1]+.
3-(5-Carbamoylfuran-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (24)
Method A:
Yield 30%; Semipreparative HPLC: Rt = 28.9 min; HPLC purity 97% (Rt = 12.47 min); 1H NMR (400 MHz, DMSO-d6) δ 9.63 (s, 1H), 8.59 (s, 1H), 8.47 (s, 1H), 8.40 (s, 1H), 8.21 (d, J = 8.16 Hz, 2H), 8.14 (broad s, 0.5 H; NH), 7.90 (d, J = 8.32 Hz, 2H), 7.55 (broad s, 0.5 H; NH), 7.35 (d, J = 3.48 Hz, 1H), 7.24 (d, J = 3.57 Hz, 1H), 6.67 (broad s, 0.5H; NH), 2.94 (m, 6H; Et3N), 1.12 (t, J = 7.20 Hz, 9H; Et3N); MS (ESI, M/Z) 443.1 [M+1]+; ESI-HRMS calcd. m/z for C21H14N4O4F3 443.0967, found 443.0964 [M+1]+.
3-(4-((3-Aminopropyl)carbamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (25)
Method C:
Yield 61%; Semipreparative HPLC: Rt = 27.3 min; HPLC purity 98% (Rt = 10.23 min); 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.68 (broad s, 1H; NH), 8.33 (s, 1H), 8.22 (s, 1H), 8.17 (d, J = 8.40 Hz, 2H), 8.16 (s, 1H), 8.13 (s, 1H), 8.07 (s, 1H), 7.84 (d, J = 8.16 Hz, 2H), 3.36 (merged with water peak), 2.90 (t, J = 6.86 Hz, 2H), 1.84 (t, J = 7.64 Hz, 2H); MS (ESI, M/Z) 516.1 [M+1]+; ESI-HRMS calcd. m/z for C24H21N5O3F3S 516.1317, found 516.1311 [M+1]+.
3-(5-((3-Aminopropyl)carbamoyl)thiophen-3-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (26)
Method C:
Yield 31%; Semipreparative HPLC: Rt = 26.8 min; HPLC purity 96% (Rt = 10.41 min); 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 9.08 (broad s, 1H; NH), 8.40 (s, 1H), 8.34 (s, 1H), 8.31 (s, 1H), 8.20 (s, 2H), 8.15 (d, J = 8.16 Hz, 2H), 7.83 (d, J = 8.32 Hz, 2H), 3.36 (merged with water peak),2.93 (t, J = 7.28 Hz, 2H), 1.91–1.87 (m, 2H); MS (ESI, M/Z) 516.1 [M+1]+; ESI-HRMS calcd. m/z for C24H21N5O3F3S 516.1317, found 516.1320 [M+1]+.
3-(6-Carbamoylpyridin-3-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (27)
Method A:
Yield 52%; HPLC purity 96% (Rt = 12.16 min); 1H NMR (400 MHz, CD3OD) δ 9.21 (s, 1H), 9.19 (d, J = 1.68 Hz, 1H), 8.81 (s, 2H), 8.60 (s, 1H), 8.40 (dd, J = 2.20, 8.28 Hz, 1H), 8.20–8.16 (m, 3H), 7.80 (d, J = 8.24 Hz, 2H); MS (ESI, M/Z) 454.1 [M+1]+; ESI-HRMS calcd. m/z for C22H15N5O3F3, 454.1127, found 454.1130 [M+1]+.
3-(6-Carbamoylpyridin-3-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (28)
Method A:
Yield 44%; HPLC purity 95% (Rt = 12.81 min); 1H NMR (400 MHz, CD3OD) δ 9.24 (s,1H), 9.10 (d, J = 1.88 Hz, 1H), 8.55 (s, 1H), 8.47 (s, 1H), 8.41 (d, J = 2.36 Hz, 1H), 8.40 (s, 1H), 8.27 (d, J = 8.08 Hz, 1H), 8.19 (d, J = 8.20 Hz, 2H), 7.81 (d, J = 8.20 Hz, 2H); MS (ESI, M/Z) 454.1 [M+1]+; ESI-HRMS calcd. m/z for C22H15N5O3F3 454.1127, found 454.1124 [M+1]+.
3-(5-Carbamoylpyrazin-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoic acid (29)
Method A:
Yield 45%; HPLC purity 95% (Rt = 12.4S min); 1H NMR (400 MHz, CD3OD) δ 9.38 (s, 2H), 9.27 (s, 1H), 8.92 (s, 2H), 8.66 (s, 1H), 8.19 (d, J = 7.60 Hz, 2H), 7.81 (d, J = 7.44 Hz, 2H); MS (ESI, M/Z) 455.1 [M+1]+; ESI-HRMS calcd. m/z for C21H14N6O3F3, 455.1079, found 455.1074 [M+1]+.
4'-Carbamoyl-5-(4-(4-(trifluoromethyl)phenyl)-lH-l,2,3-triazol-l-yl)-[l,l'-biphenyl]-3-carboxylic acid (30)
Method A:
Yield 74%; Semipreparative HPLC: Rt = 26.4 min; HPLC purity 98% (Rt = 12.66 min); 1H NMR (400 MHz, CD3OD) δ 9.22 (s, 1H), 8.48 (s, 1H), 8.44 (s, 1H), 8.34 (s, 1H), 8.19 (d, J = 8.04 Hz, 2H), 8.05 (d, J = 8.24 Hz, 2H), 7.93 (d, J = 8.64 Hz, 2H), 7.81 (d, J = 7.88 Hz, 2H); MS (ESI, M/Z) 453.1 [M+1]+; 13C NMR (125 MHz, DMSO-d6) δ 138.0, 137.3, 146.4, 144.7, 142.4, 140.3, 137.0, 134.9, 128.8, 128.6, 128.0, 127.2, 126.5, 126.3, 125.9, 121.7, 120.4, 118.6; ESI-HRMS calcd. m/z for C23H16N4O3F3 453.1175, found 453.1183 [M+1]+. UV absorption (50% AcCN in PBS): λmax = 259 nm, ε = 3.0X104.
4'-(Methylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-l-yl)-[1,1'-biphenyl]-3- carboxylic acid (31)
Method A:
Yield 74%; HPLC purity 99% (Rt = 12.97 min); 1H NMR (400 MHz, CD3OD) δ 9.25 (s, 1H), 8.55 (s, 1H), 8.42 (s, 2H), 8.15 (d, J = 8.04 Hz, 2H), 7.98 (d, J = 8.12 Hz, 2H), 7.88 (d, J = 8.16 Hz, 2H), 7.79 (d, J = 8.12 Hz, 2H), 2.97 (s, 3H); MS (ESI, M/Z) 467.1 [M+1]+; ESI-HRMS calcd. m/z for C24H18N4O3F3 467.1331, found 467.1324 [M+1]+.
4'-(Ethylcarbamoyl)-5-(4-(4-(trifluoromethyl)pheny1)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3- carboxylic acid (32)
Method A:
Yield 74%; HPLC purity 99% (Rt = 13.27 min); 1H NMR (400 MHz, CD3OD) δ 9.24 (s, 1H), 8.55 (s, 1H), 8.43 (s, 1H), 8.41 (s, 1H), 8.16 (d, J = 7.76 Hz, 2H), 7.98 (d, J = 7.88 Hz, 2H), 7.97 (d, J = 7.96 Hz, 2H), 7.79 (d, J = B.G4 Hz, 2H), 3.46 (q, J = 7.3G Hz, 2H), 1.2B (t, J = 7.26 Hz, 3H); MS (ESI, M/Z) 4B1.1 [M+l]+; ESI-HRMS calcd. m/z for C25H2GN4O3F3 4B1.14BB, found 4B1.14B2 [M+l]+.
4'-(Propylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3- carboxylic acid (33)
Method A:
Yield 59%; HPLC purity 99% (Rt = 13.62 min); 1H NMR (4GG MHz, CD3OD) δ 9.25 (s, 1H), B.55 (s, 1H), B.43 (s, 2H), B.16 (d, J = 7.96 Hz, 2H), 7.9B (d, J = B.GB Hz, 2H), 7.B9 (d, J = B.G4 Hz, 2H), 7.79 (d, J = B.16 Hz, 2H), 3.39 (t, J = 7.G6 Hz, 2H), 1.74–1.65 (m, 2H), 1.G2 (t, J = 7.4G Hz, 3H); MS (ESI, M/Z) 495.2 [M+l]+; ESI-HRMS calcd. m/z for C26H22N4O3F3 495.1644, found 495.1642 [M+l]+.
4'-(tert-Butylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3- carboxylic acid (34)
Method A:
Yield 68%; HPLC purity 98% (Rt = 14.G3 min); 1H NMR (4GG MHz, CD3OD) δ 9.19 (s, 1H), 8.48 (s, 1H), 8.36 (s, 1H), 8.34 (s, 1H), B.09 (d, J = 8.00 Hz, 2H), 7.88 (d, J = 8.16 Hz, 2H), 7.80 (d, J = 8.00 Hz, 2H), 7.74 (d, J = 8.12 Hz, 2H), 1.51 (s, 9H); MS (ESI, M/Z) 509.2 [M+l]+; ESI-HRMS calcd. m/z for C27H24N4O3F3 509.1801, found 509.1802 [M+l]+.
4-(3'-(1H-Tetrazol-5-yl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-4- yl)piperidine (35)
A mixture of compound 113 in trifluoroacetic acid (0.l mL) was stirred at room temperature for 10 min. The mixture was diluted with methanol and neutralized with triethylamine. After all volatiles were evaporated under reduced pressure, the residue was purified by HPLC (10 mM triethylammonium acetate buffer:acetonitrile=20:80 to 80:20 in 40 min) to afford compound 35 (4.3 mg, 74%) as a white solid; Semipreparative HPLC: Rt = 26.5 min; HPLC purity 96% (Rt = 11.25 min); 1H NMR (400 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.55 (s, 1H), 8.39 (s, 1H), 8.23 (d, J = 8.08 Hz, 2H), 8.13 (t, J = 1.78 Hz, 1H), 7.90 (d, J = 8.24 Hz, 2H), 7.84 (d, J = 8.24 Hz, 2H), 7.43 (d, J = 8.24 Hz, 2H), 3.42 (d, J = 12.92 Hz, 2H), 3.08–3.02 (m, 2H), 2.99–2.93 (m, 1H), 2.02 (d, J = 13.08 Hz, 2H), 1.91–1.80 (m, 2H); MS (ESI, M/Z) 517.2 [M+l]+; ESI-HRMS calcd. m/z for C27H24N8F3 517.2076, found 517.2079 [M+l]+.
5-(3-(1#-Tetrazol-5-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenyl)-N-(3- aminopropyl)thiophene-2-carboxamide (36a)
Compound 114 (8 mg, 12.5 μmol) was converted to compound 36a (3 mg, 44%) as a white solid, using similar procedure used in the preparation of compound 35; Semipreparative HPLC: Rt = 28.1 min; HPLC purity 95% (Rt = 10.24 min); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.58 (s, 2H), 8.32 (s, 1H) 8.21 (d, J = 8.00 Hz, 2H), 7.82 (d, J = 8.24 Hz, 2H), 7.76 (d, J = 3.88 Hz, 1H), 7.70 (d, J = 3.88 Hz, 1H), 3.44 (t, J = 6.84 Hz, 2H), 3.17 (t, J = 6.80 Hz, 2H), 1.79 (t, J = 6.62 Hz, 2H), 1.47 (s, 9H); MS (ESI, M/Z) 540.1 [M+l]+; ESI-HRMS calcd. m/z for C24H21N9OF3S 540.1542, found 540.1547 [M+l]+.
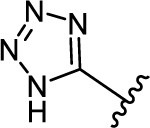
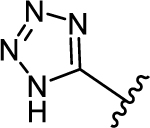
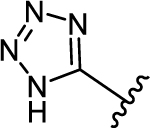
3'-(1H-Tetrazol-5-yl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-4- carboxamide (36b)
Compound 111 (5.5 mg, 11.5 μmol) was converted to compound 36b (5.8 mg, 99%) as a white solid, using similar procedure used in the preparation of compound 113; Purified by silica gel column chromatography (dichloromethane:methanol=4:l); HPLC purity 96% (Rt = 11.57 min); 1H NMR (400 MHz, CD3OD) δ 9.27 (s, 1H), 8.62 (s, 1H), 8.57 (s, 1H), 8.32 (s, 1H), 8.20 (d, J = 8.16 Hz, 2H), 8.07 (d, J = 8.40 Hz, 2H), 7.98 (d, J = 8.40 Hz, 2H), 7.82 (d, J = 8.28 Hz, 2H); MS (ESI, M/Z) 477.1 [M+l]+; ESI-HRMS calcd. m/z for C23H16N8OF3 477.1399, found 477.1391 [M+l]+.
N-(terf-Butyl)-3'-(1H-tetrazol-5-yl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-4-carboxamide (36c)
Compound 110 (10 mg, 0.020 mmol) was converted to compound 36c (10 mg, 92%) as a white solid, using similar procedure used in the preparation of compound 113; Purified by silica gel column chromatography (dichloromethane:methanol=5:1); HPLC purity 98% (Rt = 12.73 min); !H NMR (400 MHz, DMSO-d6) δ 9.77 (s, 1H), 8.33 (s, 1H), 8.22 (d, J = 7.88 Hz, 3H), 8.00 (m, 4H), 7.20–7.90 (m, 4H), 1.47 (s, 9H); MS (ESI, M/Z) 533.2 [M+1]+; ESI-HRMS calcd. m/z for C27H24N8OF3 533.2025, found 533.2030 [M+1]+.
tert-Butyl 4-(3'-(methoxycarbonyl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-4-yl)piperidine-1-carboxylate (79)
The mixture of compound 42 (74 mg, 0.156 mmol), Pd(PPh3)4 (11 mg, 9.36 μmol) and potassium carbonate (64 mg, 0.462 mmol) in dimethylformamide (2 mL) was purged with nitrogen gas for 15 min, and then compound 55 (64 mg, 0.188 mmol) was added to the mixture. The mixture was stirred at 80 °C for 2 h, and then allowed to be cooled at room temperature. This mixture was partitioned diethyl ether (10 mL) and water (20 mL). The aqueous layer was extracted with diethyl ether (5 mL x 2), and then the combined organic layer was washed with brine (3 mL), dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl actate=4:1) to afford compound 79 (42 mg, 42%) as a white solid; 1H NMR (400 MHz, CDC3) δ 8.43 (s, 1H), 8.38 (t, J = 1.54 Hz, 1H), 8.35 (s, 1H), 8.34 (s, 1H), 8.07 (d, J = 8.04 Hz, 2H), 7.75 (d, J = 8.20 Hz, 2H), 7.67 (d, J = 8.28 Hz, 2H), 7.37 (d, J = 8.24 Hz, 2H), 4.31 (s, 2H), 4.03 (s, 3H), 2.86 (d, J = 12.22 Hz, 2H), 2.78–2.72 (m, 1H), 1.89 (d, J = 12.68 Hz, 2H), 1.75–1.64 (m, 2H), 1.52 (s, 9H); MS (ESI, M/Z) 607.3 [M+1]+; ESI-HRMS calcd. m/z for C33H34N4O4F3 607.2532, found 607.2538 [M+1]+.
General procedure (Suzuki reaction) for 80–102
Method A:
A mixture of compound 42 (1 eq), aryl bromide (1.2 eq) and PdCl2(dppf) (0.1 eq) in dimethoxyethane:2 M Na2CO3 aqueous solution (10:1) was stirred at 50 °C. After cooling at room temperature, this mixture was partitioned ethyl acetate and water. The aqueous layer was extracted with ethyl acetate twice, and then the combined organic layer was washed with brine, dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl actate) to afford the condensed compounds as white solids.
Method B:
A mixture of compound 42 (1 eq) and aryl bromide (1.2 eq) in DMF:H2O (20:1; 20 mM) was purged with nitrogen gas for 30 min, and then Pd(PPh3)4 (0.05 eq) and Na2CO3 (3 eq) were added to the mixture, which was stirred for 1 h at 40–120 °C. The aqueous layer was extracted with ethyl acetate twice, and then the combined organic layer was washed with brine, dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl actate) to afford the condensed compounds as white solids.
Methyl 3-(5-(N-(2-((tert-butoxycarbonyl)amino)ethyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoate (80)
Method A;
Yield 44%; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.37 (t, J = 1.2 Hz, 1H), 8.35 (t, J = 2.0 Hz, 1H), 8.34 (t, J = 1.4 Hz, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 4.0 Hz, 1H), 7.47 (d, J = 3.6 Hz, 1H), 5.64 (broad s, 1H), 4.93 (broad s, 1H), 4.05 (s, 3H), 3.33 (t, J = 5.8 Hz, 2H), 3.27 (t, J = 5.6 Hz, 2H), 1.45 (s, 9H).
Methyl 3-(5-(N-(3-((tert-butoxycarbonyl)amino)propyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoate (81)
Method A;
Yield 44%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.37 (s, 1H), 8.35 (s, 1H), 8.34 (s, 1H), 8.08 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 8.0 Hz, 2H), 7.63 (d, J = 3.6 Hz, 1H), 7.46 (d, J = 4.0 Hz, 1H), 6.03 (broad s, 1H), 4.71 (broad s, 1H), 4.05 (s, 3H), 3.26 (q, J = 6.2 Hz, 2H), 3.16 (q, J = 6.2 Hz, 2H), 1.73–1.70 (m, 2H), 1.41 (s, 9H); MS (ESI, M/Z) 566.1 [M+1-Boc]+, 666.2 [M+1]+; ESI-HRMS calcd. m/z for C29H31N5O6F3S 666.1668, found 666.1676 [M+1]+.
Methyl 3-(5-(N-(4-((fert-butoxycarbonyl)amino)butyl)sulfamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoate (82)
Method A;
Yield 62%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.38 (s, 1H), 8.37 (s, 1H), 8.35 (s, 1H), 8.08 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 7.6 Hz, 2H), 7.64 (d, J = 3.2 Hz, 1H), 7.47 (d, J = 3.2 Hz, 1H), 5.01 (broad s, 1H), 4.58 (broad s, 1H), 4.09 (s, 3H), 3.16–3.14 (m, 4H), 1.63 (m, merged with water peak), 1.45 (s, 9H); MS (ESI, M/Z) 792.0 [M-l+TFA]−, 678.2 [M-l]'; ESI-HRMS calcd. m/z for C30H31N5O6F3S2 678.1668, found 678.1666 [M-H]−.
Methyl 3-(5-(N-(4-hydroxybutyl)sulfamoyl)thiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H- 1,2,3-triazol-1-yl)benzoate (83)
Method A;
Yield 65%; 1H NMR (400 MHz, CDCl3) δ 9.30 (s, 1H), 8.57 (s, 1H), 8.51 (s, 1H), 8.39 (s, 1H), 8.17 (d, J = 8.08 Hz, 2H), 7.81 (d, J = 8.24 Hz, 2H), 7.70 (d, J = 3.92 Hz, 1H), 7.65 (d, J = 3.96 Hz, 1H), 4.04 (s, 3H), 3.56 (t, J = 7.84 Hz, 2H), 3.06 (t, J = 6.58 Hz, 2H), 1.63–1.57 (m, 4H); MS (ESI, M/Z) 581.1 [M+1]+; ESI-HRMS calcd. m/z for C25H24N4O5F3S2 581.1140, found 581.1143 [M+H]+.
tert-Butyl 4-((5-(3-(methoxycarbonyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)phenyl)thiophen-2-yl)sulfonyl)piperazine-1-carboxylate (84)
Method A;
Yield 63%; 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.39–8.38 (m, 2H), 8.37 (s, 1H), 8.09 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 3.6 Hz, 1H), 7.54 (d, J = 3.6 Hz, 1H), 4.06 (s, 3H), 3.60 (t, J = 4.8 Hz, 4H), 3.14 (t, J = 4.8 Hz, 4H), 1.45 (s, 9H); MS (ESI, m/z) 622.1 [M+l− isobutyl]+, 678.2 [M+1]+; ESI-HRMS calcd. m/z for C30H31N5O6F3S 678.1668, found 678.1671 [M+1]+. Methyl 3-(5-sulfamoylthiophen-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoate (85)
Method A;
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.37 (s, 2H), 8.36 (s, 1H), 8.08 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 4.0 Hz, 1H), 7.47 (d, J = 4.0 Hz, 1H), 5.11 (s, 2H), 4.05 (s, 3H); MS (ESI, M/Z) 509.0 [M+1]+; ESI-HRMS calcd. m/z for C21H16N4O4F3S2 509.0565, found 509.0565 [M+1]+.
tert-Butyl 4-((3'-(methoxycarbonyl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-4-yl)sulfonyl)piperazine-1-carboxylate (86)
Method A;
Yield 40%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.45–8.44 (m, 1H), 8.42–8.40 (m, 2H), 8.09 (d, J = 8.04 Hz, 2H), 7.92 (d, J = 8.60 Hz, 2H), 7.88 (d, J = 8.60 Hz, 2H), 7.77 (d, J = 8.16 Hz, 2H), 4.06 (s, 3H), 3.56 (t, J = 4.90 Hz, 4H), 3.07 (t, J = 4.70 Hz, 4H), 1.43 (s, 9H); MS (ESI, M/Z) 616.1 [M+1-isobutyl]+, 672.2 [M+1]+; ESI-HRMS calcd. m/z for C32H33N5O6F3S 672.2104, found 672.2098 [M+1]+.
Methyl 4'-sulfamoyl-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3- carboxylate (87)
Method A;
Yield 44%; 1H NMR (400 MHz, CD3OD) δ 9.31 (s, 1H), 8.63 (s, 1H), 8.52 (s, 1H), 8.45 (s, 1H), 8.17 (d, J = 7.92 Hz, 2H), 8.08 (d, J = 8.32 Hz, 2H), 8.00 (d, J = 8.36 Hz, 2H), 7.80 (d, J = 8.08 Hz, 2H), 4.04 (s, 3H); MS (ESI, M/Z) 503.1 [M+1]+; ESI-HRMS calcd. m/z for C23H18N4O4F3S 503.1001, found 503.0998 [M+1]+.
tert-Butyl 4-((5-(3-(methoxycarbonyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)phenyl)thiophen-2-yl)methyl)piperazine-1-carboxylate (88)
Method A;
Yield 61%; 1H NMR (400 MHz, CDCh) δ 8.41 (s, 1H), 8.35 (t, J = 1.46 Hz, 1H), 8.31 (t, J =1.86 Hz, 1H), 8.26 (m, 1H), 8.08 (d, J = 8.08 Hz, 2H), 7.76 (d, J = 8.24 Hz, 2H), 7.38 (d, J = 3.60 Hz, 1H), 6.98 (s, 1H), 4.03 (s, 3H), 3.79–3.63 (m, 2H), 3.51 (s, 4H), 2.53 (s, 4H), 1.48 (s, 9H); MS (ESI, M/Z) 572.2 [M+1-isobutyl]+, 628.2 [M+1]+; ESI-HRMS calcd. m/z for C31H33N5O4F3S 628.2205, found 628.2200 [M+1]+.
Methyl 3-(5-((3-((tert-butoxycarbonyl)amino)propyl)carbamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl) phenyl)-1H-1,2,3-triazol-1-yl)benzoate (89)
Method A;
Yield 48%; 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.40 (s, 1H), 8.37 (s, 1H), 8.36 (s, 1H), 8.09 (d, J = 8.0 Hz, 2H), 7.77 (d, J = 8.0 Hz, 2H), 7.64 (s, 1H), 7.54 (s, 1H), 4.87 (s, 0.5H; NH), (s, 3H), 3.54 (s, 2H), 3.32 (s, 2H), 1.76 (s, 2H), 1.50 (s, 9H); MS (ESI, M/Z) 530.1 [M+l-Boc]+, 574.1 [M+l-isobutyl]+, 652.2 [M+Na]+; ESI-HRMS calcd. m/z for C30H30N5O5F3SN 652.1817, found 652.1818 [M+Na]+.
tert-Butyl 4-(5-(3-(methoxycarbonyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)phenyl)thiophene-2-carbonyl)piperazine-1-carboxylate (90)
Method A;
Yield 67%; 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 1H), 8.37 (s, 2H), 8.34 (s, 1H), 8.08 (d, J = 8.04 Hz, 2H), 7.76 (d, J = 8.00 Hz, 2H), 7.46 (d, J = 3.56 Hz, 1H), 7.36 (d, J = 3.56 Hz, 1H), 4.05 (s, 3H), 3.79 (t, J = 4.84 Hz, 4H), 3.56 (t, J = 5.00 Hz, 4H), 1.51 (s, 9H); MS (ESI, M/Z) 586.1 [M+l- isobutyl]+, 642.2 [M+l]+; ESI-HRMS calcd. m/z for C31H31N5O5F3S 642.1998, found 642.2GGG [M+l]+.
Methyl 3-(5-((3-((tert-butoxycarbonyl)amino)propyl)carbamoyl)furan-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)- 1H- 1,2,3-triazol-1-yl)benzoate (91)
Method A;
Yield 25%; 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 2H), 8.44 (s, 1H), 8.42 (s, 1H), 8.06 (d, J = 8.08 Hz, 2H), 7.75 (d, J = 8.16 Hz, 2H), 7.48 (broad s, 1H), 7.25 (d, J = 3.48 Hz, 1H), 7.00 (d, J = 3.48 Hz, 1H), 4.04 (s, 3H), 3.59–3.56 (m, 2H), 3.36–3.33 (m, 2H), 1.82–1.76 (m, 2H), 1.40 (s, 9H); MS (ESI, M/Z) 514.1 [M+l-Boc]+; ESI-HRMS calcd. m/z for C25H23N5O4F3 514.17G2 found 514.1703 [M+l-Boc]+.
Methyl 3-(5-carbamoylfuran-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzoate (92)
Method B;
Yield 57%; 1H NMR (400 MHz, CD3OD) δ 9.24 (s, 1H), 8.64 (s, 1H), 8.56 (s, 1H), 8.53 (s, 1H), 8.15 (d, J = 8.12 Hz, 2H), 7.80 (d, J = 8.20 Hz, 2H), 7.69–7.54 (m, POPh3), 7.29 (d, J = 3.64 Hz,1H), 7.23 (d, J = 3.64 Hz, 1H), 4.03 (s, 3H); MS (ESI, M/Z) 457.1 [M+1]+; ESI-HRMS calcd. m/z for C22H16N4O4F3 457.1124 found 457.1125 [M+1]+.
Methyl 3-(4-((3-((tert-butoxycarbonyl)amino)propyl)carbamoyl)thiophen-2-yl)-5-(4-(4- (trifluoromethyl)phenyl)- 1H- l,2,3-triazol-l-yl)benzoate (93)
Method A;
Yield 73%; 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.38 (s, 1H), 8.34 (s, 2H), 8.08 (d, J = 7.00 Hz, 2H), 8.00 (s, 1H), 7.95 (s, 1H), 7.76 (d, J = 6.96 Hz, 2H), 4.04 (s, 3H), 3.55 (s, 2H), 3.32 (s, 2H), 1.76 (s, 2H), 1.49 (s, 9H); MS (ESI, M/Z) 530.1 [M+1-Boc]+; ESI-HRMS calcd. m/z for C25H23N5O3F3S 530.1474, found 530.1476 [M+1-Boc]+.
Methyl 3-(5-((3-((tert-butoxycarbonyl)amino)propyl)carbamoyl)thiophen-3-yl)-5-(4-(4- (trifluoromethyl)phenyl)- 1H- l,2,3-triazol-l-yl)benzoate (94)
Method A;
Yield 38%; 1H NMR (400 MHz, CD3OD) δ 9.26 (s, 1H), 8.60 (s, 1H), 8.49 (s, 1H), 8.40 (s, 1H), 8.18 (d, J = 8.24 Hz, 3H), 7.92 (s, 1H), 7.81 (d, J = 8.08 Hz, 2H), 4.03 (s, 3H), 3.46–3.40 (m, 2H), 3.17–3.14 (m, 2H), 1.80–1.75 (m, 2H), 1.44 (s, 9H); MS (ESI, M/Z) 530.1 [M+1-Boc]+, 652.2 [M+Na]+; ESI-HRMS calcd. m/z for C30H30N5O5F3SNa 652.1817, found 652.1825 [M+Na]+.
Methyl 3-(5-carbamoylpyridin-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-lH-l,2,3-triazol-l- yl)benzoate (95)
Method B;
Yield 26%; 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 8.89 (s, 1H), 8.79 (s, 1H), 8.59 (s, 1H), 8.52 (s, 1H), 8.36 (dd, J = 1.68, 8.28 Hz, 1H), 8.09 (d, J = 8.08 Hz, 2H), 8.05 (d, J = 8.36 Hz, 1H), 7.76 (d, J = 8.16 Hz, 2H), 7.71–7.47 (m, POPh3), 4.06 (s, 3H); MS (ESI, M/Z) 468.1 [M+1]+; ESI- HRMS calcd. m/z for C23H17N5O3F3 468.1283, found 468.1282 [M+1]+.
Methyl 3-(6-carbamoylpyridin-3-yl)-5-(4-(4-(trifluoromethyl)phenyl)-lH-l,2,3-triazol-l- yl)benzoate (96)
Method B;
Yield 11%; 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.46 (s, 2H), 8.45 (s, 1H), 8.42 (s, 1H), 8.39 (d, J = 7.84 Hz, 1H), 8.21 (dd, J = 1.84, 8.08 Hz, 1H), 8.09 (d, J = 8.08 Hz, 2H), 7.77 (d, J = 8.28 Hz, 2H), 7.73–7.49 (m, POPh3), 4.07 (s, 3H); MS (ESI, M/Z) 468.1 [M+l]+; ESI-HRMS calcd. m/z for C23H17N5O3F3 468.1283, found 468.1275 [M+l]+.
Methyl 3-(5-carbamoylpyrazin-2-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)benzoate (97)
Method B;
Yield 33%; 1H NMR (400 MHz, CDCl3) δ 9.43 (s, 1H), 9.25 (s, 1H), 8.92 (s, 2H), 8.86 (s, 1H), 8.67 (s, 1H), 8.08 (d, J = 8.00 Hz, 2H), 7.74 (d, J = 8.04 Hz, 2H), 7.75–7.46 (m, POPh3), 4.05 (s, 3H); MS (ESI, M/Z) 469.1 [M+l]+; ESI-HRMS calcd. m/z for C22H16N6O3F3 469.1236, found 469.1232 [M+l]+.
Methyl 4'-carbamoyl-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3- carboxylate (98)
Method A;
Yield 61%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.41 (m, 3H), 8.09 (d, J = 8.04 Hz, 2H), 8.00 (d, J = 8.24 Hz, 2H), 7.82 (d, J = 8.20 Hz, 2H), 7.77 (d, J = 8.16 Hz, 2H), 4.05 (s, 3H); MS (ESI, M/Z) 467.1 [M+l]+; ESI-HRMS calcd. m/z for C24H18N4O3F3 467.1331, found 467.1332 [M+l]+.
Methyl 4'-(methylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-3-carboxylate (99)
Method B;
Yield 33%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.38 (s, 2H), 8.36 (d, J = 1.60 Hz, 1H), 8.07 (d, J = 8.12 Hz, 2H), 7.92 (d, J = 8.20 Hz, 2H), 7.77–7.74 (m, 4H), 7.70–7.46 (m, POPh3), 6.36 (d, J = 4.24 Hz, 1H; NH), 4.04 (s, 3H), 3.08 (d, J = 4.80 Hz, 3H); MS (ESI, M/Z) 481.1 [M+l]+; ESI-HRMS calcd. m/z for C25H20N4O3F3 481.1488, found 481.1487 [M+l]+.
Methyl 4'-(ethylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)- 1H- 1,2,3-triazol-1-yl)-[1,1'-biphenyl]-3-carboxylate (100)
Method B;
Yield 31%; 1H NMR (400 MHz, CDCl3) δ 8.45 (s, 1H), 8.38 (s, 2H), 8.37 (s, 1H), 8.07 (d, J = 8.08 Hz, 2H), 7.92 (d, J = 8.12 Hz, 2H), 7.77 (d, J = 7.28 Hz, 2H), 7.75 (d, J = 7.40 Hz, 2H), 6.25 (s, 1H), 4.04 (s, 3H), 3.60–3.53 (m, 2H), 1.32 (t, J = 7.24 Hz, 3H); MS (ESI, M/Z) 495.2 [M+l]+; ESI- HRMS calcd. m/z for C26H22N4O3F3 495.1644, found 495.1650 [M+l]+.
Methyl 4'-(propylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-3-carboxylate (101)
Method B;
Yield 43%; 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.37 (s, 2H), 8.35 (d, J = 1.56 Hz, 1H), 8.06 (d, J = 8.12 Hz, 2H), 7.91 (d, J = 8.20 Hz, 2H), 7.76 (d, J = 4.36 Hz, 2H), 7.74 (d, J = 4.32 Hz, 2H), 6.31 (t, J = 5.24 Hz, 1H), 4.04 (s, 3H), 3.48 (q, J = 6.69 Hz, 2H), 1.75–1.63 (m, 2H), 1.04 (t, J = 7.38 Hz, 3H); MS (ESI, M/Z) 509.2 [M+l]+; ESI-HRMS calcd. m/z for C27H24N4O3F3 509.1801, found 509.1795 [M+l]+.
Methyl 4'-(tert-butylcarbamoyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-3-carboxylate (102)
Method B;
Yield 45%; 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.39–8.37 (m, 3H), 8.08 (d, J = 7.88 Hz, 2H), 7.88 (d, J = 8.16 Hz, 2H), 7.77–7.75 (m, 4H), 4.04 (s, 3H), 1.26 (s, 9H); MS (ESI, M/Z) 523.1 [M+l]+; ESI-HRMS calcd. m/z for C28H26N4O3F3 523.1957, found 523.1967 [M+l]+.
4'-(1-(tert-Butoxycarbonyl)piperidin-4-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)- [1,1'-biphenyl]-3-carboxylic acid (103)
To a solution of 79 (52 mg, 0.086 mmol) in methanol (2 mL) was added potassium hydroxide (24 mg, 0.429 mmol), and then this reaction mixture was stirred at 50 °C for 15 h. After solvent was evaporated under reduced pressure, the residue was purified by silica gel column chromatography (dichloromethane:methanol=l0:1) to afford as a white solid 103 (47 mg, 93%); 1H NMR (400 MHz, CD3OD) δ 9.22 (s, 1H), 8.48 (s, 1H), 8.35 (s, 2H), 8.14 (d, J = 7.76 Hz, 2H), 7.78 (d, J = 7.92 Hz, 2H), 7.71 (d, J = 7.72 Hz, 2H), 7.39 (d, J = 7.60 Hz, 2H), 4.25 (d, J = 12.8 Hz, 2H), 2.91 (s, 2H), 2.80 (t, J = 11.78 Hz, 1H), 1.88 (d, J = 12.2 Hz, 2H), 1.69–1.60 (m, 2H), 1.51 (s, 9H); MS (ESI, M/Z) 537.1 [M+1- isopropyl]+; ESI-HRMS calcd. m/z for C28H24N4O4F3 537.1750, found 537.1760 [M+1-isopropyl]+.
tert-Butyl 4-(3'-carbamoyl-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-4-yl)piperidine-1-carboxylate (104)
To a solution of compound 103 (47 mg, 0.079 mmol) in dimethylformamide (3 mL) were added NH4Q (8.5 mg, 0.159 mmol), HATU (45 mg, 0.119 mmol) and N,N-diisopropylethylamine (20 mg, 28 μl, 0.159 mmol), and then this reaction mixture was stirred at room temperature for 1 h. This mixture was partitioned ethyl acetate (6 mL) and water (3 mL). The aqueous layer was extracted with ethyl acetate (5 mL x 2), and then the combined organic layer was washed with brine (3 mL), dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl actate=1:1) to afford compound 104 (48 mg, 99%) as a white solid; 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.27 (s, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 8.07 (d, J = 8.04 Hz, 2H), 7.76 (d, J = 8.16 Hz, 2H), 7.66 (d, J = 8.24 Hz, 2H), 7.38 (d, J = 8.20 Hz, 2H), 4.31 (d, J = 13.68 Hz, 2H), 2.89– 2.81 (m, 2H), 2.80–2.73 (m, 1H), 1.89 (d, J = 12.00 Hz, 2H), 1.67 (merged with water peak), 1.51 (s, 9H); MS (ESI, M/Z) 536.1 [M+1-isopropyl]+, 592.2 [M+1]+; ESI-HRMS calcd. m/z for C28H25N5O3F3 536.1909, found 536.1911 [M+1-isopropyl]+.
tert-Butyl (3-(5-(3-carbamoyl-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)phenyl)thiophene-2-carboxamido)propyl)carbamate (105)
Compound 19 (55 mg, 0.089 mmol) was converted to compound 105 (44 mg, 80%) as a white solid, using similar procedure used in the preparation of compound 104; 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.29 (s, 1H), 8.23 (s, 1H), 8.13 (s, 1H), 8.08 (d, J = 8.00 Hz, 2H), 7.77 (d, J = 8.28 Hz, 2H), 7.63 (d, J = 3.88 Hz, 1H), 7.49 (d, J = 3.84 Hz, 1H), 3.53 (dd, J = 6.2, 12.04 Hz, 2H), 3.33–3.30 (m, 2H), 1.76–1.73 (m, 2H), 1.50 (s, 9H); MS (ESI, M/Z) 515.1 [M+l-Boc]+, 559.1 [M+l-isopropyl]+; ESI- HRMS calcd. m/z for C25H22N6O4F3S 559.1375, found 559.1383 [M+l-isopropyl]+.
N4,-(tert-Butyl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-y1)-[1,1,-biphenyl]-3,4'- icarboxamide (106)
Compound 34 (19 mg, 37.3 μmol) was converted to compound 106 (16 mg, 86%) as a white solid, using similar procedure used in the preparation of compound 104; 1H NMR (400 MHz, CDCL3) δ 8.46 (s, 1H), 8.27 (s, 2H), 8.13 (s, 1H), 8.06 (d, J = 7.32 Hz, 2H), 7.85 (d, J = 6.96 Hz, 2H), 7.75 (d, J = 8.44 Hz, 2H), 7.73 (d, J = 8.40 Hz, 2H), 1.53 (s, 9H); MS (ESI, M/Z) 508.1 [M+l]+; ESI-HRMS calcd. m/z for C27H25N5O2F3 508.1960, found 508.1962 [M+l]+.
3-Bromo-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzamide (107)
Compound 37b (82 mg, 0.199 mmol) was converted to compound 107 (83 mg, 100%) as a white solid, using similar procedure used in the preparation of compound 104; 1H NMR (400 MHz, CDCL3) δ 8.43 (s, 1H), 8.38 (s, 0.5H; NH), 8.33–8.32 (m, 0.5H; NH), 8.28 (s, 1H), 8.25 (s, 1H), 8.06–8.04 (m, 3H), 7.75 (d, J = 8.20 Hz, 2H); MS (ESI, M/Z) 411.0, 413.0 [M+l]+; ESI-HRMS calcd. m/z for C16H11N4OF379Br 411.0068, found 411.0067 [M+l]+.
tert-Butyl 4-(3'-cyano-5'-(4-(4-(trifluoromethyl)phenyl)- 1H- 1,2,3-triazol-1-yl)-[1,1'-biphenyl]-4- yl)piperidine-1-carboxylate (108)
To a solution of compound 104 (41 mg, 0.069 mmol) in dichloromethane (2 mL) were added trifluoroacetic anhydride (97 mg, 64 μl, 0.462 mmol) and triethylamine (50 mg, 69 μl, 0.494 mmol) at 0 °C, and then this reaction mixture was stirred at room temperature for 1 h. This mixture was partitioned dichloromethane (6 mL) and water (3 mL). The aqueous layer was extracted with dichloromethane (5 mL x 2), and the organic layer was washed with brine (3 mL), dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane;ethyl actate=4:1) to afford compound 108 (30 mg, 76%) as a white solid; 1H NMR (400 MHz, CDCL3) δ 8.38 (s, 1H), 8.31 (t, J = 1.84 Hz, 1H), 8.07 (d, J = 8.08 Hz, 2H), 8.06–8.04 (m, 1H), 7.96 (t, J = 1.42 Hz,1H), 7.77 (d, J = 8.20 Hz, 2H), 7.62 (d, J = 8.28 Hz, 2H), 7.39 (d, J = 8.20 Hz, 2H), 4.31 (d, J = 12.84 Hz, 2H), 2.89–2.83 (m, 2H), 2.79–2.72 (m, 1H), 1.89 (d, J = 12.04 Hz, 2H), 1.75–1.65 (m, 2H), 1.52 (s, 9H); MS (ESI, M/Z) 518.1 [M+1-isopropyl]+; ESI-HRMS calcd. m/z for C28H23N5O2F3 518.1804, found 518.1801 [M+1-isopropyl]+.
tert-Butyl (3-(5-(3-cyano-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)phenyl)thiophene- 2-carboxamido)propyl)carbamate (109)
Compound 105 (40 mg, 0.065 mmol) was converted to compound 109 (28 mg, 72%) as a white solid, using similar procedure used in the preparation of compound 108; 1H NMR (400 MHz, CDCL3) δ 8.39 (s, 1H), 8.31 (t, J = 3.44 Hz, 1H), 8.08 (d, J = 8.20 Hz, 2H), 8.07–8.06 (m, 1H), 7.99 (s, 1H), 7.77 (d, J = 8.20 Hz, 2H), 7.65 (d, J = 3.76 Hz, 1H), 7.49 (d, J = 3.88 Hz, 1H), 4.85 (s, 1H; NH), 3.53 (dd, J = 6.28, 11.96 Hz, 2H), 3.32 (dd, J = 6.34, 12.06 Hz, 2H), 1.75 (t, J = 5.34 Hz, 2H), 1.51 (s, 9H); MS (ESI, M/Z) 497.2 [M+1-Boc]+, 541.1 [M+1-isopropyl]+; ESI-HRMS calcd. m/z for C24H20N6OF3S 497.1371, found 497.1375 [M+1-Boc]+.
N-(tert-Butyl)-3'-cyano-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'-biphenyl]-4- carboxamide (110)
Compound 106 (16 mg, 31.5 μmol) was converted to compound 110 (11 mg, 71%) as a white solid, using similar procedure used in the preparation of compound 108 ; 1H NMR (400 MHz, CDCL3) δ 8.41 (s, 1H), 8.35 (s, 1H), 8.11 (s, 1H), 8.07 (d, J = 8.08 Hz, 2H), 7.99 (s, 1H), 7.91 (d, J = 8.28 Hz, 2H),7.77 (d, J = 8.24 Hz, 2H), 7.72 (d, J = 8.24 Hz, 2H), 1.54 (s, 9H); MS (ESI, M/Z) 490.2 [M+1]+; ESI- HRMS calcd. m/z for C27H23N5OF3 490.1855, found 490.1860 [M+1]+.
3'-Cyano-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,r-biphenyl]-4-carboxamide (111)
To a solution of compound 110 (7.8 mg, 15.9 pmol) in anisole (1 mL) was added trimethylsilyl trifluoromethanesulfonate (6 μl, 31.9 μmol), and this reaction mixture was stirred at 140 °C for 1 h in microwave. The reaction mixture was filtered through silica gel and washed with hexane to remove anisole. The desired product was eluted with ethyl acetate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (dichloromethane:methanol=30:1) to afford as compound 111 (5.5 mg, 80%) as a white solid; 1H NMR (400 MHz, CD3OD) δ 9.32 (s, 1H), 8.61 (t, J = 1.72 Hz, 1H), 8.43 (s, 1H), 8.26 (s, 1H), 8.18 (d, J = 8.36 Hz, 2H), 8.07 (d, J = 8.36 Hz, 2H), 7.94 (d, J = 8.36 Hz, 2H), 7.82 (d, J = 8.28 Hz, 2H); MS (ESI, M/Z) 434.1 [M+1]+; ESI-HRMS calcd. m/z for C23H15N5OF3 434.1229, found 434.1230 [M+1]+.
3-Bromo-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)benzomtrile (112)
Compound 107 (83 mg, 0.199 mmol) was converted to compound 112 (51 mg, 64%) as a white solid, using similar procedure used in the preparation of compound 108; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 8.30 (t, J = 1.82 Hz, 1H), 8.11 (t, J = 1.56 Hz, 1H), 8.05 (d, J = 8.08 Hz, 2H), 7.91 (t, J = 1.40 Hz, 1H), 7.76 (d, J = 8.20 Hz, 2H); MS (ESI, M/Z) 393.0, 395.0 [M+1]+; ESI-HRMS calcd. m/z for C16H9N4F379Br 392.9963, found 392.9957 [M+1]+.
tert-Butyl 4-(3'-(1H-tetrazol-5-yl)-5'-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)-[1,1'- biphenyl]-4-yl)piperidine-1-carboxylate (113)
To a solution of compound 108 (13 mg, 22.7 μmol) in dimethylsulfoxide (1 mL) were added sodium azide (3.2 mg, 49.8 μmol) and copper sulfate hydrate (1.4 mg, 5.6 μmol), and then this reaction mixture was heated at 140 °C for 6 h. After the solvent removed, the residue was purified by silica gel column chromatography (dichloromethane:methanol=20:1) to afford compound 113 (10 mg, 69%) as a white solid; 1H NMR (400 MHz, CD3OD) δ 9.26 (s, 1H), 8.59 (s, 1H), 8.47 (s, 1H), 8.31 (s, 1H), 8.18 (d, J = 7.00 Hz, 2H), 7.82–7.80 (m, 4H), 7.44–7.43 (m, 2H), 4.27–4.24 (m, 2H), 2.92–2.83 (m, 3H), 1.92–1.88 (m, 2H), 1.77–1.62 (m, 2H), 1.59 (s, 9H); MS (ESI, M/Z) 561.2 [M+1-isopropyl]+; ESI-HRMS calcd. m/z for C28H24N8O2F3 561.1974, found 561.1969 [M+1-isopropyl]+.
tert-Butyl (3-(5-(3-(1H-tetrazol-5-yl)-5-(4-(4-(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1- yl)phenyl)thiophene-2-carboxamido)propyl)carbamate (114)
Compound 109 (24 mg, 0.065 mmol) was converted to compound 114 (20 mg, 78%) as a white solid, using similar procedure used in the preparation of compound 113; Semipreparative HPLC (10 mM triethylammonium acetate buffer:acetonitrile=20:80 to 80:20 in 40 min): Rt = 35.9 min; 1H NMR (400 MHz, CD3OD) δ 9.27 (s, 1H), 8.58 (s, 2H), 8.32 (s, 1H) 8.21 (d, J = 8.00 Hz, 2H), 7.82 (d, J = 8.24 Hz, 2H), 7.76 (d, J = 3.88 Hz, 1H), 7.70 (d, J = 3.88 Hz, 1H), 3.44 (t, J = 6.84 Hz, 2H), 3.17 (t, J = 6.80 Hz, 2H), 1.79 (t, J = 6.62 Hz, 2H), 1.47 (s, 9H); MS (ESI, M/Z) 540.1 [M+1-Boc]+, 584.2 [M+1- isopropyl]+; ESI-HRMS calcd. m/z for C25H21N9O3F3S 584.1440, found 584.1435 [M+1-isopropyl]+.
tert-Buty1 (6-hydroxyhexyl)carbamate (116)
To a solution of 6-amino-1-hexanol (115, 200 mg, 1.71 mmol) in dichloromethane (2.5 mL) was added dropwise a solution of di-tert-butyl dicarbonate (410 mg, 1.88 mmol) in dichloromethane (2.5 mL). After stirring for 30 min, the reaction mixture was partitioned dichloromethane (10 mL) and water (5 mL). The aqueous layer was extracted with dichloromethane (5 mL x 2), and then the organic layer was dried over MgSO4, filtered and evaporated under reduced pressure to afford compound 116 (296 mg, 80%) as a white solid; 1H NMR (400 MHz, DMSO-d6) δ 6.75 (s, 1H; NH), 4.32 (t, J = 5.16 Hz, 1H; OH), 3.37 (q, J = 5.24 Hz, 2H), 2.88 (q, J = 6.76 Hz, 2H), 1.43–1.30 (s, 13H), 1.29–1.21 (m, 4H).
tert-Butyl (6-azidohexyl)carbamate (117)
To a solution of compound 116 (150 mg, 0.69 mmol) in toluene (5 mL) were added triethylamine (96 μl, 70 mg, 0.69 mmol) and methanesulfonyl chloride (53 μl, 70 mg, 0.69 mmol) at 0 °C. After the mixture stirring at 0 °C for 30 min, tetrabutylammonium bromide (20 mg, 0.069 mmol) and a solution of sodium azide (269 mg, 4.14 mmol) in water (2.5 mL) were added to the reaction mixture, which was stirred at 100 °C for 15 h. The mixture was partitioned diethyl ether (10 mL) and water (5 mL), and the organic layer was washed with brine (3 mL), dried over MgSO4, filtered and evaporated to afforded compound 117 (99 mg, 59%) as yellow syrup; 1H NMR (400 MHz, CDCl3) δ 4.52 (s, 1H; NH), 3.28 (t, J = 6.88 Hz, 2H), 3.13 (q, J = 6.12 Hz, 2H), 1.66–1.58 (m, 2H), 1.54–1.46 (m, 2H), 1.46 (s, 9H), 1.43–1.35 (m, 4H); MS (ESI, M/Z) 243.2 [M+1]+; ESI-HRMS calcd. m/z for C11H23N4O2 243.1821, found 243.1821 [M+1]+.
tert-Buty1 4-(4-(3-(ethoxycarbonyl)-6-(4-(trifluoromethyl)phenyl)naphthalen-1- yl)phenyl)piperidine-1-carboxylate (120)
A mixture of compound 119 (150 mg, 0.319 mmol), which was synthesized according to literature procedures reported17, and compound 55 (123 mg, 0.361 mmol) in dimethylformamide (5 mL) was purged with nitrogen gas for 30 min, and then Pd(PPh3)4 (22 mg, 0.019 mmol) and potassium carbonate (132 mg, 0.957 mmol) were added to the mixture, which was stirred at 80 °C for 2 h. The reaction mixture was partitioned ethyl acetate (20 mL) and water (10 mL), and then the aqueous layer was extracted with ethyl acetate (10 mL x 2). The combined organic layer was washed with brine (5 mL), dried (MgSO4), filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:ethyl actate=10:1) to afford compound 120 (113 mg, 59%) as a white solid; 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 8.25 (d, J = 1.68 Hz, 1H), 8.09–8.07 (m, 2H), 7.86 (d, J = 8.24 Hz, 2H), 7.81–7.77 (m, 3H), 7.50 (d, J = 8.12, 2H), 7.38 (d, J = 8.08 Hz, 2H), 4.48 (q, J = 7.12 Hz, 2H), 4.32 (d, J = 13.4 Hz, 2H), 2.92–2.86 (m, 2H), 2.82–2.76 (m, 1H), 1.96 (d, J = 12.4 Hz, 2H), 1.80–1.68 (m, 2H), 1.52 (s, 9H), 1.47 (t, J = 7.12 Hz, 3H); MS (ESI, M/Z) 548.2 [M+1-isopropyl]+,504.2 [M+1-Boc]+.
Ethyl 4-(4-(piperidin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (121)
A mixture of compound 120 (160 mg, 0.265 mmol) in trifluoroacetic acid (2 mL) and tetrahydrofuran (2 mL) was stirred at room temperature for 1 h. The solvent was evaporated with methanol under reduced pressure. The residue was purified by silica gel column chromatography(dichloromethane:methanol=10:1) to afford compound 121 (153 mg, 93%; TFA salt form) as a white solid; 1H NMR (400 MHz, CD3OD) δ 8.76 (s, 1H), 8.46 (d, J = 1.64 Hz, 1H), 8.04–8.01 (m, 3H), 7.98–7.94 (m, 2H), 7.82 (d, J = 8.12 Hz, 2H), 7.56–7.49 (m, 4H), 4.47 (q, J = 7.12 Hz, 2H), 3.58 (d, J = 12.32 Hz, 2H), 3.26–3.20 (m, 2H), 3.15–3.05 (m, 1H), 2.22 (d, J = 13.24 Hz, 2H), 2.09–1.98 (m, 2H), 1.47 (t, J = 7.12 Hz, 3H); MS (ESI, M/Z) 504.2 [M+1]+; ESI-HRMS calcd. m/z for C31H29NO2F3 504.2150, found 504.2157 [M+1]+.
Ethyl 4-(4-(1-(Hex-5-yn-1-yl)piperidin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (122)
Potassium carbonate (68 mg, 0.24 mmol) was added to a solution of compound 121 (100 mg, 0.16 mmol) in dry N,N-dimethylformamide (5 mL), and the reaction mixture was stirred for 20 min. 6- Bromohexyne-1 (0.12 mL, 0.40 mmol) was subsequently added, and the reaction mixture was first stirred at room temperature for 2 h and then at 50 °C for 2.5 h. The reaction mixture was partitioned saturated NaHCO3 solution (5 mL) and ethyl acetate (10 mL). The aqueous layer was extracted with ethyl acetate (5 mL x 2), and then the combine organic layer was washed with brine (5 mL), dried over MgSO4, filtered, and evaporated under reduced pressure. The residue was purified by silica column chromatography (hexane:ethyl acetate=6:1) to afford compound 122 (66 mg, 70%) as a white solid; 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 8.24 (d, J = 1.80 Hz, 1H), 8.05 (d, J = 1.60 Hz, 1H), 8.02 (d, J = 8.84 Hz, 1H), 7.84 (d, J = 8.16 Hz, 2H), 7.81–7.76 (m, 3H), 7.52 (d, J = 8.20 Hz, 2H), 7.47 (d, J = 8.20 Hz, 2H), 4.48 (q, J = 7.08 Hz, 2H), 3.77 (d, J = 10.36 Hz, 2H), 3.10–3.04 (m, 2H), 2.92–2.79 (m, 4H), 2.36–2.32 (m, 2H), 2.20–2.14 (m, 4H), 2.03 (t, J = 2.62 Hz, 1H), 1.72–1.65 (m, 2H), 1.47 (t, J = 7.12 Hz, 3H); MS (ESI, M/Z) 584.3 [M+l]+; ESI-HRMS calcd. m/z for C37H37NO2F3 584.2776, found 584.2783 [M+l]+.
Ethyl 4-(4-(1-(4-(1-(6-((tert-butoxycarbonyl)amino)hexyl)-1H-1,2,3-triazol-4-yl)butyl)piperidin-4- yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (123)
To a solution of compound 122 (40 mg, 68.5 μmol) in CH2Cl2:tert-BuOH:H2O (1:1:1; 2 mL), compound 117 (50 mg, 0.203 mmol) was added, followed by copper(II) sulfate pentahydrate (2.4 mg, 10.3 μmol) and sodium ascorbate (6.4 mg, 30.9 prnol). The reaction mixture was stirred at room temperature for 24 h. The solvents were removed under vacuum and the residue rinsed with 37% ammonia solution (5 mL) and extracted with ethyl acetate (10 mL x 2). The combined organic layer was washed with brine (5 mL), dried over MgSO4, filtered, and the evaporated under reduced pressure. The residue was purified by silica gel column chromatography (dichloromethane:methanol=20:l) to afford compound 123 (33 mg, 58%) as a white solid; 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 1H), 8.24 (d, J = 1.68 Hz, 1H), 8.06 (dd, J = 3.58, 5.18 Hz, 2H), 7.84 (d, J = 8.20 Hz, 2H), 7.81–7.76 (m, 3H), 7.49 (d, J = 8.12 Hz, 2H), 7.43 (d, J = 8.12 Hz, 2H), 7.38 (s, 1H), 4.56 (s, 1H), 4.47 (q, J = 7.12 Hz, 2H), 4.34 (t, J = 7.18 Hz, 2H), 3.39 (s, 1H), 3.14–3.09 (m, 2H), 2.83–2.75 (m, 4H), 2.53–2.15 (m, 2H), 2.06–2.04 (m, 2H), 1.94– 1.81 (m, 6H), 1.49–1.46 (m, 16H), 1.38–1.36 (m, 6H); MS (ESI, M/Z) 826.4 [M+l]+; ESI-HRMS calcd. m/z for C48H59N5O4F3 826.4519 found 826.4528 [M+l]+.
Pharmacological assays
Cell Culture:
Chinese hamster ovary cells stably expressing the hP2Yl4-R (CHO-hP2Yl4R) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) / Ham’s F12 (F12) 1:1 supplemented with 10% FBS, 100 units/mL penicillin, 100 mg/mL streptomycin, 2mM L-glutamine and 0.500 mg/mL G418 Sulfate (Geneticin). Cells were maintained in a humidified atmosphere and sterile incubation conditions held at 37 °C and 5% CO2 (g).
Competitive Assay:
Competitive fluorescent assays were performed on a BD FACSCalibur flow cytometer in conjunction with the softwares BD Bioscience PlateManager and CellQuest. All cell culture growth and assays for this procedure were conducted on flat-bottom 96-well plates. CHO- hP2Y14R cells were grown to approximately 80–90% confluency prior to assays. The 96-well plate format enabled four compounds to be analyzed in triplicate per run. All unlabeled ligand compounds are stored as 5 mM stock solutions in dimethyl sulfoxide (DMSO). Serial dilutions of each compound were prepared in complete medium. Cells were initially incubated with unlabeled compounds for 30 min at 37 °C and 5% CO2 (g). Cells were then incubated with the fluorescent labeled (AlexaFluor 488) ligand MRS4174 for 30 min at a final concentration of 20 nM. After three consecutive washes in sterile 1X Dulbecco’s Phosphate Buffered Saline (DPBS) minus Ca2+/Mg2+, cells were detached from the plate using Corning Cellstripper™ to reduce damaging the hP2Y14R protein. Final cell suspensions for flow cytometry was in DPBS minus Ca2+/Mg2+.
IC50 values were determined from the gathered data with the program GraphPad Prism version7.0.
Calculation of pharmacokinetic properties
cLogP was calculated using http://bleoberis.bioc.cam.ac.uk/pkcsm/prediction.46 cLogS and cLogD were calculated as a function of pH using https://disco.chemaxon.com/apps/demos/. Pharmacokinetic properties were calculated using http://bleoberis.bioc.cam.ac.uk/pkcsm/prediction, as described by Pires et al.47
The in vivo pharmacokinetics and in vitro preclinical testing of compound 30 was measured by GVK Biosciences Private Limited, Hyderabad, India using a protocol we described previously.42 The vehicle used was 15%DMSO (in which the compound was initially dissolved, containing 10 equivalents of aq. NaHCO3), 15% Kolliphor® ELP (Sigma 30906, CAS 61791–12-6), 70% PBS, at a max. concentration of 2 mg/mL for the highest p.o. dose of 10 mg/kg.
Supplementary Material
Acknowledgement
We acknowledge support from the NIDDK Intramural Research Program (ZIA DK031116–29) We thank Bryan L. Roth, National Institute of Mental Health's Psychoactive Drug Screening Program (Univ. North Carolina at Chapel Hill, Contract # HHSN-271–2008-00025-C) for screening data. We thank Rolf Swenson and Zhen-Dan Shi (Imaging Probe Development Center, NHLBI, HHSN261200800001E) for reagent synthesis and Young-Hwan Jung (NIDDK) for NMR spectra.
Abbreviations:
- AF488
Alexa Fluor 488
- CHO
Chinese hamster ovary
- DCM
dichloromethane
- DIPEA
diisopropylethylamine
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
N,N-dimethylformamide
- DPPF
1,1'-bis(diphenylphosphino)ferrocene
- ECL
extracellular loop
- EDC
N-ethyl-N’- dimethylaminopropylcarbodiimide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- IE
interaction energy
- MD
molecular dynamics
- PDSP
Psychoactive Drug Screening Program
- PLC
phospholipase C
- PPTN
4-(4-(piperidin-4-yl)-phenyl)-7- (4-(trifluoromethyl)-phenyl)-2-naphthoic acid
- RMSD
root-mean-square deviation
- SAR
structure- activity relationship
- UDPG
uridine-5'-diphosphoglucose
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TM
transmembrane helix
- TSTU
N,N,N',N'-tetramethyl-O-(N-succinimidyl)-uronium tetrafluoroborate
Footnotes
Supporting Information Available:
The Supporting Information is available free of charge on the ACS Publications website at http://pubs.acs.org.
3D coordinates of hP2Y14R in complex with 7 and 20 (PDB)
Videos (AVI) of MD simulations of hP2Y14R complexes with 9 compared to 20 (Video S1) and with 30 (Video S2) and with 2a (Video S3).
Supplementary file containing synthesis of compounds 55-78, off-target binding inhibition curves, pharmacological data, molecular modeling data, including: Figure S1, Study of the interactions with ELs; Figure S2, Selection of the optimal spacer geometry; Figure S3, Lowest IE structure; Table S1, structure and docking scores of designed compounds; Table S2, replicate analysis of MD simulations; Table S3, Predicted pharmacokinetic parameters; Figure S4, Predicted LogD and LogS (PDF).
NMR and mass spectra of triazole derivatives (PDF).
References
- 1.Burnstock G Purinergic signalling: Therapeutic developments. Front. Pharmacol. 8, 661. doi: 10.3389/fphar.2017.00661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cekic C; Linden J Purinergic regulation of the immune system. Nature Rev. Immunol. 2016, 16, 177–192. [DOI] [PubMed] [Google Scholar]
- 3.Abbracchio MP; Burnstock G; Boeynaems JM; Barnard EA; Boyer JL; Kennedy C; Fumagalli M; King BF; Gachet C; Jacobson KA; Weisman GA International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazarowski ER; Harden TK Signalling and pharmacological properties of the P2Y14 receptor. Mol. Pharmacol. 2015, 88, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sesma JI; Kreda SM; Steinckwich-Besancon N; Dang H; García-Mata R; Harden TK; Lazarowski ER The UDP-sugar-sensing P2Y14 receptor promotes Rho-mediated signaling and chemotaxis in human neutrophils. Am. J. Physiol. - Cell Physiol. 2012, 303, C490–C498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett MO; Sesma JI; Ball CB; Jayasekara PS; Jacobson KA; Lazarowski ER; Harden TK A selective high-affinity antagonist of the P2Y14 receptor inhibits UDP-glucose- stimulated chemotaxis of human neutrophils. Mol. Pharmacol. 2013, 84, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Z-G; Ding Y; Jacobson KA UDP-glucose acting at P2Y14 receptors is a mediator of mast cell degranulation. Biochem. Pharmacol. 2010, 79, 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azroyan A; Cortez-Retamozo V; Bouley R; Liberman R; Ruan YC; Kiselev E; Jacobson KA; Pittet MJ; Brown D; Breton S Renal intercalated cells sense and mediate sterile inflammation via the P2Y14 receptor. PLoS ONE 2015, 10(3), e0121419. doi: 10.1371/journal.pone.0121419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J; Morinaga H; Oh D; Li P; Chen A; Talukdar S; Lazarowski E; Olefsky JM; Kim JJ GPR105 Ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J. Immunol. 2012, 189, 1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinoshita M; Nasu-Tada K; Fujishita K; Sato K; Koizumi S Secretion of matrix metalloproteinase-9 from astrocytes by inhibition of tonic P2Y14-receptor-mediated signal(s). Cell. Mol. Neurobiol. 2013, 33, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi K; Yamanaka H; Yanamoto F; Okubo M; Noguchi K Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 2012, 60, 1529–1539. [DOI] [PubMed] [Google Scholar]
- 12.Sesma JI; Weitzer CD; Livraghi-Butrico A; Dang H; Donaldson S; Alexis NE; Jacobson KA; Harden TK; Lazarowski ER UDP-glucose promotes neutrophil recruitment in the lung. Purinergic Signalling 2016, 12, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stachon P; Geis S; Peikert A; Heidenreich A; Michel NA; Ünal F; Hoppe N; Dufner B; Schulte L; Marchini T; Cicko S; Ayata K; Zech A; Wolf D; Hilgendorf I; Willecke F; Reinöhl J; von Zur Mühlen C.; Bode C; Idzko M; Zirlik A Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1577–1586. doi: 10.1161/ATVBAHA.115.307397 Epub 2016 Jun 23. [DOI] [PubMed] [Google Scholar]
- 14.Idzko M; Ferrari D; Eltzschig HK Nucleotide signalling during inflammation. Nature 2014,509, 310–317, doi: 10.1038/nature13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K; Zhang J; Gao ZG; Zhang D; Zhu L; Han GW; Moss SM; Paoletta S; Kiselev E; Lu W; Fenalti G; Zhang W; Müller CE; Yang H; Cherezov V; Katritch V; Han GW; Jacobson KA; Stevens RC; Wu B; Zhao Q Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 2014, 509, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K; Zhang J; Gao ZG; Paoletta S; Zhang D; Han GW; Li T; Ma L; Zhang W; Müller CE; Yang H; Jiang H; Cherezov V; Katritch V; Jacobson KA; Stevens RC; Wu B; Zhao Q Agonist-bound structure of the human P2Y12R receptor. Nature 2014, 509, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiselev E; Barrett M; Katritch V; Paoletta S; Weitzer CD; Hammes E; Yin AL; Zhao Q; Stevens RC; Harden TK; Jacobson KA Exploring a 2-naphthoic acid template for the structure- based design of P2Y14 receptor antagonist molecular probes. ACS Chem. Biol. 2014, 9, 2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trujillo K; Paoletta S; Kiselev E; Jacobson KA Molecular modeling of the human P2Y14 receptor: A template for structure-based design of selective agonist ligands. Bioorg. Med. Chem. 2015, 23, 4056–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gauthier JY; Belley M; Deschênes D; Fournier J-F; Gagné S; Gareau Y; Hamel M; Hénault M; Hyjazie H; Kargman S; Lavallée G; Levesque JF; Li L; Mamane Y; Mancini J; Morin N; Mulrooney E; Robichaud J; Thérien M; Tranmer G; Wang Z; Wu J; Black WC The identification of 4,7-disubstituted naphthoic acid derivatives as UDP-competitive antagonists of P2Y14. Bioorg. Med. Chem. Lett. 2011, 21, 2836–2839. [DOI] [PubMed] [Google Scholar]
- 20.Junker A; Balasubramanian R; Ciancetta A; Uliassi E; Kiselev E; Martiriggiano C; Trujillo K; Mtchedlidze G; Birdwell L; Brown KA; Harden TK; Jacobson KA Structure-based design of 3-(4-aryl-1H-1,2,3-triazol-1-yl)-biphenyl derivatives as P2Y14 receptor antagonists. J. Med. Chem. 2016, 59, 6149–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ballesteros JA; Weinstein H Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- 23.Charifson PS; Walters WP Acidic and basic drugs in medicinal chemistry: A perspective. J. Med. Chem. 2014, 57, 9701–9717. [DOI] [PubMed] [Google Scholar]
- 24.Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB; Huryn DM; Ballatore C Structure property relationships of carboxylic acid isosteres. J. Med. Chem. 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- 26.Conroy S; Kindon N; Kellam B; Stocks MJ Drug-like Antagonists of P2Y Receptors—From Lead Identification to Drug Development. J. Med. Chem. 2016, 59, 9981–10005. [DOI] [PubMed] [Google Scholar]
- 27.Robichaud J; Fournier JF; Gagné S; Gauthier JY; Hamel M; Han Y; Hénault M; Kargman S; Levesque J-F; Mamane Y; Mancini J; Morin N; Mulrooney E; Wu J; Black WC Applying the pro-drug approach to afford highly bioavailable antagonists of P2Y14. Bioorg. Med. Chem. Lett. 2011, 21, 4366–4368. [DOI] [PubMed] [Google Scholar]
- 28.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments J. Comput. Aided Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 29.LigPrep, Schrödinger, LLC, New York, NY, 2017.
- 30.Glide, Schrödinger, LLC, New York, NY, 2017.
- 31.Doerr S; Harvey MJ; Noé F; De Fabritiis G HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [DOI] [PubMed] [Google Scholar]
- 32.Humphrey W; Dalke A; Schulten K VMD - Visual molecular dynamics. J. Mol. Graphics, 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- 34.Harvey M; Giupponi G; De Fabritiis G ACEMD: Accelerated molecular dynamics simulations in the microseconds timescale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- 35.Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi 1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B, 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kräutler V; Van Gunsteren WF; Hünenberger PH A Fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001,22, 501–508. [Google Scholar]
- 40.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LGA Smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- 41.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tosh DK; Ciancetta A; Warnik E; Crane S; Gao Z-G; Jacobson KA; Structure-based scaffold repurposing for GPCRs: Transformation of adenosine derivatives into selective 5HT2B/5HT2C serotonin receptor antagonists. J. Med. Chem. 2016, 59, 11006–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Innocenti P; Cheung K-MJ; Solanki S; Mas-Droux C; Rowan F; Yeoh S; Boxall K; Westlake M; Pickard L; Hardy T; Baxter JE; Aherne GW; Bayliss R; Fry AM; Hoelder S Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: Structure-activity relationship, structural biology, and cellular activity. J. Med. Chem. 2012, 55, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akhlaghinia B; Rezazadeh S A novel approach for the synthesis of 5-substituted-1H-tetrazoles. J. Braz. Chem. Soc. 2012, 23, 2197–2203. [Google Scholar]
- 45.Amison RT; Arnold S; O'Shaughnessy BG; Cleary SJ; Ofoedu J; Idzko M; Page CP; Pitchford SC Lipopolysaccharide (LPS) induced pulmonary neutrophil recruitment and platelet activation is mediated via the P2Y1 and P2Y14 receptors in mice. Pulm. Pharmacol. Ther. 2017, 45, 62–68. [DOI] [PubMed] [Google Scholar]
- 46.Tetko IV; Tanchuk VY Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program, J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [DOI] [PubMed] [Google Scholar]
- 47.Pires DE; Blundell TL; Ascher DB pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.