

Abstract
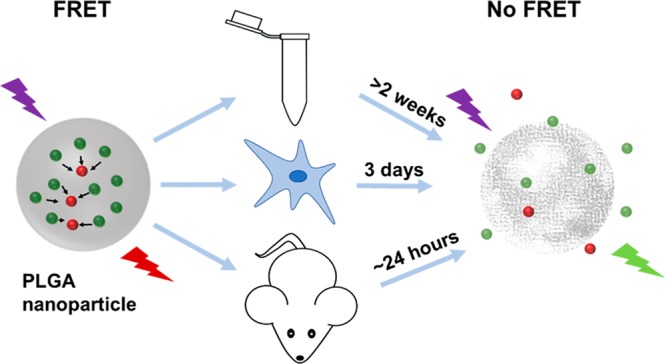
The knowledge of in vitro and in vivo stability of polymeric nanoparticles is vital for the development of clinical formulations for drug delivery and cell labeling applications. Förster resonance energy transfer (FRET)-based fluorescence labeling approaches are promising tools to study nanoparticle stability under different physiological conditions. Here, we present the FRET-based stability assessment of poly(lactic-co-glycolic acid) (PLGA) nanoparticles encapsulating BODIPY-FL12 and Nile Red as the donor and acceptor, respectively. The stability of PLGA nanoparticles is studied via monitoring the variations of fluorescence emission characteristics along with colloidal characterization. Accordingly, PLGA nanoparticles are colloidally stable for more than 2 weeks when incubated in aqueous buffers in situ, whereas in vitro particle degradation starts in between 24 and 48 h, reaching a complete loss of FRET at 72 h as shown with fluorescence microscopy imaging and flow cytometry analysis. PLGA nanoparticles systemically administered to mice predominantly accumulate in the liver, in which FRET no longer takes place at time points as early as 24 h postadministration as determined by ex vivo organ imaging and flow cytometry analysis. The results of this study expand our knowledge on drug release and degradation behavior of PLGA nanoparticles under different physiological conditions, which will prove useful for the rational design of PLGA-based formulations for various applications that can be translated into clinical practice.
Keywords: FRET, polymeric nanoparticles, PLGA, in vivo stability, drug delivery
1. Introduction
Drug delivery systems utilizing polymeric nanocarriers have emerged as versatile tools with tremendous potential in clinical applications.1−4 Entrapment of drugs within polymeric nanoparticles (NPs) offers the advantages of reduced toxicity and systemic side effects, as well as improved stability and targeted delivery of the drug.5 Furthermore, colloidal (e.g., size, composition, loading efficiency) and functional (e.g., release profile) properties of nanoparticles can be adjusted to achieve the desired therapeutic effects.6 An optimally designed drug delivery system should ensure the availability of the drug at a specific location for a required period of time.7 Despite recent advances in the development of drug delivery nanoparticles with precisely controlled colloidal and functional properties, their translation to clinical use remains challenging. This is largely because the nanoparticles developed and tested in vitro do not necessarily reflect the in vivo performance, especially when drug release is also dependent on polymer degradation.8−10 Furthermore, complex biological environments, including blood components such as serum proteins, lipids, enzymes, or cells, can interfere with the colloidal and functional stability of nanoparticles, causing drug leakage or premature release.11−13 Therefore, the rational design of drug delivery nanoparticles with an improved therapeutic efficacy requires the development of robust techniques to monitor the functional stability of nanoparticles in different physiological conditions.
Fluorescence labeling strategies are commonly employed to evaluate drug retention and cellular uptake of nanoparticles.14 Typically, nanoparticles that encapsulate a single type of fluorescent label are tracked using fluorescence microscopy or flow cytometry analysis.15 However, the colloidal stability of nanoparticles cannot be assessed with this approach because the fluorescence signals originating from the released and retained dyes cannot be distinguished.16 Alternatively, two-color labeled nanoparticles can be designed using fluorescent dyes that exhibit a Förster resonance energy transfer (FRET).17 FRET is a nonradiative transfer of energy from an excited donor molecule (D) to a nearby acceptor (A), displaying spectral overlap via dipole–dipole coupling.18 The strong dependence of FRET on the D–A separation distance can provide useful information on the release13 and degradation19 profile of nanoparticles encapsulating D–A dyes. When dyes are in close proximity within the intact nanoparticle, the energy is transferred from the excited D to A, resulting in D quenching and A sensitization. Upon dye release and nanoparticle degradation, the D–A distance becomes too large for an efficient energy transfer. Therefore, release and degradation profiles of nanoparticles can be determined by monitoring the recovery of D emission and loss of A sensitization using fluorescence spectroscopy and imaging.20,21
FRET-based assessment of dye release and particle degradation have been reported for various types of polymeric carriers formulated as self-assembled micelles11,22−24 and nanoparticles.10,25−28 Among the polymeric carriers, poly(lactic-co-glycolic acid) (PLGA) has received particular attention due to its excellent biocompatibility, tunable degradation characteristics, and long clinical history.29 Drug delivery systems based on PLGA particles are already available on the market (e.g., Lupron Depot by Abbott Laboratories, USA, and Trelstar by Watson Pharmaceuticals, USA), and new formulations are under continuous development for various applications. Although the development of PLGA particles of varying size,30,31 surface functionality,32−34 and encapsulated cargo35−37 have been shown in several studies, reports on the colloidal and functional stability of these particles in vivo are scarce.38 We have previously shown that PLGA nanoparticles of ∼200 nm diameter are suitable for cell loading, do not affect cell viability, and can specifically deliver their cargo to desired cell populations39−42 with a relatively higher intracellular uptake compared to micron-sized particles.43
In this study, we present a detailed stability assessment of PLGA nanoparticles of ∼200 nm diameter in situ, in vitro, and in vivo, exploiting a FRET-based fluorescence labeling approach. We study both the functional (i.e., release profile) and colloidal stability (i.e., degradation) of the PLGA nanoparticles using FRET. PLGA nanoparticles were loaded with BODIPY-FL and Nile Red dyes that acted as D and A, respectively. After optimization of the dye loading ratio, we studied the dye release and particle degradation profile in different buffers in situ. In vitro assessment of particle uptake and stability was done using human-derived peripheral blood mononuclear cells and mouse-derived bone marrow dendritic cells. Finally, we tracked nanoparticles systemically administered to mouse by extracting and analyzing organs for the evaluation of in vivo particle stability. Monitoring the recovery of quenched D fluorescence upon dye release and particle degradation revealed striking differences under different physiological conditions. The results demonstrated here will prove useful for rational design of PLGA nanoparticles for applications, such as drug delivery or cell loading for therapy monitoring with an improved therapeutic efficacy.
2. Materials and Methods
2.1. Materials
PLGA (Resomer RG 502H), with a 50:50 ratio of lactic acid–glycolic acid and MW 7000–17 000 Da, was obtained from Evonik Nutrition & Care GmbH (Germany). Poly(vinyl alcohol) (PVA, 9000–10 000 Mw, 80%, hydrolyzed) and Nile Red were obtained from Sigma-Aldrich (USA). CholEsteryl BODIPY-FL C12 was purchased from Thermo Fisher Scientific (USA) and acetonitrile was from VWR (The Netherlands). Ultrapure Milli-Q water (18.2 MΩ cm) was used where necessary (Merck, USA). RPMI-1640 medium, Anti-Anti (AA), and ß-mercaptoethanol were obtained from Gibco. X-Vivo medium and ultraglutamine were from Lonza. Fetal bovine serum (FBS) was purchased from Hyclone (GE Healthcare, USA).
2.2. Nanoparticle Formulation
Dye-loaded PLGA nanoparticles were prepared via a nanoprecipitation method. Briefly, the organic phase containing 4 mg of PLGA in 72 μL of acetonitrile was mixed with the appropriate amounts of fluorescent dyes followed by dropwise addition to 1 mL of 2% aqueous PVA solution under constant stirring at 450 rpm. Particles were washed three times with ultrapure water by centrifugation at 15 000 rpm for 35 min after evaporation of the organic solvent overnight at 4 °C. Finally, particles were lyophilized. For the determination of optimal dye concentration without inducing dye quenching, BODIPY-FL green was loaded within PLGA nanoparticles at concentrations of 0.1, 0.5, 1.0, 1.3, 1.5, 1.6, and 2.0% (w/w). For the Nile Red encapsulation, the dye concentration was varied as 0.1, 0.3, 0.7, 1.0, and 1.3% (w/w). Dye loading ratios determined in the previous optimization steps were used for coencapsulation of donor (1.0%) and acceptor (0.2%) dyes to prepare PLGA nanoparticles that exhibit FRET.
2.3. Colloidal and Optical Characterization of Nanoparticles
The size distribution of nanoparticles was measured via dynamic light scattering (DLS) (Nanotrac Flex, Microtrac). Briefly, 50 μL of the particle suspension was diluted to 1 mL using filtered Milli-Q water to avoid multiple scattering events. The measurements were obtained at room temperature (25 °C ± 1 °C), at which the corresponding water viscosity and refractive index were 0.872 and 1.330 cP, respectively. An average of three measurements was used to report the Z-average values for each sample. The ζ potential of nanoparticles was measured using a Zetasizer Nano ZS (Malvern Instruments). Particles were dispersed in 5 mM NaCl solution for the measurements, and an average of three measurements was used to report the ζ potential. Atomic force microscopy (AFM) images of nanoparticles were obtained with a Catalyst BioScope (Bruker) coupled to a confocal microscope (TCS SP5II, Leica). 100 μL of the particle suspension was dried on clean glass substrates, and particles were imaged in peak-force tapping mode using silicon nitride cantilevers with nominal spring constants of 0.4 N/m (Bruker). AFM images were analyzed using NanoScope analysis software (Bruker).
Optical characterization of fluorescent nanoparticles was done by measuring the steady-state fluorescence of diluted samples (1 mg/mL) using Eppendorf semimicro Vis Cuvettes on an LS 55 Fluorescence spectrometer (PerkinElmer). A xenon lamp was used as the excitation source. Samples were excited at 488 nm, and the emission was recorded between 500 and 800 nm. The width of excitation and detection slits and other data collection parameters were kept the same for each measurement.
2.4. Stability Assessment of PLGA Nanoparticles in Situ
PLGA nanoparticles encapsulating D and A dyes (will be referred to as “FRET NP”) were studied in situ to determine dye release and particle integrity. A suspension of FRET NP in Milli-Q water was incubated at 37 °C for a period of 2 weeks. Emission spectra and size distribution of particles were measured at different time points. PLGA nanoparticles encapsulating the only donor BODIPY-FL green dye (will be referred to as “green NP”) and acceptor Nile Red dye (will be referred to as “red NP”) were studied in parallel as a control. The recovery of FRET-quenched D emission was monitored using fluorescence spectroscopy and was compared to variations of the green NP emission; thus, the influence of direct Nile Red excitation was excluded from the analysis. This process was repeated for nanoparticles incubated in PBS buffers at pH 7.4 and at pH 5.8 for a period of 1 week.
2.5. Cell Culture
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats of healthy individuals after informed consent, using ficoll density centrifugation (Lymphoprep, STEMCELL Technologies, Vancouver, Canada). Adherent monocytes were cultured in X-VIVO 15 media supplemented with 2% human serum and in the presence of interleukin-4 (300 U/mL) and granulocyte-monocyte colony stimulating factor (GM-CSF, 450 U/mL) to obtain immature dendritic cells (DCs).
Bone marrow-derived DCs (BMDCs) were isolated from the femur and tibia of donor mice. Isolated cells were then cultured in RPMI-1640 medium (supplemented with 10% FBS, 1% AA, 1% Ultraglutamine, and β-mercaptoethanol) in the presence of murine GM-CSF (20 ng/mL).
2.6. Cell Labeling
Uptake and intracellular trafficking of nanoparticles were studied on a monocyte-derived DC (moDC) culture. Day-3 immature moDCs were collected from the Costar flasks and counted using Trypan Blue. The cell suspension was diluted to the desired concentration (0.1 × 106/mL). Then, 0.2 × 105 cells were plated on sterile coverslips in 24-well plates and were labeled with green NP at a concentration 1 mg of nanoparticles per million cells. The cells were then incubated for 6 and 24 h at 37 °C. At each time point, the medium was removed and the coverslips were washed carefully with PBS. The cells were then fixed by adding 300 μL of 2% paraformaldehyde (PFA) and permeabilized with PBA + 0.1% Saponin. LAMP1 (Sigma-Aldrich)- or EEA1 (BD Bioscience)-specific primary antibodies (Ab) were incubated with cells followed by staining with isotype-specific 568AlexaFluor-conjugated secondary Ab. The cell nuclei were stained with DAPI using 4 μL droplets of a Mowiol–DAPI mixture. Coverslips were transferred to the slides, kept overnight in the dark, and then examined with an Olympus FV1000 confocal laser scanning microscope. An argon laser (488 nm) was used as the excitation source, and images were collected with an UPLSAPO 60× objective at a 502–538 nm range. Images were then processed with ImageJ software.
2.7. Stability Assessment of PLGA NP in Vitro
Day-3 human moDCs and day-14 murine CD103+ BMDCs were labeled with the particles at a concentration of 1 mg of nanoparticles per million cells and were incubated for different time points (0.5, 1, 3, 6, 48, and/or 72 h). At each time point, the cells were analyzed with flow cytometry (BD FACS Verse, BD Biosciences). FlowJo analysis software was used to determine the mean fluorescence intensity of cells. BMDCs were also imaged with a Leica DMI6000 epi-fluorescence microscope equipped with a 63× 1.4 NA oil immersion objective, a metal halide EL6000 lamp for excitation, a DFC365FX CCD camera, and GFP filter sets (all from Leica) to measure the variations of intracellular intensity at the green emission channel for green NP and FRET NP at 24 and 72 h of incubation.
2.8. Stability Assessment of PLGA NP in Vivo
Albino C57BL/6J female mice (B6 (Cg)-Tyrc-2J) were obtained from Jax Laboratory and maintained under specific pathogen-free conditions at the Central Animal Laboratory in Nijmegen, The Netherlands. Experiments were performed according to the guidelines for animal care of the Nijmegen Animal Experiments Committee (DEC 2016-0045). Mice were injected intravenously (i.v.) with 200 μL of either FRET NP or green NP at a concentration of 10 mg/mL. The organs (liver, spleen, kidneys, lungs) were isolated at 2 and 24 h postinjection after sacrificing the mice and were imaged with an IVIS LUMINA in vivo imaging system with the excitation set to 465 nm, and the emission was detected at GFP channel (5 s exposure time). Finally, the cells isolated from the liver were analyzed with flow cytometry.
3. Results and Discussion
3.1. Formulation and Characterization of Dye-Loaded PLGA Nanoparticles
PLGA nanoparticles (PLGA NPs) encapsulating fluorescent dyes were prepared via a nanoprecipitation method.6 A water-miscible organic solvent (acetonitrile) containing PLGA and fluorescent dyes was mixed with the aqueous phase containing 2% PVA solution under constant stirring. The hydrophobic nature of both dyes allowed for their encapsulation within PLGA NPs during the particle formation. PLGA NPs encapsulating only donor (green nanoparticles), only acceptor (red nanoparticles), and a combination of donor/acceptor dyes (FRET nanoparticles) were prepared (Figure 1A). Particles with an average diameter of ∼200 nm (PDI < 0.2) and a slightly negative ζ potential (−2.74 ± 0.377 mV for FRET nanoparticles) were obtained using this preparation method. A representative atomic force microscopy image of FRET nanoparticles is shown in Figure 1B. BODIPY-FL and Nile Red were selected as the D–A pair, respectively, due to their excellent spectral overlap44 (Figure 1C).
Figure 1.

(A) Schematic illustration for the preparation of PLGA NPs encapsulating only donor (green NPs), only acceptor (red NPs), and coencapsulating donor and acceptor dyes (FRET NPs). (B) A representative atomic force microscopy height image of FRET NPs coencapsulating 1% donor and 0.2% acceptor (w/w) dyes. Scan size: 750 nm × 750 nm. Scale bar: Z-axis. (C) Normalized excitation (dashed curves) and emission (solid curves) spectra of BODIPY-FL (green) and Nile Red (red) donor–acceptor pair.
The large spectral overlap integral, determined as J = 2.25 × 1015 nm4 M–1 cm–1, ensured an efficient energy transfer between the BODIPY-FL and Nile Red. The D–A separation distance, at which the FRET efficiency is 50% (i.e., the Förster distance, R0) was calculated using eq 1.
| 1 |
The FRET orientation factor κ2 = 2/3, the refractive index of PLGA n = 1.46, and the BODIPY-FL quantum yield ΦD = 0.9 led to a Förster distance of R0 = 5.5 nm. Therefore, an efficient FRET distance of approximately 2.75–11 nm that corresponded to ca. 0.5 to 2-fold R0 was obtained using this FRET pair.
An ideal FRET probe to assess particle stability should be robust and efficient, which largely depends on the photophysical properties of the fluorescent dyes used as FRET pairs. The robustness ensures that observed changes in the emission characteristics are solely due to dye release and particle degradation rather than modified emission properties of fluorescent dyes due to, e.g., temperature. In this respect, BODIPY dyes with an excellent stability are good candidates to be utilized as FRET probes. However, previously reported FRET pairs constructed with BODIPY dyes as both donors and acceptors displayed smaller Förster distances.20 The Förster distance is a measure of the donor–acceptor separation distances that can be probed efficiently using FRET. The ability to detect FRET at larger molecular separation distances results in improved precision even at larger length scales. By pairing the BODIPY donor with a Nile Red acceptor, we drastically improved the Förster distance as 5.5 nm, which enabled an efficient energy transfer between donor and acceptor molecules at separation distances as large as 11 nm (2-fold of Förster distance). Therefore, compared to previously reported studies, the FRET system we developed displayed both a high robustness and a more efficient energy transfer at larger distances.
Prior to preparation of FRET NPs, loading ratios of both dyes were optimized. A series of dye concentrations in the 0.1–2.0% (w/w) range were used for encapsulation within PLGA NPs. Self-quenching of dyes was observed at concentrations above 1.3% and 0.3% (w/w) for the donor (Figure 2A) and acceptor dyes (Figure 2B), respectively. Therefore, loading ratios of 1.3% D and 0.3% A were used for coencapsulation to prepare FRET NPs, for which an efficient quenching of D emission was observed. Lowering the dye fractions to 1.0% D and 0.2% A was observed to increase the efficiency of D quenching further. Figure 2C shows the emission spectra of FRET NPs with different D–A fractions normalized at the acceptor emission wavelength (∼615 nm). The size distribution analysis of FRET NPs showed a mean diameter of ∼200 nm with a small PDI (<0.2) for all particles (Figure 2D). Other tested loading ratios displayed either a low FRET efficiency (Figure S1A) or larger average particle size (Figure S1B). Therefore, the formulation containing 1.0% D and 0.2% A was selected for further studies. For this loading ratio, the FRET efficiency (E) was calculated as ∼70% using eq 2.
| 2 |
IDA and ID are the total donor fluorescence intensities in the presence and absence of the acceptor, respectively.
Figure 2.

(A) Fluorescence intensity of green NPs prepared with various concentrations of BODIPY-FL (from 0.1% to 2.0%). (B) Fluorescence intensity of red NPs prepared with various concentrations of Nile Red (from 0.1% to 1.3%). (C) Emission spectra of FRET NPs loaded with 1.0% D–0.2% A (black), 1.0% D–0.3% A (red), and 1.3% D–0.3% A (blue) normalized at the acceptor emission wavelength (λexcitation 488 nm). (D) Size distribution of FRET NPs with the depicted D–A loading ratios. (E) Emission spectra of green NPs loaded with 1.0% BODIPY-FL (green), red NPs loaded with 0.2% Nile Red (red), and FRET NPs coencapsulating 1.0% BODIPY-FL and 0.2% Nile Red (orange). (F) Emission spectra of intact FRET NPs dispersed in water (orange) and disassembled FRET NPs dispersed in acetonitrile (green).
Steady-state fluorescence spectroscopy analysis revealed quenching of D emission accompanied by a sensitized A emission for FRET NPs compared to the emission spectra of green NPs containing only 1.0% BODIPY-FL and red NPs containing only 0.2% of Nile Red (Figure 2E). Disassembly of FRET NPs upon resuspension in acetonitrile resulted in the recovery of D emission and loss of A sensitization (Figure 2F), indicating the relevance of emission characteristics to structural integrity. Relying on these observations, the spectral changes of FRET NPs were monitored under different physiological conditions to obtain information on the structural changes of NPs that result in an increase in the D–A separation distance, such as dye release and particle degradation.
3.2. Assessment of Dye Release and Particle Stability in Situ
The optical and colloidal analysis of FRET NPs incubated in an aqueous solution at 37 °C enabled the particle stability assessment in situ. Fluorescence emission spectra and size distribution of NPs were measured at different time points up to 2 weeks. Figure 3A shows the emission spectra of FRET NPs normalized at the emission peak of the acceptor dye. A gradual recovery of D emission was observed at longer incubation times, which indicated a lower FRET efficiency at these time points. The FRET ratios were calculated by dividing the emission intensity of A by the emission intensity of D at each measurement point (Figure 3B). As a control, green NPs containing only 1.0% D were studied in parallel. Calculation of FRET ratios is a useful way to analyze FRET as it involves the contribution of both D and A emission intensities simultaneously. A decrease in FRET ratio would indicate a less efficient energy transfer between the D/A pair due to dye release or particle degradation. Indeed, a sharp decrease of FRET ratio was observed at day 1, which was due to an initial burst release of dyes. The biphasic release pattern that involves this initial burst followed by a more sustained release is typically observed for PLGA NPs.45 The gradual decrease of FRET ratio at the following time points corresponded to the sustained release phase.
Figure 3.
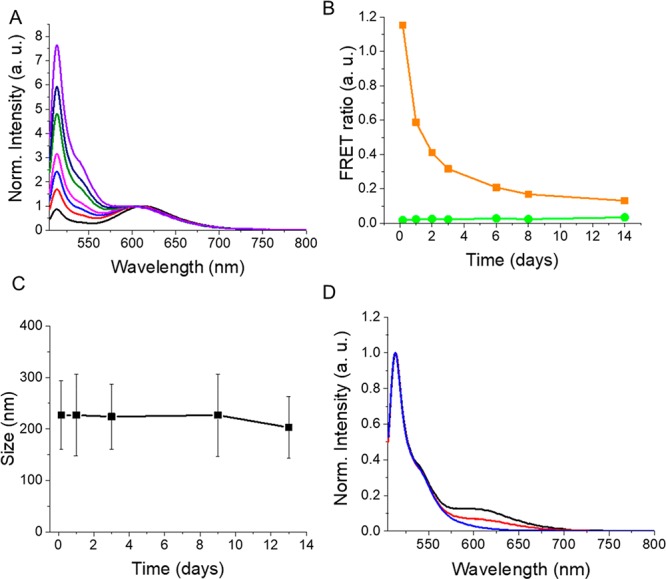
Stability assessment of PLGA NPs in situ. (A) Fluorescence emission spectra of FRET NPs normalized at the acceptor emission peak measured at 4 h (black), day 1 (red), day 2 (blue), day 3 (magenta), day 6 (green), day 8 (navy), and day 14 (purple). Excitation wavelength: 488 nm. (B) FRET ratio plot for FRET NPs (orange) and green NPs (green) at different measurement time points. (C) Average particle size of FRET NPs measured at different incubation times. (D) Normalized emission spectra of FRET NPs (black), green NPs (blue), and disassembled FRET NPs (red) on day 14.
Monitoring the green NPs in parallel ensured that the observed changes in the FRET ratio were not due to the influence of other factors (such as temperature) on the photophysical properties of the BODIPY-FL. As shown in Figure 3B, the variations in the FRET ratio values of green NPs were negligible compared to FRET NPs due to the remarkable stability of BODIPY dyes.46
Here, the decrease of FRET efficiency was mainly the result of dye release, particle degradation, or a combination of both. A nearly complete degradation of PLGA NPs with an average size of 230 nm was previously reported to take place within 10 weeks of incubation.47 Interestingly, these nanoparticles maintained their size until they were totally degraded, despite the decrease in their molecular weight. In our study, the particle size measurements showed that the average size of nanoparticles remained within the 200 nm range during the incubation period as well (Figure 3C). Although a certain degree of particle degradation possibly took place during the 2 weeks of incubation,48 it can be concluded that dye release was the major mechanism that resulted in a decreased FRET efficiency.
On the last day of incubation, FRET NPs were redispersed in acetonitrile in order to induce particle disassembly. These “broken” FRET NPs served as “no-FRET controls”. The emission spectra of FRET NPs dispersed in water and acetonitrile in comparison to green NPs are shown in Figure 3D. The comparison of the emission spectra of FRET NPs to their “broken” counterparts on day 14 revealed the presence of FRET still on week two, which was evident by the A sensitization peak for the FRET NPs dispersed in water. The small shoulder observed around 620 nm for the “broken” FRET particles was due to the direct excitation of acceptor dye.
The release profile of FRET NPs was also studied in neutral and slightly acidic conditions as pH can influence cargo release mechanisms and particle degradation characteristics.49 FRET NPs dispersed in PBS buffer with pH 5.8 (Figure 4A,B) or pH 7.4 (Figure 4C,D) were incubated at 37 °C for a period of 7 days. Fluorescence emission spectra and particle size distribution (Figure S2) were recorded at different time points. While a higher initial burst release was observed at pH 7.4 (Figure 4D), the release was faster at pH 5.8 in the following release period (Figure 4B). The burst release takes place during the initial water uptake and swelling of the lyophilized nanoparticles. The PLGA used for the preparation of nanoparticles was terminated with carboxylic acid groups. Therefore, it is very likely that a higher degree of interactions took place between the cationic species present in the buffer and the carboxylic acid groups on the polymer, which resulted in the facilitated diffusion and a higher burst release of dyes at pH 7.4. A higher burst release at pH 7.4 compared to more acidic conditions was reported for PLGA NPs also in previous studies.45
Figure 4.
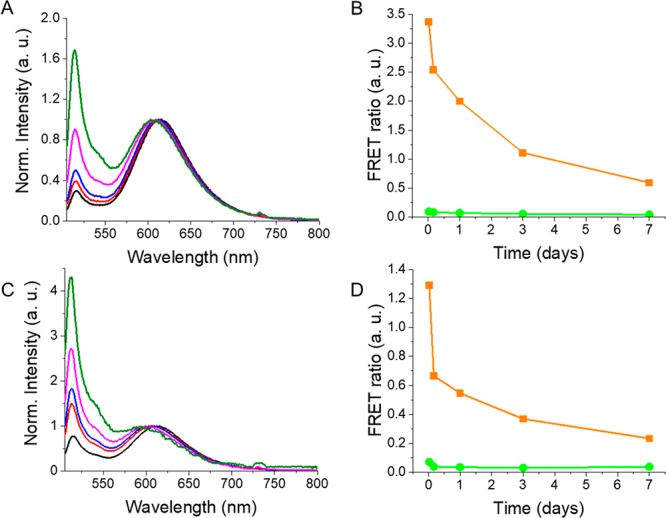
Normalized fluorescence emission spectra of FRET NPs incubated at pH 5.8 (A) and pH 7.4 (C) at 0.5 h (black), 4 h (red), day 1 (blue), day 3 (magenta), and day 7 (green). Excitation wavelength: 488 nm. FRET ratio plot for FRET NPs (orange) and green NPs (green) dispersed in PBS buffer with pH 5.8 (B) and pH 7.4 (D).
In addition to burst release, the pH has been shown to influence the degradation mechanism of PLGA NPs as well.50 However, an incubation period of 7 days is probably too short to observe the influence of polymer degradation on the release profile at different pH values. In one of our previous studies, we also have shown that the release of a hydrophobic dye was faster in a more acidic environment.39 Thus, in situ release behavior of the formulations that we reported previously is in good agreement with the results presented in this work.
3.3. Assessment of Cellular Uptake and Particle Stability in Vitro
The intracellular fate of nanoparticles has a strong influence on their therapeutic efficacy particularly when the encapsulated cargo is unstable at specific intracellular locations due to, e.g., organelle’s pH.51 Therefore, it is necessary to study the cellular uptake, intracellular trafficking, and in vitro stability of polymeric drug delivery nanoparticles.
Monocyte-derived DCs (moDCs) obtained from healthy donors, known to have a high phagocytic capacity, were used for the analysis of uptake and intracellular trafficking of NPs. Green NPs containing only 1.0% D were used to examine the intracellular trafficking. MoDCs incubated with green NPs were fixed at 6 and 24 h of incubation for confocal microscopy (Figure 5A). In order to monitor the colocalization of nanoparticles with early endosomes and lysosomes, EEA1 and LAMP1 staining were performed, respectively. The intracellular BODIPY-FL signal was observed to increase at 24 h compared to 6 h of incubation. In addition, while the NPs colocalized with the early endosomes at 6 h, only a partial overlap with the early endosomes and lysosomes was observed at 24 h. This is in accordance with the capability of PLGA nanoparticles to escape endolysosomal pathways as has been shown also in previous studies.43,52,53 The mechanism behind the nanoparticle escape is likely due to reversal of the surface charge of nanoparticles from anionic to cationic in acidic endolysosomal pH. This reversal leads to the interaction of nanoparticles with endolysosomal membrane and escape to cytosol.53 Thus, the fact that we observe only partial colocalization with an endolysosomal compartment could be explained by this escape mechanism.
Figure 5.

(A) Confocal microscopy images of moDCs incubated with green NPs for 6 and 24 h. Cells were stained with DAPI for nuclei (blue) and either with EEA1 for early endosomes or with LAMP1 for lysosomes (red). The fluorescence emission of green NPs is presented in green. The overlap between green NPs and EEA1 or LAMP1 is shown in yellow. (B) Mean fluorescence intensity values obtained with flow cytometry at 4 and 24 h for moDCs incubated at 4 °C (light gray) and 37 °C (dark gray) with green NPs. (C) Mean fluorescence intensity plot for moDCs incubated with green NPs (green) and FRET NPs (orange) for various time points up to 48 h.
In order to evaluate the energy dependence of NP uptake, cells were incubated with green NPs at 4 °C and mean fluorescence intensities were measured at 4 and 24 h using flow cytometry. The intracellular BODIPY-FL signal was negligible at 4 °C compared to 37 °C at both incubation times (Figure 5B), which demonstrated that NPs were mainly taken up via energy-dependent endocytic pathways. The low level of intracellular fluorescence at 4 °C can be attributed to the passive entry of leaked dye molecules via diffusion39 or adsorption of nanoparticles to the cell surface.54 Dye leakage is indeed a common problem for polymeric nanoparticles, which can result in misinterpretation of their cellular uptake and intracellular distribution.16
Cellular uptake and in vitro stability of nanoparticles was evaluated by flow cytometry analysis of moDCs incubated with green NPs and FRET NPs for a period of 48 h (Figure 5C). The intracellular fluorescence intensity increased rapidly up to ca. 6 h, and then reached a plateau at 24 h for both particle types. A slight decrease of the mean fluorescence intensity was observed for green NPs at 48 h, which can be an indication of dye release as free dyes show lower emission intensities in an aqueous environment than in the NPs.20 Nevertheless, the emission intensity difference between green NPs and FRET NPs revealed the presence of FRET still at 48 h.
In vitro particle stability assessment was also performed on murine CD103+ BMDCs. Cells incubated with FRET NPs and green NPs were analyzed with fluorescence microscopy imaging and flow cytometry at 24 and 72 h of incubation (Figure 6). Fluorescence microscopy images were obtained using excitation and emission filters suitable for the detection of BODIPY-FL. An efficient donor quenching was observed at 24 h of incubation as indicated by the dimmer intracellular fluorescence intensity for FRET NPs compared to green NPs (Figure 6A). On the other hand, similar intracellular intensities of donor emission observed at 72 h revealed that there was no longer an efficient energy transfer at this time point between donor and acceptor dyes. These findings were confirmed by the flow cytometry analysis as well (Figure 6B). The mean fluorescence intensity values of FRET NPs measured at the green emission channel was comparable to that of green NPs at 72 h, which confirmed the recovery of donor emission at this time of incubation.
Figure 6.

(A) Fluorescence microscopy images of murine BMDCs incubated with FRET NPs and green NPs for 24 and 72 h collected at the green emission channel. Cell nuclei are stained with DAPI (blue). (B) Mean fluorescence intensity value BMDCs incubated with green NPs (light gray) and FRET NPs (dark gray) measured at the green emission channel using flow cytometry. A complete recovery of donor quenching is observed at 72 h.
Overall, fluorescence microscopy imaging and flow cytometry analyses revealed a significantly faster donor recovery in vitro, reaching a complete loss of FRET at 72 h probably due to both dye release and particle degradation. It should be noted that cell culture media can alter the colloidal and chemical properties of NPs due to the presence of proteins and high ion content.55 Furthermore, during their uptake through the endocytic pathway, nanoparticles are exposed to varying physiological conditions such as acidic pH of late endosomes55 and high levels of hydrolytic enzymes in lysosomes.56 All of these factors affect nanoparticle stability and degradation, resulting in a faster release of the encapsulated cargo in vitro when compared to in situ settings.
3.4. Assessment of Particle Stability in Vivo
In vivo stability of particles systemically administered to mice was studied via ex vivo organ imaging and flow cytometry (Figure 7). Green NPs and FRET NPs were injected intravenously (i.v.) for liver imaging, and cells were isolated at 2 and 24 h after injection. In accordance with the biodistribution studies reported for PLGA NPs after i.v. administration,8,57,58 NPs were detected mainly in liver as confirmed by flow cytometry analysis and ex vivo organ imaging (Figure S3). Therefore, cells isolated from the liver were used in the further analyses. A higher fluorescence emission intensity was detected in the liver of mice injected with green NPs (Figure 7A) compared to those injected with FRET NPs (Figure 7B). For both experimental groups, a significant decrease of fluorescence emission was observed at the 24 h time point. The decrease in emission probably resulted from the particle degradation or enhanced release, accompanied by fast clearance of the dyes from the liver. A similar observation was also reported by Simon et al., who showed a significant decrease of nanoparticle concentration in various tissues over 24 h period.57 Therefore, the observed decrease in emission could be also attributed to a rapid clearance of the nanoparticles from the liver as well.
Figure 7.

Ex vivo imaging of the liver 2 and 24 h after i.v. injection of (A) Green NPs and (B) FRET NPs. (Excitation wavelength = 465 nm, emission filter = GFP, exposure time = 5 s.) (C) Flow cytometry histograms of cells isolated from the liver at 2 and 24 h postinjection. (Excitation wavelength = 488 nm, detection filter = FITC). Red: unlabeled control. Green: Green NPs. Orange: FRET NPs.
Next, we isolated the cells from this organ and analyzed them with flow cytometry. FRET was still detectable at 2 h after administration as depicted by a lower mean fluorescence intensity at the donor emission channel compared to the cells obtained from the liver of a mouse injected with green NPs (Figure 7C). On the other hand, a reduced mean fluorescence intensity was observed for the cells isolated at the 24 h time point for both FRET NPs and green NPs (Figure 7C). Furthermore, intensity peaks for both conditions showed an overlap, indicating the loss of FRET at this time point. Additional analysis of the cells shown in Figure 7C revealed an almost complete loss of FRET in vivo within 24 h after injection (Figure S4).
A previous study by Mohammad et al. showed degradation of PLGA nanoparticles with a slight reduction in molecular weight already within the first 24 h after i.v. administration and predominant accumulation in the liver.8 In that study, the degradation profile of 200 nm PLGA nanoparticles showed differences between in vitro and in vivo conditions, as well as between various types of tissues. In our study, a faster recovery of the donor emission was observed in vivo. However, here we used a lower molecular weight polymer (7000–17 000 Da), which can lead to faster polymer degradation and a more rapid drug release.59 The impact of enzymes on the PLGA degradation is unclear; nevertheless, due to the fact that liver has a high concentration of esterases,8 we cannot exclude a possible influence of these enzymes on the faster release and degradation observed in our study. The presence of proteins in blood and variations of pH in different tissue types could also affect the degradation profile of the nanoparticles. Therefore, our in vivo study suggests the involvement of additional parameters, e.g., enzymes, on release and nanoparticle degradation.
4. Conclusion
It is of major importance to monitor the stability of polymeric nanoparticles under different physiological conditions for the development of clinically applicable formulations. In this study, we demonstrated that FRET is a powerful tool to assess the stability of PLGA nanoparticles by encapsulating BODIPY-FL green and Nile Red dyes as the donor–acceptor pair, respectively. These FRET NPs of ∼200 nm in size showed ∼70% FRET efficiency. Optical and colloidal analysis of nanoparticles revealed that PLGA nanoparticles were stable for at least 2 weeks in situ, while in vitro particle degradation started in between 24 and 48 h, reaching a complete recovery of donor emission at 72 h. The nanoparticles were readily taken up by human-derived moDCs mainly via energy-dependent endocytic pathways, but could escape the endolysosomal routes after uptake. In vivo studies showed the accumulation of systemically administered PLGA nanoparticles in the liver and particle degradation within 24 h. The results of our study will have a major impact on research focusing on the applications such as drug delivery and cell therapy monitoring using PLGA nanoparticles, as they provide a better understanding of the degradation processes and an attractive way to monitor the uptake of the nanoparticles and release of encapsulated cargo.
Acknowledgments
The authors of this study would like to thank Dr. Yusuf Dölen and Dr. Olga Ilina for providing cells and for help with the in vivo experiments. The authors acknowledge the financial support by the EU grant PRECIOUS (686089). C.F. received the NWO Spinoza grant, ERC Advanced grant Pathfinder (269019), and Dutch cancer society award 2009-4402. M.S. received a European Research Council (ERC) starting grant (CoNQUeST, grant 336454) and grant 14716 from The Netherlands organization for scientific research NWO-TTW.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsabm.8b00754.
Emission spectra of loaded FRET NPs, size distribution of FRET NPs with other D–A ratios, particle size distribution at different time points obtained at pH 5.8 and pH 7.4, ex vivo analysis of collected organs after intravenous administration of green NPs, and MFI ratio of cells isolated from the liver at 2 and 24 h after the injection of FRET NPs and green NPs (PDF)
Author Contributions
§ S.M. and K.H. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Banik B. L.; Fattahi P.; Brown J. L. Polymeric Nanoparticles: The Future of Nanomedicine. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2016, 8 (2), 271–299. 10.1002/wnan.1364. [DOI] [PubMed] [Google Scholar]
- Chan J. M.; Valencia P. M.; Zhang L.; Langer R.; Farokhzad O. C.. Polymeric Nanoparticles for Drug Delivery. In Cancer Nanotechnology: Methods and Protocols; Grobmyer S. R., Moudgil B. M., Eds.; Humana Press: Totowa, NJ, 2010; pp 163–175. [DOI] [PubMed] [Google Scholar]
- El-Say K. M.; El-Sawy H. S. Polymeric Nanoparticles: Promising Platform for Drug Delivery. Int. J. Pharm. 2017, 528 (1), 675–691. 10.1016/j.ijpharm.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Masood F. Polymeric Nanoparticles for Targeted Drug Delivery System for Cancer Therapy. Mater. Sci. Eng., C 2016, 60, 569–578. 10.1016/j.msec.2015.11.067. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Yeo Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. 10.1016/j.ces.2014.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swider E.; Koshkina O.; Tel J.; Cruz L. J.; de Vries I. J. M.; Srinivas M. Customizing Poly(Lactic-Co-Glycolic Acid) Particles for Biomedical Applications. Acta Biomater. 2018, 73, 38–51. 10.1016/j.actbio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Charrueau C.; Zandanel C.. Drug Delivery by Polymer Nanoparticles: The Challenge of Controlled Release and Evaluation. In Polymer Nanoparticles for Nanomedicines: A Guide for their Design, Preparation and Development; Vauthier C., Ponchel G., Eds.; Springer International Publishing: Cham, 2016; pp 439–503. [Google Scholar]
- Mohammad A. K.; Reineke J. J. Quantitative Detection of PLGA Nanoparticle Degradation in Tissues Following Intravenous Administration. Mol. Pharmaceutics 2013, 10 (6), 2183–2189. 10.1021/mp300559v. [DOI] [PubMed] [Google Scholar]
- Liu K. C.; Yeo Y. Extracellular Stability of Nanoparticulate Drug Carriers. Arch. Pharmacal Res. 2014, 37 (1), 16–23. 10.1007/s12272-013-0286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P.; Chen H.; Paholak H. J.; Sun D. Noninvasive Fluorescence Resonance Energy Transfer Imaging of in Vivo Premature Drug Release from Polymeric Nanoparticles. Mol. Pharmaceutics 2013, 10 (11), 4185–4194. 10.1021/mp4002393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Fay F.; Hak S.; Manuel Perez-Aguilar J.; Sanchez-Gaytan B. L.; Goode B.; Duivenvoorden R.; de Lange Davies C.; Bjørkøy A.; Weinstein H.; Fayad Z. A.; Pérez-Medina C.; Mulder W. J. M. Augmenting Drug–carrier Compatibility Improves Tumour Nanotherapy Efficacy. Nat. Commun. 2016, 7, 11221. 10.1038/ncomms11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel A. E.; Mädler L.; Velegol D.; Xia T.; Hoek E. M. V; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano–bio Interface. Nat. Mater. 2009, 8, 543–557. 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- Chen H.; Kim S.; He W.; Wang H.; Low P. S.; Park K.; Cheng J.-X. Fast Release of Lipophilic Agents from Circulating PEG-PDLLA Micelles Revealed by in Vivo Förster Resonance Energy Transfer Imaging. Langmuir 2008, 24 (10), 5213–5217. 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]
- Claudia M.; Kristin Ö.; Jennifer O.; Eva R.; Eleonore F. Comparison of Fluorescence-Based Methods to Determine Nanoparticle Uptake by Phagocytes and Non-Phagocytic Cells in Vitro. Toxicology 2017, 378, 25–36. 10.1016/j.tox.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Gullotti E.; Tong L.; Highley C. B.; Errabelli D. R.; Hasan T.; Cheng J.-X.; Kohane D. S.; Yeo Y. Intracellular Drug Delivery by Poly(Lactic-Co-Glycolic Acid) Nanoparticles, Revisited. Mol. Pharmaceutics 2009, 6 (1), 190–201. 10.1021/mp800137z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snipstad S.; Hak S.; Baghirov H.; Sulheim E.; Mørch Ý.; Lélu S.; von Haartman E.; Bäck M.; Nilsson K. P. R.; Klymchenko A. S.; de Lange Davies C.; Åslund A. K. O. Labeling Nanoparticles: Dye Leakage and Altered Cellular Uptake. Cytometry, Part A 2017, 91 (8), 760–766. 10.1002/cyto.a.22853. [DOI] [PubMed] [Google Scholar]
- Chen N.-T.; Cheng S.-H.; Liu C.-P.; Souris J.; Chen C.-T.; Mou C.-Y.; Lo L.-W. Recent Advances in Nanoparticle-Based Förster Resonance Energy Transfer for Biosensing, Molecular Imaging and Drug Release Profiling. Int. J. Mol. Sci. 2012, 13 (12), 16598. 10.3390/ijms131216598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer B. W.Förster Theory. In FRET—Förster Resonance Energy Transfer: From Theory to Applications. Medintz I., Hildebrandt N., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA, 2014; pp 23–62. [Google Scholar]
- Li Y.; Budamagunta M. S.; Luo J.; Xiao W.; Voss J. C.; Lam K. S. Probing of the Assembly Structure and Dynamics within Nanoparticles during Interaction with Blood Proteins. ACS Nano 2012, 6 (11), 9485–9495. 10.1021/nn302317j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin A.; Tagit O.; Sobot D.; Lepetre-Mouelhi S.; Mougin J.; Martens T. F.; Braeckmans K.; Nicolas V.; Desmaële D.; De Smedt S. C.; Hildebrandt N.; Couvreur P.; Andrieux K. Transport Mechanisms of Squalenoyl-Adenosine Nanoparticles across the Blood-Brain Barrier. Chem. Mater. 2015, 27 (10), 3636–3647. 10.1021/acs.chemmater.5b00267. [DOI] [Google Scholar]
- Charron D. M.; Zheng G. Nanomedicine Development Guided by FRET Imaging. Nano Today 2018, 18, 124–136. 10.1016/j.nantod.2017.12.006. [DOI] [Google Scholar]
- Chen H.; Kim S.; Li L.; Wang S.; Park K.; Cheng J.-X. Release of Hydrophobic Molecules from Polymer Micelles into Cell Membranes Revealed by Förster Resonance Energy Transfer Imaging. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (18), 6596–6601. 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diezi T. A.; Bae Y.; Kwon G. S. Enhanced Stability of PEG-Block-Poly(N-Hexyl Stearate l-Aspartamide) Micelles in the Presence of Serum Proteins. Mol. Pharmaceutics 2010, 7 (4), 1355–1360. 10.1021/mp100069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T.; Rachel R.; Besheer A.; Uezguen S.; Weigandt M.; Goepferich A. Comparative Investigations on In Vitro Serum Stability of Polymeric Micelle Formulations. Pharm. Res. 2012, 29 (2), 448–459. 10.1007/s11095-011-0555-x. [DOI] [PubMed] [Google Scholar]
- Chen T.; Li C.; Li Y.; Yi X.; Wang R.; Lee S. M.-Y.; Zheng Y. Small-Sized MPEG–PLGA Nanoparticles of Schisantherin A with Sustained Release for Enhanced Brain Uptake and Anti-Parkinsonian Activity. ACS Appl. Mater. Interfaces 2017, 9 (11), 9516–9527. 10.1021/acsami.7b01171. [DOI] [PubMed] [Google Scholar]
- Yu D.; Zou G.; Cui X.; Mao Z.; Estrela-Lopis I.; Donath E.; Gao C. Monitoring the Intracellular Transformation Process of Surface-Cleavable PLGA Particles Containing Disulfide Bonds by Fluorescence Resonance Energy Transfer. J. Mater. Chem. B 2015, 3 (45), 8865–8873. 10.1039/C5TB01687H. [DOI] [PubMed] [Google Scholar]
- Wagh A.; Jyoti F.; Mallik S.; Qian S.; Leclerc E.; Law B. Polymeric Nanoparticles with Sequential and Multiple FRET Cascade Mechanisms for Multicolor and Multiplexed Imaging. Small 2013, 9 (12), 2129–2139. 10.1002/smll.201202655. [DOI] [PubMed] [Google Scholar]
- Conte C.; Mastrotto F.; Taresco V.; Tchoryk A.; Quaglia F.; Stolnik S.; Alexander C. Enhanced Uptake in 2D- and 3D- Lung Cancer Cell Models of Redox Responsive PEGylated Nanoparticles with Sensitivity to Reducing Extra- and Intracellular Environments. J. Controlled Release 2018, 277, 126–141. 10.1016/j.jconrel.2018.03.011. [DOI] [PubMed] [Google Scholar]
- Operti M. C.; Fecher D.; van Dinther E. A. W.; Grimm S.; Jaber R.; Figdor C. G.; Tagit O. A Comparative Assessment of Continuous Production Techniques to Generate Sub-Micron Size PLGA Particles. Int. J. Pharm. 2018, 550 (1), 140–148. 10.1016/j.ijpharm.2018.08.044. [DOI] [PubMed] [Google Scholar]
- Ding D.; Zhu Q. Recent Advances of PLGA Micro/Nanoparticles for the Delivery of Biomacromolecular Therapeutics. Mater. Sci. Eng., C 2018, 92, 1041–1060. 10.1016/j.msec.2017.12.036. [DOI] [PubMed] [Google Scholar]
- Mohammadi-Samani S.; Taghipour B. PLGA Micro and Nanoparticles in Delivery of Peptides and Proteins; Problems and Approaches. Pharm. Dev. Technol. 2015, 20 (4), 385–393. 10.3109/10837450.2014.882940. [DOI] [PubMed] [Google Scholar]
- Jose S.; Sowmya S.; Cinu T. A.; Aleykutty N. A.; Thomas S.; Souto E. B. Surface Modified PLGA Nanoparticles for Brain Targeting of Bacoside-A. Eur. J. Pharm. Sci. 2014, 63, 29–35. 10.1016/j.ejps.2014.06.024. [DOI] [PubMed] [Google Scholar]
- Kiss É.; Gyulai G.; Pénzes C. B.; Idei M.; Horváti K.; Bacsa B.; Bösze S. Tuneable Surface Modification of PLGA Nanoparticles Carrying New Antitubercular Drug Candidate. Colloids Surf., A 2014, 458, 178–186. 10.1016/j.colsurfa.2014.05.048. [DOI] [Google Scholar]
- Kocbek P.; Obermajer N.; Cegnar M.; Kos J.; Kristl J. Targeting Cancer Cells Using PLGA Nanoparticles Surface Modified with Monoclonal Antibody. J. Controlled Release 2007, 120 (1), 18–26. 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Jose S.; Juna B. C.; Cinu T. A.; Jyoti H.; Aleykutty N. A. Carboplatin Loaded Surface Modified PLGA Nanoparticles: Optimization, Characterization, and in Vivo Brain Targeting Studies. Colloids Surf., B 2016, 142, 307–314. 10.1016/j.colsurfb.2016.02.026. [DOI] [PubMed] [Google Scholar]
- Dölen Y.; Kreutz M.; Gileadi U.; Tel J.; Vasaturo A.; van Dinther E. A. W.; van Hout-Kuijer M. A.; Cerundolo V.; Figdor C. G. Co-Delivery of PLGA Encapsulated Invariant NKT Cell Agonist with Antigenic Protein Induce Strong T Cell-Mediated Antitumor Immune Responses. OncoImmunology 2016, 5 (1), e1068493 10.1080/2162402X.2015.1068493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harguindey A.; Domaille D. W.; Fairbanks B. D.; Wagner J.; Bowman C. N.; Cha J. N. Synthesis and Assembly of Click-Nucleic-Acid-Containing PEG–PLGA Nanoparticles for DNA Delivery. Adv. Mater. 2017, 29 (24), 1700743. 10.1002/adma.201700743. [DOI] [PubMed] [Google Scholar]
- Zou P.; Stern S. T.; Sun D. PLGA/Liposome Hybrid Nanoparticles for Short-Chain Ceramide Delivery. Pharm. Res. 2014, 31 (3), 684–693. 10.1007/s11095-013-1190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swider E.; Staal A. H. J.; Riessen N. K. van; Jacobs L.; White P. B.; Fokkink R.; Janssen G.-J.; Dinther E. van; Figdor C. G.; Vries I. J. M. de; Koshkina O.; Srinivas M. Design of Triphasic Poly(Lactic-Co-Glycolic Acid) Nanoparticles Containing a Perfluorocarbon Phase for Biomedical Applications. RSC Adv. 2018, 8 (12), 6460–6470. 10.1039/C7RA13062G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas M.; Cruz L. J.; Bonetto F.; Heerschap A.; Figdor C. G.; de Vries I. J. M. Customizable, Multi-Functional Fluorocarbon Nanoparticles for Quantitative in Vivo Imaging Using 19F MRI and Optical Imaging. Biomaterials 2010, 31 (27), 7070–7077. 10.1016/j.biomaterials.2010.05.069. [DOI] [PubMed] [Google Scholar]
- Srinivas M.; Tel J.; Schreibelt G.; Bonetto F.; Cruz L. -J.; Amiri H.; Heerschap A.; Figdor C. G.; de Vries I. J. M. PLGA-encapsulated perfluorocarbon nanoparticles for simultaneous visualization of distinct cell populations by 19F MRI. Nanomedicine 2015, 10 (15), 2339–2348. 10.2217/nnm.15.76. [DOI] [PubMed] [Google Scholar]
- Cruz L. J.; Tacken P. J.; Fokkink R.; Joosten B.; Stuart M. C.; Albericio F.; Torensma R.; Figdor C. G. Targeted PLGA Nano- but Not Microparticles Specifically Deliver Antigen to Human Dendritic Cells via DC-SIGN in Vitro. J. Controlled Release 2010, 144 (2), 118–126. 10.1016/j.jconrel.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Panyam J.; Labhasetwar V. Biodegradable Nanoparticles for Drug and Gene Delivery to Cells and Tissue. Adv. Drug Delivery Rev. 2003, 55 (3), 329–347. 10.1016/S0169-409X(02)00228-4. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Wi Y.; Yoon Y.-M.; Verwilst P.; Jang J. H.; Kim T. W.; Kang C.; Kim J. S. BODIPY/Nile-Red-Based Efficient FRET Pair: Selective Assay of Endoplasmic Reticulum Membrane Fluidity. Chem. - Asian J. 2016, 11 (4), 527–531. 10.1002/asia.201501060. [DOI] [PubMed] [Google Scholar]
- Guo L.-Y.; Yan S.-Z.; Li Q.; Xu Q.; Lin X.; Qi S.-S.; Yu S.-Q.; Chen S.-L. Poly(Lactic-Co-Glycolic) Acid Nanoparticles Improve Oral Bioavailability of Hypocrellin A in Rat. RSC Adv. 2017, 7 (67), 42073–42082. 10.1039/C7RA04748G. [DOI] [Google Scholar]
- Vyšniauskas A.; López-Duarte I.; Duchemin N.; Vu T.-T.; Wu Y.; Budynina E. M.; Volkova Y. A.; Peña Cabrera E.; Ramírez-Ornelas D. E.; Kuimova M. K. Exploring Viscosity, Polarity and Temperature Sensitivity of BODIPY-Based Molecular Rotors. Phys. Chem. Chem. Phys. 2017, 19 (37), 25252–25259. 10.1039/C7CP03571C. [DOI] [PubMed] [Google Scholar]
- Zweers M. L. T.; Engbers G. H. M.; Grijpma D. W.; Feijen J. In Vitro Degradation of Nanoparticles Prepared from Polymers Based on Dl-Lactide, Glycolide and Poly(Ethylene Oxide). J. Controlled Release 2004, 100 (3), 347–356. 10.1016/j.jconrel.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Hussein A. S.; Ahmadun F.-R.; Abdullah N. In Vitro Degradation of Poly (D, L-Lactide-Co-Glycolide) Nanoparticles Loaded with Linamarin. IET Nanobiotechnol. 2013, 7 (2), 33–41. 10.1049/iet-nbt.2012.0012. [DOI] [PubMed] [Google Scholar]
- Li J.; Jiang G.; Ding F. The Effect of PH on the Polymer Degradation and Drug Release from PLGA-MPEG Microparticles. J. Appl. Polym. Sci. 2008, 109 (1), 475–482. 10.1002/app.28122. [DOI] [Google Scholar]
- Baiti R. N.; Ardhyananta H.; El Kirat K. Effect of Acidic and Basic Environment to the Degradation Behavior of PLGA Nanocapsules for Biomedical Application. Adv. Mater. Res. 2015, 1123, 213–216. 10.4028/www.scientific.net/AMR.1123.213. [DOI] [Google Scholar]
- Cho E. J.; Holback H.; Liu K. C.; Abouelmagd S. A.; Park J.; Yeo Y. Nanoparticle Characterization: State of the Art, Challenges, and Emerging Technologies. Mol. Pharmaceutics 2013, 10 (6), 2093–2110. 10.1021/mp300697h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartiera M. S.; Johnson K. M.; Rajendran V.; Caplan M. J.; Saltzman W. M. The Uptake and Intracellular Fate of PLGA Nanoparticles in Epithelial Cells. Biomaterials 2009, 30 (14), 2790–2798. 10.1016/j.biomaterials.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyam J.; Zhou W.-Z.; Prabha S.; Sahoo S. K.; Labhasetwar V. Rapid Endo-Lysosomal Escape of Poly(Dl -Lactide- Co -Glycolide) Nanoparticles: Implications for Drug and Gene Delivery. FASEB J. 2002, 16 (10), 1217–1226. 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- Chen Y.-S.; Alany R. G.; Young S. A.; Green C. R.; Rupenthal I. D. In Vitro Release Characteristics and Cellular Uptake of Poly(D,L-Lactic- Co -Glycolic Acid) Nanoparticles for Topical Delivery of Antisense Oligodeoxynucleotides. Drug Delivery 2011, 18 (7), 493–501. 10.3109/10717544.2011.589088. [DOI] [PubMed] [Google Scholar]
- Moore T. L.; Rodriguez-Lorenzo L.; Hirsch V.; Balog S.; Urban D.; Jud C.; Rothen-Rutishauser B.; Lattuada M.; Petri-Fink A. Nanoparticle Colloidal Stability in Cell Culture Media and Impact on Cellular Interactions. Chem. Soc. Rev. 2015, 44 (17), 6287–6305. 10.1039/C4CS00487F. [DOI] [PubMed] [Google Scholar]
- Soenen S. J.; Parak W. J.; Rejman J.; Manshian B. (Intra)Cellular Stability of Inorganic Nanoparticles: Effects on Cytotoxicity, Particle Functionality, and Biomedical Applications. Chem. Rev. 2015, 115 (5), 2109–2135. 10.1021/cr400714j. [DOI] [PubMed] [Google Scholar]
- Simon L. C.; Sabliov C. M. Time Analysis of Poly(Lactic-Co-Glycolic) Acid Nanoparticle Uptake by Major Organs Following Acute Intravenous and Oral Administration in Mice and Rats. Ind. Biotechnol. 2013, 9 (1), 19–23. 10.1089/ind.2012.0032. [DOI] [Google Scholar]
- Wagh A.; Qian S. Y.; Law B. Development of Biocompatible Polymeric Nanoparticles for in Vivo NIR and FRET Imaging. Bioconjugate Chem. 2012, 23 (5), 981–92. 10.1021/bc200637h. [DOI] [PubMed] [Google Scholar]
- Ansary R. H.; Awang M. B.; Rahman M. M. Biodegradable Poly(D,L-Lactic-Co-Glycolic Acid)-Based Micro/Nanoparticles for Sustained Release of Protein Drugs - A Review. Trop. J. Pharm. Res. 2014, 13 (7), 1179. 10.4314/tjpr.v13i7.24. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.